

Abstract
The intracellular protein B‐cell‐lymphoma‐2 (BCL2) has been considered an attractive target for cancer therapy since the discovery of its function as a major promoter of cell survival (an anti‐apoptotic) in the late 1980s. However, the challenges of targeting a protein‐protein interaction delayed the discovery of fit‐for‐purpose molecules until the mid‐2000s. Since then, a series of high affinity small organic molecules that inhibits the interaction of BCL2 with the apoptotic machinery, the so‐called BH3‐mimetics, have been developed. Venetoclax (formerly ABT‐199) is the first to achieve US Food and Drug Administration approval, with an indication for treatment of patients with previously treated chronic lymphocytic leukemia (CLL) bearing deletion of the long arm of chromosome 17. Here, we review key aspects of the science underpinning the clinical application of BCL2 inhibitors and explore both our current knowledge and unresolved questions about its clinical utility, both in CLL and in other B‐cell malignancies that highly express BCL2.
Apoptosis and the biology of B‐cell malignancies
The B‐cell‐lymphoma‐2 (BCL2) gene was discovered as a partner in the recurring t(14;18) chromosomal translocation that is the hallmark of follicular lymphoma.1 Initially of unknown function, several years later its oncogenic potential was revealed by the discovery of its unique role in promoting cell survival.2 The finding that overexpression of BCL2 protected cells from apoptosis seeded impetus for research into cell death regulation and its relationship to cancer formation. Recapitulation of the t(14;18) translocation in murine B cells revealed its capacity to expand B lymphogenesis3 and thereby accelerate lymphomagenesis when other co‐operating oncogenic events (such as MYC dysregulation) occurred.4 Evasion of apoptosis is now recognized as one of the hallmarks of cancer and is a prominent feature of many B‐cell malignancies.
B‐cell‐lymphoma‐2 regulates the intrinsic apoptosis pathway
There are two major pathways to apoptosis—an extrinsic pathway that is triggered by ligation of so‐called death receptors on the cell surface (e.g., tumor necrosis factor‐α to its cognate receptor) and the intrinsic pathway that is triggered by diverse cellular stresses, such as loss of survival signals, DNA damage, or uncontrolled cellular proliferation. Key to understanding how BCL2 has been able to be successfully targeted is detailed knowledge of how the intrinsic pathway to apoptosis is normally regulated in healthy cells. This has been elucidated in detail over the last 30 years, and been reviewed extensively elsewhere.5, 6, 7 Also referred to as the mitochondrial pathway to apoptosis, this is a series of protein‐protein interactions in the cytosol and predominantly on the outer mitochondrial membrane, which culminates in permeabilization of the outer mitochondrial membrane leading to mitochondrial depolarization, release of cytochrome C, and activation of caspases that drive cellular demolition.
The intrinsic pathway is regulated by a large family of proteins named after its founding member, BCL2 (see Figure 1).7 All contain at least one of four BCL2 homology (BH) domains and fall into three functional subfamilies. BAX and BAK are the two key death effector proteins that homodimerize or heterodimerize to permeabilized mitochondria. These two proteins are normally held inactive through direct binding by the prosurvival proteins: BCL2, MCL1, BCLxL (also known as BCL2L1), BCLW, A1 (also known as BFL‐1), and BCLB. Antagonizing their function are the pro‐apoptotic BH3‐only proteins: BIM, BID, NOXA, p53 upregulated modulator of apoptosis, BAD, HRK, BMF, and BIK. These pro‐apoptotic proteins are distant relatives of BCL2 and share only one BH domain with the other two subfamilies. Hence, they are referred to as the BH3‐only proteins.6
Figure 1.
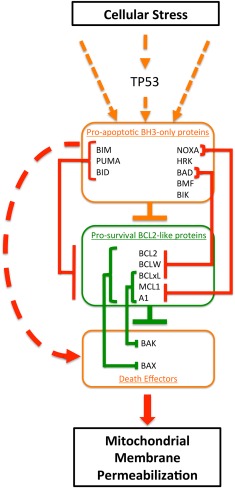
Overview of the regulation of the intrinsic pathway to apoptosis by B‐cell‐lymphoma‐2 (BCL2) family members. Within the cytoplasm of normal cells, apoptosis is regulated by highly specific interactions between three subfamilies of the BCL2 protein family. The BCL2 homology (BH)3‐only proteins integrate a multitude of stress‐induced signals, and apoptosis is unleashed when the net BH3‐only pro‐apoptotic activity exceeds the activity of the prosurvival proteins, most prominent of which is BCL2. In healthy cells, BCL2 and structurally and functionally related proteins, such as MCL1 or BCLxL, bind and repress the activity of the third subfamily of BCL2‐like proteins, the death effectors (mediators) BAX and BAK. When sufficient stress signals are applied, prosurvival proteins are displaced from BAX/BAK by interaction with BH3‐only proteins, allowing BAX and BAK to oligomerize on the outer membrane of mitochondria, triggering its permeabilization, depolarization, cytochrome C release, caspase activation, and cell death, morphologically recognizable as apoptosis. Stresses related to DNA damage from chemotherapy and from oncogenic signaling typically induce BH3‐only protein activity via the TP53 pathway. Interactions between BH3‐only proteins and prosurvival proteins can be specific (e.g., BAD only binds BCL2, BCLxL, and BCLW with high affinity; and BCL2 preferentially binds and inhibits BAX), or more promiscuous (e.g., BIM will bind and inhibit all prosurvival proteins, and MCL1 will bind and inhibit both BAX and BAK).7 Orange boxes and orange lines represent apoptosis inducing proteins and actions. The red lines indicate the pro‐apoptotic action of BH3‐only proteins. Green boxes and lines represent survival promoting proteins and their actions. Lines with arrows indicate signals that enhance activity, whereas lines headed with bars indicate repressive actions.
The BCL2 family of proteins acts to prevent or induce apoptosis by integrating diverse prosurvival or pro‐apoptotic intracellular signals generated within a cell.7 In healthy cells, the death mediators BAX and BAK are directly repressed by BCL2 and other prosurvival relatives (Figure 1).7 Cellular stress signals, such as DNA‐damage‐induced TP53 activation, trigger pro‐apoptotic BH3‐only proteins (such as p53 upregulated modulator of apoptosis) to neutralize the prosurvival BCL2 proteins by binding to the same hydrophobic pocket used to bind BAX and BAK, or by directly activating BAX or BAK, thereby initiating apoptosis. Apoptosis is normally under tight control and this is achieved through specificity of interactions between prosurvival and BH3‐only proteins,8 differential induction and post‐translational modulation of BH3‐only protein expression,9 and cell‐type dependent expression of family members. Although BIM, p53 upregulated modulator of apoptosis, and BID can neutralize the function of all prosurvival proteins, BAD only binds and inhibits BCL2, BCLxL, and BCLW, and NOXA preferentially binds and inhibits MCL1 and A1.7, 8 This multilayered regulatory network allows exquisite control such that apoptosis can be triggered for any cell type in the appropriate physiological context. This implies that the threshold for each cell type to undergo apoptosis is varied and subject to control by the complex network of protein‐protein interactions.
Dysregulation of apoptosis in B‐cell malignancies
The intrinsic pathway is commonly perturbed in B‐cell malignancies. Table 1 summarizes the different patterns of expression of prosurvival proteins in the cancers that are the focus of this review. Importantly, high BCL2 expression is almost universal in chronic lymphocytic leukemia (CLL), follicular lymphoma, mantle cell lymphoma (MCL), and Waldenstrom macroglobulinemia. In contrast, the levels of BCL2 expression are somewhat more variable among myeloma and substantially more variable among diffuse large B‐cell lymphoma (DLBCL) and B‐lineage acute lymphoblastic leukemia. Even where BCL2 expression is enforced by oncogenic events (e.g., loss of repression of BCL2 in CLL due to deletion of genetic loci for miRNA‐15 and miRNA‐1610 or t(14;18) in follicular lymphoma),1 the malignant cells will still respond to external stimuli (e.g., cytokines and cell‐cell interactions) to modulate expression of other prosurvival proteins. In particular, BCLxL and MCL1 expression are upregulated by cytokine signaling and CD40 ligation in CLL cells in lymph nodes11, 12 and in myeloma cells under conditions that mimic the bone marrow microenvironment.13
Table 1.
Patterns of expression of B‐cell‐lymphoma‐2 family prosurvival proteins in selected B‐cell malignancies
| B‐cell malignancy | Major prosurvival proteins expressed | Level of BCL2 expression | Variability | Comment on mechanism |
|---|---|---|---|---|
| CLL | BCL2 > MCL >> BCLxL12, 14 | High | Some variability, but always high | BCL2: loss of repression by miRNA 15/1610; MCL1 & BCLxL induced by CD40 ligation and microenvironmental stimuli12, 15 |
| Follicular lymphoma | BCL21; BCLxL16; MCL117, 18 | High | Rare to not be expressed | t(14;18) leads to constitutive expression from IgH promotor1; CD40L stimulates BCLxL expression16; MCL1 in centroblasts17 |
| Diffuse large B‐cell lymphoma | BCL2 or MCL118 | High in GC type; low in many ABC type | High; where MYC‐driven MCL1 > BCL2 | Varies: including gene amplification,19 t(14;18) in double‐hit lymphoma, consequence of MYC dysregulation20 |
| MCL | BCL2 > MCL118, 21 | High | Minor | Consequence of Cyclin D1 dysregulation22; MCL1 high in blastoid21 |
| Myeloma | BCL2, MCL1, BCLxL13, 23 | High, especially t(11;14) | Moderate | BCL2 consequence of Cyclin dysregulation24; MCL1 constitutively expressed by plasma cells25; BCLxL increased through microenvironmental stimulation13 |
| ALL | BCL2, MCL1, BCLxL26, 27 | Variable | Significant26 | Appears to mimic expression pattern of precursor cell; patterned by oncogenic driver |
ABC, activated B‐cell or origin subtype; ALL, acute lymphoblastic leukemia; BCL2, B‐cell‐lymphoma‐2; GC, germinal center cell of origin subtype; MCL, mantle cell lymphoma.
As portrayed in Figure 1,7 apoptosis can also be perturbed by loss of BH3‐only protein function. A common mechanism for dampening apoptotic activity in CLL is loss of TP53 function through either gene deletion (loss of the long arm of chromosome 17; del(17p)) or mutation of the TP53 gene, or both. In CLL, myeloma, aggressive lymphomas, and B‐lineage acute lymphoblastic leukemia, del(17p) is associated with poor prognosis and resistance to (or propensity to early relapse after) DNA‐damaging chemotherapy regimens. Failure of TP53 pathway signaling in response to cellular stress, particularly DNA damage, diminishes induction of BH3‐only protein expression and activity. Less frequently, genetic loss28 or epigenetic silencing29 of the BIM locus is observed in some B‐cell malignancies, and these can often impair the normal apoptosis responses to stress signals.
Development of BH3‐mimetics to target B‐cell‐lymphoma‐2
What are the characteristics of a true BH3‐mimetic?
Having established that BCL2 function is normally inhibited by binding of selective BH3‐only proteins into a grooved pocket that also serves as the binding site for BAX, a logical approach to drug design was to identify small molecules that would bind this same pocket with high affinity. Such molecules would act as mimics of BH3‐only proteins, and the term “BH3‐mimetic” was coined. Key features of chemical compounds that allow them to be classified as acting as BH3‐mimetics have been enunciated by Lessene et al.30 and include (i) the biological activity of the agent must be to induce apoptosis via mitochondrial disruption by BAX and BAK; (ii) the agent must bind to at least one BCL2 family member with high affinity; (iii) the activity of the agent must correlate with the expression of the relevant BCL2 family members in the cell of interest; and (iv) relevant biomarkers should be affected by the agent in animal models. Earlier attempts to identify BH3‐mimetics falsely identified a number of cytotoxic compounds that were active against BCL2‐overexpressing cancer cell lines, but which lacked the specific mechanisms of action that mimicked BH3‐only protein activity31 (reviewed in refs. 32 and 33). None of these compounds have been successfully developed into a registered therapeutic. Part of the problem lay in the lack of precedent for successful design of drugs targeting protein‐protein interactions. Ultimately, however, use of technical innovations, such as structure‐activity relationship‐by‐nuclear magnetic resonance34 and state of the art combinatorial chemistry, coupled with the aforementioned biological understanding, enabled the generation of potent and specific BH3‐mimetics.35
Early B‐cell‐lymphoma‐2‐inhibiting BH3‐mimetics
The first such BH3‐mimetic was ABT‐737, a small organic molecule tool compound that bound BCL2, BCLxL, and BCLW with high affinity.35 ABT‐737 mimicked the action of the BH3‐only protein BAD and displayed low nanomolar activity in cell‐based assays against cell lines dependent on each of the three prosurvival proteins. Further, it was inactive when cell lines expressed high levels of MCL1,31 and the most sensitive cell lines were those with high BCL2 expression.35 In vitro, follicular lymphoma and CLL cells from patients were killed when incubated with <100 nM ABT‐737.14, 35, 36
Despite providing major insights into how to clinically develop a BCL2 inhibitor in preclinical models, ABT‐737 was not sufficiently orally bioavailable to take into clinical trials. Navitoclax (ABT‐263), an analog with similar target specificity and activity, was developed for this purpose.37, 38 As predicted preclinically, navitoclax showed single agent antitumor activity against CLL39, 40 and follicular lymphoma39 in early phase clinical trials, but full exploration of its potential as a BCL2 inhibitor was compromised by on‐target toxicity related to BCLxL inhibition. BCLxL is a key survival factor for platelets41 and, during the development of navitoclax, it was recognized that pharmacological inhibition of BCLxL would lead to apoptosis of platelets and acute thrombocytopenia.42 In patients, highly reproducible dose‐proportional reductions in platelet counts were observed within the first 48–72 h of commencement of daily dosing.39, 40 This proved dose‐limiting in patients with CLL.40 The thrombocytopenia could be mitigated by starting at low doses and building to the maximal tolerated dose of 250 mg/day, allowing increases in bone marrow megakaryopoiesis to partially compensate for the increased peripheral destruction of platelets. Nevertheless, it was recognized that it was desirable to develop a BCL2‐specific BH3‐mimetic to avoid this complication. In spite of limitations posed by reductions in platelet counts, navitoclax did induce partial responses in 35% of heavily pretreated patients with CLL, and many of these responses proved durable with ongoing therapy (median progression‐free survival of 25 months).40
Venetoclax
The first potent BCL2‐selective BH3‐mimetic to be reported was venetoclax (previously known as ABT‐199 or GDC‐0199), 4‐(4‐{[2‐(4‐chlorophenyl)‐4,4‐dimethylcyclohex‐1‐en‐1‐yl]methyl}piperazin‐1‐yl)‐N‐({3‐nitro‐4‐[(tetrahydro‐2H‐pyran‐4‐ylmethyl)amino]phenyl}sulfonyl)‐2‐(1H‐pyrrolo[2,3‐b]pyridin‐5‐yloxy)benzamide).43 Like navitoclax, its in vitro cytotoxicity was dependent upon the presence of BAX and BAK, and cells died by apoptosis. Unlike navitoclax, venetoclax had strong affinity only for BCL2, with >100‐fold less affinity for BCLxL or BCLW.43 Consistent with this, CLL cells were highly sensitive to venetoclax (median LC50 3 nM), whereas platelets were relatively resistant (median LC50 >5,000 nM) in vitro.43, 44 Indeed, venetoclax seemed more potent than navitoclax against CLL, suggesting that BCL2 inhibition rather than BCL2 and BCLxL inhibition underpinned the activity observed for navitoclax in patients.
Mechanism of action of venetoclax in vivo
Based on promising preclinical data and the prior experience with navitoclax, the first clinical testing occurred in patients with CLL (and the closely related condition, small lymphocytic lymphoma) refractory to standard therapies.45 Venetoclax induced dose‐dependent reductions in lymphocyte counts within 8–24 h of first exposure.43, 45 Consistent with its expected mechanism‐of‐action, the cell death triggered by venetoclax was apoptotic.46 Circulating CLL cells collected 6 and 24 h after the first oral dose displayed caspase activation and cell membrane phosphatidylserine exposure.46 A prediction arising from the model of regulation of apoptosis depicted in Figure 1,7 is that BH3‐mimetics should be cytotoxic irrespective of whether the TP53 pathway is fully functional. This prediction was confirmed by analyzing the responses of CLL that lacked TP53 function through deletion of one allele of TP53 and mutation of the other. Both in vitro sensitivity and in vivo initial responses to venetoclax were indistinguishable between subgroups of CLL with normal or dysregulated TP53 function.46
Clinical data for venetoclax
Venetoclax commenced clinical trials in June 2011, and formally peer‐reviewed data began emerging in 2016. To date, phase I45 and II47 trials of venetoclax monotherapy in patients with relapsed or refractory CLL have been published. Some mature data for monotherapy in lymphoma48 and combination therapy in relapsed CLL49 have also been presented at international meetings.
Efficacy of venetoclax monotherapy in previously treated patients with chronic lymphocytic leukemia
The first‐in‐human dose‐escalation phase I study enrolled 116 patients, the majority of whom were heavily pretreated and refractory to their most recent prior therapy. This trial identified 400 mg taken once daily as the recommended phase II dose for venetoclax, and reported marked anti‐CLL activity across a wide range of doses (50–1,200 mg).45 Subsequently, a phase II trial of continuous venetoclax 400 mg/day in patients with relapsed or refractory del(17p) CLL was initiated.
Venetoclax displayed substantial cytotoxic activity against CLL in all tissue compartments. As assessed using International Workshop on CLL 2008 criteria, 79% of all patients in phase I achieved an objective response, regardless of the dose of venetoclax. Complete remissions were observed in 20% of patients. Although not systematically performed, negativity for minimal residual disease (MRD) in the bone marrow by sensitive flow cytometry was observed in 5% of all patients. Objective responses were as common among patients who manifest clinical features typically associated with poor response to chemo‐immunotherapy, including older age, fludarabine‐refractoriness, and deletion of either chromosome 17p or chromosome 11q (see Figure 2).45, 47, 49, 65 Similarly, in the phase II trial conducted exclusively in patients with del(17p) CLL, independent review committee‐assessed objective responses were documented in 79% of all patients.47 Again response rates were similar regardless of whether the CLL was refractory to prior fludarabine‐based therapy or had a deleterious mutation in the remaining allele of TP53. Documentation of complete remissions was less frequent in phase II, but this may well be a function of the shorter duration of follow‐up. In the phase I trial, the median time to complete remission was 6 months, with several patients only achieving this depth of response after >18 months of continuous treatment. The median follow‐up for the phase II trial when published was only 12 months. As for the phase I trial, MRD was not systematically collected from the outset of the phase II trial. Eighteen of 45 patients tested for MRD in the peripheral blood were negative, 6 of whom were also negative for MRD in the bone marrow.
Figure 2.
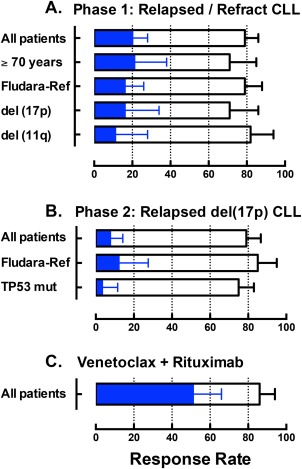
The graph summarizes previously published or presented overall response and complete remission rates for patients with relapsed or refractory chronic lymphocytic leukemia (CLL) treated with venetoclax on early phase clinical trials. Open bars represent the overall response rate and the blue bars indicate the complete remission rate, as assessed against International Workshop on CLL 2008 criteria. The error bars indicate the upper 95% confidence interval for the response rates. (a) Pooled data across all dose cohorts for 116 patients entering the first‐in‐human phase I study.45 (b) Independent review committee‐assessed data for the 107 patients with del(17p) CLL entering the phase II trial at the now‐approved 400 mg/day dose.47 (c) Pooled data across all dose cohorts for 49 patients treated with the combination of venetoclax and rituximab (6 doses only) on the phase Ib trial.49, 65 Fludara‐Ref, refractory to previous fludarabine‐containing therapy.
Collectively, these data indicate that venetoclax monotherapy induces response in the majority of patients with previously treated CLL/small lymphocytic lymphoma, and that up to one fifth of patients may achieve a complete remission with ongoing treatment. Responses in a small minority are MRD‐negative. Formal determination of the durability of responses is hampered by the range of doses tested in the more mature phase I trial, and the immaturity of follow‐up for phase II. For patients receiving 400 mg/day in the phase I trial, the 15‐month estimate for durability of response is 75% (95% confidence interval = 58–86),45 whereas the 12‐month estimate in phase II is 85% (95% confidence interval = 75–91).47 In both studies, patients achieving a complete remission were less likely to progress than patients whose best response was partial. In partial responders and nonresponders, the risk of progression was continuous, and, in some patients, manifest as Richter transformation to aggressive lymphoma. The latter phenomenon was more common in the phase I study (16% of all study entrants) than in the phase II trial (10%).
Toxicity of venetoclax monotherapy in previously treated patients with chronic lymphocytic leukemia
The most important safety finding in the phase I trial was the occurrence of tumor lysis in some patients after the first dose of venetoclax.45 Early in the study, rapid reductions in circulating lymphocyte counts associated with biochemical changes of tumor lysis (rises in serum phosphate, serum lactate dehydrogenase, and, in some patients, serum potassium; or falls in serum calcium) were observed. Three patients experienced clinically significant tumor lysis syndrome, including two patients with serious clinical complications (one acute renal failure, and one sudden death). Among the first 56 patients, 7 others experienced tumor lysis syndrome based solely on biochemical markers. The principle risk factor for development of tumor lysis syndrome with venetoclax was recognized to be high tumor burden. Following the introduction of a slow ramp‐up in dosing from 20 mg/day in the first week, through 50, 100, 200 mg/day to 400 mg/day in weekly steps, and strict monitoring for biochemical evidence of tumor lysis, no clinically significant tumor lysis syndrome was observed in the final 60 patients, and only 1 patient had an adverse event related to biochemical changes indicative of tumor lysis.45 This mitigation strategy proved effective in the phase II trial. Laboratory tumor lysis syndrome was reported in 5 of 107 patients during the ramp‐up period (4 patients within the first 2 days of treatment; 1 patient at week 3 of treatment) and resolved without clinical sequelae, and with minimal or no interruption of dosing.47
Common toxicities attributable to venetoclax were mild gastrointestinal side effects and neutropenia.45, 47, 50 Approximately half of all patients experienced mild nausea and/or vomiting, especially soon after commencing the treatment. This was typically self‐limiting, but some patients required anti‐emetics. Similarly, mild diarrhea manifest as one or two extra bowel actions/day was common (50% in phase I and 29% in phase II). As for nausea, some patients required antidiarrheal therapy. Discontinuation due to gastrointestinal side effects was rare in both the phase I and II trials. Whether the mechanism of the gastrointestinal toxicity is an on‐target effect of BCL2 inhibition or a consequence of the excipient is unknown at this time.
Neutropenia below 500 cells/μL was also common, but complications of neutropenia were not. In phase I, grade 4 neutropenia was observed in 28% of patients and 23% in phase II. Grade 4 neutropenia responded to intermittent use of granulocyte colony‐stimulating factors or to dose reduction of venetoclax and was most common in patients who entered the trials with pre‐existing neutropenia. The pathophysiology of the neutropenia relates to selective toxicity against granulocytic progenitor cells,51 and its frequency is likely to have been increased by the extent of prior stem cell damaging chemotherapy in the trial population and the degree of marrow infiltration by the CLL. Consistent with this, the incidence of grade 3 or 4 neutropenia was lower in patients with lymphoma receiving venetoclax monotherapy—12%.48 Febrile neutropenia occurred in 6% and 5% of patients with CLL in the phase I and II trials, respectively. Serious infections (≥ grade 3) were infrequent, occurring in 17% of patients in phase I and 20% in phase II (1% fatal). The exposure‐adjusted rate for serious infections was 1.4 per 100 patient months in the phase I trial. The most common significant infections were respiratory tract infections, as is typical for any population of patients with CLL requiring therapy. Infection prophylaxis was not mandated in either trial. At our center, use of prophylactic valaciclovir and co‐trimoxazole for herpetic and Pneumocystis infections was usual for patients who were heavily immunocompromised at study entry.
Efficacy of venetoclax monotherapy in other B‐cell lineage hematological malignancies
To date, results of phase I monotherapy trials in relapsed or refractory lymphoma48 and multiple myeloma52 have not been published in the peer‐reviewed literature, but have been presented at major conferences. Patients in both studies were treated in dose escalation cohorts (up to 1,200 mg/day). The preliminary results of these ongoing studies are summarized in Table 2.48, 52 Among the common B‐cell lymphomas, only MCL seemed to be as sensitive to venetoclax as CLL.48 Responses were less frequent in follicular lymphoma and DLBCL, and the doses required to achieve responses were typically higher (600–1,200 mg/day) than required in CLL. Although durable benefits were observed in responding patients with MCL and follicular lymphoma, with median progression‐free survival estimates of 14 and 11 months, respectively, this was not the case in aggressive DLBCL. BCL2 expression by the tumor cells was not a criterion for study entry, and, as yet, the relationship between BCL2 expression and clinical responses in patients with lymphoma is unknown.
Table 2.
Preliminary efficacy of venetoclax monotherapy for lymphoma and myeloma in phase I clinical trials
| B‐lineage malignancy | No. | Response rate | Durability of benefit (progression‐free survival) | Reference | |
|---|---|---|---|---|---|
| Overall | CR | ||||
| Follicular lymphoma | 29 | 38% | 14% | Median 11 mo | 48, a |
| MCL | 28 | 75% | 21% | Median 14 mo | 48, a |
| DLBCL | 34 | 18% | 12% | Median 1 mo | 48, a |
| Waldenstrom macroglobulinemia | 4 | 100% | 0% | NR | 48, a |
| Myeloma | |||||
| All | 43 | 12% | 5% | NR | 52 |
| t(11;14) | 17 | 24% | 12% | NR | |
CR, complete response; DLBCL, diffuse large B‐cell lymphoma; MCL, mantle cell lymphoma; NR, not reported.
Data as updated at ASH 2015 meeting presentation.
In heavily pretreated patients with relapsed or refractory myeloma, venetoclax has some single agent activity,52 but complete responses are rare and seem confined to the subgroup of t(11;14) myeloma, as predicted by preclinical testing.24 BCL2 expression is generally readily observed by immunohistochemistry in myeloma, and, therefore, these data suggest that BCL2 expression alone is insufficient as a biomarker for response to venetoclax in this disease.
Unanswered questions arising from initial clinical applications
What are the mechanisms of resistance?
To date, the molecular mechanisms of acquired resistance to venetoclax in patient samples have not been defined. In contrast, there is a wealth of preclinical data addressing the issue that provide insight to the question. From early in the development of BCL2 inhibitors, it was recognized that MCL1 expression was likely to provide cells with an alternative way to suppress apoptosis, even when BCL2 function was blocked.31 However, MCL1 expression is common in CLL,14 and the clinical experience with navitoclax treatment indicated that objective responses were achievable despite MCL1 expression.40 Several groups have described the importance of BIM to the response of CLL cells in vitro to BCL2 inhibitors.36, 53 Analyses of limited biospecimens from patients treated with navitoclax suggested that it was the balance between levels of expression of pro‐apoptotic BIM and prosurvival MCL1 that influenced response. A low BIM:MCL1 ratio was associated with lack of response to navitoclax.40
Figure 3, 53 highlights the importance of the relevant abundance of different prosurvival proteins to the cytotoxicity of venetoclax. Normal cells from many tissues express a variety of BCL2‐like prosurvival proteins, and so BCL2 is largely redundant (Figure 3 a). BCL2‐overexpressing cancer cells (e.g., CLL) have a gross imbalance between the levels of BCL2 and other prosurvival proteins, leading to an apparent dependency on BCL2 for survival. This creates a “primed for death” situation in which the high levels of BCL2 sequester high levels of BIM in the viable leukemic cell.54 In these cells, once BCL2 is inhibited, most of the signal maintaining survival is lost, and BIM, which has been displaced from BCL2, is available to neutralize any residual prosurvival activity from less abundant molecules of MCL1 or BCLxL molecules53 (Figure 3 b). Therefore, resistance is expected when expression of non‐BCL2 prosurvival proteins is dominant. Figure 3 also highlights how this may arise—through heightened expression of MCL1 or BCLxL in response to extracellular signals from the microenvironment11, 12, 15 (Figure 3 c), or through dysregulation of expression through genetic aberration (typically copy number increase) or epigenetic upregulation of upstream signaling pathways, such as AKT55 (Figure 3 d).
Figure 3.
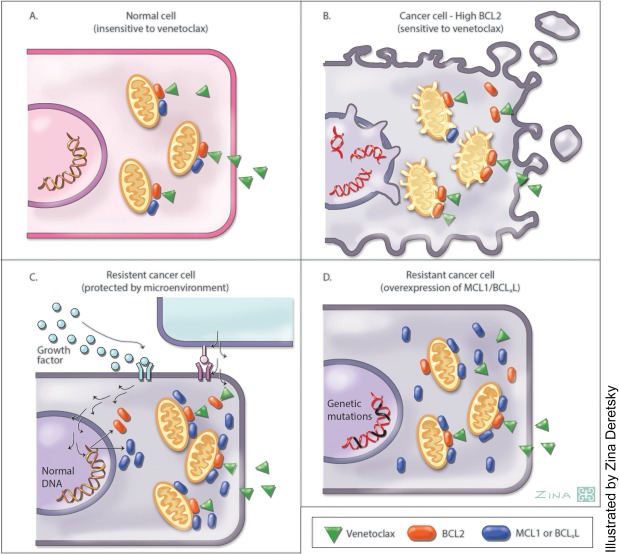
The figure depicts current understanding of cellular sensitivity or resistance to cytotoxicity induced by venetoclax. (a) Normal cells express a mix of prosurvival proteins, and are not sensitive to venetoclax (green arrowheads). (b) Malignant cells that highly express B‐cell‐lymphoma‐2 (BCL2) (red) and have minimal expression of other prosurvival proteins (blue), depend predominantly on BCL2 for survival. Chronic lymphocytic leukemia (CLL) cells and mantle cell lymphoma are examples. In these cells, venetoclax directly inhibits the prosurvival activity of BCL2, and any residual prosurvival activity related to low level MCL1 or BCLxL expression is inhibited by the BCL2 homology (BH)3‐only activity of BIM after it has been displaced from BCL2 by venetoclax.53 Cells die by apoptosis after the initiation of depolarization of mitochondria by BAX and BAK. This is recognized morphologically by cytoplasmic blebbing. (c) Malignant cells that overexpress BCL2 may be protected from venetoclax cytotoxicity by the induction of high level MCL1 or BCLxL expression by extracellular stimuli from the tumor microenvironment (e.g., CLL cells in very bulky lymph node masses or follicular lymphoma cells in the lymph nodes). (d) Malignant cells may constitutively express high levels of MCL1 or BCLxL due to gene amplification, chromosomal translocation, or endogenous activation of upstream signaling pathways (e.g., AKT that mimic microenvironmental signals).
Why is venetoclax highly effective in chronic lymphocytic leukemia, but apparently less so in follicular lymphoma?
Both CLL and follicular lymphoma routinely express BCL2 in high levels in most malignant cells. However, early clinical data indicate that venetoclax is substantially more effective in CLL than in follicular lymphoma, at least in patients with disease previously treated with chemo‐immunotherapy.45, 48 Further, it seems that higher doses of venetoclax are required in follicular lymphoma to achieve substantial cytoreduction. It is tempting to speculate that the differences relate to the predominantly nodal nature of follicular lymphoma, presumably providing a microenvironment that induces expression of sufficient BCLxL and MCL to impair the susceptibility of the lymphoma cells to venetoclax.
How should venetoclax be combined with other therapies?
From the above discussion, the rationale for combining BCL2 inhibitors with other agents is clear. The BH3‐mimetic BCL2 inhibitors ABT‐737,35 navitoclax,37 and venetoclax43 consistently demonstrate augmentation of efficacy for DNA damaging chemotherapy,22, 43, 56 monoclonal antibodies,38 tyrosine kinase inhibitors,57, 58, 59 steroids,60 and proteasome inhibitors13, 61 both in vitro 14, 51, 57, 58, 59 and in vivo 13, 37, 43, 51, 56, 61 model systems. To date, the majority of preclinical models suggest that for a given tumor type (e.g., CLL, lymphoma, and myeloma), combination with an agent with established efficacy in that tumor is likely to increase efficacy.
In the clinic, venetoclax is being combined with all the aforementioned classes of agents. The most mature combination data relate to trials of venetoclax plus rituximab in patients with relapsed CLL,49 and venetoclax plus bortezomib and dexamethasone in relapsed refractory myeloma.62 Building on earlier experiences with navitoclax, in which combination with rituximab increased response rates in CLL63 and follicular lymphoma,64 six doses of rituximab at monthly intervals have been combined with continuous venetoclax in patients with relapsed or refractory CLL.49 Preliminary reports indicate that this combination achieves an ∼50% complete remission rate without additional toxicity49, 65 (Figure 2 c). Importantly, >50% of patients have been reported to have clearance of MRD from the bone marrow, and some patients in remission have now ceased all therapy and experienced durable responses. Two patients have relapsed after 2 years off‐treatment and both were successfully re‐treated with venetoclax.49 Although early, this experience suggests that the combination increases the depth of response achievable with BCL2 inhibition alone, and thereby opens the possibility for abbreviated rather than continuous therapy in patients with CLL. An ongoing phase III trial is testing this combination and strategy in comparison with standard chemo‐immunotherapy in patients with relapsed CLL (NCT02005471).
Previous experience with navitoclax in combination with cytotoxic chemotherapy revealed that myelosuppression (both neutropenia and thrombocytopenia) was significant.66 Trials of chemotherapy plus venetoclax and anti‐CD20 monoclonal antibody regimens in lymphoma are ongoing (e.g., NCT02187861). In myeloma, the addition of venetoclax to standard bortezomib plus dexamethasone seems tolerable,62 and a phase III trial has commenced (NCT02755597). In CLL and MCL, preclinical data indicate synergy between Bruton tyrosine kinase inhibition and BCL2 inhibition.58, 59 Phase II clinical trials in each condition are underway (e.g., NCT02756897 and NCT02471391). Early data in patients with relapsed MCL suggest that such a combination is tolerable and can achieve complete responses, including on positron‐emission tomography scanning.67
Will targeting of other prosurvival proteins prove to be clinically tractable?
An important question that arises from the successful application of a BCL2‐inhibiting BH3‐mimetics (venetoclax) to treatment of a BCL2‐dependent cancer (CLL), is whether BH3‐mimetics, which target other prosurvival proteins, can also succeed clinically. Conceptually, the answer is yes, if (i) similarly potent and specific inhibitors can be generated with adequate pharmacological properties, if (ii) malignancies with dependency on the target protein can be identified, as they were for BCL2 inhibitors in CLL and some lymphomas, and (iii) if normal cells have sufficient redundancy for target protein function to generate a therapeutic window. For BCLxL as a target, the navitoclax experience suggests that the acute reductions in platelet counts consequent upon the role of BCLxL in regulating platelet survival are a barrier to application in hematological malignancies, either as a single agent or in combination with cytotoxics, in which severe neutropenia was also observed. Whether drugs that solely target BCLxL rather than both BCL2 and BCLxL will have less toxicity in patients when used in combination with other classes of therapeutics is unknown. MCL1 is an attractive therapeutic target, particularly in multiple myeloma23 and MYC‐driven lymphomas20 among the B‐cell malignancies. Normal plasma cells require MCL1 for development,25 and the protein is widely and highly expressed in primary myeloma samples. Emerging genetic and chemical biology data indicate MCL1 dependence in a majority of myeloma cell lines, and susceptibility of the same cell lines to MCL1 inhibition using various preclinical peptidyl and chemical tools.23, 68, 69 Clinical candidate compounds have recently been identified, and a therapeutic index identified in early preclinical models.70
Early conclusions about B‐cell‐lymphoma‐2 inhibition as cancer therapy
Sufficient experience now has accrued to draw some general conclusions about the introduction of BH3‐mimetic BCL2 inhibitors into the therapeutic armamentarium. First, the US Food and Drug Administration approval reflects the phase I and II clinical data indicating that venetoclax monotherapy is an effective and tolerable treatment for patients with previously treated del(17p) CLL. Second, once the biology of how BCL2 and related molecules regulate apoptosis was unraveled and potent and specific BH3‐mimetics subsequently generated, translation into clinical application was rapid. Third, preclinical data has proved predictive of the experience in the clinic, at least for the diseases most studied to date—B‐cell malignancies, including CLL, lymphomas, and multiple myeloma. This suggests that further development of BCL2 inhibitors and indeed inhibitors of other prosurvival proteins, such as MCL1, can be guided with some confidence by appropriate laboratory models. Fourth, high level BCL2 expression is necessary, but not sufficient, for prediction of susceptibility to venetoclax. A major focus of ongoing research should be identification of biomarkers that allow discrimination between BCL2‐expressing tumor cells, which have a dependency on BCL2 that can be exploited by treatment with venetoclax, and ostensibly phenotypically similar cells that are resistant to BCL2 inhibition alone. Logically, this leads to the final conclusion that rational combination therapies hold the key to maximizing the impact of BCL2 inhibition in patients with currently incurable B‐cell malignancies. Again, preclinical observations of additive and synergistic benefit are being supported in emerging preliminary data from early phase clinical trials. With over 20 registered clinical trials of venetoclax‐containing regimens currently ongoing, we anticipate the need for an updated review of the clinical application of venetoclax within 3–5 years.
SOURCE OF FUNDING
Research by A.W.R. and D.C.S.H. is supported by fellowships and grants from the National Health and Medical Research Council of Australia, the Cancer Council of Victoria, the Leukemia and Lymphoma Society, the Victorian Cancer Agency, and the Australian Cancer Research Foundation.
CONFLICT OF INTEREST
A.W.R. has received research support from AbbVie and Genentech for conduct of translational research associated with clinical trials of venetoclax. A.W.R. and D.C.S.H. are employees of the Walter and Eliza Hall Institute of Medical Research that receives milestone and royalty payments related to venetoclax.
AUTHOR CONTRIBUTIONS
A.W.R. and D.C.S.H. co‐wrote the manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of a large number of scientists in many laboratories around the world to this field. In particular, we thank the clinical and laboratory research colleagues at Walter and Eliza Hall Institute, Royal Melbourne Hospital, and Peter MacCallum Cancer Centre for helpful discussions; and our former PhD students Andrew Wei, Mark van Delft, Kylie Mason, Seong Lin Khaw, and Mary Ann Anderson for their pioneering research into BH3‐mimetics.
References
- 1. Tsujimoto, Y. , Cossman, J. , Jaffe, E. & Croce, C.M. Involvement of the bcl‐2 gene in human follicular lymphoma. Science 228, 1440–1443 (1985). [DOI] [PubMed] [Google Scholar]
- 2. Vaux, D.L. , Cory, S. & Adams, J.M. Bcl‐2 gene promotes haemopoietic cell survival and cooperates with c‐myc to immortalize pre‐B cells. Nature 335, 440–442 (1988). [DOI] [PubMed] [Google Scholar]
- 3. McDonnell, T.J. et al bcl‐2‐immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57, 79–88 (1989). [DOI] [PubMed] [Google Scholar]
- 4. Strasser, A. , Harris, A.W. , Bath, M.L. & Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl‐2. Nature 348, 331–333 (1990). [DOI] [PubMed] [Google Scholar]
- 5. Cory, S. & Adams, J.M. The Bcl2 family: regulators of the cellular life‐or‐death switch. Nat. Rev. Cancer 2, 647–656 (2002). [DOI] [PubMed] [Google Scholar]
- 6. Adams, J.M. et al Subversion of the Bcl‐2 life/death switch in cancer development and therapy. Cold Spring Harb. Symp. Quant. Biol. 70, 469–477 (2005). [DOI] [PubMed] [Google Scholar]
- 7. Strasser, A. , Cory, S. & Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 30, 3667–3683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen, L. et al Differential targeting of prosurvival Bcl‐2 proteins by their BH3‐only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403 (2005). [DOI] [PubMed] [Google Scholar]
- 9. Kutuk, O. & Letai, A. Regulation of Bcl‐2 family proteins by posttranslational modifications. Curr. Mol. Med. 8, 102–118 (2008). [DOI] [PubMed] [Google Scholar]
- 10. Cimmino, A. et al miR‐15 and miR‐16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. U S A 102, 13944–13949 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vogler, M. et al Concurrent up‐regulation of BCL‐XL and BCL2A1 induces approximately 1000‐fold resistance to ABT‐737 in chronic lymphocytic leukemia. Blood 113, 4403–4413 (2009). [DOI] [PubMed] [Google Scholar]
- 12. Herishanu, Y. et al Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL‐1. Leuk. Lymphoma 52, 1758–1769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Punnoose, E.A. et al Expression profile of BCL‐2, BCL‐XL, and MCL‐1 predicts pharmacological response to the BCL‐2 selective antagonist venetoclax in multiple myeloma models. Mol. Cancer Ther. 15, 1132–1144 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Mason, K.D. et al The BH3 mimetic compound, ABT‐737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia 23, 2034–2041 (2009). [DOI] [PubMed] [Google Scholar]
- 15. Thijssen, R. et al Resistance to ABT‐199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica 100, e302–e306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghia, P. et al Unbalanced expression of bcl‐2 family proteins in follicular lymphoma: contribution of CD40 signaling in promoting survival. Blood 91, 244–251 (1998). [PubMed] [Google Scholar]
- 17. Michels, J. et al Immunohistochemical analysis of the antiapoptotic Mcl‐1 and Bcl‐2 proteins in follicular lymphoma. Br. J. Haematol. 132, 743–746 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Cho‐Vega, J.H. et al MCL‐1 expression in B‐cell non‐Hodgkin's lymphomas. Hum. Pathol. 35, 1095–1100 (2004). [DOI] [PubMed] [Google Scholar]
- 19. Wessendorf, S. et al Hidden gene amplifications in aggressive B‐cell non‐Hodgkin lymphomas detected by microarray‐based comparative genomic hybridization. Oncogene 22, 1425–1429 (2003). [DOI] [PubMed] [Google Scholar]
- 20. Grabow, S. et al Critical B‐lymphoid cell intrinsic role of endogenous MCL‐1 in c‐MYC‐induced lymphomagenesis. Cell Death Dis. 7, e2132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khoury, J.D. , Medeiros, L.J. , Rassidakis, G.Z. , McDonnell, T.J. , Abruzzo, L.V. & Lai, R. Expression of Mcl‐1 in mantle cell lymphoma is associated with high‐grade morphology, a high proliferative state, and p53 overexpression. J. Pathol. 199, 90–97 (2003). [DOI] [PubMed] [Google Scholar]
- 22. Touzeau, C. et al ABT‐737 induces apoptosis in mantle cell lymphoma cells with a Bcl‐2high/Mcl‐1low profile and synergizes with other antineoplastic agents. Clin. Cancer Res. 17, 5973–5981 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Gong, J.N. et al Hierarchy for targeting pro‐survival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood (2016); e‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 24. Touzeau, C. et al The Bcl‐2 specific BH3 mimetic ABT‐199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia 28, 210–212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peperzak, V. et al Mcl‐1 is essential for the survival of plasma cells. Nat. Immunol. 14, 290–297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Findley, H.W. , Gu, L. , Yeager, A.M. & Zhou, M. Expression and regulation of Bcl‐2, Bcl‐xl, and Bax correlate with p53 status and sensitivity to apoptosis in childhood acute lymphoblastic leukemia. Blood 89, 2986–2993 (1997). [PubMed] [Google Scholar]
- 27. Khaw, S.L. et al Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL‐rearranged leukemia. Blood 128, 1382–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mestre‐Escorihuela, C. et al Homozygous deletions localize novel tumor suppressor genes in B‐cell lymphomas. Blood 109, 271–280 (2007). [DOI] [PubMed] [Google Scholar]
- 29. Bachmann, P.S. et al Epigenetic silencing of BIM in glucocorticoid poor‐responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood 116, 3013–3022 (2010). [DOI] [PubMed] [Google Scholar]
- 30. Lessene, G. , Czabotar, P.E. & Colman, P.M. BCL‐2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 7, 989–1000 (2008). [DOI] [PubMed] [Google Scholar]
- 31. van Delft, M.F. et al The BH3 mimetic ABT‐737 targets selective Bcl‐2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl‐1 is neutralized. Cancer Cell 10, 389–399 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davids, M.S. & Letai, A. Targeting the B‐cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 30, 3127–3135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anderson, M.A. , Huang, D. & Roberts, A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin. Hematol. 51, 219–227 (2014). [DOI] [PubMed] [Google Scholar]
- 34. Shuker, S.B. , Hajduk, P.J. , Meadows, R.P. & Fesik, S.W. Discovering high‐affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534 (1996). [DOI] [PubMed] [Google Scholar]
- 35. Oltersdorf, T. et al An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005). [DOI] [PubMed] [Google Scholar]
- 36. Del Gaizo Moore, V. , Brown, J.R. , Certo, M. , Love, T.M. , Novina, C.D. & Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT‐737. J. Clin. Invest. 117, 112–121 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tse, C. et al ABT‐263: a potent and orally bioavailable Bcl‐2 family inhibitor. Cancer Res. 68, 3421–3428 (2008). [DOI] [PubMed] [Google Scholar]
- 38. Park, C.M. et al Discovery of an orally bioavailable small molecule inhibitor of prosurvival B‐cell lymphoma 2 proteins. J. Med. Chem. 51, 6902–6915 (2008). [DOI] [PubMed] [Google Scholar]
- 39. Wilson, W.H. et al Navitoclax, a targeted high‐affinity inhibitor of BCL‐2, in lymphoid malignancies: a phase 1 dose‐escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11, 1149–1159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberts, A.W. et al Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 30, 488–496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mason, K.D. et al Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 (2007). [DOI] [PubMed] [Google Scholar]
- 42. Zhang, H. et al Bcl‐2 family proteins are essential for platelet survival. Cell Death Differ. 14, 943–951 (2007). [DOI] [PubMed] [Google Scholar]
- 43. Souers, A.J. et al ABT‐199, a potent and selective BCL‐2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013). [DOI] [PubMed] [Google Scholar]
- 44. Vogler, M. , Dinsdale, D. , Dyer, M.J. & Cohen, G.M. ABT‐199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br. J. Haematol. 163, 139–142 (2013). [DOI] [PubMed] [Google Scholar]
- 45. Roberts, A.W. et al Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 374, 311–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anderson, M.A. et al The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53‐independent mechanism. Blood 127, 3215–3224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stilgenbauer, S. et al Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open‐label, phase 2 study. Lancet Oncol. 17, 768–778 (2016). [DOI] [PubMed] [Google Scholar]
- 48. Gerecitano, J.F. et al A phase 1 study of venetoclax (ABT‐199 / GDC‐0199) monotherapy in patients with relapsed/refractory non‐Hodgkin lymphoma. Blood 126, 254 (2015). [Google Scholar]
- 49. Brander, D. et al Durable treatment‐free remission and effective retreatment in patients with relapsed/refractory chronic lymphocytic leukemia who achieved a deep response with venetoclax combined with rituximab. Haematologica 101, 58 (2016). [Google Scholar]
- 50. Davids, M.S. et al Safety, efficacy and immune effects of venetoclax 400 mg daily in patients with relapsed chronic lymphocytic leukemia (CLL). J. Clin. Oncol. 34, abstract 7527 (2016). [Google Scholar]
- 51. Leverson, J.D. et al Exploiting selective BCL‐2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 7, 279ra40 (2015). [DOI] [PubMed] [Google Scholar]
- 52. Kumar, S. et al Phase 1 study of venetoclax monotherapy for relapsed/refractory multiple myeloma. J. Clin. Oncol. 34, abstract 8032 (2016). [Google Scholar]
- 53. Merino, D. et al Bcl‐2, Bcl‐x(L), and Bcl‐w are not equivalent targets of ABT‐737 and navitoclax (ABT‐263) in lymphoid and leukemic cells. Blood 119, 5807–5816 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Letai, A.G. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat. Rev. Cancer 8, 121–132 (2008). [DOI] [PubMed] [Google Scholar]
- 55. Choudhary, G.S. et al MCL‐1 and BCL‐xL‐dependent resistance to the BCL‐2 inhibitor ABT‐199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 6, e1593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mason, K.D. et al In vivo efficacy of the Bcl‐2 antagonist ABT‐737 against aggressive Myc‐driven lymphomas. Proc. Natl. Acad. Sci. USA 105, 17961–17966 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuroda, J. et al Bim and Bad mediate imatinib‐induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc. Natl. Acad. Sci. USA 103, 14907–14912 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cervantes‐Gomez, F. et al Pharmacological and protein profiling suggests venetoclax (ABT‐199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin. Cancer Res. 21, 3705–3715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chiron, D. et al Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl‐2 to overcome CD40‐induced ABT‐199 resistance in mantle cell lymphoma. Oncotarget 6, 8750–8759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matulis, S.M. et al Dexamethasone treatment promotes Bcl‐2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia 30, 1086–1093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vandenberg, C.J. & Cory, S. ABT‐199, a new Bcl‐2‐specific BH3 mimetic, has in vivo efficacy against aggressive Myc‐driven mouse lymphomas without provoking thrombocytopenia. Blood 121, 2285–2288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moreau, P. et al Phase 1b study of venetoclax combined with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Haematologica 101, 81–82 (2016). [Google Scholar]
- 63. Kipps, T.J. et al A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT‐263) with or without rituximab, in previously untreated B‐cell chronic lymphocytic leukemia. Leuk. Lymphoma 56, 2826–2833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roberts, A.W. et al Phase 1 study of the safety, pharmacokinetics, and antitumour activity of the BCL2 inhibitor navitoclax in combination with rituximab in patients with relapsed or refractory CD20+ lymphoid malignancies. Br. J. Haematol. 170, 669–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roberts, A.W. et al Impact of adding rituximab to venetoclax on the rate, quality, and duration of response in patients with relapsed/refractory chronic lymphocytic leukaemia: a cross‐study multivariable analysis. Haematologica 101, 51–52 (2016). [Google Scholar]
- 66. Vlahovic, G. et al A phase I safety and pharmacokinetic study of ABT‐263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Invest. New Drugs 32, 976–984 (2014). [DOI] [PubMed] [Google Scholar]
- 67. Tam, C. et al The combination of ibrutinib and venetoclax (Abt‐199) rapidly achieves complete remissions in patients with relapsed/refractory mantle cell lymphoma: preliminary results of the phase II aim study. Haematologica 101, 103 (2016). [Google Scholar]
- 68. Leverson, J.D. et al Potent and selective small‐molecule MCL‐1 inhibitors demonstrate on‐target cancer cell killing activity as single agents and in combination with ABT‐263 (navitoclax). Cell Death Dis. 6, e1590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Touzeau, C. et al BH3 profiling identifies heterogeneous dependency on Bcl‐2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 30, 761–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kotschy, A. et al The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482 (2016). [DOI] [PubMed] [Google Scholar]