

Abstract
Glucose-stimulated insulin secretion [GSIS] involves interplay between metabolic and cationic events. Seminal contributions from multiple laboratories affirm essential roles for small G-proteins [Rac1, Cdc42, Arf6, Rab27A] in GSIS. Activation of these signaling proteins promotes cytoskeletal remodeling, transport and docking of insulin granules on the plasma membrane for exocytotic secretion of insulin. Evidence in rodent and human islets suggests key roles for lipidation [farnesylation and geranylgeranylation] of these G-proteins for their targeting to appropriate cellular compartments for optimal regulation of effectors leading to GSIS. Interestingly, however, inhibition of prenylation appears to cause mislocalization of non-prenylated, but [paradoxically] activated G-proteins, in “inappropriate” compartments leading to activation of stress kinases and onset of mitochondrial defects, loss in GSIS and apoptosis of the islet β-cell. This review highlights our current understanding of roles of G-proteins and their posttranslational lipidation [prenylation] signaling networks in islet function in normal health, metabolic stress [glucolipotoxicity and ER stress] and diabetes. Critical knowledge gaps that need to be addressed for the development of therapeutics to halt defects in these signaling steps in β cells in models of impaired insulin secretion and diabetes are also highlighted and discussed.
Keywords: G-proteins, Rac1, farnesylation, geranylgeranylation, islet β-cell, NADPH oxidase
1. Introduction
Glucose-stimulated insulin secretion [GSIS] from pancreatic β-cells is mediated largely via the generation of soluble second messengers including cyclic nucleotides, biologically-active hydrolytic products of phospholipases [A2, C, and D], and adenine nucleotides.1–4 However, the precise molecular and cellular mechanisms underlying GSIS remain only partially understood. Following Glut-2 mediated entry into the β-cell, glucose is metabolized via the glycolytic and tricarboxylic acid cycles with a resultant increase in intracellular ATP, which, in turn, mediates closure of ATP-sensitive K+ channels localized on the plasma membrane resulting in membrane depolarization. These signaling events promote influx of extracellular calcium through the voltage-gated calcium channels. Increase in intracellular calcium has been shown to be needed for the transport of insulin-laden secretory granules to the plasma membrane for fusion and release of insulin into circulation. It is noteworthy that, in addition to adenine nucleotides, the guanine nucleotides [e.g., GTP) have been shown to play major regulatory roles in GSIS. For example, using selective inhibitors of the GTP biosynthetic pathway [e.g., mycophenolic acid], Metz and associates provided the first evidence for a permissive role for GTP in GSIS.5 Although the precise mechanisms underlying the regulatory role[s] of GTP in GSIS remain elusive, emerging evidence indicates that they might involve activation of one [or more] GTP-binding proteins [G-proteins]6,7. At least two major groups of G-proteins have been described in pancreatic β-cells. The first group is trimeric in nature, which is comprised of α [39–43 kDa]-, β [35–37 kDa]-, and γ [6–8 kDa]-subunits. These signaling proteins are involved in coupling of various G-protein coupled receptors [GPCRs] to their intracellular effectors, such as adenylate cyclase, phosphodiesterase, or phospholipases. The second group of G-proteins [the main focus of this review] is comprised of small molecular weight [20–25 kDa] monomeric G-proteins, which are involved in protein sorting as well as trafficking of secretory vesicles.6,7
2. Identification and regulation of small G-proteins and their regulatory factors in the islet β-cell
It is well established now that both trimeric and small G-proteins play critical roles in islet β-cell function including cytoskeletal remodeling, vesicular transport and GSIS. The reader is referred to seminal contributions from numerous laboratories, which are highlighted in.6–11 Briefly, based on available evidence on G-protein mediated regulation of islet function, the small G-protein family can be divided into three subfamilies. The first one is comprised of Rho, Rac1, Cdc42, and Arf-6. Published evidence implicates these proteins in the cytoskeletal remodeling and vesicle fusion in the pancreatic β-cell. Rap1, Rab3A and Rab27 belong to the second subfamily of small G-proteins. The Rab GTPases are associated with the secretory granules and play regulatory roles in the priming and docking of insulin-laden secretory granules at the plasma membrane. Rap1 plays significant roles in β-cell functions including GSIS. Recent studies by Kelly et al demonstrated that Rap1 promotes β-cell proliferation through mammalian target of rapamycin complex 1.12 The third group of small GTPases [e.g., Rab2, Rhes, and Rem2] is relatively less studied in the islet. It is noteworthy that RalA, a small G protein, appears to elicit direct regulatory effects in exocytosis via its direct interaction with the exocyst complex. Over the years, several regulatory factors/proteins have been identified in the islet β-cell that precisely regulate small G-proteins, which cycle between their inactive [GDP-bound] and active [GTP-bound] conformations. Three major types of such regulatory proteins/factors have been described for small G proteins. The first group is comprised of guanine nucleotide exchange factors [GEFs], which facilitate the conversion of the GDP-bound [inactive] forms to their GTP-bound [active] forms. Tiam1 and Vav2 have been identified as GEFs for Rac1 in the islet β-cell. Potential roles of these GEFs in the regulation of Rac1 function has been tested recently using novel small molecule inhibitors for Tiam1 [NSC23766] and Vav2 [Ehop-016] [Figure 1]13.
Figure 1. Activation-deactivation of Rac1: Roles of Tiam1 and Vav2 as GEFs for Rac1 activation.
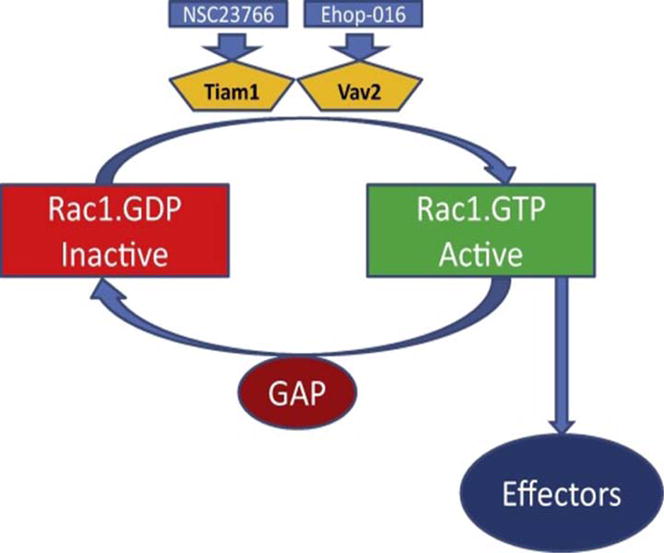
Rac1 undergoes activation [GTP-bound conformation] and deactivation [GDP-bound conformation] cycle. Inactive Rac1 is converted to its active form by at least two GEFs, namely Tiam1 and Vav2. Published evidence suggests that both of these GEFs are expressed in a variety of insulin-secreting cells including clonal INS-1 832/13 cells, rodent islets and human islets [see text for additional details]. As depicted in the figure, NSC23766 and Ehop-016 inhibit Tiam1 and Vav2, respectively. GTP-bound Rac1, in turn, regulates functions for multiple effector proteins, including Nox2. Following transmission of necessary signals, the active form of Rac1 is converted back to its inactive conformation mediated by the GTPase activity that is intrinsic to Rac1. Additional regulatory proteins/factors [e.g., GTPase-activating proteins; GAPs], further facilitate the conversion of the active G protein to its inactive form. Potential identity of GAPs for Rac1 is an understudied area in the islet β-cell. Using NSC23766 and Ehop-016 and siRNA approaches, we have demonstrated novel regulatory roles for Tiam1 and Vav2 in glucose-induced cytoskeletal remodeling and insulin secretion. [Reproduced with permission from Elsevier].13
Kepner and associates identified Cool-1/βPIX as a GEF for Cdc42 in regulation of physiological insulin secretion.14 Detailed studies by Jayaram and associates provided evidence for key modulatory roles for Arf nucleotide-binding site opener [ARNO] in GSIS.15 The small G-protein Rap1 has been implicated in cAMP-sensitive signaling pathways involving exchange proteins directly activated by cAMP [Epac]-like GEFs. Epacs have also been shown to play key regulatory roles in various cellular signaling steps involved in β-cell function, including calcium-induced calcium mobilization, insulin secretion, β-cell growth, and proliferation.16 The second group of regulatory factors are the GDP-dissociation inhibitors [GDIs], which prevent the dissociation of GDP from G proteins, and hence are considered “inhibitory” in the G protein activation cascade. Rho-GDI and caveolin-1 have been implicated to subserve the roles of GDIs for regulation of Rho, Rac1 and Cdc42 functions in the islet β-cell.6,7,14 The third group represents the GTPase-activating proteins [GAPs]; these proteins promote the conversion of the active G-proteins to their respective inactive conformations by activating the intrinsic GTPase function of candidate G-proteins to complete the GTP hydrolytic cycle.
3. Small G-proteins undergo post-translational modifications
As depicted in Figure 2, the first of a four-step modification includes incorporation of a 15-carbon farnesyl pyrophosphate [FPP] or 20-carbon geranylgeranylpyrophosphate [GGPP], which are derived from mevalonic acid [MVA], onto the carboxyl terminal cysteine of the candidate G-proteins. The farnesylation or geranylgeranylation steps are catalyzed by farnesyltransferase [FTase] or geranylgeranyltransferase [GGTase], respectively. This is followed by proteolysis of several amino acids [up to a maximum of three] by Ras converting enzyme 1 [Rce1], a protease of microsomal origin. A carboxylmethylation [CML] step then modifies the newly exposed carboxylate anion of the prenylated cysteine. This step is catalyzed by isoprenylcysteine carboxylmethyl-transferase [ICMT].
Figure 2. Schematic representation of post-translational prenylation of G-proteins.
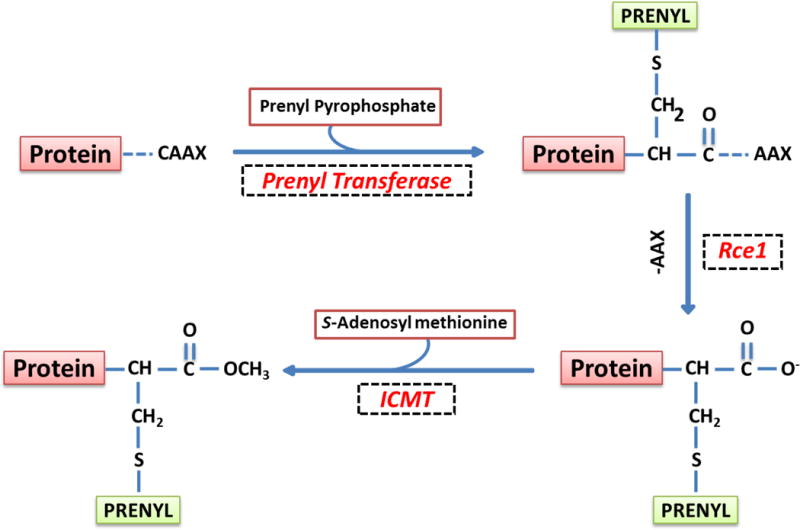
The first of the four-step modification is incorporation of either a farnesyl or a geranylgeranyl derivative of MVA into the carboxy terminal cysteine [CAAX motif] via a thioether linkage. Farnesylation and geranylgeranylation steps are catalyzed by the FTase and the GGTase, respectively. Collectively, they are referred to as prenyl transferase. Following prenylation, the three amino acids after the prenylated cysteine [AAX] are removed by a protease [Rce1] of microsomal origin, thereby exposing the carboxylate anion. This site is then methylated by carboxyl methyl transferase [ICMT], which transfers a methyl group onto the carboxylate group using S-adenosyl methionine [SAM] as the methyl donor. The CML of G-proteins increases their hydrophobicity and translocation to the membrane fraction. In addition to these, certain G-proteins [H-Ras and Rac1] have been shown to undergo palmitoylation at a cysteine residue, which is upstream to the prenylated cysteine. It is thought that palmitoylation provides a “firm” anchoring for the modified protein into the cell membrane for optimal interaction with its respective effector proteins. [Reproduced with permission from Elsevier].19
In some cases [not depicted in Figure 2], the covalent addition of a long-chain fatty acid, typically palmitate, at cysteine residues, which are upstream to the CAAX motif, completes the cascade.7,17–19 It is widely accepted that such modification[s] would render the modified G-proteins more hydrophobicity thereby enabling them to associate with membranes for interaction with their respective effectors. Because the prenylation of GTPases occurs shortly after their synthesis, and due to the fact that half-lives of prenylated proteins are rather long, this is not likely to be an acute regulatory step; however, in many cases, prenylation is necessary to allow candidate G proteins to intercalate into the relevant membrane compartment. In contrast, the CML and palmitoylation steps are subject to acute regulation via addition or deletion of methyl or palmitoyl groups. The addition and removal of methyl groups are catalyzed by ICMT and esterase, respectively. Likewise, addition and deletion of palmitoyl groups are facilitated by palmitoyl transferase and esterase, respectively.7,17–19 Several previous studies have demonstrated critical roles for posttranslational modifications of these proteins in GSIS.19 They are briefly discussed below.
4. G-protein prenylation in islet β-cell function and dysfunction
4.1 Identification and functional regulation of prenyltransferases in islet β-cells
Earlier studies from our laboratory have identified at least three distinct prenyl transferases in the islet β-cell [Figure 3]19. The FTase and GGTase-I are referred to as CAAX prenyltransferases because their candidate substrate proteins share a conserved CAAX motif at the C-terminal region. In contrast, the GGTase-II [also termed as Rab geranylgeranyl transferase; RGGT] geranylgeranylates Rab GTPases at the CXC or CC motif, and hence referred to as the non-CAAX prenyltransferase. Structurally, FTase, GGTase-I and GGTase-II are heterodimeric consisting of α-and β-subunits. The FTase and GGTase-I share a common α-subunit [FTase/GGTase-α], but distinct β-subunits. Experimental evidence suggests that the α-subunit is the regulatory subunit whereas the β-subunit confers substrate specificity. Known examples of farnesylated proteins include Ras, lamins A/B and certain γ subunits of trimeric G proteins. GGTase I prenylates Cdc42, Rac1, Rap1 and certain γ subunits of trimeric G proteins, whereas Rab GTPases [Rab3A, Rab27A] undergo GGTase II-mediated prenylation.7,19
Figure 3. Schematic representation of protein prenylation pathway.
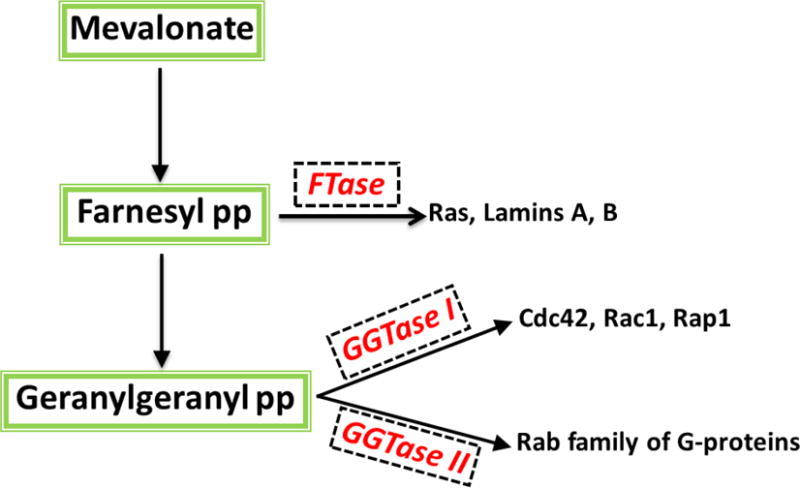
Mevalonic acid [MVA] is the precursor for the biosynthesis of farnesyl pyrophosphate [FPP] and geranylgeranyl pyrophosphate [GGPP]. FTase farnesylates nuclear lamins (A and B), Ras and γ-subunits of trimeric G proteins. GGTase I geranylgeranylates Cdc42, Rac1 and Rap1. GGTase II catalyzes prenylation of Rab subfamily of G proteins. Numerous studies have characterized these enzymes in the islet β-cell in relation to their roles in various cellular events including vesicular transport, cytoskeletal remodeling, proliferation and insulin secretion [see text for additional details]. [Reproduced with permission from Elsevier].19
4.2 G-protein prenylation is necessary for physiological insulin secretion
Original studies by Metz et al.20 and Li et al.21 utilized inhibitors of cholesterol biosynthesis [e.g., lovastatin] to assess roles of protein prenylation in insulin secretion. Statins inhibit the synthesis of MVA, a precursor for the biosynthesis of FPP and GGPP [Figure 2]. These studies in clonal β-cells and normal rodent islets have demonstrated inhibition of nutrient-induced insulin secretion by lovastatin. Coprovision of MVA reversed the inhibitory effect of statins on GSIS. They also demonstrated significant alterations in the subcellular distribution of G-proteins following inhibition of prenylation with statins. Based on data accrued in these studies it was concluded that inhibition of protein prenylation in β-cells results in selective accumulation of unprenylated G proteins in the soluble compartment, possibly interfering with the interaction of these proteins with their respective effector proteins, which may be required for GSIS. Follow-up investigations from our laboratory have utilized selective inhibitors of FTase and GGTases including allyl- or vinyl-farnesols or geranylgeraniols, FTI-277, GGTI-2147 to inhibit islet endogenous FTase and GGTases.7,19 Data from these studies provided compelling evidence to implicate that protein prenylation is essential for GSIS to occur. Observations from these pharmacological investigations implicating key functional roles of protein prenylation in GSIS were further confirmed by molecular biological approaches. They included siRNA-mediated knockdown of FTase-β subunit and overexpression of an inactive mutant of FTase/GGTase-α; these experimental manipulations resulted in marked suppression of nutrient-, but not KCl-induced insulin secretion from pancreatic β-cells.22
Published evidence from multiple laboratories implicates novel roles for Rab GTPases in physiological insulin secretion from the islet β-cell.6,7,9,23–25 In a manner akin to Ras and Rho GTPases, prenylation has been shown to regulate cellular functions of Rab GTPases in the islet.25 The FTase catalyzes incorporation of a 15-carbon farnesyl group, while the GGTase I and GGTase II mediate incorporation of a 20-carbon geranylgeranyl group into the C-terminal cysteines of candidate G-proteins [Figure 3].
In contrast to considerable evidence suggesting FTase/GGTase I-mediated regulation of GSIS, little is known about GGTase II [also referred to as Rab GGTase; RGGT] and its regulatory proteins in the cascade of events leading to GSIS. Recent investigations from our laboratory have provided the first immunological evidence to suggest expression of α- and β-subunits of RGGT in clonal INS-1 832/13 β-cells, normal rat islets and human islets.25 It is noteworthy that, in contrast to Ras and Rho GTPases, the prenylation of Rab GTPases has been shown to be regulated by additional regulatory factors, such as the Rab escort proteins [REPs], which complex with RGGT [REP-RGGT complex]. In this context, we were able to demonstrate expression of REP-1 in INS-1 832/13 cells, normal rat islets and human islets. Furthermore, we demonstrated significant inhibition of GSIS in INS-1 832/13 cells following siRNA-mediated knockdown of α- or β-subunits of RGGT and REP1. Taken together, our findings provided compelling evidence in support of key roles for RGGT and its regulatory proteins in GSIS.25 Thus, the data from pharmacological and molecular biological approaches further validated our original hypothesis that protein prenylation represents a key signaling step in the cascade of events leading to GSIS.
4.3 How does glucose regulate prenyltransferase activities?
The next question that we sought answers for was how physiological concentrations of glucose promote prenylation of candidate G-proteins leading to insulin secretion? There are at least three potential mechanisms that might underlie glucose-mediated regulation of these signaling events. First, recent investigations by Lorenz and associates in INS-1 832/13 cells suggested transient increase in intracellular FPP levels following glucose stimulation in a time course that mirrors first phase of GSIS.26 We propose that such an increase in FPP, a substrate for FTase [Figure 3], would favor prenylation of specific GTPases that are necessary for GSIS to occur. Second, Goalstone et al. recently quantified the levels of expression and catalytic activity of FTase and GGTase-I in INS-1 832/13 cells and normal rodent islets exposed to stimulatory glucose concentrations.27 They reported increased expression, by glucose, of the FTase/GGTase-α, but not the β-subunits of FTase or GGTase-1 in INS-1 832/13 cells and rat islets. Compatible with these findings, they also observed a substantial increase (2.5- to 4.0-fold over basal) in the activities of FTase and GGTase-1 in pancreatic β-cells exposed to glucose.27 A third potential possibility that remains to be verified in the islet β-cell is regulation of FTase/GGTase-α activation via phosphorylation–dephosphorylation mechanisms. Published evidence suggests that FTase/GGTase-α undergoes phosphorylation, which, in turn, results in the functional activation of the enzyme.28 Together, findings from above described studies suggest that glucose-induced activation of the endogenous prenyltransferases and prenylation of specific proteins is necessary for GSIS to occur.
4.4 Some examples of prenylation-sensitive signaling events in pancreatic β-cells
Emerging evidence in pancreatic islet β-cell clearly implicates novel regulatory roles for protein prenylation in islet function including tonic generation of reactive oxygen species [ROS] generated via activation of phagocyte-like NADPH oxidase [Nox2]29,30. Along these lines, earlier studies from our laboratory have suggested that acute exposure of normal rodent islets or clonal INS-1 832/13 cells to stimulatory glucose or a mixture of mitochondrial fuels [mono-methylsuccinate plus α-ketoisocaproic acid] led to elevated intracellular accumulation of ROS; this signaling step was sensitive to selective inhibitors of Nox2 [diphenyleneiodonium chloride or apocynin] or siRNA-p47phox, one of the subunits of Nox2. In addition, inhibitors of protein prenylation [FTI-277 or GGTI-2147] significantly suppressed nutrient-induced ROS generation, suggesting that activation of one [or more] prenylated small G-proteins and/or γ-subunits of trimeric G proteins is involved in this signaling axis. It was also noted that depletion of endogenous GTP levels with mycophenolic acid significantly reduced glucose-induced activation of Rac1 and ROS generation in these cells. Other immunosuppressants, like cyclosporine A or rapamycin, which do not deplete endogenous GTP levels, failed to affect glucose-induced ROS generation, suggesting that endogenous GTP is necessary for glucose-induced Nox2 activation and ROS generation. Treatment of INS-1 832/13 cells or rat islets with pertussis toxin [Ptx], which ADP ribosylates and inhibits inhibitory class of trimeric G proteins [Gi or Go], significantly attenuated glucose-induced ROS generation in these cells, implicating activation of a Ptx-sensitive G protein in these signaling cascade. Together, our findings suggest a prenylated Ptx-sensitive signaling step couples Rac1 activation in the signaling steps necessary for glucose-mediated generation of ROS in the pancreatic β-cells. These and other relevant observations including contributory roles of protein prenylation in cytoskeletal remodeling are reviewed in.30
Further, we recently identified a novel pertussis toxin [Ptx]-sensitive farnesylated G-protein [termed Probin] that appears to suppress Akt phosphorylation in INS-1 832/13 cells and normal rat islets.31 Specifically, these studies investigated putative regulatory roles of protein farnesylation in cell survival signaling pathways in insulin-secreting INS-1 832/13 cells and normal rodent islets, especially at the level of protein kinase-B/Akt phosphorylation induced by insulin-like growth factor [IGF-1]. It was noted that selective FTase inhibitors [FTI-277 or FTI-2628] or knockdown of the β-subunit of FTase by siRNA significantly increased Akt activation under basal and IGF-1-stimulated conditions. Under these conditions, the relative abundance of phosphorylated Foxo1 and Bad were increased implicating inactivation of critical components of the cell death machinery. Furthermore, FTI-induced Akt activation was suppressed by LY294002, a known inhibitor of PI-3-kinase. Interestingly, exposure of INS-1 832/13 cells to Ptx potentiated Akt phosphorylation suggesting involvement of a Ptx-sensitive G-protein in this signaling axis. Lastly, prostaglandin E₂, a known agonist of inhibitory G-proteins, significantly attenuated FTI-induced Akt phosphorylation. Taken together, these studies identified a novel farnesylated G-protein in INS-1 832/13 cells and normal rat islets, which appears to suppress Akt activation and subsequent cell survival signaling steps.31
Based on the findings reviewed above, we propose a Working Model, which implicates protein prenylation in physiological insulin secretion from the pancreatic β-cell. Glucose metabolism leads to increases in the levels of soluble second messenger molecules including GTP, and intermediates of isoprenoid pathway [FPP and GGPP]. Published evidence from our laboratory also suggests acute regulation [activation] of FTase/GGTase activities in a glucose-stimulated β-cell.27 These signaling steps result in increased prenylation of key G proteins culminating in their targeting to appropriate cellular compartment [plasma membrane and secretory granule] for optimal interaction and regulation of their respective effector proteins (Raf-1, ERK1/2, Nox2) to promote conditions (e.g., ROS generation, cytoskeletal remodeling) conducive for secretory granule fusion with the plasma membrane leading to exocytotic secretion of insulin.30,32
4.5 Defects in prenylation signaling networks lead to human disease
At least 300 prenylated proteins are identified in the human genome; the majority of which are implicated in a variety of cellular processes including cell growth, differentiation, cytoskeletal function and vesicle trafficking. It has been shown that alterations in prenylation of Ras GTPases and nuclear lamins are involved in multiple human diseases including cancer, neurodegenerative disorders, retinitis pigmentosa and premature ageing syndromes.33,34 Evidence is also emerging to suggest that defects in protein prenylation lead to aberrant activation and mislocalization of unprenylated proteins. For example, Khan et al. have recently reported hyperactivation of macrophages and associated induction of erosive arthritis in mice lacking GGTase-1, thus raising potential for regulatory roles of geranylgeranylation in inflammatory cell signaling.35 It is noteworthy that GGTase deficiency in these cells is manifested by significantly elevated levels of active [GTP-bound] Rac1, Cdc42 and RhoA. These findings suggest alternate prenylation-independent mechanisms for the activation of these unprenylated G proteins under pathological conditions.35 Along these lines, Dunford et al have demonstrated constitutive activation of Rac, Cdc42 and Rho G-proteins in J774 macrophages following inhibition of prenylation by nitrogen-containing bisphosphonates.36 Along these lines published evidence suggests significant alterations in subcellular distribution [mistargeting] of G-proteins under conditions in which their prenylation is suppressed via mutation of prenylatable cysteine in the CAAX motif.37 Taken together, these findings demonstrate inhibition of prenylation results in sustained activation and translocation of G-proteins to inappropriate cellular compartments, thus raising an important question if such a signaling step leads to cellular pathology including islet dysfunction.
4.6 Potential mechanism[s] underlying the inactivation of prenyltransferases culminating in cell dysfunction and demise
In 2001, Kim and associates reported caspase-3-mediated cleavage of FTase/GGTase-α during apoptosis in Rat-2/H-ras, W4 and Rat-1 cells treated with LB42708 [an FTase inhibitor], anti-Fas antibody and etoposide, respectively.38 They also provided evidence to indicate significant inhibition of degradation of FTase/GGTase-α by specific inhibitors of caspase-3. Interestingly, over-expression of caspase-resistant D59A FTase/GGTase-α mutant significantly desensitized cells to etoposide-induced death, thus suggesting that cleavage of FTase/GGTase-α by caspase-3 contributes to the progression of apoptosis. We arrived at similar conclusions in pancreatic β-cells undergoing apoptosis following exposure to genotoxic agents, such as etoposide.39 Briefly, exposure of INS-1 832/13 cells or normal rodent islets to etoposide significantly increased activation of caspase-3 and subsequent degradation of FTase/GGTase-α, which was prevented by Z-DEVD-FMK, a known inhibitor of caspase-3. Treatment of cell lysates with recombinant caspase-3 also caused FTase/GGTase α-subunit degradation further affirming our conclusions. Based on these findings, it was concluded that etoposide induces loss in cell viability by inducing mitochondrial dysfunction, caspase-3 activation and degradation of FTase/GGTase α-subunit.39 It is noteworthy that evidence from recent investigations appears to provide additional insights into potential defects in protein prenylation under conditions of metabolic stress and diabetes. They are reviewed below.
Recently, Chen et al. investigated potential alterations in the functional status of enzymes in the MVA pathway in vascular smooth muscle cells (VSMC) from the streptozotocin [Stz]-induced diabetic BALB/c mice.40 They reported a temporal relationship between the onset of atherogenesis [8–16 weeks following induction of diabetes] and the expression [mRNA and protein] levels of key enzymes of MVA pathway, including HMG CoA-reductase, FPP synthase, GGPP synthase, FTase and GGTase in VSMC. Expression levels of squalene synthase and phospho-HMG CoA reductase remained unchanged in diabetic VSMC. The data accrued in diabetic animal models were replicated in VSMC incubated under glucotoxic conditions [22.2 mM glucose; 48 h]. Taken together, findings from these studies provide additional clues with regard to potential pathophysiological roles of this signaling pathway in the induction of atherosclerosis, specifically under glucotoxic/diabetic conditions.
More recent studies from our laboratory have examined the functional status of protein prenylation pathway in a variety of insulin-secreting cells exposed to metabolic stress [glucotoxicity, lipotoxicity and endoplasmic reticulum stress] conditions.41 We documented that metabolic stress activates caspase-3 and induces degradation of FTase/GGTase-α in INS-1 832/13 cells, normal rat islets and human islets. Furthermore, we observed significant inhibition in FTase and GGTase activities in INS-1 832/13 cells exposed to these conditions. Consistent with these findings, we also noted increases in caspase-3 activation and FTase/GGTase-α degradation in islets derived from the ZDF rat, a model for obesity and type-2 diabetes. Consequential to the defects in FTase/GGTase signaling, we demonstrated that inhibition of prenyltransferases results in accumulation of unprenylated G-proteins [e.g., Rap1] in INS-1 832/13 cells and normal rat islets.41 Lastly, it was observed that a caspase-resistant mutant of FTase/GGTase-α restored high glucose-induced FTase/GGTase-α degradation and loss in cell viability in INS-1 832/13 cells. Together, these data suggest that defective prenylation, due to loss in FTase/GGTase activities, could contribute to loss in metabolic cell viability and dysfunction in pancreatic β-cells. Additional investigations are needed to further validate this model.
4.7 Inhibition of prenylation results in sustained activation of unprenylated G-proteins in the islet β-cell
As discussed above, the observations from the laboratories of Khan35 and Dunford36 have suggested that inhibition of protein prenylation leads to activation of G-proteins. Compatible with above observations are our findings in the islet β-cell to indicate increased abundance of GTP-bound [active] Rac1 in β-cell models of glucolipotoxicity, ER stress and diabetes.42 Furthermore, hyperactivation of Rac1 was demonstrable in human islets exposed to glucotoxic conditions and in islets derived from the ZDF rat.43 These findings have led us to propose that Rac1 plays both positive and negative modulatory roles in the regulation of β-cell function in the sense that while it is critical for GSIS, Rac1 also exerts damaging roles under pathological conditions by inducing Nox2 activity to create excessive oxidative stress, mitochondrial damage and cell demise.30,42 In support of our findings implicating sustained activation of Rac1 in islet β-cell in in vitro and in vivo models of glucolipotoxicity and T2DM are recent observations by Zhou and associates demonstrating significant increase in the expression and activation of Rac1 in pancreatic tissue from ob/ob mice, a model for obesity; these findings were replicated in insulin-secreting NIT-1β cells under hyperglycemic and hyperlipidemic conditions in vitro.44 Based on the above discussion, it can be concluded that post-translational prenylation plays a regulatory role in GSIS in normal and healthy β-cell. However, metabolic stress conditions suppress protein prenyltransferase activity, which may, in part, be due to degradation of FTase/GGTase-α. Recent evidence in pancreatic β-cells suggests that inactivation of protein prenylation leads to sustained activation of Rho GTPases [e.g., Rac1] leading to activation of various stress kinases [p38MAPK, p53, ATM Kinase, JNK1/2] leading to mitochondrial dysfunction and demise of the islet β-cell. These observations are reviewed below.
5. Sustained activation of Rac1 results in stress kinase activation in the islet β-cell under conditions of metabolic stress and diabetes
We30 and others29 have recently reviewed the evidence suggesting that Nox2 contributes to the generation of ROS under glucotoxic conditions, resulting in mitochondrial dysregulation and loss of islet β-cell function. As a logical extension to these studies, Sidarala and associates investigated potential functional consequences of accelerated Nox2 signaling pathway in mediating p38MAPK, a stress kinase, activation under glucotoxic conditions in normal rodent islets and INS-1 832/13 cells.45 Their findings suggested that gp91-ds-tat, a specific inhibitor of Nox2, but not its inactive analog, significantly suppressed high glucose-induced Nox2 activation, ROS generation and p38MAPK activation, thus suggesting that Nox2 activation couples with p38MAPK activation under high glucose exposure conditions. Furthermore, inhibition of Rac1 using EHT 1864, NSC23766 and Ehop-016, markedly reduced high glucose-induced p38MAPK activation in isolated β-cells. In addition, 2-Bromopalmitate, a known inhibitor of protein [Rac1] palmitoylation, significantly reduced HG-induced p38MAPK phosphorylation. Based on these observations, we concluded that Rac1-Nox2 signaling module plays novel regulatory roles promoting p38MAPK activation and loss of GSIS under conditions of glucotoxicity.45
In a more recent study, Sidarala et al examined roles of Rac1 in promoting activation of p53, a known apoptotic factor, in pancreatic β-cells exposed to glucotoxic conditions.46 Data accrued from these studies demonstrated significant stimulatory effects of high glucose on p53 activation in INS-1 832/13 cells, normal rodent and human islets. Pharmacological inhibition of Rac1 [EHT1864 or NSC23766; Figure 1] significantly suppressed HG-induced p53 activation in INS-1 832/13 cells and rat islets, suggesting novel roles for Rac1 in the activation of p53. Interestingly, high glucose-induced p53 activation was also suppressed by SB203580, a known inhibitor of p38MAPK. Together, these findings suggested that sustained activation of Rac1-p38MAPK signaling axis leads to activation of p53 leading to β-cell dysfunction under the duress of chronic hyperglycemic conditions.46
Lastly, studies from our laboratory have confirmed above findings [i.e., FTase/GGTase-α degradation, sustained activation of Rac1-Nox2 signaling module, and stress kinase activation] in islets from the ZDF rat, a model for type 2 diabetes.41–43 We noted that levels of phosphorylated p47phox, active Rac1, Nox2 activity, ROS generation, JNK 1/2 phosphorylation, and caspase-3 activity were significantly higher in the ZDF islets compared to the islets from lean rats. Furthermore, chronic exposure of INS-1 832/13 cells to glucolipotoxic conditions [as above] resulted in increased JNK1/2 phosphorylation and caspase-3 activity; such effects were largely reversed by SP600125, a selective inhibitor of JNK. Incubation of normal human islets with high glucose also increased the activation of Rac1 and Nox2. More importantly, in a manner akin to the ZDF rat islets, Rac1 expression, JNK1/2, and caspase-3 activation were also significantly increased in diabetic human islets.43
Based on the findings from above studies we conclude that glucotoxic/diabetic conditions promote aberrant activation of Rac1, which, in turn, leads to activation of Nox2-ROS-p38MAPK/p53/JNK1/2 signaling pathway in the islet β-cell leading to the onset of mitochondrial dysregulation and apoptotic demise of the islet β-cell. These findings are summarized in Figure 4.
Figure 4. A model for Rac1-mediated, Nox2-dependent activation of p38MAPK in pancreatic β-cells under glucotoxic conditions.
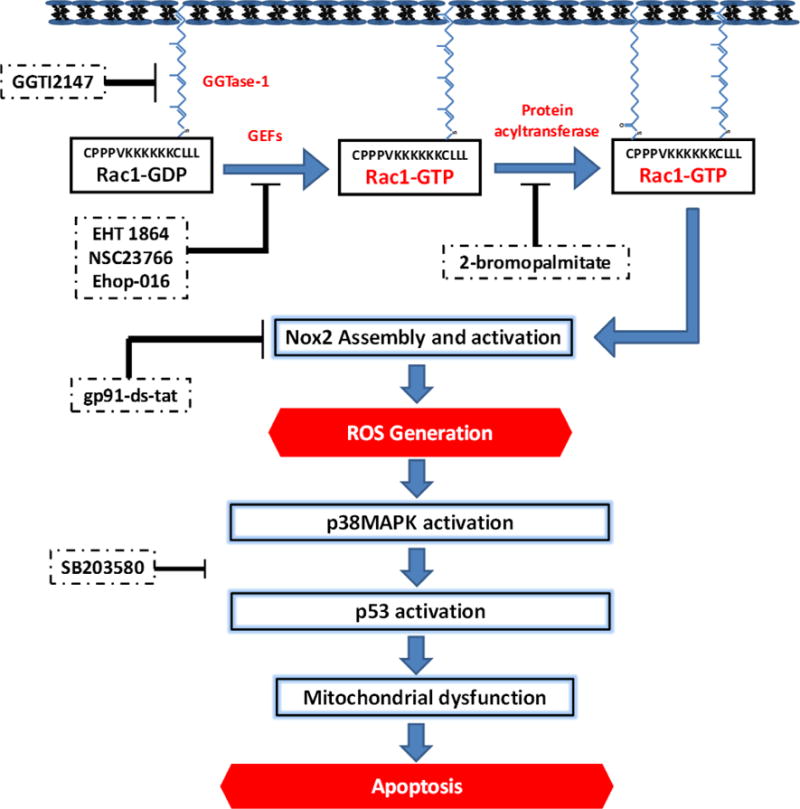
Based on the findings reviewed in this article, we propose a model to implicate Rac1 in Nox2-mediated activation of stress kinase [p38MAPK and p53] in pancreatic β-cells under the duress of glucotoxic conditions. Inhibition of Rac1 activation or function at the level of GEFs [EHT 1864, NSC23766 and Ehop-016] suppresses high glucose-induced activation of stress kinases. Furthermore, it appears that palmitoylation [sensitive to 2-brompalmitate; 2-BP], but not geranylgeranylation [inhibited by GGTI-2147] is necessary for high glucose-induced effects on stress kinase activation. Lastly, inhibition of Nox2 [with gp91-ds-tat peptide] markedly attenuated HG-induced Nox2 activation, ROS generation and p38MAPK activation. Together, our findings provide the first evidence to support our hypothesis that high glucose exposure conditions promote activation of Rac1, a key member of Nox2 holoenzyme, which, in turn, promotes the activation of Nox2 culminating in the generation of excessive ROS. An increase inROS leads to the onset of mitochondrial dysfunction as evidenced by caspase-3 activation, loss in metabolic function, including impaired GSIS. SB203580, a known inhibitor of p38MAPK, markedly attenuated high glucose-induced p53 activation, thus suggesting that p38MAPK activation is upstream to p53 activation. This figure is reproduced [with permission from Elsevier].45 Note that data on Rac1-mediated activation of p53 in pancreatic β-cells under the duress of glucotoxicity, as indicated in the figure, are published recently.46
6. Diabetes-induced alterations in prenylation of Rab G-proteins and associated signaling pathways in the islet β-cell
Recent studies by Jiang et al further confirmed novel roles for prenylation of Rab G-proteins in islet function, including GSIS.47 Using a variety of experimental approaches they demonstrated essential roles for geranyl geranyl pyrophosphate synthase [GGPPS], the enzyme that converts isopentenyl pyrophosphate to GGPP, in islet β-cell function during the onset of T2DM. They reported a significant increase in the catalytic activation of GGPPS in islets derived from db/db mice during the initial compensatory period followed by a significant reduction during the onset of insulin secretory abnormality. Furthermore, conditional deletion of GGPPS in the islet β-cell resulted in depletion of intracellular GGPP, geranylgeranylation and membrane targeting of Rab27A. They also noted a marked reduction in the number of insulin-containing secretory granules beneath the plasma membrane suggesting significant defects in granule docking in GGPPS-null mice culminating in inhibition of GSIS. Over-expression of GGPPS or provision of exogenous GGPP significantly restored GSIS in islets from GGPPS-null mice, affirming the postulation that defects in GGPPS pathway (generation of GGPP, geranylgeranylation of Rab27A and granule docking) cause islet β-cell dysfunction in diabetes.47 Indeed, these observations will form the basis for immediate investigations in the field, which might fill the knowledge gaps in our current understanding of regulatory roles of G proteins in islet biology. More importantly, these findings identify three important avenues for future investigations in the field.48 First, it is likely that decreased levels of GGPP and defective catalytic function of GGPPS under diabetic conditions could result in impaired prenylation of other key signaling proteins mediated by GGTase-I [Cdc42, Rac1, Rho and Rap1] since GGPP is necessary for GGTase I-mediated prenylation of these proteins. Thus, functional inactivation of these proteins consequential to defective GGPPS could contribute to islet dysregulation. Second, geranylgeranylation of other Rab G proteins, which are substrates for GGTase-II could also be affected resulting in the dysregulation of the islet β-cell in diabetes. Third, while these investigations have focused on GGPPS, it would be interesting to critically evaluate the kinetics of FPPS activity and the intracellular concentrations of FPP in normal and diabetic islet β-cells to determine contributory roles of these signaling proteins in islet function in health and diabetes. It should be also kept in mind that decreased GGPPS activity, as shown in the diabetic islet β-cell, could result in increased levels [accumulation] of FPP, which, in turn could lead to farnesylation of proteins.48 It is notable that the studies of Jiang and colleagues have identified no significant defects in farnesylation and membrane association of H-Ras, a farnesylated protein, in GGPPS-depleted islet β-cells.47 In conclusion, data accrued from the investigations of Jiang et al provide compelling evidence to implicate defects in roles for G-protein geranylgeranylation in islet function, including translocation and docking of insulin-laden secretory granules to the plasma membrane for their fusion and secretion of insulin. They further affirm roles of geranylgeranylated Rab27A in these cellular events. More importantly, these findings also suggest significant defects in these signaling mechanisms in islets derived from an animal model of T2DM. Future investigations along these lines should provide valuable insights in not only identifying novel targets that regulate islet function under normal physiological conditions, but also potential abnormalities in the functions of those proteins that could lead to dysregulation of insulin secretion and islet function under the duress of metabolic stress and diabetes.
7. Roles of trimeric G-proteins and regulators of G-protein signaling [RGS] in islet function in health and diabetes
As stated above, trimeric G-proteins regulate cell function via coupling extracellular ligands to intracellular effector proteins through GPCRs. Studies from multiple laboratories identified these G-proteins in the islet β-cell including Gi, Go, Gq, Gz6,8,10,49,50. It is becoming increasingly clear that termination of GPCR signaling is facilitated by regulators of G-protein signaling [RGS]. In contrast to regulatory roles of small G-proteins and their regulatory factors in islet β-cell function, including insulin secretion, relatively less is known with regard to trimeric G-protein-mediated regulation of islet function in health and diabetes. This may, in part, be due to complexities of the system including a large number of α, β and γ subunits and specific mechanisms/signals that are required for the assembly of the trimer. In addition, each of these subunits is regulated by post-translational modification steps for optimal function. For example, the α-subunits undergo ADP-ribosylation, myristoylation, palmitoylation.7 Gβ subunits have been shown to be regulated via histidine phosphorylation50 and Gγ subunits are modified by farnesylation/geranylgeranylation.7,17 These modification steps not only control the association and stability of the αβγ trimer, but also their catalytic function. Besides these signaling events, several regulatory factors/proteins for the GPCR-G-protein coupling, activation [by GEFs] and termination and function [by RGS] have been identified in the islet β-cells. Original contributions from the author’s laboratory provided evidence for acute regulation of histidine phosphorylation of Gβ subunits and farnesylation/geranylgeranylation of Gγ subunits under conditions favorable for GSIS50. Follow-up studies have also demonstrated significant reduction in histidine kinase activities in islets derived from animal models of type-2 diabetes [ZDF rat and GK rat].50 Specific examples of trimeric G-proteins and RGSs in islet β-cells in health and in models of impaired insulin secretion are provided in Table I. Clearly, additional studies are needed to provide clarity of contributory roles of these proteins in islet function.
Table I.
Examples of roles of heterotrimeric G-proteins and regulators of G-protein signaling [RGS] in islet β-cells in health and diabetes
| Observations |
|---|
| Stimulatory glucose or a membrane depolarizing concentration of KCl transiently increased the carboxylmethylation [CML] of the Gγ-subunits of heterotrimeric G-proteins in HIT-T15 cells, rat islets and human islets. Mastoparan, a known activator of G-proteins also stimulated the CML of Gγ subunits. Pertussis toxin [Ptx] markedly attenuated the stimulatory effects of glucose, KCl or mastoparan.52 |
| Fluoride, a known activator of trimeric G-proteins, induced apoptosis in RINm5F cells and normal rat islets. Potential involvement of G-proteins was confirmed by culture of β-cells with Ptx prior to exposure to fluoride.53 |
| The β-subunit of trimeric G-proteins underwent phosphorylaton at a histidine residue in the membrane and secretory granule fraction in pancreatic β-cells. Incubation of phosphorylated β-subunit with Gα.GDP accelerated the dephosphorylation of the β-subunit, accompanied by the formation of GαGTP. This is the first evidence for a non-receptor-mediated activation of trimeric G-proteins by glucose in the pancreatic islet.54 |
| Overexpression of G11α and PLCβ1 or PLCβ3 did not affect glucose- or carbachol-stimulated insulin secretion in INS-1 and βG40/110 cells. These findings suggested no correlation between inositol triphosphate accumulation and insulin secretion.55 |
| Insulin secretion elicited by glucose, KCl and mitochondrial fuels was significantly blunted in islets from the GK rat. Mastoparan, a global activator of trimeric G-proteins circumvented such a secretory defect. These findings suggested a defect late in stimulus-secretion coupling is responsible for insulin secretory defects in this animal model.56 |
| Glucotoxic conditions reduced the ADP ribosylation of Gαs and Gαolf subunits in the membrane fraction in HIT-T15 cells and [fed or fasted] rodent islets. It was concluded that defects in cAMP-dependent insulin secretion under the duress of glucotoxicity may, in part, be due to reduced ADP ribosylation of specific Gα subunits.57 |
| Expression levels of Gαs and Gαolf, adenylyl cyclases 1 and 3 are significantly higher in GK rat islets compared to their control counterparts.58 |
| Gαz-null mice exhibited increased glucose clearance following intraperitoneal and oral glucose challenge as compared with WT controls. Islets isolated from these mice also exhibited increased GSIS compared to those of WT mice. It was concluded that Gαz is negative regulator of insulin secretion.59 |
| siRNA-Regulator of G-protein Signaling4 [RGS4] expression in MIN6 cells resulted in significantly higher M3 muscarinic receptor-mediated GSIS and calcium release. These findings were replicated in islets derived from RGS4-deficient mice. Based on these and additional supporting evidence, it was concluded that RGS4 plays a negative modulatory role in in vitro and in vivo models of insulin secretion.60 |
| Whole pancreas or β-cell-specific Gαs deficiency leads to early-onset insulin-deficient diabetes with a severe defect in β-cell proliferation. These findings provide compelling evidence in support of critical roles of Gαs signaling in β-cell growth and function.61 |
| Gαo2 deficient mice, but not Gαo1 or Gαi deficient mice exhibited higher glucose clearance more efficiently than WT mice via increased GSIS.62 |
| Gαz-deficient mice are resistant to developing glucose intolerance following a high fat diet. These mice also exhibited increased β-cell proliferation and β-cell mass.63 |
| Regulator of G-protein Signaling16 [RGS16] is abundantly expressed in pancreatic β-cells. Overexpression of RGS16 augmented GSIS in mouse and human islets suggesting a critical regulatory role for this protein in physiological insulin secretion. siRNA-RGS16 inhibited GSIS. Based on additional experimental evidence it was concluded that RGS16 plays a positive modulatory role of β-cell function including insulin secretion and proliferation by suppressing inhibitory signals elicited by somatostatin derived from the δ-cell of the islet.64 |
8. Conclusions and future directions
Based on the published evidence from multiple laboratories it can be concluded that small G-proteins play significant roles in normal islet function including cytoskeletal remodeling, vesicular transport and fusion, insulin secretion and β-cell proliferation. Several regulatory molecules for these G-proteins [GEFs and GDIs] have been identified in the β-cells. Post-translational prenylation of small G-proteins [Ras, Rho, Rac1, Cdc42, Rap1 and Rabs] is critical for GSIS to occur. It should be noted that tonic increase in Nox2-mediated generation of ROS appears to favor cytoskeletal remodeling and activation of ERK1/2 signaling modules while suppressing stress kinases such as JNK1/2 [Figure 5]51. Interestingly, however, metabolic stress condition [glucotoxicity, lipotoxicity and ER stress] promotes sustained activation of Rac1, which, in turn, accelerates the Nox2-ROS-stress kinase signaling pathway leading to mitochondrial dysfunction, loss in GSIS and apoptotic demise of the islet β-cell [Figure 5].
Figure 5. Proposed model for ROS-dependent activation of MAPKinases.
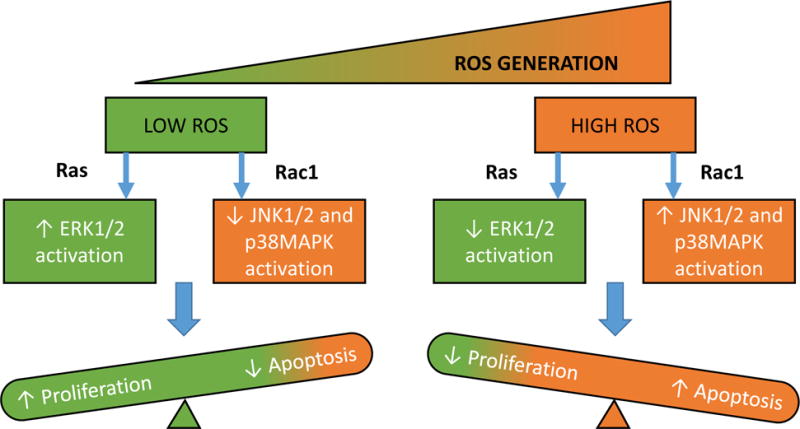
We propose that a tonic increase in Nox2-mediated ROS generation favors cytoskeletal remodeling, insulin secretion and cell proliferation events. Low-levels of ROS activate proliferative ERK signaling under the regulatory control of Ras. In contrast, sustained activation of Nox2 results in increased levels of ROS, which, in turn, promote Rac1-induced activation of apoptotic JNK1/2, and inhibit ERK1/2 signaling pathways. Additional studies are needed to further validate this model. [Reproduced with permission from Bentham-Science].51
Additional studies are needed to further validate this model. Key findings from the above studies in clonal β-cells, rodent islets and human islets are replicated in diabetic islets from animal models and humans. Data accrued in Pharmacological investigations involving Rac1 inhibitors identify Tiam1 [NSC23766] and Vav2 [Ehop-016] as GEFs involved in sustained activation of Rac1. It is important to note that other pathological conditions including lipotoxicity [ceramide-mediated?] or chronic inflammation [proinflammatory cytokines, such as IL-1β, TNFα and IFNγ] could mediate their cytotoxic effects via activation of Tiam1/Vav2-Rac1-Nox2 pathway, thus suggesting that this signaling axis might be “druggable” for prevention or halting metabolic defects induced by various pathological conditions.
While evidence reviewed above provides support to the above formulation, some aspects remain speculative and needed to be confirmed experimentally. They include, but not limited to the following. First, evidence is needed to verify potential cross-talk and/or inter-dependence, if any, between Tiam1 and Vav2 in mediating their effects on Rac1 activation. Second, potential regulatory mechanisms upstream to activation of these two GEFs need to be determined. Third, it is critical to determine the subcellular compartmentalization of these two GEFs as well as Rac1 under acute stimulatory conditions leading to GSIS and chronic exposure conditions [mistargeting] culminating in cellular dysfunction and demise. It should be kept in mind that, Rap1, which represents one of the components of the membranous core of Nox2, could contribute significantly in the subcellular-localization [e.g., plasma membrane] of Tiam1 and Vav2 in the steps leading to activation of Rac1 and optimal assembly of the Nox2 complex. In this context, findings from many laboratories [Table II] have utilized NSC23766 to validate novel regulatory roles for Tiam1-Rac1 signaling pathway in NoX2-mediated generation of ROS and induction of oxidative stress culminating in cell dysfunction and pathology.
Table II.
Regulation of cell dysfunction by Tiam1-Rac1 signaling axis under conditions of metabolic stress and diabetes
| Observations |
|---|
| Streptozotocin [Stz]-induced diabetes induced Rac1-Nox2 signaling pathway resulting in intracellular accumulation of ROS and apoptosis of cardiomyocytes. These findings are replicated in myocytes cultured in the presence of high glucose. NSC23766 significantly attenuated Nox2 activity and cell apoptosis and slightly improved loss in cardiomyocytes under these conditions.65 |
| Exposure of INS-1 832/13 cells to lipotoxic conditions or to a cell-permeable ceramide led to increased levels of superoxides and lipid peroxides resulting in loss of mitochondrial membrane potential. Coprovision of NSC23766 significantly attenuated palmitate or ceramide-induced Rac1 activation, ROS generation and mitochondrial defects.66 |
| Rac1 is significantly activated in diabetic hearts following ischemia-reperfusion injury compared to the non-diabetic hearts. Perfusion with NSC23766 prevented diabetes-induced effects. Inhibition of Rac1 also prevented activation of Nox2 and associated ROS production and protein carbonyl accumulation.67 |
| Incubation of INS-1 832/13 cells with a mixture of pro-inflammatory cytokines [IL-1β+IFNγ+TNFα] resulted in a significant activation of Rac1 and Nox2 leading to a loss in mitochondrial membrane potential. siRNA-p47phox or apocynin, a known inhibitor of Nox2, markedly suppressed cytokine-induced effects. Coprovision of NSC23766 alleviated cytokine-induced Rac1 activation and mitochondrial defects.68 |
| Rac1-Nox2 signaling pathway is accelerated in retina derived from Stz-diabetic rats and mice leading to increased ROS generation and mitochondrial damage. Similar increases in Rac1 activation and p47phox expression are noted in retinal microvasculature from human donors with established diabetic retinopathy. Treatment of diabetic mice with NSC23766 prevented retinal Rac1 activation and ROS generation.69 |
| NSC23766 prevented post-ischemic neuronal apoptosis in Stz-induced diabetic rats. NSC23766 also prevented cerebral ischemia-induced mitochondrial p53 translocation and expression of p53-upregulated modulator of apoptosis.70 |
| Exendin-4 significantly alleviated angiotensin II-induced H2O2 generation, expression of p53, p21 and associated senescence in VSMC. NSC23766 abrogated the suppressive effects of Exendin-4 on angiotensin-II-induced H2O2 generation and premature senescence of VSMC.71 |
| Glucotoxic conditions promoted activation of Rac-Nox2 signaling pathway leading to activation of downstream signaling events including activation of p38MAPK in normal rat islets and INS-1 832/13 cells. NSC23766 markedly suppressed high glucose-induced p38MAPK activation.45 |
| Glucotoxic conditions promoted activation of p38MAPK in retinal endothelial cells. Consistent with these observations, a significant increase in p38MAPK was seen in retina from Stz-induced mice. NSC23766 inhibited high glucose-induced p38MAPK in cultured retinal endothelial cells.72 |
| Administration of NSC23766 significantly prevented the development of spontaneous diabetes in the NOD mouse model. Furthermore, NSC23766 suppressed expression of Rac1 and CHOP [a marker for endoplasmic reticulum stress] in islets from the diabetic NOD mice.73 |
| Glucotoxic conditions induced activation of Rac1-Nox2 signaling axis resulting in increased expression of CD36 [a fatty acid transporter] culminating in apoptosis in INS-1 cells and human β-cells [1.1B4]. NSC23766 significantly attenuated high glucose-induced Rac1-Nox2 activation, CD36 expression and mitochondrial dysfunction in these cells.74 |
| NSC23766 markedly suppressed high glucose-induced p53 activation in normal rat islets and INS-1 832/13 cells.46 |
It is noteworthy that recent investigations by Elumalai and associates provided additional insights into contributory roles of Tiam1-Rac1-Nox2 signaling axis in pancreatic islet β-cell dysfunction under the duress of glucotoxicity74. Compatible with earlier observations from our laboratory7,13,30,42,45,46, they reported protection against high glucose-induced dysfunction of INS-1 and human 1.1b4 cells following inhibition of Tiam1-mediated Rac1 activation. More importantly, they demonstrated inhibition of high glucose-induced expression of Cluster Differentiating 36 [CD36], a fatty acid transporter, by NSC23766, a known inhibitor of Tiam1-Rac1 module. These data suggest that CD36 expression in the membrane fraction [and functional activation?] is under the control of Tiam1-Rac1-Nox2 signaling pathway. Based on these observations it was concluded that Rac1/Nox2-dependent CD36 expression contributes to β-cell dysfunction and demise under the duress of hyperglycemia.74 Interestingly, recent studies by Gharib and associates reported significant reduction in obesity-induced oxidative stress in the heart in a CD36 deficient animal model. Specifically, their findings suggested that CD36 deficiency reduced NADPH oxidase activity and associated ROS generation. Furthermore, CD36 deficiency suppressed palmitate-induced normalized NADPH oxidase activity and ROS generation. While studies of Elumalai74 and Gharib75 provide evidence for regulatory roles of CD36 in high glucose- [and palmitate] induced metabolic dysfunction of the cell, their findings place CD36 either upstream75 or downstream74 to NADPH oxidase signaling step. Reasons for such differences remain unclear at this time. Additional studies are needed for further clarity and potential utility of CD36 as a therapeutic target to prevent islet β-cell dysfunction in diabetes.
Earlier studies by Vicent and associates76 have presented evidence to indicate significantly high expression of geranylgeranyl pyrophosphate synthase [GGPPS] in various tissues from the ob/ob mouse. Using Northern blot analysis, they detected three mRNAs [4.3, 3.2, and 1.7 kb] for GGPPS. Interestingly, GGPPS mRNA expression was increased [5 to 20-fold] in skeletal muscle, liver, and fat of ob/ob mice. Compatible with these data, Western blot analysis indicated a 2-fold increase in the expression of GGPS protein in muscle and fat, but not in liver. More importantly, GGPPS expression increased significantly [20-fold] during differentiation of 3T3-L1 fibroblasts into adipocytes. Together, these findings suggested that the expression of GGPPS is regulated in multiple tissues in obesity and is induced during adipocyte differentiation.76 Potential alterations in the regulation of isoprenoid biosynthesis could, in turn, determine the ability of cells to respond to stimulation by specific ligands or agonists [hormones], which require precisely defined GPCR-effector signaling networks, including activation of trimeric G-proteins [see below].
Lastly, significant knowledge gaps exist on potential defects in prenylation [farnesylation/geranyl geranylation] prenylation of Gγ subunits under conditions of metabolic stress/diabetic conditions. At least 12 Gγ genes are encoded in the human genome, and all are either farnesylated or geranylgeranylated. As reviewed by Hildebrandt77 prenylation of Gγ is critical for optimal cell function, including membrane attachment of prenylated proteins, intracellular membrane trafficking of prenylated proteins, reversible trafficking between plasma membrane and endomembranes and protein-protein interactions. Therefore, it is conceivable that defects in FTase/GGTase activities that we reported in the islet β-cell under the duress of metabolic stress could result in defects in Gγ prenylation status in the islet β-cell. Potential defects in this signaling pathway could lead to defects in the activation-deactivation cycle of trimeric G-proteins leading to alterations in coupling of GPCR signaling pathways under these pathological conditions. This remains to be verified in in vitro and in vivo model systems of metabolic stress and diabetes.
Callouts.
Inhibition of protein prenylation in β-cells results in selective accumulation of unprenylated G-proteins in the soluble compartment, possibly interfering with the interaction of these proteins with their respective effector proteins, which may be required for GSIS.
Glucose acutely stimulates FTase/GGTases and prenylation of key G-proteins culminating in their trafficking to appropriate cellular compartments for optimal regulation of their respective effector proteins to promote conditions conducive for secretory granule fusion with the plasma membrane leading to insulin exocytosis.
Metabolic stress promotes inactivation of FTase/GGTase and aberrant activation of Rac1, which, in turn, leads to accelerated Nox2-ROS-p38MAPK/p53/JNK1/2 signaling pathway in the islet β-cell leading to the onset of mitochondrial dysregulation and apoptotic demise of the islet β-cell.
Significant knowledge gaps exist on potential defects in prenylation of Gγ subunits under conditions of metabolic stress/diabetic conditions. In a maker akin to small G-proteins, potential alterations in this signaling pathway could lead to defects in the assembly of the αβγ trimer and alterations in coupling of ligand/GPCR/G-protein/effector signaling pathways under these pathological conditions.
Acknowledgments
The author thanks his former and current laboratory associates and collaborators for their contributions in the studies reviewed herein. This research was supported in part by a MERIT Review Award from the US Department of Veterans Affairs [1BX000469], the National Institutes of Health [DK-74921 and EY-022230], and the Juvenile Diabetes Research Foundation [52012257]. AK also received a Senior Research Career Scientist Award [13S-RCS-006] from the Department of Veterans Affairs. The author thanks the School of Biomedical Sciences, Curtin University, Perth, Western Australia for a Visiting Professorship. The author acknowledges work of other investigators in the field that has not been cited in this review due to space limitations.
Abbreviations used
- Arf6
ADP ribosylation factor 6
- ARNO
ARF nucleotide-binding site opener
- CD36
cluster differentiating 36
- Cdc42
cell division control protein 42
- CML
carboxylmethylation
- ER-stress
endoplasmic reticulum stress
- ERK1/2
Extracellular signal-regulated kinases 1/2
- FPP
farnesylpyrophosphate
- FTase
farnesyltransferase
- FTI
farnesyltransferase inhibitor
- FTase/GGTase-α
α-subunit of farnesyl- and geranylgeranyltransferase
- GAPs
GTPase-activating proteins
- GEFs
guanine nucleotide exchange factor
- GGTase
geranylgeranyltransferase
- GGTI
geranylgeranyltransferase inhibitor
- GK rat
Goto-Kakizaki rat
- G-proteins or GTPases
GTP-binding proteins
- GGPP
geranylgeranylpyrophosphate
- GGPPS
geranylgeranylpyrophosphate synthase
- GPCRs
G-protein coupled receptors
- GSIS
glucose-stimulated insulin secretion
- ICMT
isoprenylcysteine methyl transferase
- IGF-1
insulin-like growth factor-1
- JNK1/2
Jun NH2-terminal kinases 1/2
- MVA
mevalonic acid
- Nox2
phagocyte-like NADPH oxidase
- Ptx
Pertussis toxin
- Rac1
Ras-related C3 botulinum toxin substrate-1
- Rce1
Ras converting enzyme 1
- REP-1
Rab escort protein-1
- RGGT
Rab geranylgeranyl transferase
- RGS
Regulators of G-protein signaling
- ROS
reactive oxygen species
- Stz
streptozotocin
- VSMC
vascular smooth muscle cells
- ZDF rat
Zucker diabetic fatty rat
Footnotes
Conflict of interests: The author declares no conflict of interest
References
- 1.Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signaling. Diabetologia. 2010;53:1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Berggren PO, Leibiger IB. Novel aspects on signal transduction in the pancreatic beta cell. Nutr Metab Cardiovasc Dis. 2006;16(suppl 1):S7–S10. doi: 10.1016/j.numecd.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Komatsu M, Takei M, Ishii H, Sato Y. Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Investig. 2013;4:511–516. doi: 10.1111/jdi.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metz SA, Rabaglia ME, Pintar TJ. Selective inhibitors of GTP synthesis impede exocytotic insulin release from intact rat islets. J Biol Chem. 1992;267:12517–12527. [PubMed] [Google Scholar]
- 6.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis- roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:896–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev. 2010;31:52–78. doi: 10.1210/er.2009-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robertson RP, Seaquist ER, Walseth TF. G proteins and modulation of insulin secretion. Diabetes. 1991;40:1–6. doi: 10.2337/diab.40.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Regazzi R, Ravazzola M, Iezzi M, Lang J, Zahraoui A, Andereggen E, Morel P, Takai Y, Wollheim CB. Expression, localization and functional role of small GTPases of the Rab3 family in insulin-secreting cells. J Cell Sci. 1996;109:2265–2273. doi: 10.1242/jcs.109.9.2265. [DOI] [PubMed] [Google Scholar]
- 10.Sharp GW. Mechanisms of inhibition of insulin release. Am J Physiol. 1996;271:C1781–1799. doi: 10.1152/ajpcell.1996.271.6.C1781. [DOI] [PubMed] [Google Scholar]
- 11.Kowluru A, Li G, Rabaglia ME, Segu VB, Hoffmann F, Aktories K, Metz SA. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta-cells. Biochem Pharmacol. 1997;54:1097–1108. doi: 10.1016/s0006-2952(97)00314-6. [DOI] [PubMed] [Google Scholar]
- 12.Kelly P, Bailey CL, Fueger PT, Newgard CB, Casey PJ, Kimple ME. Rap1 promotes multiple pancreatic islet functions and signals through mammalian target of rapamycin complex 1 to enhance proliferation. J Biol Chem. 2010;285:15777–15785. doi: 10.1074/jbc.M109.069112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kowluru A. Tiam1/Vav2-Rac1 axis: A tug-of-war between islet function and dusfunction. Biochem Pharmacol. 2017 doi: 10.1016/j.bcp.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kepner EM, Yoder SM, Oh E, Kalwat MA, Wang Z, Quilliam LA, Thurmond DC. Cool-1/βPIX functions as a guanine nucleotide exchange factor in the cycling of Cdc42 to regulate insulin secretion. Am J Physiol Endocrinol Metab. 2011;301:E1072–1080. doi: 10.1152/ajpendo.00312.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayaram B, Syed I, Kyathanahalli CN, Rhodes CJ, Kowluru A. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem Pharmacol. 2011;81:1016–1027. doi: 10.1016/j.bcp.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leech CA, Holz GG, Chepurny O, Habener JF. Expression of cAMP-regulated guanine nucleotide exchange factors in pancreatic beta-cells. Biochem Biophys Res Commun. 2000;278:44–47. doi: 10.1006/bbrc.2000.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casey PJ, Seabra MC. Protein Prenyltransferases. J Biol Chem. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 18.McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63:255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kowluru A, Kowluru RA. Protein prenylation in islet beta-cell function in health and diabetes: Putting the pieces of the puzzle together. Biochem Pharmacol. 2015;98:363–370. doi: 10.1016/j.bcp.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metz SA, Rabaglia ME, Stock JB, Kowluru A. Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem J. 1993;295:31–40. doi: 10.1042/bj2950031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G, Regazzi R, Roche E, Wollheim CB. Blockade of mevalonate production by lovastatin attenuates bombesin and vasopressin potentiation of nutrient-induced insulin secretion in HIT-T15 cells. Probable involvement of small GTP-binding proteins. Biochem J. 1993;289(Pt 2):379–385. doi: 10.1042/bj2890379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veluthakal R, Kaur H, Goalstone M, Kowluru A. Dominant-negative alpha-subunit of farnesyl- and geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes. 2007;56:204–210. doi: 10.2337/db06-0668. [DOI] [PubMed] [Google Scholar]
- 23.Iezzi M, Escher G, Meda P, Charollais A, Baldini G, Darchen F, Wollheim CB, Regazzi R. Subcellular distribution and function of Rab3A, B, C and D isoforms in insulin-secreting cells. Mol Endocrinol. 1999;13:202–212. doi: 10.1210/mend.13.2.0228. [DOI] [PubMed] [Google Scholar]
- 24.Aizawa T, Komatsu M. Rab27a: a new face in beta cell metabolism-secretion coupling. J Clin Invest. 2005;115:227–230. doi: 10.1172/JCI24269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arora DK, Syed I, Machhadieh B, Mckenna CE, Kowluru A. Rab-geranylgeranyl transferase regulates glucose-stimulated insulin secretion from pancreatic β-cells. Islets. 2012;4:354–358. doi: 10.4161/isl.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorenz MA, El Azzouny MA, Kennedy RT, Burant CF. Metabolome response to glucose in the beta-cell line INS-1 832/13. J Biol Chem. 2013;288:10923–10935. doi: 10.1074/jbc.M112.414961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goalstone M, Kamath V, Kowluru A. Glucose activates prenyltransferases in pancreatic islet beta-cells. Biochem Biophys Res Commun. 2010;391:895–898. doi: 10.1016/j.bbrc.2009.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goalstone M, Carel K, Leitner JW, Draznin B. Insulin stimulates the phosphorylation and activity of farnesyltransferase via the Ras-mitogen-activated protein kinase pathway. Endocrinology. 1997;138:5119–5124. doi: 10.1210/endo.138.12.5621. [DOI] [PubMed] [Google Scholar]
- 29.Newsholme P, Morgan D, Rebelato E, Oliveira-Emillo HC, Procopio J, Curi R, et al. Insights into the critical role of NADPH oxidases(s) in the normal and dysregulated pancreatic beta cell. Diabetologia. 2009;52:2489–2498. doi: 10.1007/s00125-009-1536-z. [DOI] [PubMed] [Google Scholar]
- 30.Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucotoxicity and diabetes. Biochem Pharmacol. 2014;88:275–283. doi: 10.1016/j.bcp.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyathanahalli CN, Kowluru A. A farnesylated G-protein suppresses Akt phosphorylation in INS 832/13 cells and normal rat islets: regulation by pertussis toxin and PGE2. Biochem Pharmacol. 2011;81:1237–1247. doi: 10.1016/j.bcp.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic beta-cells. Diabetes. 2010;59:967–977. doi: 10.2337/db09-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novelli G, D’Apice MR. Protein farnesylation and disease. J Inherit Metab Dis. 2012;35:917–926. doi: 10.1007/s10545-011-9445-y. [DOI] [PubMed] [Google Scholar]
- 34.Resh MD. Targeting protein lipidation in disease. Trends Mol Med. 2012;18:206–214. doi: 10.1016/j.molmed.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan OM, Ibrahim MX, Jonsson IM, Karlsson C, Liu M, Sjogren AK, et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J Clin Invest. 2011;121:628–639. doi: 10.1172/JCI43758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunford JE, Rogers MJ, Ebetino FH, Phipps RJ, Coxon FP. Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J Bone Miner Res. 2006;21:684–694. doi: 10.1359/jbmr.060118. [DOI] [PubMed] [Google Scholar]
- 37.Reddy JM, Samuel FG, McConnell JA, Reddy CP, Beck BW, Hynds DL. Non-prenylatable, cytosolic Rac1 alters neurite outgrowth while retaining the ability to be activated. Cell Signal. 2015;27:630–637. doi: 10.1016/j.cellsig.2014.11.033. [DOI] [PubMed] [Google Scholar]
- 38.Kim KW, Chung HH, Chung CW, Kim IK, Miura M, Wang S, et al. Inactivation of farnesyltransferase and geranylgeranyltransferase I by caspase-3: cleavage of the common alpha subunit during apoptosis. Oncogene. 2001;20:358–366. doi: 10.1038/sj.onc.1204099. [DOI] [PubMed] [Google Scholar]
- 39.Arora DK, Mohammed AM, Kowluru A. Nifedipine prevents etoposide-induced caspase-3 activation, prenyl transferase degradation and loss in cell viability in pancreatic beta-cells. Apoptosis. 2013;18:1–8. doi: 10.1007/s10495-012-0763-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G-P, Zhang X-Q, Wu T, Li L, Han J, Du CQ. Alteration of mevalonate pathway in proliferated vascular smooth muscle from diabetic mice: possible role in high-glucose-induced atherogenic process. J Diab Res. 2015:379287. doi: 10.1155/2015/379287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veluthakal R, Arora DK, Goalstone ML, Kowluru RA, Kowluru A. Metabolic stress induces caspase-3 mediated degradation and inactivation of farnesyl and geranylgeranyl transferase activities in pancreatic beta-cells. Cell Physiol Biochem. 2016;39:2110–2120. doi: 10.1159/000447907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81:965–975. doi: 10.1016/j.bcp.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, et al. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60:2843–2852. doi: 10.2337/db11-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou S, Yu D, Ning S, Zhang H, Jiang L, He L, et al. Augmented Rac1 expression and activity are associated with oxidative stress and decline of beta cell function in obesity. Cell Physiol Biochem. 2015;35:2135–2148. doi: 10.1159/000374019. [DOI] [PubMed] [Google Scholar]
- 45.Sidarala V, Veluthakal R, Syeda K, Vlaar C, Newsholme P, Kowluru A. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1) Biochem Pharmacol. 2015;95:301–310. doi: 10.1016/j.bcp.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sidarala V, Kowluru A. Exposure to chronic hyperglycemic conditions results in Ras-related C3 botulinum toxin substrate 1 (Rac1)-mediated activation of p53 and ATM kinase in pancreatic β-cells. Apoptosis. 2017 doi: 10.1007/s10495-017-1354-6. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang S, Shen D, Jia WJ, Han X, Shen N, Tao W, Gao X, Xue B, Li CJ. GGPPS-mediated Rab27A geranylgeranylation regulates beta-cell dysfunction during type 2 diabetes development by affecting insulin granule docked pool formation. J Pathol. 2016;238:109–119. doi: 10.1002/path.4652. [DOI] [PubMed] [Google Scholar]
- 48.Kowluru A. A lack of ‘glue’ misplaces Rab27A to cause islet dysfunction in diabetes. J Pathol. 2016;238:375–377. doi: 10.1002/path.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lang J, Nishimoto I, Okamoto T, Regazzi R, Kiraly C, Weller U, Wollheim CB. Direct control of exocytosis by receptor-mediated activation of the heterotrimeric GTPases Gi and G(o) or by the expression of their active G alpha subunits. EMBO J. 1995;14:3635–3644. doi: 10.1002/j.1460-2075.1995.tb00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kowluru A. Emerging roles for protein histidine phosphorylation in cellular signal transduction: lessons from the islet beta-cell. J Cell Mol Med. 2008;12:1885–1908. doi: 10.1111/j.1582-4934.2008.00330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sidarala V, Kowluru A. The regulatory roles of mitogen-activated protein kinase (MAPK) pathways in health and diabetes: Lessons learned from the pancreatic beta cell. Recent Pat Endocri Metab Immune Drug Discov. 2016 doi: 10.2174/1872214810666161020154905. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kowluru A, Li G, Metz SA. Glucose activates the carboxyl methylation of gamma subunits of trimeric GTP-binding proteins in pancreatic beta cells. Modulation in vivo by calcium, GTP and pertussis toxin. J Clin Invest. 1997;100:1596–1610. doi: 10.1172/JCI119684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loweth AC, Williams GT, Scarpello JH, Morgan NG. Hetrotrimeric G-proteins are implicated in the regulation of apoptosis in pancreatic beta-cells. Exp Cell Res. 1996;229:69–76. doi: 10.1006/excr.1996.0344. [DOI] [PubMed] [Google Scholar]
- 54.Kowluru A, Seavey SE, Rhodes CJ, Metz SA. A novel regulatory mechanism for trimeric GTP-bonding proteins in the membrane and secretory granule fractions of human and rodent beta cells. Biochem J. 1996;313(Pt 1):97–107. doi: 10.1042/bj3130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gasa R, Trinh KY, Yu K, Wilkie TM, Newgard CB. Overexpression of G11alpha and isoforms of phospholipase C in the islet beta-cells reveals a lack of correlation between inositol phosphate accumulation and insulin secretion. Diabetes. 1999;48:1035–1044. doi: 10.2337/diabetes.48.5.1035. [DOI] [PubMed] [Google Scholar]
- 56.Metz SA, Meredith M, Vadakekalam J, Rabaglia ME, Kowluru A. A defect late in stimulus-secretion coupling impairs insulin secretion in Goto-Kakizaki diabetic rats. Diabetes. 1999;48:1754–1762. doi: 10.2337/diabetes.48.9.1754. [DOI] [PubMed] [Google Scholar]
- 57.Phan NH, Boissard C, Pessah M, Regnauld K, Emami S, Gespach C, Rosselin G. Decreased ADP-ribosylation of the Gaplha(olf) and Galpha(s) subunits by high glucose in pancreatic β-cells. Biochem Biophys Res Commun. 2000;27:86–90. doi: 10.1006/bbrc.2000.2580. [DOI] [PubMed] [Google Scholar]
- 58.Portela-Gomes GM, Abdel-Halim SM. Overexpression of Gs proteins and adenylyl cyclase in normal and diabetic islets. Pancreas. 2002;25:176–181. doi: 10.1097/00006676-200208000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Kimple ME, Joseph JW, Bailey CL, Fueger PT, Hendry IA, Newgard CB, Casey PJ. Galphaz negatively regulates insulin secretion and glucose clearance. J Biol Chem. 2008;283:4560–4567. doi: 10.1074/jbc.M706481200. [DOI] [PubMed] [Google Scholar]
- 60.Ruiz de Azua I, Scarselli M, Rosemond E, Gautam D, Jou W, Gavrilova O, Ebert PJ, Levitt P, Wess J. RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:7999–8004. doi: 10.1073/pnas.1003655107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie T, Chen M, Weinstein LS. Pancreas-specific Gsalpha deficiency has divergent effects on pancreatic alpha- and beta-cell proliferation. J Endocrinol. 2010;206:261–269. doi: 10.1677/JOE-10-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, Park S, Bajpayee NS, Nagaoka Y, Boulay G, Birnbaumer L, Jiang M. Augmented glucose-induced insulin release in mice lacking G(o2), but not G(o1) or G(i) proteins. Proc Natl Acad Sci U S A. 2011;108:1693–1698. doi: 10.1073/pnas.1018903108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kimple ME, Moss JB, Brar HK, Rosa TC, Truchan NA, Pasker RL, Newgard CB, Casey PJ. Deletion of GαZ protein protects against diet-induced glucose intolerance via expansion of β-cell mass. J Biol Chem. 2012;287:20344–20355. doi: 10.1074/jbc.M112.359745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vivot K, Moulle VS, Zarrouki B, Tremblay C, Mancini AD, Maachi H, Ghislain J, Poitout V. The regulator of G-protein signaling RGS16 promotes insulin secretion and β-cell proliferation in rodent and human islets. Mol Metab. 2016;5:988–996. doi: 10.1016/j.molmet.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, Arnold JM, Peng T. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009;58:2386–2395. doi: 10.2337/db08-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Syed I, Jayaram B, Subasinghe W, Kowluru A. Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol. 2010;80:874–883. doi: 10.1016/j.bcp.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shan L, Li J, Wei M, Ma J, Wan L, Zhu W, Li Y, Zhu H, Arnold JM, Peng T. Disruption of Rac1 signaling reduces ischemia-reperfusion injury in the diabetic heart by inhibiting calpain. Free Radic Biol Med. 2010;49:1804–1814. doi: 10.1016/j.freeradbiomed.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 68.Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol. 2011;300:R12–20. doi: 10.1152/ajpregu.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kowluru RA, Kowluru A, Veluthakal R, Mohammad G, Syed I, Santos JM, Mishra M. TIAM1-RAC1 signaling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia. 2014;57:1047–1056. doi: 10.1007/s00125-014-3194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liao J, Ye Z, Huang G, Xu C, Guo Q, Wang E. Delayed treatment with NSC23766 in streptozotocin-induced diabetic rats ameliorates post-ischemic neuronal apoptosis through suppression of mitochondrial p53 translocation. Neuropharmacology. 2014;85:508–516. doi: 10.1016/j.neuropharm.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 71.Zhao L, Li AQ, Zhou TF, Zhang MQ, Qin XM. Exendin-4 alleviates angiotensin II-induced senescence in vascular smooth muscle cells by inhibiting Rac1 activation via a cAMP/PKA-dependent pathway. Am J Physiol Cell Physiol. 2014;307:C1130–1141. doi: 10.1152/ajpcell.00151.2014. [DOI] [PubMed] [Google Scholar]
- 72.Veluthakal R, Kumar B, Mohammad G, Kowluru RA. Tiam1-Rac1 axis promotes activation of p38 MAP Kinase in the development of diabetic retinopathy: Evidence for a requisite role for protein palmitoylation. Cell Physiol Biochem. 2015;36:208–220. doi: 10.1159/000374065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Veluthakal R, Sidarala V, Kowluru A. NSC23766, a known inhibitor of Tiam1-Rac1 signaling module, prevents the onset of type 1 diabetes in the NOD mouse model. Cell Physiol Biochem. 2016;39:760–767. doi: 10.1159/000445666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elumalai S, Karunakaran U, Lee IK, Moon JS, Won KC. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2016;11:126–134. doi: 10.1016/j.redox.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gharib M, Tao H, Fungwe TV, Hajri T. Cluster differentiating 36 (CD36) deficiency attenuates obesity-associated oxidative stress in the heart. PloS One. 2016;11:e0156611. doi: 10.1371/journal.pone.0155611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vicent D, Maratos-Flier E, Kahn CR. The branch point enzyme of the mevalonate pathway for protein prenylation is overexpressed in the ob/ob mouse and induced by adipogenenesis. Mol Cell Biol. 2000;20:2158–2166. doi: 10.1128/mcb.20.6.2158-2166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hildebrandt JD. Heterogeneous prenyl processing of the heterotrimeric G protein gamma subunits. In: Tamanoi F, Hrycyna CA, Bergo MO, editors. The Enzymes. Vol. 39. Academic Press; 2011. pp. 97–124. [Google Scholar]