

Abstract
Renal parenchymal injury predisposes to salt-sensitive hypertension, but how this occurs is not known. Here we tested whether renal tubular angiotensin converting enzyme (ACE), the main site of kidney ACE expression, is central to the development of salt sensitivity in this setting. Two mouse models were used: it-ACE mice in which ACE expression is selectively eliminated from renal tubular epithelial cells; and ACE 3/9 mice, a compound heterozygous mouse model that makes ACE only in renal tubular epithelium from the ACE 9 allele, and in liver hepatocytes from the ACE 3 allele. Salt sensitivity was induced using a post L-NAME salt challenge. While both wild-type and ACE 3/9 mice developed arterial hypertension following three weeks of high salt administration, it-ACE mice remained normotensive with low levels of renal angiotensin II. These mice displayed increased sodium excretion, lower sodium accumulation, and an exaggerated reduction in distal sodium transporters. Thus, in mice with renal injury induced by L-NAME pretreatment, renal tubular epithelial ACE, and not ACE expression by renal endothelium, lung, brain, or plasma, is essential for renal angiotensin II accumulation and salt-sensitive hypertension.
Keywords: angiotensin II, angiotensin-converting enzyme, hypertension, renal sodium transporters
Approximately 1 billion people worldwide have high blood pressure, a disease that is a major risk factor for stroke, atherosclerosis, heart failure, and end-stage renal disease.1,2 While the cause of hypertension is multifactorial, excessive dietary salt and a positive correlation between sodium intake and blood pressure, termed “salt sensitivity,” are key factors. Indeed, half of hypertensive patients are salt-sensitive.3 Further, besides its high prevalence, salt sensitivity remains a major health challenge due to limited diagnostic tests, lack of consensus definition, excessive dietary salt intake, and above all, a very poor understanding of its etiology.
There is mounting evidence indicating a role of the reninangiotensin system (RAS) in the etiology of salt-sensitive hypertension.4 However, the general consensus is that patients with salt-sensitive hypertension have lower blood pressure response rates to RAS blockers, in particular to angiotensin-converting enzyme (ACE) inhibitors, than patients who are not salt-sensitive.5,6 While this seems to be the case for African-Americans and the elderly, patients with diabetes and chronic kidney disease, who benefit from salt restriction, respond well to ACE inhibitors.7,8 Thus, for unknown reasons, the role of ACE in human salt sensitivity seems to be more dominant in certain populations than others.
In our view, the answer to this clinical conundrum lies in the expression of ACE within the kidneys. Specifically, our hypothesis is that the ACE in renal tubules, the site of highest ACE expression in the kidney, is an important locus for salt sensitivity in conditions where there is renal injury, such as diabetes and chronic kidney disease. This concept builds on recent advances in the understanding of the renal RAS. First, renal inflammation is associated with increased expression of ACE and other RAS components along the tubules.9–13 This occurs even in the presence of systemic RAS suppression. Second, in many cases, renal injury leads to proteinuria and an increased presence of angiotensinogen, the angiotensin II precursor, in the renal ultra-filtrate.14 Third, our previous work indicates that renal ACE plays a unique role in the regulation of sodium reabsorption by the kidneys.15
Using 2 genetic models, we present data showing in unequivocal terms that tubular ACE plays a pivotal role in the development of salt-sensitive hypertension in the context of renal inflammation. Further, our results indicate that this effect is independent and nonredundant with ACE expression in other tissues. Finally, we show that manipulating tubular ACE activity and expression can be used both to prevent the development of salt-sensitive hypertension and to treat this condition after its establishment.
RESULTS
The it-ACE mice specifically lack ACE in renal tubular epithelial cells
ACE in the kidney is made by many different cell types including podocytes, mesangial cells, endothelial cells, and in large amounts by tubular epithelium.16,17 We showed that mice lacking ACE in kidney, endothelium, brain, and several other tissues were protected against different forms of hypertension, including salt-sensitive hypertension.15,18,19 These studies raised the question of where in the kidney and where systemically the lack of ACE was important for the protection observed in these mouse models. In particular the lack of ACE by lung endothelium (the tissue associated with physiologic conversion of angiotensin I to angiotensin II) or by the brain may have had effects, as these organs are important in blood pressure control.20 To pinpoint the cell types involved, we developed a new strain of mice termed it-ACE (inducible tubular ACE). The Methods section contains a detailed description of how these mice were made. Briefly, the it-ACE mice were made transgenic for constructs that generate short hairpin RNA (shRNA) in tubular epithelial cells that specifically silence ACE expression in tubular epithelium (Figure 1). This was investigated by several means including immunohistochemistry (Figure 2a and Supplementary Figure S1A); wild-type (WT) kidney expresses abundant ACE in the renal tubular epithelial brush border, whereas an equivalent section of kidney from it-ACE mice shows almost no ACE in the brush border. In contrast to the dissimilar distribution in the tubules, renal endothelial and glomerular ACE expression was similar in mutant and WT mice (Figure 2a and Supplementary Figure S1A). Importantly, the it-ACE mice were designed to restore renal epithelial ACE after doxycycline administration (200 μg/ml in the drinking water). After 6 days of doxycycline treatment, it-ACE mice displayed levels of renal tubular ACE equivalent to those in WT mice (Figure 2a, rightmost panel). Moreover, we studied renal ACE expression using flow cytometric analysis of single-cell suspensions from the kidneys of WT and it-ACE mice with or without doxycycline administration. For this assay, cells were stained with antibodies to megalin (a marker of proximal tubular cells), CD31 (a marker of endothelial cells), and ACE. In it-ACE mice, peak ACE fluorescence intensity in megalin+ cells was reduced to 20% ± 3% of WT levels in males and to 29% ± 6% in females (P < 0.01, n = 3–4). The expression of ACE in CD31+ cells was equivalent to WT (P = NS, n = 3–4). In contrast, after doxycycline treatment for 6 days, ACE expression in megalin+ cells from it-ACE mice was essentially restored to WT levels in both male and female mice (P = NS compared with WT; Figure 2b).
Figure 1. Schematic description of it-ACE mice.
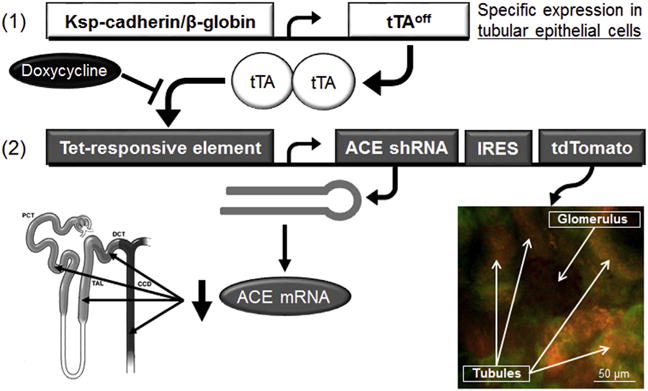
Two types of transgenic constructs were used to make these mice. Construct 1 is the kidney specific (Ksp)-cadherin/β-globin promoter driving the transcription factor tetracycline transactivator (tTAoff) only in renal tubular epithelial cells. Construct 2 possesses a modified cytomegalovirus promoter that, in the presence of tTAoff, produces the tdTomato fluorescent protein and a short hairpin RNA (shRNA) designed to silence angiotensin converting enzyme (ACE) mRNA. Renal confocal microscopy shows a glomerulus (negative) surrounded by tdTomato-positive tubules (orange). Green color is normal tubular autofluorescence. Oral administration of doxycycline (200 μg/ml in the drinking water) inactivates the tTAoff transcription factor, prevents shRNA production, and restores ACE expression. IRES, internal ribosome entry site.
Figure 2. The it-ACE mice specifically lack renal tubular epithelial angiotensin converting enzyme (ACE).
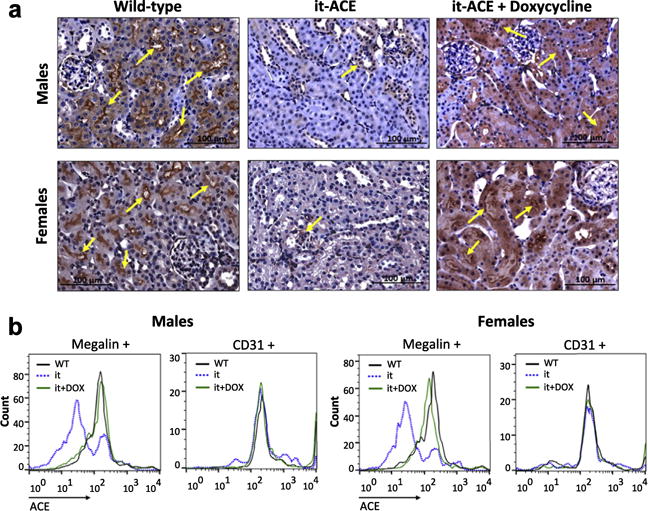
(a) Renal ACE was evaluated by immunohistochemistry. Flow cytometric analysis was performed using a single-cell suspension obtained from kidneys of wild type (WT), it-ACE (it), and it-ACE treated with 6 days of doxycycline (it+DOX). (b) Megalin and CD31 were used as markers of proximal tubular cells and endothelial cells, respectively. See also Supplementary Figure S1.
We also measured ACE activity in individual organs. Total kidney homogenates from it-ACE mice possessed 39% ± 7% and 37% ± 9% of the ACE catalytic activity found in equivalent preparations from male and female WT mice, respectively (Figure 3a, P < 0.001, n = 5–10). ACE activity measured in all other organs of it-ACE mice was indistinguishable from that in WT mice (Figure 3a). These findings were confirmed by Western blot analysis (Supplementary Figure S1B). The it-ACE mice treated with doxycycline for 6 days displayed renal ACE activity equivalent to WT mice (Figure 3b). In toto, these data demonstrate that it-ACE mice have a selective depletion of ACE in renal tubular epithelial cells that can be restored to WT levels by doxycycline treatment.
Figure 3. The it-ACE mice display lower levels of renal angiotensin converting enzyme (ACE) activity.
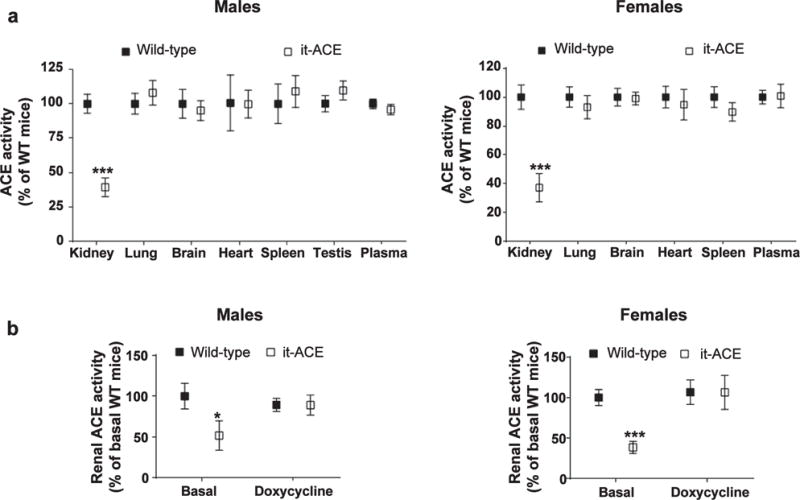
(a) ACE activity was assessed in total tissue homogenates from male and female mice in nontreated mice. (b) Renal ACE expression was restored after a 6-day treatment with doxycycline (200 μg/ml in the drinking water). Values are expressed as mean SEM; *P < 0.05 and ***P < 0.001 versus nontreated wild type (WT); n = 5–10. See also Supplementary Figure S1.
We characterized basal kidney function in it-ACE mice. Specifically, we measured the glomerular filtration rate (Supplementary Figure S2A) and renal concentrating ability after 24-hour water deprivation (Supplementary Figure S2B). No significant differences were observed between nontreated it-ACE and WT mice. Similarly, both prorenin/renin (Supplementary Figure S2C) and angiotensin II (Supplementary Figure S2D) levels were preserved in it-ACE mice. These data indicate that basal kidney function and basal levels of renal angiotensin II are normal and that they can be achieved with the reduced renal ACE as in the it-ACE mice.
Using telemetry assessment of blood pressure, we confirmed that in the absence of prior kidney injury, WT and it-ACE mice displayed no changes in blood pressure in response to a sodium load. In WT mice, the mean arterial pressure was 101 ± 3 mm Hg at baseline and 105 ± 2 mm Hg after 2 weeks of salt loading (P = 0.83). In it-ACE, mean arterial pressure went from 99 ± 3 to 103 ± 2 mm Hg (P = 0.89; Supplementary Figure S3A). No significant differences in sodium balance during the first 3 days of high-salt diet were observed between WT and it-ACE mice (Supplementary Figure S3B–D).
The it-ACE mice do not develop salt-sensitive hypertension
We exposed both WT and it-ACE mice to the post–Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME) model of salt sensitive hypertension: L-NAME (4 weeks) → washout (1 week) → high-salt diet (NaCl 4%, 3 weeks). As shown by several groups including us, the initial exposure to L-NAME induces salt sensitivity in previously salt-resistant mice.15,21,22 As expected, WT mice developed salt-sensitive hypertension after L-NAME (0.5 mg/ml in the drinking water, Figure 4a). The standard dose of L-NAME did not induce hypertension in the it-ACE mice (Supplementary Figure S4). This was similar to the protection from L-NAME–induced hypertension observed in other mouse models lacking renal ACE.18 Because of this, the L-NAME dose was tripled in it-ACE mice to 1.5 mg/ml to induce a similar systolic blood pressure increase as in WT mice (Figure 4a). However, even after equivalent levels of hypertension at week 4, it-ACE mice were still protected against the development of salt sensitivity. Indeed, it-ACE mice had a systolic blood pressure that was 27 mm Hg lower than WT mice after 3 weeks of salt loading (Figure 4a; P < 0.001).
Figure 4. The absence of renal tubular angiotensin converting enzyme (ACE) prevents the development of salt-sensitive hypertension.
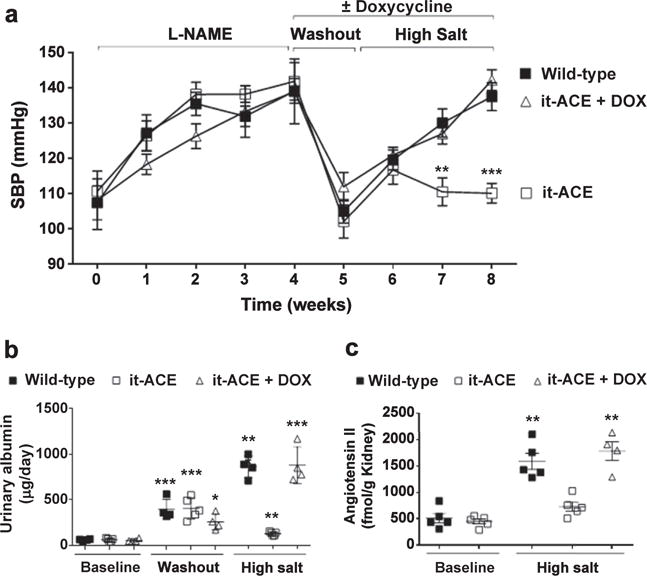
Wild-type (WT), it-ACE, and it-ACE mice treated with doxycycline (it-ACE + DOX) were exposed to 4 weeks of L-NAME, a 1-week washout period, and then 3 weeks of a high-salt diet (4% NaCl in food). Doxycycline was administered during the washout and the high-salt phase. L-NAME was given to WT (0.5 mg/ml) and mutant mice (1.5 mg/ml) in the drinking water. Systolic blood pressure (SBP) was assessed by tail-cuff plethysmography. (a) Values represent mean SEM, n = 5–7, **P < 0.01, ***P < 0.001 versus other groups. Urinary albumin was assessed by an enzyme immunoassay in nontreated mice (i.e., before L-NAME treatment), after the washout and at the end of the protocol. (b) Data were expressed as μg of albumin per 24 hours. Renal angiotensin II content was measured by an enzyme immunoassay in nontreated mice and at the end of the protocol. (c) Data were expressed as fmol of angiotensin II per gram of kidney; n = 4–6; *P < 0.05, **P < 0.01, and ***P < 0.001 versus the corresponding nontreated group.
To further investigate the role of tubular ACE in the development of salt sensitivity, we studied an additional group of it-ACE mice treated with high-dose L-NAME (1.5 mg/ml). But now, to restore tubular ACE during the sodium load, doxycycline was giving during the 1-week washout and the 3-week high-salt diet period. When these mice were exposed to high salt, they displayed a blood pressure increase similar to WT mice (Figure 4a). Thus, restoring tubular ACE rendered the it-ACE mice salt-sensitive. Finally, we performed the opposite experiment of giving doxycycline (and restoring ACE) only during the 4-week L-NAME phase. Thus, doxy-cycline was not given during the washout and high-salt phase. In such mice, only the standard dose of L-NAME (0.5 mg/ml) was required to induce hypertension during the initial 4 weeks of the experiment (Supplementary Figure S5). Even more important, without ACE expression during the high-salt diet, these mice did not develop hypertension. We did not include a group of WT mice exposed to the post–L-NAME model in the presence of doxycycline. However, we confirmed that doxycycline does not alter their eating, drinking behavior, urine excretion, or sodium balance (Supplementary Figure S6). No significant differences were observed in body weight among experimental groups (Supplementary Figure S7A).
In all experimental groups, the analysis of urinary albumin after the washout period revealed that the hypertension induced by the initial 4-week treatment with L-NAME was associated with significant levels of albuminuria (Figure 4b). After the 3-week high-salt diet, WT and it-ACE mice treated with doxycycline during salt loading evidenced further and equivalent increases of urinary albumin. In contrast, there was no further increase in the albuminuria of the it-ACE mice (Figure 4b).
Renal tubular ACE is the main source of renal angiotensin II synthesis during salt sensitivity
Previously, we demonstrated that in WT mice, the development of salt sensitivity is associated with significant accumulation of renal angiotensin II and that this is blunted in mice lacking renal ACE.15 The kidneys from it-ACE mice exposed to the high-salt protocol were assessed for angiotensin II levels. The absence of salt sensitivity in it-ACE mice was associated with significantly lower levels of renal angiotensin II as compared with WT mice (Figure 4c). The opposite is also true; the development of salt sensitivity observed after restoring ACE with doxycycline during salt loading resulted in significant accumulation of renal angiotensin II (Figure 4c). These data indicate that during salt sensitivity, renal accumulation of angiotensin II is primarily mediated through the catalytic activity of ACE expressed by renal tubular epithelial cells. Interestingly, in WT and it-ACE mice treated with doxycycline, renal renin levels were dramatically suppressed by salt loading, and to a greater extent than in it-ACE mice (Supplementary Figure S7B). This study was performed by Western blot, which is indicative of renin production mostly by the juxtaglomerular apparatus and reflects plasma levels of renin. Thus, in WT or it-ACE mice treated with doxycycline exposed to the post–L-NAME model, local accumulation of angiotensin II seems to be mostly independent from the systemic RAS.
Renal tubular ACE blunts natriuresis during high sodium intake
Renal sodium handling was studied in mice exposed to the post–L-NAME model. During the 1-week washout, sodium and urine excretion were similar in both it-ACE and WT strains (Figure 5a and 5b). As expected, when mice were switched to a high-salt diet, both WT and it-ACE mice increased their sodium excretion. However, it-ACE mice displayed an increased natriuretic response compared with WT mice (Figure 5a). Urinary volume output was equivalent between both genotypes (Figure 5b). Sodium balance calculations revealed that both WT and it-ACE mice developed a positive daily sodium balance after commencement of the sodium load (Figure 5c). However, the average sodium accumulation during the first 72 hours (h) of the high-salt diet was significantly lower in it-ACE compared with WT mice (271 ± 48 vs. 527 ± 43 μmol Na+/24h; P < 0.01). At the end of the protocol, sodium input and output were in equilibrium in both it-ACE and WT mice but at the expense of hypertension in the latter group (Figure 5c). Further, it-ACE mice treated with doxycycline during salt loading displayed reduced natriuresis with sodium retention that was indistinguishable from WT mice (Figure 5a–c). Of note, the different sodium handling observed in it-ACE and WT mice was not a reflection of different sodium intake, as this was similar between all analyzed groups (Figure 5d).
Figure 5. The it-ACE mice display a greater natriuretic response and less sodium accumulation when exposed to a sodium load.
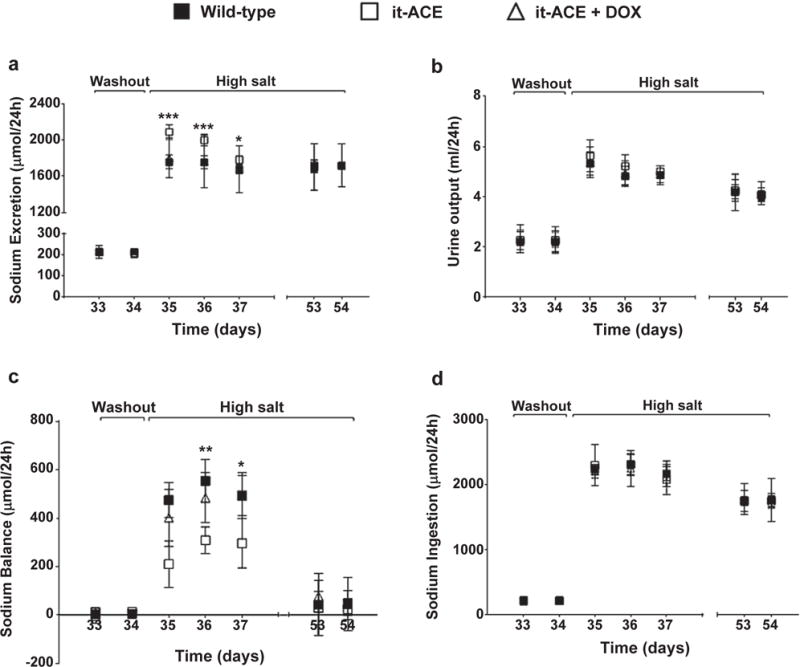
Wild-type (WT), it-ACE, and it-ACE mice treated with doxycycline (it-ACE + DOX) were housed individually in metabolic cages with free access to food and water before and during the high-salt diet. Doxycycline was administered during the washout and the high-salt phase. (a) Sodium excretion, (b) urine output, and (d) sodium ingestion were used to calculate (c) sodium balance. Values were expressed as μmol sodium per day. L-NAME was previously given to wild-type (0.5 mg/ml) and it-ACE (1.5 mg/ml) mice in the drinking water. Values represent mean ± SEM; n = 5; *P < 0.05, **P < 0.01, and ***P < 0.001 versus other groups.
Lower levels of distal sodium transporters in high salt–fed it-ACE mice
To understand the molecular mechanisms underlying the enhanced natriuretic response of it-ACE mice, a sodium transporter profile was performed by immunoblot analysis. For this, we measured the abundance, phosphorylation, and processing of several key sodium transporters: Na+/H+ exchanger 3, Na+-K+-2Cl+ cotransporter, NaCl cotransporter (NCC), and the epithelial sodium channel (ENaC). Before L-NAME treatment (i.e., in nontreated mice), the sodium transporter profile was similar between WT and it-ACE mice (Figure 6a). As previously demonstrated, mice respond to hypertensive stimuli with renal adaptations that facilitate natriuresis.15,18 In line with these findings, our current data showed that, after exposing mice to the post–L-NAME model, both WT and it-ACE mice displayed significantly lower levels and phosphorylation of Na+-K+-2Cl+ cotransporter and NCC, and lower levels of the proteolytic active product of the α subunit of ENaC (αENaC-CL; Figure 6a). However, in it-ACE mice greater natriuretic adaptations occurred. These included a more substantial decrease in NCC abundance and phosphorylation, and lower levels of αENaC as compared with WT (Figure 6b).
Figure 6. The absence of renal tubular angiotensin converting enzyme (ACE) amplifies reductions in abundance and phosphorylation of NaCl cotransporter (NCC) and cleaved (CL) a subunit of the epithelial sodium channel (aENaC) after 3 weeks of a high-salt diet.
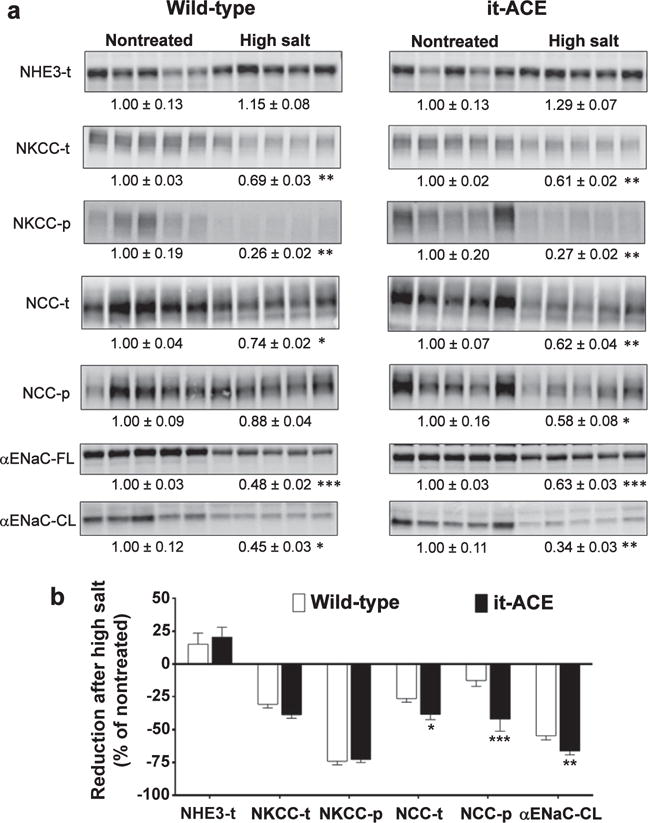
Renal sodium transporter expression was analyzed in total kidney homogenates from nontreated mice and after 3 weeks of high-salt diet. L-NAME was given to wild-type (0.5 mg/ml) and mutant (1.5 mg/ml) mice in the drinking water. Immunoblots of renal cortical homogenates were performed with a constant amount of protein per lane. Relative abundance from each group is displayed below the corresponding blot as mean SEM. (a) n = 5 per group; *P < 0.05, **P < 0.01, and ***P < 0.001 versus corresponding nontreated mice. (b) Bars represent the relative reduction versus nontreated mice (set as 0); *P < 0.05, **P < 0.01, and ***P < 0.001. NHE3, Na+/H+ exchanger 3; NKCC, Na+-K+-2Cl+ cotransporter.
Expressing renal ACE exclusively in tubular epithelial cells is sufficient to develop salt sensitivity
We described a mouse model in the past, ACE 9/9, that made ACE exclusively in renal tubular epithelium.20 These mice lack endothelial and circulating ACE and were very hypotensive with varying degrees of renal pathology. We also studied ACE 3/3 mice that expressed ACE in liver hepatocytes.23 Such mice have normal blood pressure and renal morphology and function, though renal ACE levels were very low. To provide additional evidence concerning the role of tubular epithelial ACE, we developed a new mouse model, ACE 3/9. These compound heterozygous mice make ACE only in renal tubular epithelium due to the ACE 9 allele, and in liver hepatocytes (Figure 7a). In nontreated ACE 3/9 mice, blood pressure, glomerular filtration rate (1256 ± 47 μl/min/100g b.w.,) and kidney morphology are normal. In this model, plasma ACE was 63% ± 5% of WT mice (Figure 7a, P < 0.001). In contrast, these mice have no or very little ACE activity in the lungs (i.e., endothelium, 1.0% ± 0.1% of WT) and brain (15% ± 1% of WT; Figure 7a). In total kidney homogenate, ACE activity was significantly decreased (Figure 7a, 47% ± 6% of WT, P < 0.001). Although this reduction is similar to that observed in it-ACE mice, a detailed immunohistochemistry analysis of ACE distribution revealed that renal ACE is expressed by tubular epithelium with undetectable ACE expression in endothelium, the glomeruli, and the tubulointerstitial space (Figure 7b). We further confirmed these results using flow cytometry. For this, single-cell suspensions from WT and ACE 3/9 mice were stained with an antibody against ACE and markers of endothelial cells and proximal tubular cells. While ACE expression is equivalent in megalin+ tubular epithelium from ACE 3/9 and WT mice, the levels of endothelial ACE in ACE 3/9 were dramatically reduced compared with WT mice (Figure 7c). Thus, in the context of renal epithelial and endothelial ACE expression, the ACE 3/9 mouse is the reverse of the it-ACE mouse; the ACE 3/9 mouse expresses renal ACE only where ACE was absent in the it-ACE mouse, namely, in tubular epithelium. We observed that, during the 4-week L-NAME phase, ACE 3/9 mice developed hypertension equivalent to WT when exposed to the conventional 0.5 mg/ml dose of L-NAME. Also, ACE 3/9 mice developed hypertension when challenged with a high salt load (Figure 7d). These data show that, in this model, renal tubular ACE, and not endothelial or brain ACE, is necessary for salt sensitivity. Thus, the expression of ACE solely in renal tubular epithelium was sufficient to generate a response to the post–L-NAME model indistinguishable from that of WT mice (Figure 7d).
Figure 7. In ACE 3/9 mice, the only source of renal ACE are the tubular epithelial cells.
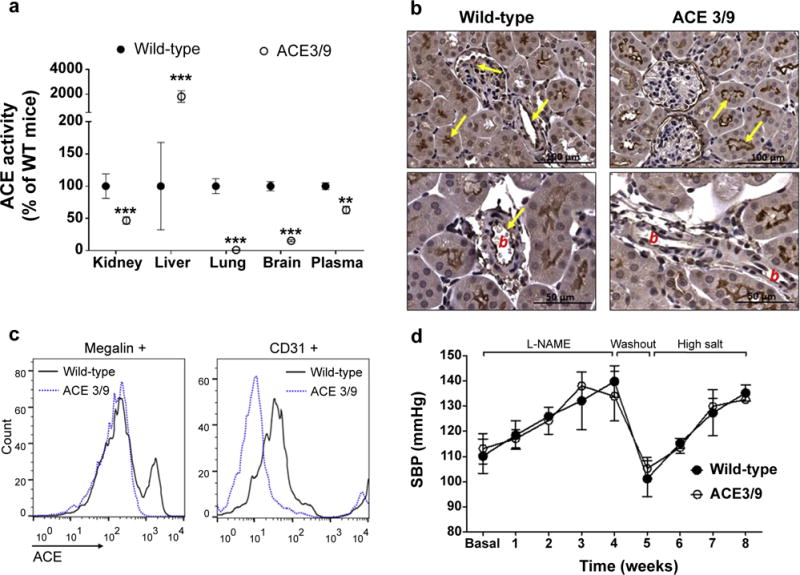
ACE activity was assessed in different tissues. (a) Values represent mean ± SEM; n = 5 per group, **P < 0.01 and ***P < 0.001 versus wild-type mice. Renal ACE expression was evaluated by (b) immunohistochemistry, where b indicates blood vessel, and by (c) flow cytometry. Megalin and CD31 were used as markers of proximal tubular cells and endothelial cells, respectively. (d) Wild-type and ACE 3/9 mice were exposed to the post–L-NAME protocol (n = 5).
DISCUSSION
Salt-sensitive hypertension is a multifactorial disorder. Notwithstanding the importance of genetics in its etiology, salt-sensitive hypertension can also result from acquired conditions, including renal inflammation.24 However, exactly how renal inflammation causes renal sodium retention is not well understood. Here, we present evidence showing that tubular ACE is a pivotal factor impairing the ability of the injured kidney to respond appropriately to high salt intake.
While the consensus is that salt-sensitive individuals respond poorly to ACE inhibition, several lines of evidence led us to question this paradigm. First, as discussed in the introduction, some subsets of salt-sensitive patients benefit from ACE inhibition, including those with renal disease.7,8 Second, the renal inflammatory response is often associated with increased expression of ACE and other RAS component along the tubules.9–13 Third, previous work established the importance of local angiotensin II production within the kidney for the antinatriuresis of many forms of experimental hypertension.15,18,19 Based on these considerations, we decided to assess the role of tubular ACE, the main source of kidney ACE, in salt-sensitive hypertension.
We developed 2 new strains of mice, it-ACE and ACE 3/9, with opposing phenotypes in terms of renal ACE expression. The it-ACE mice lack ACE exclusively in the tubules; the ACE 3/9 mice produce ACE only in the tubules. We exposed our mutant mice to the post–L-NAME model of hypertension, a 4-week treatment with L-NAME that induces significant renal angiotensin II accumulation with plasma angiotensin II suppression, kidney injury, and ultimately salt sensitivity. Our results show that mice lacking ACE in the tubules (i.e., it-ACE) are resistant to salt-sensitive hypertension. In contrast, ACE 3/9 mice, in which tubular ACE is the only source of renal ACE, resemble WT in their response to the post–L-NAME salt loading. The ACE 3/9 mice lack ACE in the brain, lung, and renal endothelium. Nevertheless, they develop salt sensitivity following L-NAME. Both ACE 3/9 and it-ACE have normal plasma levels of ACE. We have not formally evaluated the role of plasma ACE in our model. However, the protection against hypertension observed in it-ACE mice, and not in ACE 3/9 mice, suggests that tubular ACE is central to experimental hypertension while plasma ACE plays a secondary role in blood pressure elevation. Thus, the consistency of data from both strains, including the restoration of tubular epithelial ACE with doxycycline, is compelling and strongly supports the conclusion that renal tubular epithelial ACE expression is responsible for the antinatriuretic state and the development of salt sensitivity during renal injury.
Mechanistic insight into why removing tubular ACE protects against salt sensitivity was obtained by careful analysis of the renal response to a sodium load. In accordance with previous publications, WT mice respond to the post–L-NAME model with increases in renal angiotensin II content, as well as sodium and fluid retention that induces high blood pressure in response to a sodium load.15 We now demonstrate that removing ACE from renal tubular epithelium blunts the sodium retention and prevents the development of salt sensitivity in it-ACE mice. Indeed, the studies with doxycycline indicate that intrarenal ACE is only necessary during the high-salt diet, not during the L-NAME period of the protocol. These findings further confirm that tubular ACE is essential to establish the antinatriuretic state associated with the salt-driven hypertension observed in WT mice exposed to the post–L-NAME model.
Normally, without exposure to L-NAME, the kidneys of rodents respond to a dietary salt load with compensatory reductions of sodium transporters along the nephron that results in a higher rate of sodium excretion, matching the higher sodium intake.25–29 Our data show that, after the 3-week salt loading phase of the post–L-NAME model, most sodium transporters were significantly reduced in both WT and it-ACE mice compared to nontreated expression levels. However, when comparing the relative reductions between genotypes, we observed that the distal transporters NCC and αENaC-CL were significantly more reduced in it-ACE mice than in WT mice. Thus, the L-NAME pretreatment blunted the normal reduction of distal sodium transporters in WT mice but not in mice lacking renal tubular ACE. We hy-pothesize that the different response to the post–L-NAME salt load between WT and it-ACE mice is determined by the absence of intrarenal angiotensin II accumulation in the latter. Angiotensin II stimulates virtually all sodium transporters along the nephron.30–34 Thus, we surmise that, in WT mice, angiotensin II produced by renal tubular ACE opposes the compensatory natriuretic response to a sodium load. The angiotensin II may arise from either proximal tubular cells and travel downstream to modify the activity of distal sodium transporters or be the result of distal nephron production of renin and ACE, which then process angiotensinogen.35 Whether 1 or both of these scenarios is correct, removing ACE from renal tubular epithelial cells totally prevented the renal accumulation of angiotensin II following the post–L-NAME salt challenge, and this was associated with greater downregulation of distal sodium transporters and less sodium retention during the sodium load.
There are several limitations to our study. First, we did not perform a time-based analysis of sodium transporters to show that they precede the establishment of the hypertension. Second, we did not measure blood pressure by telemetry, which is more accurate than the tail-cuff method. Third, we did not pursue other potential explanations to our findings. For instance, it is possible that tubular ACE has a role in the regulation of glomerular hemodynamics. Finally, it is also possible that the genetic manipulation somehow affects sodium retention in extrarenal tissues such as the subcutaneous interstitium.36 However, none of these observations should detract from the central finding of this study, which is that without tubular ACE, there is no hypertension in the post–L-NAME model.
In summary, we show that removing ACE from renal tubular epithelial cells blunts the antinatriuretic state driving the salt retention during renal parenchymal inflammation. These data provide further evidence to understand the better therapeutic outcome of ACE inhibitors in controlling salt sensitivity associated with renal injury, such as in diabetes or chronic kidney disease. Thus, although ACE is ubiquitously expressed, our data indicate that in mice with renal injury induced by L-NAME pretreatment, renal tubular epithelial ACE and not ACE expression by renal endothelium, lung, brain, or plasma is obligatory for the induction of salt-sensitive hypertension during renal injury.
METHODS
Animal models
The it-ACE mouse was made transgenic for the Tet-Off Advanced Inducible Gene System (Clontech Laboratories, Inc., Mountain View, CA). Briefly, using standard DNA microinjection of ovocytes, 3 constructs were integrated at a single genetic site. One construct is the kidney-specific (Ksp)-cadherin/β-globin promoter driving the transcription factor tetracycline transactivator (tTAoff). As a result, tTAoff is made only by renal tubular epithelial cells.37 The other 2 constructs each possess a modified cytomegalovirus promoter that, in the presence of the transcription factor tTAoff and the absence of doxycycline, produces the tdTomato fluorescent protein and 2 different shRNAs. These are complementary to portions of the ACE C-domain and silence ACE mRNA (Figure 1). The shRNAs were selected to target sequences localized at the beginning (cDNA position 1995–2015: 5′-CCA AGT GTT GTT GAA CGA GTA C-3′) and at the end (cDNA position 3600–3620: 5′-CGC CAT GAT GAA TTA CTT CAA-3′) of the ACE C-domain. Importantly, ACE expression can be regulated by doxycycline administration. In nontreated mice, tubular ACE expression is low. However, when mice are fed doxycycline (200 μg/ml in the drinking water), the tTAoff factor is inactivated, blocking the expression of the shRNA and restoring normal ACE expression. These mice were named inducible tubular ACE (it-ACE). Genotyping was done by polymerase chain reaction of ear DNA using primers: 5′ – GAA GCC GCT TGG AAT AAG GC – 3′ and 5′ – CTT TGA TGA CCT CCT CGC CC – 3′. The it-ACE mice were raised in a pure C57BL/6J background. ACE 3/9 mice were generated after crossing the ACE 3/3 with the ACE 9/9 mice. In ACE 3/3 mice, embryonic stem cell gene targeting was used to replace the endogenous ACE promoter with the albumin promoter. In these mice, the main source of ACE is hepatocytes, with only a small amount of ACE produced in the kidney (14% of WT mice).23 No ACE is made by endothelium or the brain. In ACE 9/9 mice, the endogenous ACE promoter was replaced with the Ksp-cadherin/β-globin promoter restricting ACE expression to renal tubular epithelium.20,37 The genetic background of ACE 3/9 mice is mixed between 129j and C57BL/6. All mice were selected to have only 1 renin gene.
Salt-sensitive hypertension
The experimental protocol to induce salt sensitivity includes 3 successive phases: a 4-week nitric oxide synthesis inhibition with L-NAME (0.5 mg/ml or 1.5 mg/ml in the drinking water), a 1-week washout, and a 3-week exposure to a high-salt diet (4% NaCl in the food; Harlan Laboratories, Placentia, CA).21,24 In a previous publication, we did an extensive characterization of this model showing that mice exposed to L-NAME showed significant levels of renal parenchymal inflammation that persisted through the washout phase and predisposed to salt-sensitive hypertension. Blood pressure was monitored weekly by tail-cuff plethysmography using a Visitech BP2000 system (Visitech Systems Inc., Apex, NC) in previously trained mice. Experiments were conducted in 8- to 12-week-old male mice and WT mice of the corresponding genetic background. All studies were approved by the Cedars-Sinai Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Metabolic studies
Mice were individually housed in metabolic cages for urine sampling and water and food consumption monitoring. To avoid urine contamination with food, mice were fed a gelled diet containing all necessary nutrients plus 0.4% NaCl (Nutra-gel; Bio-Serv, French-town, NJ; Cat: S4798) or 4% NaCl (Nutra-gel customized diet; Bio-Serv; Cat: F6835). NaCl content is expressed on a dry-weight basis. Gel diet contains 70% water. Animals had free access to water at all times. Urinary albumin was assessed by ELISA (Exocell, Philadelphia, PA) Urinary sodium was determined by flame photometry (Cole-Parmer, Vernon Hills, IL). For urine concentration experiments, mice were deprived of water for 24 hours. After that, a urine drop was collected. Urinary osmolality was assessed using a vapor pressure osmometer (Wescor, Inc., Logan, UT). All values were expressed as total daily excretion.
Glomerular filtration rate measurements
Glomerular filtration rate was determined in conscious, unrestrained mice using the excretion kinetics of a single i.v. bolus of FITC-sinistrin assessed by a miniaturized fluorescence detector attached to the back of the mouse.18 Glomerular filtration rate was calculated using the half-life derived from the rate constant (α2) of the single exponential phase of the FITC-sinistrin excretion curve and a semi-empirical conversion factor, as validated in rats and mice.18,38 The miniaturized device (NIC-Kidney; Mannheim Pharma & Diagnostics, Mannheim, Germany) is equipped with two light-emitting diodes that transcutaneously excite FITC-sinistrin at 480 nm and a photodiode to detect the emitted light signal at 521 nm.18,38
Renal sodium transporters
At the end of the protocol, mice were killed and both kidneys were quickly excised. Transporter profiling was assessed by immunoblot as described previously.15,18,19 For this, whole kidney protein extracts (20 μg) were denatured, resolved by SDS-PAGE, and transferred into polyvinylidene difluoride membranes (Millipore Immobilon-FL; EMD Millipore, Billerica, MA), blocked (Odyssey blocking buffer, Lincoln, NE), and then probed with specific antibodies against Na+/H+ exchanger 3 (1:2000; McDonough laboratory), Na+-K+-2Cl+ cotransporter (1:1000; C. Lytle, UCR), phosphorylated Thr96/Thr101–Na+-K+-2Cl+ cotransporter (1:2000; Forbush, Yale), NCC (1:2000; McDonough laboratory), phosphorylated Ser71-NCC, and αENaC (1:1000; J. Loffing, U. Zurich) as described.18,19 After washing, membranes were incubated with the appropriate fluorochrome-labeled secondary antibody. To verify uniform loading, loading gels were run (5 mg/lane) and stained with Coomassie blue, and random bands were quantified. Linearity of signal intensity was established by loading 1 and 1/2 amounts of each sample on each gel to verify halving of signal intensity. Signals on immunoblots were detected and quantitated with the Odyssey Infrared Imaging System (Li-COR, Lincoln, NE) and accompanying software. Values were normalized to mean intensity of the corresponding nontreated group defined as 1.0.
Renal RAS assessment
Renal ACE activity was assessed in total kidney homogenates as previously described.39 To confirm the assay specificity, an aliquot of each sample was assessed in the presence of the ACE inhibitor captopril. Only the hydrolytic activity inhibited by captopril was considered for calculations. For Western blot analysis, whole kidney protein extracts (20 μg) were denatured, resolved by SDS-PAGE, and transferred into polyvinylidene difluoride membranes and then probed with antibodies against renin (1:1000 dilution; Anaspec, Fremont, CA) and ACE (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Signals on immunoblots were detected as described earlier. For ACE assessment by immunohistochemistry, tissue preparation, including paraffin embedding, antigen extracting, and staining were conducted as previously described.18 Slides were incubated with a goat anti–ACE antibody (1:300; Santa Cruz Biotechnology) at room temperature for 1 hour. Negative controls consisted of histological sections incubated with phosphate-buffered saline rather than the primary antibody. Immunostaining was carried out with an avidin-biotin-peroxidase complex and counter-stained with hematoxylin. Stained slides were scanned with a Leica SCN400 Slide Scanner (Buffalo Grove, IL), and digital images of whole cross-sections of the kidneys were saved for analysis. A total of 15 to 20 random fields from each kidney were viewed at X400 magnification. Tubular ACE expression was also assessed by flow cytometry in a single-cell suspension obtained from a renal cortex.40 Briefly, kidney cortices were isolated and minced into 1 to 2 mm3. Tissue pieces were resuspended in 1500 U of collagenase type IV dissolved in 5 ml of DMEM/F12 medium and incubated at 37°C in a gentle shaking water bath for 40 minutes. The suspension was homogenized by pipetting 10 times through a sterile transfer pipette. Digestion was stopped by adding 20 ml of fresh ice-cold DMEM/F12. Larger tissue fragments were allowed to settle, and the supernatant containing the single-cell suspension was collected and centrifuged at 300 g for 5 minutes. The supernatant was discarded and the pellet was resuspended in 1 ml of phosphate-buffered saline supplemented with 1% fetal bovine serum. Cells were then incubated with the following antibodies: anti-ACE (made by Dr. Kenneth Bernstein) followed by a secondary APC-conjugated anti-rabbit antibody (1:500; Southern Biotech, Birmingham, AL), FITC-conjugated anti-CD31 (1:100; ebioscience, San Diego, CA), and Cy7-conjugated anti-Megalin (1:100; Bioss, Woburn, MA) for 20 minutes at 4°C. This protocol allowed the quantification of ACE expression in proximal tubular cells (Megalin+ cells) versus endothelial cells (CD31+ cells). Renal angiotensin II levels were measured using a commercially available enzyme immunoassay (EIA; Peninsula Laboratories International Inc., San Carlos, CA). Briefly, snap-frozen kidney was homogenized in ice-cold methanol and centrifuged at 12,000 ×g for 10 minutes at 4°C. The supernatant fluid was collected and dried by centrifugal evaporation. Dried samples were reconstituted with the EIA buffer provided by the manufacturer and assayed for angiotensin II as previously described.41 Renal angiotensin II content was expressed as fmol of angiotensin II per g of kidney.
Statistical analyses
Data are presented as individual measurements along with mean ± SEM. Two-way ANOVA with Bonferroni’s post-test was used to analyze changes in data collected over time. One-way ANOVA and unpaired t-test were used to analyze differences between nontreated and treated mice within the same genotype or to assess the differences between wild-type and mutant mice when appropriate. All statistical tests were calculated using GraphPad Prism 5.00 (Graph-Pad Software, San Diego, CA). P < 0.05 denoted statistically significant differences.
Supplementary Material
Figure S1. The kidney of it-ACE mice displays lower levels of angiotensin converting enzyme (ACE) expression. Renal ACE was evaluated by immunohistochemistry. (A) Arrows indicate glomerular and endothelial ACE. Arrowheads indicate tubular epithelial ACE. (B) Tissue samples obtained from male and female wild-type (WT) and it- ACE (it) mice were assessed for ACE expression by Western blot.
Figure S2. Basal kidney function and renal renin-angiotensin system of it-ACE mice are undistinguishable from those of wild-type mice. (A) Glomerular filtration rate (GFR) was calculated using the excretion kinetics of a single i.v. bolus of FITC-sinistrin assessed by a miniaturized fluorescence detector attached to back of the mouse. (B) Urine osmolality was measured at baseline and after 24-hour water deprivation (Water depriv). (C) Prorenin/renin was assessed by Western blot in kidney homogenates. (D) Renal angiotensin II levels in total kidney homogenates were measured using an enzyme immunoassay. Values represent mean ± SEM, n = 4–5.
Figure S3. In the absence of prior kidney injury, wild-type and it-ACE mice display no changes in blood pressure after 2 weeks of high-salt diet (4% NaCl in food). (A) Mean arterial pressure (MAP) was measured by telemetry. To assess (B) sodium ingestion, (C) sodium excretion, and (D) sodium balance, wild-type and it-ACE mice were housed individually in metabolic cages. After 2 days of basal measurements, mice were exposed to a high-salt diet for 3 additional days. Results are expressed as daily average. Values represent mean ± SEM, n = 4–5.
Figure S4. The it-ACE mice are protected against L-NAME–induced hypertension. Wild-type and it-ACE mice were treated with 0.5 mg/ml L-NAME in the drinking water for 4 weeks. Systolic blood pressure (SBP) was measured by tail-cuff plethysmography. Values represent mean ± SEM, *P < 0.05, **P < 0.01, n = 5.
Figure S5. The absence of tubular angiotensin converting enzyme (ACE_ during the high-salt phase of the post–L-NAME salt challenge is associated with protection from salt-sensitive hypertension. Wild-type and it-ACE treated with doxycycline (it-ACE DOX) were exposed to 4 weeks of L-NAME, a 1-week washout period, + and then 3 weeks of a high-salt diet (4% NaCl in food). Doxycycline was administered only during the L-NAME phase. L-NAME was given to wild-type and it-ACE mice (0.5 mg/ml) in the drinking water. Values represent mean ± SEM, **P < 0.01, ***P < 0.001 versus wild-type, n = 4.
Figure S6. Doxycycline administration does not alter the food and drinking behavior of wild-type mice. Wild-type mice receiving either tap water or doxycycline were housed individually in metabolic cages with free access to food and water. Doxycycline was administered in the drinking water (0.2 mg/ml) for 3 days. Ingestion of (A) water and (B) food as well as (C) urine excretion and (D) sodium balance were measured. Values were expressed as mean per day ± SEM, n = 5.
Figure S7. (A) Body weight was measured before L-NAME treatment (nontreated), after the washout period, and after 3 weeks of salt load (high salt) in wild-type, it-ACE, and it-ACE treated with doxycycline (it-ACE + DOX). Doxycycline was administered during the washout and the high-salt phase. L-NAME was given to wild-type (0.5 mg/ml) and it-ACE mice (1.5 mg/ml) in the drinking water. (B) Prorenin and renin were evaluated by Western blot in total kidney homogenate of wild-type, it-ACE, and it-ACE + DOX. Values represent mean SEM, *P < 0.05, **P < 0.01, ***P < 0.001 versus the corresponding nontreated mice, n = 4–5.
Acknowledgments
This study was supported by a postdoctoral fellowship from AHA 15POST22520015 and an AHA Scientist Development Grant 16SDG30130015 to JFG; NIH grants R03DK101592 to RAG, R01HL110353, R21AI114965, and R01DK098382 to KEB, and R01DK083785 to AAM; and a grant from the University Kidney Research Organization to AAM. The authors thank K. Wawrowsky and F. Chung for their assistance in immunohistochemistry studies and B. Taylor for his assistance in preparing this manuscript.
Footnotes
DISCLOSURE
RAGV is an employee and a stockholder of Pfizer, Inc. All the other authors declared no competing interests.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.kidney-international.org.
References
- 1.Wolf-Maier K, Cooper RS, Banegas JR, et al. Hypertension prevalence and blood pressure levels in 6 European countries, Canada, and the United States. JAMA. 2003;289:2363–2369. doi: 10.1001/jama.289.18.2363. [DOI] [PubMed] [Google Scholar]
- 2.Felder RA, White MJ, Williams SM, Jose PA. Diagnostic tools for hypertension and salt sensitivity testing. Curr Opin Nephrol Hypertens. 2013;22:65–76. doi: 10.1097/MNH.0b013e32835b3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimosawa T, Mu S, Shibata S, Fujita T. The kidney and hypertension: pathogenesis of salt-sensitive hypertension. Curr Hypertens Rep. 2012;14:468–472. doi: 10.1007/s11906-012-0284-5. [DOI] [PubMed] [Google Scholar]
- 4.Izzo JL, Jr, Weir MR. Angiotensin-converting enzyme inhibitors. J Clin Hypertens. 2011;13:667–675. doi: 10.1111/j.1751-7176.2011.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson JA. Ethnic differences in cardiovascular drug response: potential contribution of pharmacogenetics. Circulation. 2008;118:1383–1393. doi: 10.1161/CIRCULATIONAHA.107.704023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weir MR, Chrysant SG, McCarron DA, et al. Influence of race and dietary salt on the antihypertensive efficacy of an angiotensin-converting enzyme inhibitor or a calcium channel antagonist in salt-sensitive hypertensives. Hypertension. 1998;31:1088–1096. doi: 10.1161/01.hyp.31.5.1088. [DOI] [PubMed] [Google Scholar]
- 7.Williams SF, Nicholas SB, Vaziri ND, Norris KC. African Americans, hypertension and the renin angiotensin system. World J Cardiol. 2014;6:878–889. doi: 10.4330/wjc.v6.i9.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arauz-Pacheco C, Parrott MA, Raskin P, American Diabetes Association Treatment of hypertension in adults with diabetes. Diabetes Care. 2003;26(Suppl 1):S80–S82. doi: 10.2337/diacare.26.2007.s80. [DOI] [PubMed] [Google Scholar]
- 9.Graciano ML, Cavaglieri RdC, Delle H, et al. Intrarenal Renin-angiotensin system is upregulated in experimental model of progressive renal disease induced by chronic inhibition of nitric oxide synthesis. J Am Soc Nephrol. 2004;15:1805–1815. doi: 10.1097/01.asn.0000131528.00773.a9. [DOI] [PubMed] [Google Scholar]
- 10.Satou R, Gonzalez-Villalobos RA, Miyata K, et al. IL-6 augments angiotensinogen in primary cultured renal proximal tubular cells. Mol Cell Endocrinol. 2009;311:24–31. doi: 10.1016/j.mce.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prieto MC, Gonzalez AA, Navar LG. Evolving concepts on regulation and function of renin in distal nephron. Pflugers Arch. 2013;465:121–132. doi: 10.1007/s00424-012-1151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57:355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension. 2002;39:316–322. doi: 10.1161/hy0202.103821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobori H, Navar LG. Urinary angiotensinogen as a novel biomarker of intrarenal renin-angiotensin system in chronic kidney disease. Int Rev Thromb. 2011;6:108–116. [PMC free article] [PubMed] [Google Scholar]
- 15.Giani JF, Bernstein KE, Janjulia T, et al. Salt sensitivity in response to renal injury requires renal angiotensin-converting enzyme. Hypertension. 2015;66:534–542. doi: 10.1161/HYPERTENSIONAHA.115.05320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alhenc-Gelas F, Baussant T, Hubert C, et al. The angiotensin converting enzyme in the kidney. J Hypertens Suppl. 1989;7:S9–S13. doi: 10.1097/00004872-198909007-00003. discussion S14. [DOI] [PubMed] [Google Scholar]
- 17.Liebau MC, Lang D, Bohm J, et al. Functional expression of the renin-angiotensin system in human podocytes. Am J Physiol Renal Physiol. 2006;290:F710–F719. doi: 10.1152/ajprenal.00475.2004. [DOI] [PubMed] [Google Scholar]
- 18.Giani JF, Janjulia T, Kamat N, et al. Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition. J Amer Soc Nephrol. 2014;25:2752–2763. doi: 10.1681/ASN.2013091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, et al. The absence of intrarenal ACE protects against hypertension. J Clin Invest. 2013;123:2011–2023. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Villalobos RA, Billet S, Kim C, et al. Intrarenal angiotensin-converting enzyme induces hypertension in response to angiotensin I infusion. J Am Soc Nephrol. 2011;22:449–459. doi: 10.1681/ASN.2010060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quiroz Y, Pons H, Gordon KL, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol. 2001;281:F38–F47. doi: 10.1152/ajprenal.2001.281.1.F38. [DOI] [PubMed] [Google Scholar]
- 22.Itani HA, Xiao L, Saleh MA, et al. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243. doi: 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole J, Quach DL, Sundaram K, et al. Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circ Res. 2002;90:87–92. doi: 10.1161/hh0102.102360. [DOI] [PubMed] [Google Scholar]
- 24.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. New Eng J Med. 2002;346:913–923. doi: 10.1056/NEJMra011078. [DOI] [PubMed] [Google Scholar]
- 25.Yang LE, Sandberg MB, Can AD, et al. Effects of dietary salt on renal Na + transporter subcellular distribution, abundance, and phosphorylation status. Am J Physiol Renal Physiol. 2008;295:F1003–F1016. doi: 10.1152/ajprenal.90235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandberg MB, Maunsbach AB, McDonough AA. Redistribution of distal tubule Na+−Cl- cotransporter (NCC) in response to a high-salt diet. Am J Physiol Renal Physiol. 2006;291:F503–F508. doi: 10.1152/ajprenal.00482.2005. [DOI] [PubMed] [Google Scholar]
- 27.Chiga M, Rai T, Yang SS, et al. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int. 2008;74:1403–1409. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- 28.Haque MZ, Ares GR, Caceres PS, Ortiz PA. High salt differentially regulates surface NKCC2 expression in thick ascending limbs of Dahl salt-sensitive and salt-resistant rats. Am J Physiol Renal Physiol. 2011;300:F1096–F1104. doi: 10.1152/ajprenal.00600.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loffing J, Pietri L, Aregger F, et al. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. Am J Physiol Renal Physiol. 2000;279:F252–F258. doi: 10.1152/ajprenal.2000.279.2.F252. [DOI] [PubMed] [Google Scholar]
- 30.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, et al. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A. 2012;109:7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.du Cheyron D, Chalumeau C, Defontaine N, et al. Angiotensin II stimulates NHE3 activity by exocytic insertion of the transporter: role of PI 3-kinase. Kidney Int. 2003;64:939–949. doi: 10.1046/j.1523-1755.2003.00189.x. [DOI] [PubMed] [Google Scholar]
- 32.Mamenko M, Zaika O, Ilatovskaya DV, et al. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. 2012;287:660–671. doi: 10.1074/jbc.M111.298919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.San-Cristobal P, Pacheco-Alvarez D, Richardson C, et al. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Lubbe N, Lim CH, Fenton RA, et al. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011;79:66–76. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 35.Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG. Collecting duct renin: a major player in angiotensin ii-dependent hypertension. J Am Soc Hypertens. 2009;3:96–104. doi: 10.1016/j.jash.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiig H, Schroder A, Neuhofer W, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. 2013;123:2803–2815. doi: 10.1172/JCI60113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao X, Johnson JE, Richardson JA, et al. A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13:1824–1836. doi: 10.1097/01.asn.0000016443.50138.cd. [DOI] [PubMed] [Google Scholar]
- 38.Schreiber A, Shulhevich Y, Geraci S, et al. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol. 2012;303:F783–F788. doi: 10.1152/ajprenal.00279.2012. [DOI] [PubMed] [Google Scholar]
- 39.Neels HM, van Sande ME, Scharpe SL. Sensitive colorimetric assay for angiotensin converting enzyme in serum. Clin Chem. 1983;29:1399–1403. [PubMed] [Google Scholar]
- 40.Yadav N, Rao S, Bhowmik DM, Mukhopadhyay A. Bone marrow cells contribute to tubular epithelium regeneration following acute kidney injury induced by mercuric chloride. Indian J Med Res. 2012;136:211–220. [PMC free article] [PubMed] [Google Scholar]
- 41.Bae EH, Konvalinka A, Fang F, et al. Characterization of the intrarenal Renin-Angiotensin system in experimental alport syndrome. Am J Pathol. 2015;185:1423–1435. doi: 10.1016/j.ajpath.2015.01.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The kidney of it-ACE mice displays lower levels of angiotensin converting enzyme (ACE) expression. Renal ACE was evaluated by immunohistochemistry. (A) Arrows indicate glomerular and endothelial ACE. Arrowheads indicate tubular epithelial ACE. (B) Tissue samples obtained from male and female wild-type (WT) and it- ACE (it) mice were assessed for ACE expression by Western blot.
Figure S2. Basal kidney function and renal renin-angiotensin system of it-ACE mice are undistinguishable from those of wild-type mice. (A) Glomerular filtration rate (GFR) was calculated using the excretion kinetics of a single i.v. bolus of FITC-sinistrin assessed by a miniaturized fluorescence detector attached to back of the mouse. (B) Urine osmolality was measured at baseline and after 24-hour water deprivation (Water depriv). (C) Prorenin/renin was assessed by Western blot in kidney homogenates. (D) Renal angiotensin II levels in total kidney homogenates were measured using an enzyme immunoassay. Values represent mean ± SEM, n = 4–5.
Figure S3. In the absence of prior kidney injury, wild-type and it-ACE mice display no changes in blood pressure after 2 weeks of high-salt diet (4% NaCl in food). (A) Mean arterial pressure (MAP) was measured by telemetry. To assess (B) sodium ingestion, (C) sodium excretion, and (D) sodium balance, wild-type and it-ACE mice were housed individually in metabolic cages. After 2 days of basal measurements, mice were exposed to a high-salt diet for 3 additional days. Results are expressed as daily average. Values represent mean ± SEM, n = 4–5.
Figure S4. The it-ACE mice are protected against L-NAME–induced hypertension. Wild-type and it-ACE mice were treated with 0.5 mg/ml L-NAME in the drinking water for 4 weeks. Systolic blood pressure (SBP) was measured by tail-cuff plethysmography. Values represent mean ± SEM, *P < 0.05, **P < 0.01, n = 5.
Figure S5. The absence of tubular angiotensin converting enzyme (ACE_ during the high-salt phase of the post–L-NAME salt challenge is associated with protection from salt-sensitive hypertension. Wild-type and it-ACE treated with doxycycline (it-ACE DOX) were exposed to 4 weeks of L-NAME, a 1-week washout period, + and then 3 weeks of a high-salt diet (4% NaCl in food). Doxycycline was administered only during the L-NAME phase. L-NAME was given to wild-type and it-ACE mice (0.5 mg/ml) in the drinking water. Values represent mean ± SEM, **P < 0.01, ***P < 0.001 versus wild-type, n = 4.
Figure S6. Doxycycline administration does not alter the food and drinking behavior of wild-type mice. Wild-type mice receiving either tap water or doxycycline were housed individually in metabolic cages with free access to food and water. Doxycycline was administered in the drinking water (0.2 mg/ml) for 3 days. Ingestion of (A) water and (B) food as well as (C) urine excretion and (D) sodium balance were measured. Values were expressed as mean per day ± SEM, n = 5.
Figure S7. (A) Body weight was measured before L-NAME treatment (nontreated), after the washout period, and after 3 weeks of salt load (high salt) in wild-type, it-ACE, and it-ACE treated with doxycycline (it-ACE + DOX). Doxycycline was administered during the washout and the high-salt phase. L-NAME was given to wild-type (0.5 mg/ml) and it-ACE mice (1.5 mg/ml) in the drinking water. (B) Prorenin and renin were evaluated by Western blot in total kidney homogenate of wild-type, it-ACE, and it-ACE + DOX. Values represent mean SEM, *P < 0.05, **P < 0.01, ***P < 0.001 versus the corresponding nontreated mice, n = 4–5.