

Abstract
A direct observation of amyloid aggregation from isolated peptides to cross-β fibrils is crucial for understanding the nucleation-dependence process, but the corresponding macroscopic timescales impose a major computational challenge. Using rapid all-atom discrete molecular dynamics simulations, we capture the oligomerization and fibrillization dynamics of the amyloid core sequences of amyloid-β (Aβ) in Alzheimer’s disease and islet amyloid polypeptide (IAPP) in type-2 diabetes, namely Aβ16–22 and IAPP22–28. Both peptides and their mixture spontaneously assemble into cross-β aggregates in silico, but follow distinct pathways. Aβ16–22 is highly aggregation-prone with a funneled free energy basin toward multi-layer β-sheet aggregates. IAPP22–28, on the other hand, features the accumulation of unstructured oligomers before the nucleation of β-sheets and growth into double-layer β-sheet aggregates. In the presence of Aβ16–22, the aggregation of IAPP22–28 is promoted by forming co-aggregated multi-layer β-sheets. Our study offers a detailed molecular insight to the long-postulated oligomerization-nucleation process in the amyloid aggregations.
Keywords: Amyloid aggregation, Oligomerization, Amyloid-β, Islet Amyloid Polypeptide, Aggregation free energy landscape
Graphical abstract
Introduction
Aggregation of proteins and peptides into amyloid fibrils is implicated in many human diseases1, including amyloid-β (Aβ) in Alzheimer’s disease (AD)2–4, α-synuclein in Parkinson’s disease5, prion protein in the prion disease6, 7, and islet amyloid polypeptide (IAPP, a.k.a. amylin) in type 2 diabetes mellitus (T2D)8–10. Experimental studies including x-ray crystallography and solid state NMR11–13 have established that amyloid fibrils formed by various proteins and peptides share the same characteristic cross-β structures, where multi-layer β-sheets with strands perpendicular to the fibril axis are forming the amyloid core. The formation of insoluble amyloid fibrils is known to be a complex multistep process, involving the formation of soluble oligomers, the nucleation of β-rich aggregates or protofibrils, and the elongation and bundling of protofibrils into mature fibrils14. Increasing evidence supports the toxic oligomer hypothesis, where soluble low molecular weight oligomers corresponding to the aggregation intermediates are found more cytotoxic than the final fibrils15,16. Therefore, understanding the detailed aggregation pathways and the structure and dynamics of various aggregation intermediates is important for the design of anti-amyloid therapeutic strategies targeting these toxic oligomers.
Due to the heterogeneous and metastable nature of the aggregation intermediate species, it is experimentally challenging to pinpoint various oligomeric species and structurally characterize them. The macroscopic timescales of amyloid aggregation, especially the rate-limiting nucleation process from pre-fibrillar oligomers to β-rich protofibrils, also make it difficult to computationally capture the assembly process at the molecular and atomic level. Computational studies of full length peptides or their aggregation-prone amyloidogenic fragments are often limited to the observation of early events of misfolding and self-association corresponding to the pre-nucleation step17 or the energetics of fibrillar oligomers with different sizes corresponding to post-nucleation step, i.e., the fibril elongation18. Using umbrella sampling simulations with the fibril structure as the template, the free energy landscape of amyloid aggregation can be inferred19–21. However, a direct observation of the nucleation process from pre-fibrillar oligomers to protofibrils in atomistic simulations is still lacking. The outstanding questions include what are the critical prefibrillar oligomers? how do these oligomers convert into protofibrils? and how do the protofibrils grow?
Here, we apply atomistic discrete molecule dynamic (DMD) simulations, an efficient and predictive molecular dynamics method22–25, to investigate the assembly dynamics of the amyloid core sequences of Aβ (16KLVFFA22E, denoted as Aβ16–22) and IAPP (22NFGAIL28S, denoted as IAPP22–28) from isolated monomers to cross-β fibril-like aggregates. The fragment Aβ16–22 has been identified as one of the β-strands constituting the fibril core of full length Aβ by solid-state NMR and H/D exchange experiments12,26–28. Numerous studies29–35 have demonstrated that Aβ16–22 could self-assemble into oligomers and amyloid fibrils with cytotoxicity. Similarly, the fragment IAPP22–28 has also been identified as one of the amyloidogenic sequences of IAPP forming the cross-β core of fibrils36–38. Short peptides with sequences around IAPP22–28 are cytotoxic to pancreatic cell lines and able to form amyloid fibrils independent of the full-length polypeptide39–41. Emerging evidence suggests that interactions between different amyloid proteins and peptides may play a critical role in amyloid diseases (e.g., Aβ–tau42, tau-synuclein43, Aβ–transthyretin44, and IAPP–Aβ45). For example, T2D has been identified as a major risk factor for AD46–48 and thus the co-aggregation of IAPP and Aβ may contribute to the cross-talk between these two diseases49,50. Therefore, we also investigate in our simulations the co-aggregation between Aβ16–22 and IAPP22–28, experimentally identified as the hotspot regions for the inter-molecular interaction between full-length Aβ and IAPP45.
With up to 16 peptides in all-atom DMD simulations, we systematically investigate the aggregation dynamics of both Aβ16–22 and IAPP22–28 and their mixture at 1:1 ratio. Both Aβ16–22 and IAPP22–28 and their mixture can form highly ordered cross-β aggregates from isolated unstructured peptides, but feature distinct different aggregation dynamics and pathways. Aβ16–22 shows a high aggregation propensity, which firstly self-assemble into small single-layer β-sheets and then further associate into multi-layer cross-β aggregates. In contrast, IAPP22–28 peptides initially form unstructured oligomers. As the oligomer size increases beyond six, a conformational transition from random coil to β-sheet take place and a large number of IAPP22–28 peptides form two-layer cross-β structures. In the mixture, the aggregation propensity of IAPP22–28 is significantly enhanced by co-aggregating with Aβ16–22. Based on the unconstrained aggregation simulations of multiple peptides from monomers to cross-β aggregates, the aggregation free energy landscape computed as the potential of mean force (PMF) with respect to the oligomer size and corresponding β-sheet contents readily capture the distinct aggregation dynamics and pathways of different peptides and mixtures.
Results and Discussion
For both Aβ16–22 and IAPP22–28, eight molecular systems with even number of peptides from 2 to 16 (denoted as (KLVFFAE)2n and (NFGAILS)2n with n from 1 to 8) were studied. Mixed at 1:1 ratio, eight co-aggregation systems with the total number of peptides from 2 to 16 (denoted as (KLVFFAE)n-(NFGAILS)n with n from 1 to 8) were also studied. In all cases, the same peptide concentration of ~26 mM was maintained by adjusting the simulation box sizes. For each molecular system, ten independent simulations starting with different inter-molecular distances and orientations and lasted 200 ns were performed (Methods).
Aβ16–22 shows a higher propensity to form β-sheet aggregates than IAPP22–28
We first examined the secondary structure properties of the pure (KLVFFAE)2n and (NFGAILS)2n systems using the last 100 ns of simulation trajectories to avoid potential biases of starting configurations (Fig. 1). Aβ16–22 displayed a high β-sheet propensity as even in the smallest (KLVFFAE)2 system an average of ~43% β-sheet propensity was observed (Fig. 1a), where the probabilities of residues V18, F19, F20 to form β-sheets were over 60% (Fig. S1a). When n increased from 2 to 8, the β-sheet propensity reached around 55%–60%, consistent with a previous computational study that three and six Aβ16–22 peptides could from β-sheet aggregates driven by only hydrogen bonds and hydrophobic interactions51. Examination of simulation trajectories indicated that the peptides in (KLVFFAE)2 simulations mainly adopted antiparallel β-sheet dimers (Fig. 1c), and multi-layered β-sheets in (KLVFFAE)16 (Fig. 1f). The time evolution of β-sheet and coil propensities (Fig. S2) showed that Aβ16–22 preferred to stay in the β-sheet conformation. The little fluctuations of β-sheet propensity in (KLVFFAE)16 simulations after saturated around 60% indicated that these β-rich aggregates were stable (Fig. S2d). These results were consistent with previous computational results where the antiparallel alignment was found to be the lowest-energy conformation for Aβ16–22 dimers52. Hence, our simulations suggested that Aβ16–22 peptides preferred to form stable β-sheet rich aggregates.
Figure 1.

The propensity of β-sheet formation. a) The average β-sheet content was computed from DMD simulations of (KLVFFAE)2n, (NFGAILS)2n, (KLVFFAE)n-(NFGAILS)n molecular systems. The probability was averaged over the last 100 ns of 10 independent simulations. b) The probability to observe pair-wise β-sheet between different types of peptides, including KLVFFAE-KLVFFAE, NFGAIL-NFGAIL and KLVFFAE-NFGAILS, was computed from simulations of (KLVFFAE)n-(NFGAILS)n mixtures. Typical β-sheet structures in simulations of 2 and 16 peptides are shown in panels (c–h). Both KLVFFAE of Aβ (blue) and NFGAILS of IAPP (pink) are shown as cartoon. The side-chains are shown as sticks and colored according to residue type (hydrophobic in white, hydrophilic in green, positive charge in blue, and negative charge in red).
Compared to Aβ16–22, IAPP22–28 displayed a weaker β-sheet propensity (Fig. 1a). Two NFGAILS peptides mainly adopted random coil structure, and the dimeric β-sheet conformation (Fig. 1d) was unstable and only transiently observed (Fig. S2b). As the number of peptides increased, the β-sheet propensity slowly increased. With sixteen peptides, the β-sheet propensity gradually increased during the first 100 ns simulations and fluctuated between 0.35 and 0.45 during the last 100 ns (Fig. S2e), also forming the β-sheet rich aggregates (Fig. 1f). While helical intermediates were found important for the aggregation of full-length IAPP53, the helical content of IAPP22–28 was very low in our simulations (less than 1%) since the 7-residue sequence was too short to form stable helixes. The analysis of the averaged β-sheet and coil propensities per residue (Fig. S1b,f) indicated that the β-sheet propensity of the central hydrophobic residues A25 and I26 were markedly enhanced when the number of peptides were larger than or equal to eight. Therefore, the formation of β-rich aggregates by IAPP22–28 depended on the system size.
The mixture of Aβ16–22 and IAPP22–28 could co-aggregate into β-sheet rich structures (Fig. 1). Compared to (NFGIALS)2n simulations, the β-sheet propensity of IAPP22–28 peptides was significantly enhanced, because the Aβ16–22 peptide could interact with IAPP22–28 and form a hetero-dimer β-sheet more stable than a β-sheet dimer of IAPP22–28 peptides (Fig. 1e, Fig. S2c). The β-sheets in the (KLVFFAE)n-(NFGAILS)n systems were mainly formed among Aβ16–22 peptides (denoted as KLVFFAE-KLVFFAE in Fig. 1b and Fig. S3), and between Aβ16–22 and IAPP22–28 (denoted as KLVFFAE-NFGAILS). The β-sheet propensities of the central hydrophobic residues A25 and I26 of IAPP22–28 in (KLVFFAE)n-(NFGAILS)n simulations (~0.55–0.77 with n larger than 1 in Fig. S1d) was much larger than those in (NFGAILS)2n simulations (Fig. S1b,d), while the β-sheet propensities of Aβ16–22 with and without IAPP22–28 peptides (Fig. S1a,c) was negligible. Therefore, Aβ16–22 promoted the amyloid aggregation of IAPP22–28.
We also analyzed the probability distribution function (PDF) of the number of inter-peptide backbone hydrogen bonds (Fig. S4, S5). Aβ16–22 peptides were mainly connected by 2, 3 and 4 hydrogen bonds in (KLVFFAE)2n simulations, with the peak at 4 hydrogen bonds (Fig. S4a). In contract, most of the IAPP22–28 peptides in (NFGAILS)2n simulations had 2 inter-peptide backbone hydrogen bonds (Fig. S4b), indicating that the β-sheet structures formed by Aβ16–22 were more stable than those of IAPP22–28. In the case of mixed (KLVFFAE)n-(NFGAILS)n simulations (Fig. S4c), the dominant number of inter-peptide backbone hydrogen bonds changed from 2 to 4 when the value of n increase from 1 to 4. By analyzing inter-peptide backbone hydrogen bonds between different types of peptides in the mixed peptide simulations, we found that the size-dependent PDF shift was mainly between Aβ16–22 and IAPP22–28 in the co-aggregates (Fig. S5). The strong inter-peptide interaction between Aβ16–22 and IAPP22–28 drove their co-aggregation (Fig. 1g).
The β-sheet aggregates of all peptide systems feature the cross-β morphology
We analyzed the parallel or antiparallel alignment propensities of adjacent β-strands in the β-sheet aggregates (Fig. S6). Aβ16–22 preferred to form antiparallel β-sheets in (KLVFFAE)2n simulations due to the electrostatic attraction interactions between the oppositely charged K16 and E22 at the termini. The formation of predominantly antiparallel β-sheets by Aβ16–22 has also been observed in previous computation and experimental studies of the same fragment sequence29,33,54, which is different from the in-registered parallel β-sheets in the fibril structures of full length Aβ1–40 or Aβ1–4212,55,56. IAPP22–28 in (NFGAILS)2n simulations had both antiparallel and parallel alignments of β-strands with a ratio ranging from 6:4 to 5:5. This result was also consistent with experimental observations for similar peptides around the amyloidogenic core of IAPP. For example, both parallel and antiparallel β-strands were observed in hIAPP20–29 fibrils using solid-state NMR and X-ray crystallography57. The β-sheets in hIAPP19–29 fibrils was found to have parallel alignment of β-strands using Micro-Electron Diffraction (MicroED)36. The antiparallel β-sheets were observed in fibrils of hIAPP23–2958 and hIPP22–2959 in other experiments. In (KLVFFAE)n-(NFGAILS)n simulations, the antiparallel β-strands were the dominant conformation (with a probability ~ 0.7–0.8) due to the antiparallel alignment preference among Aβ16–22 peptides and between Aβ16–22 and IAPP22–28 (Fig. S6d,f).
We further calculated the probability distribution of β-sheet sizes in each system (details of analysis in Methods). When the number of Aβ16–22 peptides were 2, 4 and 6, they preferred to form into a single β-sheet (Fig. 2a). As the number of peptides increased from 8 to 12, the dominant β-sheet sizes were 4, 5 and 6, respectively, which suggested that the corresponding β-sheets aggregates had a high propensity to form two-layer β-sheets. This behavior was consistent with a previous computational study30, where the most stable conformation for an Aβ16–22 octamer was two parallel β-sheets, each comprising of four antiparallel β-strands. As the number of peptides increased to 14 and 16, the β-sheet size remained ~4–6 peptides and three-layer β-sheets could be observed (Fig. 1f). The β-sheet sizes of the Aβ16–22 and IAPP22–28 mixture had a similar dependence on system size as the (KLVFFAE)2n simulations (Fig. 2a). In contrast, there were no well-defined β-sheet sizes in (NFGAILS)2n simulations, but a weak linear dependence between the β-sheet sizes and the number of peptides (Fig. 2a).
Figure 2.

The formation of multi-layer cross-beta structures. a) The probabilities of observing β-sheets with various sizes for simulations with different number of peptides, including (KLVFFAE)2n, (NFGAILS)2n, and (KLVFFAE)n-(NFGAILS)n, are shown as two-dimensional heat-maps with respect to n and β-sheet size, where the color bar indicates the color-code according to probability values. The total number of peptides equals to 2n. The analysis was carried out for the last 100 ns of all 10 independent simulations. b) The probabilities to observe different number of β-sheet layers for different number of peptides (2n) are shown as three-dimensional plots with respect to n and the number of layers.
Next, we quantified the number of β-sheet layers in the aggregates for each molecular system. For a given snapshot structure from DMD simulations, we grouped the identified β-sheets in contact by heavy atom contacts together into the β-sheet oligomers (see Methods). Using the largest β-sheet oligomer to represent the most stable aggregate, we estimated the number of β-sheet layers by dividing its size (i.e., the number of peptides) with the average mass-weighted β-sheet size of the corresponding molecular system (Fig. 2b). As expected, two or four Aβ16–22 peptides in (KLVFFAE)2n mainly aggregated into a single-layer β-sheet structure. Six Aβ16–22 peptides formed a single β-sheet structure with a probability of ~57%, and also two-layer β-sheets with a probability of ~43%. When the number of peptides increased to 8 and 10, the two-layer β-sheets became the dominant species with probabilities of ~78% and ~92%, respectively. As the number of peptides increased up to 12, 14 and 16, the peptides predominantly aggregated into two- and three-layer β-sheets. The four-layer β-sheets was also observed with a probability of ~6% in (KLVFFAE)16 simulations. The preference of Aβ16–22 peptides to aggregate into multi-layer β-sheets was due to fact that residues at both surfaces of a β-sheet - i.e., L17, F19, A21 on one side and V18, F20 on the other side - are both highly hydrophobic (Fig. 1c,f). This result was consistent with the experimental observation that KLVFFAE peptides could self-assemble into sheet-to-sheet lamination nanostructure before aggregating into nanotubes in lamination order60, indicating that the peptides could from multi-layer β-sheets.
In (NFGAILS)2n simulations, a small system with 2 or 4 peptides mainly aggregated into a single-layer β-sheet, which was usually not stable (Fig. S2). Both single- and two-layer β-sheets were observed for larger systems, but only when the number of peptides increased to 12, 14 or 16, the two-layer β-sheets became the dominant species (Fig. 2b). The probability to observe β-sheet aggregates of IAPP22–28 with more than two layers was rare. Therefore, IAPP22–28 tends to form two-layer β-sheet, corresponding to the proto-fibril building block. This feature resulted from the difference in the hydrophobicity of two surfaces in IAPP22–28 β-sheet, where the side with F23, A25, L27 is more hydrophobic than the other with L27 and G24 (Fig. 1d,g). In the mixture, the dependence of β-sheet morphology on system sizes was very similar to that of Aβ16–22, consistent with the observations that the aggregation was driven by Aβ16–22.
The dynamics of oligomerization and fibrillization
We analyzed the equilibrium distribution of oligomers with different sizes as well as the β-sheet contents per chain with respect to system sizes for each peptide system (Fig. 3). An oligomer was defined as the peptide aggregate inter-connected by at least one heavy atom contact. Aβ16–22 was highly aggregation prone as all peptides associated into a single oligomer in all simulations. Aβ16–22 peptides in these oligomers mostly formed β-sheets with an average content of ~0.6, indicating that more than 4 out of 7 residues adopted β-sheet structure besides two terminal residues always counted as coil (Fig. S1). Therefore, our results suggested that the minimal oligomer size to form β-sheet aggregation, known as the critical aggregation nucleus61, was less than or equal to 2 for Aβ16–22.
Figure 3.

The distribution of oligomer sizes and β-sheet contents. (a–c) The probabilities of observing oligomers with various sizes for simulations with different number of peptides, including (KLVFFAE)2n, (NFGAILS)2n, and (KLVFFAE)n-(NFGAILS)n are shown as the heat-maps with respect to n and oligomer size. The color bars denote the color-code according to probability values (d–f) The probability distributions of beta-sheet content for simulations with different number of peptides are also shown as heat-maps with respect to n and beta-sheet content (divided into 10 bins from 0 to 1).
In (NFGAILS)2 and (NFGAILS)4 simulations, isolated monomers were the most populated species due to the relatively weaker hydrophobicity of IAPP22–28 peptides compared to Aβ16–22 (Fig. 3b). As the system size increased, larger oligomers with their sizes equal or close to the number of peptides in simulations became the dominant species. The isolated monomers were still observe but with significantly reduced probabilities in larger systems. In terms of the β-sheet content, there was a transition with respect to system sizes (Fig. 3e). With simulations up to 6 peptides, the distribution of β-sheet content was peaked at zero; but when the system size increased to 8 and higher, the distribution peaked around 0.3–0.4 suggesting a conformational transition from random coil to β-sheet. Therefore, our results indicated that the critical aggregation nucleus of IAPP22–28 was between 6 and 8. In the mixture, a similar transition was observed between two and four peptides (Fig. 3c,f), consistent with the co-aggregation scenario driven by Aβ16–22 where two Aβ16–22 peptides nucleated the β-sheet aggregates and IAPP22–28 peptides co-aggregated with them.
We also examined the dynamics of oligomerization and fibrillization by monitoring the largest oligomer size, the largest β-sheet oligomer size, and the mass-weighted β-sheet size in large simulations of 16 peptides for each peptide system (Fig. 4). For Aβ16–22, the process of oligomerization and β-sheet formation coincided with each other with negligible differences; and the step-wise increase of the β-sheet oligomer size suggested that isolated Aβ16–22 peptides spontaneously converted into β-sheets after they associated with together in the early stage (e.g., snapshot structures at t = ~3 and 5 ns in the inset of Fig. 4a) and also that the aggregate grew by associating with smaller pre-formed β-sheet aggregates (e.g., ~77 ns). This behavior was consistent with the previously proposed "dock-lock" aggregation elongation mechanism in a computational study of Aβ16–22 aggregation34. After all peptides aggregated into a single oligomer after ~80 ns, the mass-weighted β-sheet size remained ~5, corresponding to a three-layer β-sheet aggregate (e.g., ~100 & 200 ns). The averaged root-mean-square fluctuation (RMSF) per residue computed during the last 100 ns simulations was less than 0.6 nm (Fig. S7). Together with the low RMSF values, the small fluctuations of the largest oligomer size, largest β-sheet, and mass weighted β-sheet size (especially during the last 120 ns simulation), our results indicated that these multi-layer β-sheet aggregates formed by Aβ16–22 were very stable.
Figure 4.

The dynamics of oligomerization and fibrillization. The largest oligomer size (black), largest β-sheet oligomer size (red), and mass weighted β-sheet size (blue) were plotted as the function of simulation time from typical trajectories of aggregation simulations with 16 peptides, including Aβ16–22 (a), IAPP22–28 (b), and the mixture (c). Snapshot structures at times indicated by blue arrows are shown in the inset. Aβ16–22 (blue) and IAPP22–28 (pink) are shown in cartoon representation.
Different from Aβ16–22, IAPP22–28 peptides first assembled into unstructured oligomers (e.g., ~3 ns in Fig. 4b), and then β-sheet conformations started to emerge but was not stable (e.g., ~8ns). The β-sheet conformation became relatively stable and started to increase when the oligomer grew bigger with isolated peptides directly bound to the surface of those β-rich oligomers (e.g., 22 & 30 ns), and then rearranged into a two-layer β-sheet structure (e.g., 75 & 100 ns). During the last 100 ns simulation, the gap between the largest oligomer size and the largest β-sheet size kept around 1~5, indicating that there were always unstructured peptides on the oligomer surface (e.g., 200 ns). In addition, most residues in the final aggregates displayed high conformational flexibility with RMSF values larger than 2 nm (Fig. S7), suggesting that the aggregates of IAPP22–28 were highly dynamic.
The assembly dynamics of Aβ16–22/IAPP22–28 mixture was similar to that of Aβ16–22, but with a larger gap (~1–5) between the largest oligomer size and the corresponding number of β-sheet (Fig. 4c). Aβ16–22 peptides in the early co-aggregates with IAPP22–28 (e.g., ~3 ns) initiated the β-sheet formation (e.g., ~7, 25 and 30 ns) and the β-sheet rich oligomer rearranged into a multi-layer β-sheet structure (~130 & 160 ns). In the final aggregates, most peptides featured RMSF values less than 1 nm except two IAPP22–28 peptides bound to the surface with larger fluctuations (e.g., C14 and C16 in Fig. S7). Together with small fluctuations of the largest oligomer size, number of β-sheet, and mass weighted β-sheet size, our results suggested that the co-aggregates of Aβ16–22 and IAPP22–28 were more stable than those of IAPP22–28 alone.
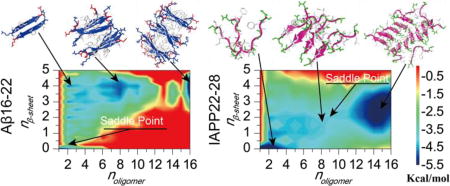
The aggregation free energy landscape
To better understand the aggregation process, we computed the potential of mean force (PMF, i.e., the effective free energy) as a function of the oligomer size (noligomer) and the number of residues in β-sheet structure per peptide (nβ-sheet) for simulations of 16 peptides (Fig. 5). All the 200ns trajectories from 10 independent simulations were included in the analysis to capture the early assembly process. Aβ16–22 featured two well-defined energy basins at (1, 0) and (16, 4), corresponding to isolated monomers at the initial aggregation stage and the multi-layer β-sheet structure in the aggregated state. There were several local basins with the oligomer size ranging from 2 to12 and nβ-sheet ~3–4.5, corresponding to β-sheet rich intermediates along the aggregation pathway (e.g., highlighted as regions 2 & 3 in Fig. 5a). IAPP22–29, on the other hand, had a deeper basin near regions with small unstructured oligomers (noligomer ~1–6 & nβ-sheet ~0–1) and another broad basin located at the region corresponding to larger oligomer size with significantly β-sheet content (n ~12–16 & nβ-sheet ~1.5–4.5) (Fig. 5b). Compared to Aβ16–22, the aggregation intermediate species, corresponding to oligomers with size around 6–8 and nβ-sheet around 1.5–2.5, were more populated. The shallow basin of large aggregates indicated that the aggregate structure of IAPP22–28 were highly dynamic with unstructured peptides at the surface (Fig. 4). The Aβ16–22/IAPP22–28 mixture displayed a similar aggregation free energy landscape (Fig. 5c) as that of Aβ16–22, including a deep basin of the large aggregates and weekly-populated intermediate states with high β-sheet content. Compared to Aβ16–22 alone (Fig. 5a), a slightly broader basin of the large aggregates (e.g., the region highlighted as 4), indicating that there were a few unstructured IAPP22–28 on the surface.
Figure 5.

The aggregation free energy landscape. The PMF (i.e., the effective free energy) is presented as the function of the oligomer size noligomer and the average number of residues adopt β-sheet conformation per chain, nβ-sheet, for the aggregation simulations of 16 peptides, including Aβ16–22 (a), IAPP22–28 (b), and their mixture (c). To capture the initial aggregation dynamics, the analysis included the whole 200 ns trajectories of 10 independent runs.
Conclusion
In summary, we investigated the assembly dynamics of amyloid core peptides of Aβ and IAPP using rapid DMD simulations without any structural bias. We found that both peptides and their mixtures can form fibril-like aggregates with the characteristic cross-β structures in our simulations, but feature distinct aggregation dynamics and pathways. Aβ16–22 peptides have a high propensity to form β-sheet aggregates, even in the dimer structure, indicating that the critical nucleus size was likely less than two. Due to their high hydrophobicity, the peptides could assemble into multi-layer β-sheet structures. The aggregation free energy landscape of Aβ16–22 features a deep energy basin toward the fibrillar aggregates. In the case of hIAPP22–28, we observed the accumulation of unstructured oligomers before the nucleation of β-sheet aggregates. Peptides in oligomers less than six mainly adopted random coil structures. When the oligomer size increased to eight or bigger, a conformational transition from random coil to β-sheet structure were observed and the β-sheet structures became stable. Our results suggested that the critical nucleus size for IAPP22–28 aggregation was approximately between 6 and 8. As a result, compare to Aβ16–22 the aggregation free energy landscape of IAPP22–28 featured a shallower basin for the amyloid aggregates with higher populations for intermediate states with small oligomers and low β-sheet content. After mixing the two types of peptides at 1:1 ratio, we observed that the aggregation of IAPP22–28 was significantly enhanced in the presence of Aβ16–22 peptides. The corresponding aggregation free energy landscape displayed a deep basin for the amyloid aggregates, where IAPP22–28 peptides were incorporated into the cross-β structure. Together, our systematic DMD simulations uncovered a complete picture of peptide oligomerization, nucleation of β-sheets, and the formation of cross-β aggregates for the amyloid core sequences of both Aβ and hIAPP and their mixture.
Material and Methods
Molecular systems used in simulations
We studied the aggregation of Aβ16–22 (Lys-Leu-Val-Phe-Phe-Ala-Glu, i.e., KLVFFAE) and IAPP22–28 (Asn-Phe-Gly-Ala-Ile-Leu-Ser, i.e., NFGAILS) as well as their co-aggregation at 1:1 ratio. For each of three cases, 8 molecular systems with the even number of peptides from 2 to 16 were investigated. For each molecular system, 10 independent DMD simulations were performed for 200 ns with different initial configurations (i.e., coordinates and velocities). All peptides started with fully extended conformations and they were initially positioned randomly (both positions and orientations) within the simulation box with any pairwise center-of-mass distances no less than 1.5 nm. In all cases, the same peptide concentration of ~26 mM was maintained by adjusting the simulation box sizes. The details of all the simulations were summarized in Table 1.
Table 1.
The details of molecule systems in our DMD simulations, including the number of peptides (Npeptide), the corresponding dimension of the cubic simulation box, the number of DMD runs (Nrun), the length of each DMD simulations, and the accumulative total simulation times.
| KLVFFAE | Npeptide | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 |
| Dimension, nm | 6.1 | 6.3 | 7.2 | 7.9 | 8.5 | 9.0 | 9.6 | 10 | |
| Nrun | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | |
| Time, ns | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | |
| Total time, µs | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| NFGAILS | Npeptide | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 |
| Dimension, nm | 6.1 | 6.3 | 7.2 | 7.9 | 8.5 | 9.0 | 9.6 | 10 | |
| Nrun | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | |
| Time, ns | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | |
| Total time, µs | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| KLVFFAE-NFGILS mixture | Npeptide | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 |
| Dimension, nm | 6.1 | 6.3 | 7.2 | 7.9 | 8.5 | 9.0 | 9.6 | 10 | |
| Nrun | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | |
| Time, ns | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | |
| Total time, µs | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
Details of DMD simulations
All simulations were carried out using the discrete molecular dynamics (DMD) algorithm22,23,62. DMD is a unique type of molecular dynamics algorithm with significantly enhanced sampling efficiency, which has been widely used by our group and other in studying protein folding22, aggregation63, small molecule/nanoparticle peptides interactions10,64. In DMD simulations, the inter-atomic interactions have similar components as conventional molecular mechanics force fields, but the potential functions are modeled by discrete step-wise functions mimicking the continuous potential functions. Bonded interactions (bonds, bond angles, and dihedrals) are modeled as infinite square wells, where covalent bonds and bond angles usually have a single well and dihedrals may feature multiple wells corresponding to cis- or trans-conformations. Non-bonded interactions (i.e., van der Waals, solvation, hydrogen bond, and electrostatic terms) are represented as a series of discrete energetic steps, decreasing in magnitude with increasing distance until reaching zero at the cutoff distance. The van der Waals parameters are adopted from the CHARMM force field65, and bonded termed are parameterized based on statistical analysis of protein structures from protein data bank (PDB). The water molecules are implicitly modeled using the EEF1 implicit solvation model developed by Lazaridis and Karplus66. A reaction-like algorithm is used to model hydrogen bond formation67. The electrostatic interactions are screened using the Debye-Hückel approximation with the Debye length set to 10 Å, which corresponds to ~100 mM of NaCl under physiological conditions.
With step-wise inter-atomic potential functions in DMD simulations, the velocity of each atom is constant until a collision occurs when an inter-atomic interaction potential changes and the new velocity is updated following the conservation laws of energy, momentum and angular momentum. The units of time, length, and energy are ~50 femtosecond, 1 Å, and 1 kcal/mol, respectively. The temperature of the system is maintained around 300K using the Anderson thermostat68. Each independent simulation was first energy minimized for 1000 DMD time units (~50 ps) with a strong heat-exchange coefficient with the virtual heat bath69, followed by equilibrium simulations carried out for four million DMD time units, which corresponds to a simulation time of ~200 ns.
Analysis methods
Secondary structure analyses were performed using the dictionary secondary structure of protein (DSSP) method70. A hydrogen bond was considered to be formed if the distance between backbone N and O atoms was ≤3.5 Å and the angle of N–H⋯O ≥120°. Following a previous work71, two chains were considered to form a β-sheet if (i) at least two consecutive residues in each chain adopted the β-strand conformation and (ii) they formed at least two backbone hydrogen bonds. Based on a vector defined as Cα atoms from N- to C-termini, two neighboring β-strands with the angle within the range of 90–180 degree were defined as antiparallel, and parallel otherwise.
The size of a β-sheet was the number of strands in a multi-strand β-sheet. The mass weighted β-sheet size, n̄β-sheet–size, was determined by the following equation
| (1) |
where nβ denoted the number of β-sheets, and ni was the size of the ith β-sheet. A β-sheet oligomer was defined as multiple β-sheets inter-connected by at least one heavy atoms pair contact, defined by a cutoff inter-atomic distance of 0.55 nm; and the total number of peptides in the complex corresponded to the β-sheet oligomer size. Two peptides inter-connected by at least one inter-molecular heavy atom contact (the cutoff of 0.55 nm) was defined to belong an oligomer. The number of peptides in an oligomer was referred to the oligomer size. The two-dimensional potential of mean force (PMF, or the effective free energy) was computed according to
| (2) |
where KB was the Boltzmann constant, T corresponded to the simulation temperature 300 K, and P(noligomer, nβ-sheet) was the probability of an oligomer with the oligomer size noligomer and the average number of residues adopt β-sheet conformation per chain, nβ-sheet.
Supporting Information
Additional data in SI include the averaged β-sheet and coil propensity per residue in every system; the time evolution of coil and β-sheet secondary structure propensities of simulations with 2 and 16 peptides; the time evolution of averaged probability of β-sheets formed between different types of peptides in the mixed peptide simulations; the PDF of the number of backbone hydrogen bonds for each peptide system; and the probability of parallel and antiparallel β-sheet observed in each molecular system.
Supplementary Material
Acknowledgments
The work is supported in part by NSF CAREER CBET-1553945 (Ding) and NIH MIRA R35GM119691 (Ding). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH and NSF.
Footnotes
The authors declare no competing financial interest
References
- 1.Ke MSPC, Ding F, Kakinen A, Javed I, Separovic F, Davis TP, Mezzenga R. Chem. Soc. Rev. 2017 doi: 10.1039/c7cs00372b. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei GH, Jewett AI, Shea JE. Phys Chem Chem Phys. 2010;12:3622–3629. doi: 10.1039/c000755m. [DOI] [PubMed] [Google Scholar]
- 3.Sun YX, Xi WH, Wei GH. J Phys Chem B. 2015;119:2786–2794. doi: 10.1021/jp508122t. [DOI] [PubMed] [Google Scholar]
- 4.Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B. Nat Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 5.Stefanis L. Csh Perspect Med. 2012;2 doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medori R, Tritschler HJ, Leblanc A, Villare F, Manetto V, Chen HY, Xue R, Leal S, Montagna P, Cortelli P, Tinuper P, Avoni P, Mochi M, Baruzzi A, Hauw JJ, Ott J, Lugaresi E, Autiliogambetti L, Gambetti P. New Engl J Med. 1992;326:444–449. doi: 10.1056/NEJM199202133260704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gambetti P, Medori R, Tritschler H, Leblanc A, Montagna P, Cortelli P, Tinuper P, Monari L, Tabaton M, Petersen R, Autiliogambetti L, Lugaresi E. J Neuropath Exp Neur. 1992;51:353–353. [Google Scholar]
- 8.Mo YX, Lei JT, Sun YX, Zhang QW, Wei GH. Sci Rep-Uk. 2016;6 doi: 10.1038/srep33076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 10.Gurzov EN, Wang B, Pilkington EH, Chen PY, Kakinen A, Stanley WJ, Litwak SA, Hanssen EG, Davis TP, Ding F, Ke PC. Small. 2016;12:1615–1626. doi: 10.1002/smll.201502317. [DOI] [PubMed] [Google Scholar]
- 11.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. P Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paravastu AK, Leapman RD, Yau WM, Tycko R. P Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stefani M, Dobson CM. J Mol Med-Jmm. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 15.Thompson LK. P Natl Acad Sci USA. 2003;100:383–385. doi: 10.1073/pnas.0337745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirkitadze MD, Bitan G, Teplow DB. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh P, Kumar A, Datta B, Rangachari V. Bmc Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-S6-S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berhanu WM, Yasar F, Hansmann UHE. Acs Chem Neurosci. 2013;4:1488–1500. doi: 10.1021/cn400141x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng WH, Tsai MY, Chen MC, Wolynes PG. P Natl Acad Sci USA. 2016;113:11835–11840. doi: 10.1073/pnas.1612362113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen MC, Wolynes PG. P Natl Acad Sci USA. 2017;114:4406–4411. doi: 10.1073/pnas.1702237114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M, Tsai M, Zheng W, Wolynes PG. J Am Chem Soc. 2016;138:15197–15203. doi: 10.1021/jacs.6b08665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding F, Tsao D, Nie HF, Dokholyan NV. Structure. 2008;16:1010–1018. doi: 10.1016/j.str.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zganec M, Zerovnik E, Urbanc B. J Chem Theory Comput. 2015;11:2355–2366. doi: 10.1021/acs.jctc.5b00067. [DOI] [PubMed] [Google Scholar]
- 24.Cheon M, Hall CK, Chang I. Plos Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emperador A, Orozco M. J Chem Theory Comput. 2017;13:1454–1461. doi: 10.1021/acs.jctc.6b01153. [DOI] [PubMed] [Google Scholar]
- 26.Colvin MT, Silvers R, Frohm B, Su YC, Linse S, Griffin RG. J Am Chem Soc. 2015;137:7509–7518. doi: 10.1021/jacs.5b03997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber M, Ovchinnikova OY, Schutz AK, Glockshuber R, Meier BH, Bockmann A. Biomol Nmr Assign. 2015;9:7–14. doi: 10.1007/s12104-013-9535-x. [DOI] [PubMed] [Google Scholar]
- 28.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. P Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie LG, Luo Y, Wei GH. J Phys Chem B. 2013;117:10149–10160. doi: 10.1021/jp405869a. [DOI] [PubMed] [Google Scholar]
- 30.Ma BY, Nussinov R. P Natl Acad Sci USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong JJ, Lu K, Lakdawala A, Mehta AK, Lynn DG. Amyloid. 2006;13:206–215. doi: 10.1080/13506120600960809. [DOI] [PubMed] [Google Scholar]
- 32.Wang JQ, Tao K, Zhou P, Pambou E, Li ZY, Xu H, Rogers S, King S, Lu JR. Colloid Surface B. 2016;147:116–123. doi: 10.1016/j.colsurfb.2016.07.052. [DOI] [PubMed] [Google Scholar]
- 33.Petty SA, Decatur SM. J Am Chem Soc. 2005;127:13488–13489. doi: 10.1021/ja054663y. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D. P Natl Acad Sci USA. 2007;104:111–116. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Y, Lynn DG, Berland KM. J Am Chem Soc. 2010;132:6306-+. doi: 10.1021/ja910964c. [DOI] [PubMed] [Google Scholar]
- 36.Krotee P, Rodriguez JA, Sawaya MR, Cascio D, Reyes FE, Shi D, Hattne J, Nannenga BL, Oskarsson ME, Philipp S, Griner S, Jiang L, Glabe CG, Westermark GT, Gonen T, Eisenberg DS. Elife. 2017;6 doi: 10.7554/eLife.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luca S, Yau WM, Leapman R, Tycko R. Biochemistry-Us. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiltzius JJW, Sievers SA, Sawaya MR, Cascio D, Popov D, Riekel C, Eisenberg D. Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. P Natl Acad Sci USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 41.Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, Merkle ML, Voelter W, Brunner H, Kapurniotu A. J Mol Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 42.Guo JP, Arai T, Miklossy J, McGeer PL. P Natl Acad Sci USA. 2006;103:1953–1958. doi: 10.1073/pnas.0509386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VMY. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 44.Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. P Natl Acad Sci USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A. Angew Chem Int Edit. 2010;49:3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]
- 46.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, Westermark P. Amyloid. 2014;21:221–224. doi: 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- 47.Li L, Hoelscher C. Brain Res Rev. 2007;56:384–402. doi: 10.1016/j.brainresrev.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT. Am J Pathol. 2015;185:834–846. doi: 10.1016/j.ajpath.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Morales R, Moreno-Gonzalez I, Soto C. Plos Pathog. 2013;9 doi: 10.1371/journal.ppat.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baram M, Atsmon-Raz Y, Ma BY, Nussinov R, Miller Y. Phys Chem Chem Phys. 2016;18:2330–2338. doi: 10.1039/c5cp03338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Favrin G, Irback A, Mohanty S. Biophys J. 2004;87:3657–3664. doi: 10.1529/biophysj.104.046839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santini S, Mousseau N, Derreumaux P. J Am Chem Soc. 2004;126:11509–11516. doi: 10.1021/ja047286i. [DOI] [PubMed] [Google Scholar]
- 53.Wiltzius JJW, Sievers SA, Sawaya MR, Eisenberg D. Protein Sci. 2009;18:1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Biochemistry-Us. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 55.Walti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Bockmann A, Guntert P, Meier BH, Riek R. Proc Natl Acad Sci U S A. 2016;113:E4976–4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao YL, Ma BY, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Nat Struct Mol Biol. 2015;22:499–U497. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madine J, Jack E, Stockley PG, Radford SE, Serpell LC, Middleton DA. J Am Chem Soc. 2008;130:14990–15001. doi: 10.1021/ja802483d. [DOI] [PubMed] [Google Scholar]
- 58.Soriaga AB, Sangwan S, Macdonald R, Sawaya MR, Eisenberg D. J Phys Chem B. 2016;120:5810–5816. doi: 10.1021/acs.jpcb.5b09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nielsen JT, Bjerring M, Jeppesen MD, Pedersen RO, Pedersen JM, Hein KL, Vosegaard T, Skrydstrup T, Otzen DE, Nielsen NC. Angew Chem Int Edit. 2009;48:2118–2121. doi: 10.1002/anie.200804198. [DOI] [PubMed] [Google Scholar]
- 60.Lu K, Jacob J, Thiyagarajan P, Conticello VP, Lynn DG. J Am Chem Soc. 2003;125:6391–6393. doi: 10.1021/ja0341642. [DOI] [PubMed] [Google Scholar]
- 61.Chen S, Ferrone FA, Wetzel R. Proc Natl Acad Sci U S A. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shirvanyants D, Ding F, Tsao D, Ramachandran S, Dokholyan NV. J Phys Chem B. 2012;116:8375–8382. doi: 10.1021/jp2114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radic S, Davis TP, Ke PC, Ding F. Rsc Adv. 2015;5:105489–105498. doi: 10.1039/C5RA20182A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nedumpully-Govindan P, Gurzov EN, Chen PY, Pilkington EH, Stanley WJ, Litwak SA, Davis TP, Ke PC, Ding F. Phys Chem Chem Phys. 2016;18:94–100. doi: 10.1039/c5cp05924k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 66.Lazaridis T, Karplus M. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 67.Ding F, Borreguero JM, Buldyrey SV, Stanley HE, Dokholyan NV. Proteins-Structure Function and Genetics. 2003;53:220–228. doi: 10.1002/prot.10468. [DOI] [PubMed] [Google Scholar]
- 68.Andersen HC. J Chem Phys. 1980;72:2384–2393. [Google Scholar]
- 69.Ramachandran S, Kota P, Ding F, Dokholyan NV. Proteins. 2011;79:261–270. doi: 10.1002/prot.22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 71.Sun YX, Qian ZY, Guo C, Wei GH. Biomacromolecules. 2015;16:2940–2949. doi: 10.1021/acs.biomac.5b00850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.