

Abstract
Background
MK-8242 is an inhibitor of MDM2 that stabilizes the tumor suppressor TP53 and induces growth arrest or apoptosis downstream of TP53 induction.
Procedures
MK-8242 was tested against the PPTP in vitro cell line panel at concentrations from 1.0 nM to 10.0 μM and against the PPTP in vivo xenograft panels using oral gavage on Days 1–5 and Day 15–19 at a dose of 125 mg/kg (solid tumors) or 75 mg/kg [acute lymphoblastic leukemia (ALL) models].
Results
The median IC50 for MK-8242 was 0.07 μM for TP53 wild-type cell lines versus > 10 μM for TP53 mutant cell lines. MK-8242 induced a two-fold or greater delay in time to event in 10 of 17 (59%) of TP53 wild-type solid tumor xenografts, excluding osteosarcoma xenografts that have very low TP53 expression. Objective responses were observed in 7 solid tumor xenografts representing multiple histotypes. For the systemic-disease ALL panel, among 8 xenografts there were 2 complete responses (CR) and 6 partial responses (PR). Two additional MLL-rearranged xenografts (MV4;11 and RS4;11) grown subcutaneously showed maintained CR and PR, respectively. The expected pharmacodynamic responses to TP53 activation were observed in TP53 wild-type models treated with MK-8242. Pharmacokinetic analysis showed that MK-8242 drug exposure in SCID mice appears to exceed that observed in adult phase 1 trials.
Conclusions
MK-8242 induced tumor regressions across multiple solid tumor histotypes and induced CRs or PRs for most ALL xenografts. This activity was observed at MK-8242 drug exposures that appear to exceed those observed in human phase 1 trials.
Keywords: Preclinical Testing, Developmental Therapeutics, PI3K inhibitor
INTRODUCTION
The transcription factor and tumor suppressor TP53 is activated in response to a variety of cellular perturbations, in particular genotoxic stress, and functions to prevent fixation of DNA damage and to prevent proliferation of cells with genetic defects (reviewed in [1]). TP53 activation induces TP53-responsive genes at the transcriptional level, resulting in cell cycle arrest, senescence, or apoptosis [2]. Disruption of the TP53 pathway is considered to occur early during oncogenesis [3].
TP53 mutations are present in approximately 50% of all cancers [2], although mutations are identified less frequently in pediatric cancers (e.g., gliomas [4], neuroblastoma [5], sarcomas [6–8], Wilms tumor [9] and leukemia [10,11]) compared with adult malignancies [12]. At relapse the frequency of TP53 mutations may increase for pediatric cancers [13,14]. TP53 is negatively regulated by MDM2, and MDM2 is a TP53 regulated gene. In most tumors with wild-type TP53, cellular levels of TP53 are low and increase in response to genotoxic damage or other cellular stress. Potentially, activation of TP53 through inhibiting the negative regulation of TP53 by MDM2 will induce proliferation arrest or induce apoptosis in cancers with functional TP53.
Small molecule inhibitors of the MDM2/TP53 interaction have been identified and have entered clinical evaluation [15]. These molecules are based on blocking the interaction between TP53 and MDM2 by the binding of the small molecule inhibitors to a deep groove on the MDM2 surface that selectively interacts with specific amino acids in the N-terminal region of TP53. The presence of the inhibitors prevents MDM2 from binding to and ubiquitinating TP53 and targeting it for proteasomal degradation. The inhibitors also prevent MDM2 from promoting nuclear export of TP53 and from blocking TP53 binding to its target DNA sequences. As a result TP53 is able to activate transcriptional programs potentially leading to cell cycle arrest and/or apoptosis.
The first MDM2 inhibitor to enter clinical evaluation was RG7112, which is an analog of Nutlin-3A, the archetypal MDM2 inhibitor that has been commonly used in preclinical studies since its discovery in 2004 [16]. A focus of development for MDM2 inhibitors has been liposarcoma, since the well-differentiated and dedifferentiated subtypes generally show MDM2 amplification [17]. An RG7112 phase 2 study for patients with well-differentiated or dedifferentiated liposarcoma (most with MDM2 amplification) showed one objective response and 14 stable disease among 20 patients treated [18]. RG7112 induced thrombocytopenia in its phase 1 and 2 evaluations [18,19], and subsequent preclinical evaluations documented thrombocytopenia as an on-target adverse effect of MDM2 inhibition [20]. Seven other MDM2 inhibitors have subsequently entered clinical evaluation [15], and thrombocytopenia is a prominent toxicity at higher doses for all for which clinical data are publicly reported [18,19,21,22].
RG7112 was previously tested against the in vitro and in vivo tumor panels by the PPTP [23]. While in vitro cell line sensitivity correlated well with TP53 mutation status, there were only five objective regressions in twenty six solid tumor models with wild-type TP53. The failure to respond, despite wild-type TP53 may be due to defects either upstream (p14ARF) or downstream (p21Cip1/WAF1, Puma) in the TP53 pathway [3,13]. Of note, tumor lines with known MDM2 amplification were not responsive to RG7112 treatment.
MK-8242 is a potent, orally bioavailable, small-molecule MDM inhibitor that has completed phase 1 evaluations for adults with leukemias and with solid tumors [22,24]. Like RG7112 and other MDM2 inhibitors, dosing of MK-8242 is limited by hematologic toxicity with thrombocytopenia being notable at the higher doses evaluated [18,19,21,22]. Using a twice-daily schedule of administration on days 1 to 7 of planned 21 day treatment cycles, the maximum tolerated dose (MTD) was 400 mg for patients with solid tumors. Objective responses were noted among two of 27 liposarcoma patients treated [22].
This report describes testing of MK-8242 as a single agent against the PPTP’s in vitro panel as well as against the in vivo tumor panel as a single agent at its maximum tolerated dose. The pharmacokinetic profile of MK-8242 in treated mice is also presented to facilitate comparison of preclinical to clinical results.
MATERIALS AND METHODS
In vitro testing
In vitro testing was performed using DIMSCAN and cells were incubated in the presence of VS-4718 for 96 hours at concentrations from 1.0 nM to 10 μM and analyzed as previously described [8,9]
In vivo tumor growth inhibition studies
Procedures for propagation of xenografts in CB17SC scid−/− for solid tumors, BALB/c nu/nu mice for glioma models, [10], and leukemia models (NOD)/scid−/− mice have been described previously [11]. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee of the appropriate consortium member. An in-depth description of the analysis methods is included in the Supplemental Response Definitions section. Statistical methods for analysis have been described previously [10].
Statistical Methods
The exact log-rank test, as implemented using Proc StatXact for SAS®, was used to compare event-free survival distributions between treatment and control groups. P-values were two-sided and were not adjusted for multiple comparisons given the exploratory nature of the studies. The Mann-Whitney test was used to compare IC50 values for TP53 wild-type and mutant cell lines.
Pharmacokinetic studies
For pharmacokinetic analysis, MK-8242 was administered at 125 mg/kg. Blood was collected via the tail vein (20–40μL) and placed on a plastic weigh boat. Ten μL was pipetted into 30 μL 0.1 M sodium citrate buffer and frozen (−20°C) until assayed at Merck & Co., Inc., Kenilworth, NJ. Samples were derived at 2, 4, 6, 8, and 24 hr.
Pharmacodynamic studies
Preparation of tumor samples and immunoblotting procedures were as described previously [23]. Rabbit antibodies and dilutions used for detecting TP53 (1:1000), p21 (1:1000), PUMA (1:1000) and GAPDH (1:8000) were from Cell Signaling Technologies (Beverly, MA) and mouse antibody against MDM2 (1:2000) was from Abcam (Cambridge, UK). Secondary anti-rabbit HRP and anti-mouse HRP conjugated antibodies were from Pierce (Rockford, IL)
Drugs and Formulation
MK-8242 was provided to the Pediatric Preclinical Testing Program by Merck & Co., Inc, (Kenilworth, NJ) through the Cancer Therapy Evaluation Program (NCI). MK-8242 was formulated as a 12.5 mg/ml suspension in methylcellulose A4C (0.5%) and sodium dodecyl sulfate (0.25%), sonicated several times (5 min each) and stirred until a homogeneous suspension was prepared. Suspensions were stored for up to 7 days at 4°C, and brought to room temperature with stirring (30 min) until administered. For solid tumor models MK-8242 was administered by oral gavage (days 1–5 and 15 –19) at 125 mg/kg (solid tumors) and 75 mg/kg (leukemia models). The lower dose for the leukemia models was based on toxicity dose-finding in NOD)/scid−/− mice performed prior to efficacy testing. MK-8242 was provided to each consortium investigator in coded vials for blinded testing.
RESULTS
In vitro testing
MK-8242 was tested against the PPTP’s in vitro cell line panel at concentrations ranging from 1.0 nM to 10.0 μM using the PPTP’s standard 96 hour exposure period. A metric used to compare the relative responsiveness of the PPTP cell lines to MK-8242 is the ratio of the median rIC50 of the entire panel to that of each cell line. Higher ratios are indicative of greater sensitivity to MK-8242 and are shown in Figure 1A by bars to the right of the midpoint line. The response of the PPTP cell lines was highly dependent upon the TP53 mutation status of each cell line (Table I). The median relative IC50 (rIC50) value for the PPTP cell lines with mutant TP53 was > 10 μM, with a range from 4.9 μM to > 10 μM. By contrast, the rIC50 for TP53 wild-type cell lines was 0.07 μM with a range from 0.03 to 0.32 μM (p< 0.0001 for TP53 mutant versus wild-type). The most sensitive cell line, MOLT-4, is an ALL cell line. Overall, the TP53 wild-type cell lines showed more than 100-fold greater sensitivity to MK-8242 in comparison to TP53-mutated cell lines. Relative I/O% values range between 100% (no treatment effect) to -100% (complete cytotoxic effect), with a Relative I/O% value of 0% being observed for a completely effective cytostatic agent. Most TP53 wild-type cell lines (indicated by hatched bars in Figure 1B) show Relative I/O% values near −100%, indicative of a near complete cytotoxic effect. By contrast, the TP53 mutant cell lines generally show Relative I/O% values > 0% indicative of a weak effect over the concentration ranges evaluated.
Figure 1.
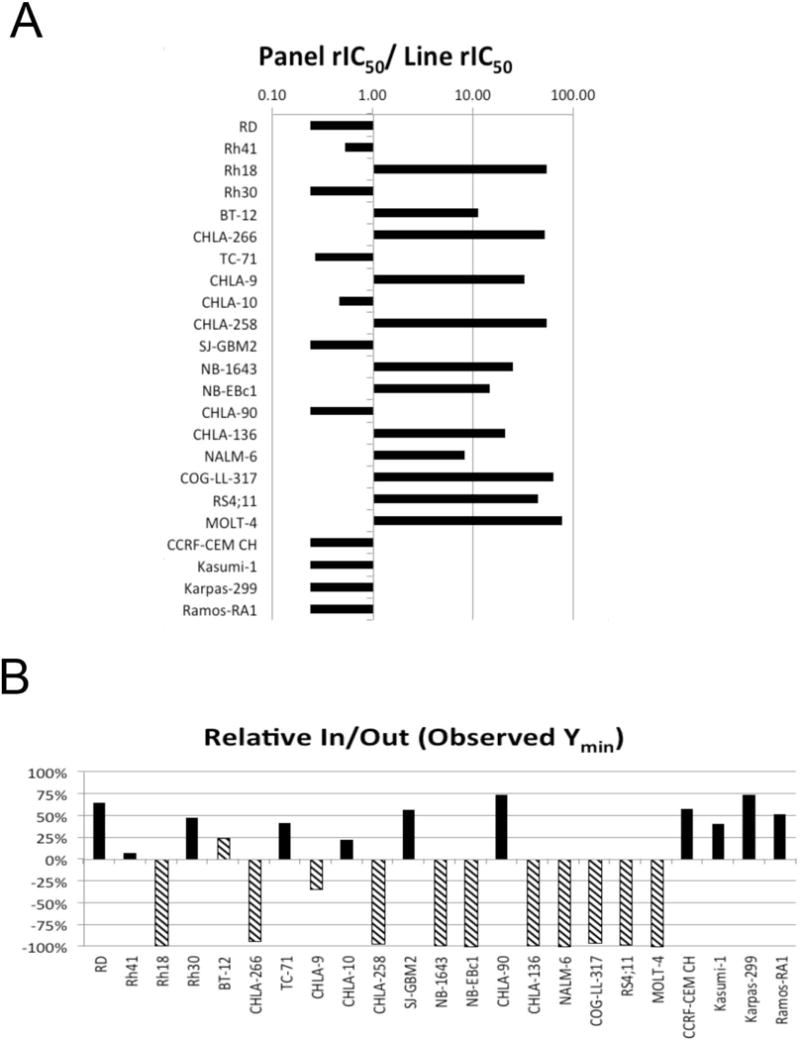
MK-8242 in vitro activity: A. The median rIC50 ratio graph shows the relative rIC50 values for the cell lines of the PPTP panel. Each bar represents the ratio of the panel rIC50 to the rIC50 value of the indicated cell line. Bars to the right represent cell lines with higher sensitivity, while bars to the left indicate cell lines with lesser sensitivity. B. Relationship between TP53 status and sensitivity (hatched bars WT p53, solid bars, Mutant p53).
Table I.
In vitro activity of MK8242 against PPTP cell lines.
| Cell Line | Histotype | TP53 Status | rIC50 (mM) | Panel rIC50/Line rIC50 | Relative In/Out (Observed Ymin) |
|---|---|---|---|---|---|
| RD | Rhabdomyosarcoma | Mutant | >10 | 0.5 | 64% |
| Rh41 | Rhabdomyosarcoma | Mutant | 4.88 | 1.1 | 7% |
| Rh18 | Rhabdomyosarcoma | WT* | 0.05 | 108.2 | −99% |
| Rh30 | Rhabdomyosarcoma | Mutant | >10 | 0.5 | 47% |
| BT-12 | Rhabdoid | WT | 0.23 | 22.7 | 24% |
| CHLA-266 | Rhabdoid | WT | 0.05 | 103.5 | −95% |
| TC-71 | Ewing sarcoma | Mutant | 9.80 | 0.5 | 42% |
| CHLA-9 | Ewing sarcoma | WT | 0.08 | 66.1 | −35% |
| CHLA-10 | Ewing sarcoma | Mutant | 5.61 | 0.9 | 22% |
| CHLA-258 | Ewing sarcoma | WT | 0.05 | 108.8 | −97% |
| SJ-GBM2 | Glioblastoma | Mutant | >10 | 0.5 | 57% |
| NB-1643 | Neuroblastoma | WT | 0.11 | 24.8 | −99% |
| NB-EBc1 | Neuroblastoma | WT | 0.18 | 29.5 | −100% |
| CHLA-90 | Neuroblastoma | Mutant | >10 | 0.5 | 73% |
| CHLA-136 | Neuroblastoma | WT | 0.13 | 41.7 | −99% |
| NALM-6 | ALL | WT | 0.32 | 16.4 | −100% |
| COG-LL-317 | ALL | WT | 0.04 | 128.4 | −96% |
| RS4;11 | ALL | WT | 0.06 | 90.5 | −98% |
| MOLT-4 | ALL | WT | 0.03 | 156.2 | −100% |
| CCRF-CEM | ALL | Mutant | >10 | 0.5 | 58% |
| Kasumi-1 | AML | Mutant | >10 | 0.5 | 41% |
| Karpas-299 | ALCL | Mutant | >10 | 0.5 | 73% |
| Ramos-RA1 | NHL | Mutant | >10 | 0.5 | 52% |
| Median TP53 WT | 0.07 | −98% | |||
| Minimum TP53 WT | 0.03 | −100% | |||
| Maximum TP53 WT | 0.32 | 24% | |||
| Median TP53 Mut | >10 | 52% | |||
| Minimum TP53 Mut | 4.88 | 7% | |||
| Maximum TP53 Mut | >10 | 73% |
MDM2 amplified cell line.
In vivo testing
MK-8242 was tested against the PPTP solid tumor xenografts using a dose of 125 mg/kg administered orally on Days 1–5 and Days 15–19. MK-8242 was formulated as a 12.5 mg/ml suspension in Methylcellulose A4C (0.5%) and sodium dodecyl sulfate (0.25%). For the solid tumor panels and for the subcutaneous leukemia xenografts (MV4;11 and RS4;11) treated at 125 mg/kg, the toxicity rate was 29 of 353 (8.2%), and for the ALL panel utilizing NOD-SCID mice and treated at 75 mg/kg, the toxicity rate was 3 of 87 (3.4%).
Forty-three of 46 tested xenograft models were considered evaluable for efficacy, with three models being inevaluable due to <75% of treated animals for these models being able to be assessed for efficacy. The inevaluable xenografts included a rhabdoid tumor (KT-14), a rhabdomyosarcoma (Rh28), and a neuroblastoma model (NB-1691). Complete details of testing are provided in Supplemental Table I including total numbers of mice, number of mice that died (or were otherwise excluded), numbers of mice with events and average times to event, tumor growth delay, as well as numbers of responses and treated to control (T/C) values. The TP53 status for each tumor line is shown in Table II, with the osteosarcoma lines being classified as TP53 “low expression” and considered to lack an intact TP53 pathway [23] (Supplemental Figure 1).
Table II.
Summary of in Vivo Activity of MK-8242
| Line | Tumor Type | Median Time to Event | P-value | EFS T/C | Median RTV/CD45 at End of Study | Median Group Response | EFS Activity | Response Activity | TP53 Mutation |
|---|---|---|---|---|---|---|---|---|---|
| BT-29 | Rhabdoid | > EP | <0.001 | > 1.8 | 3.1 | PD2 | NE | Int | |
| KT-16 | Rhabdoid | 35.0 | <0.001 | 4.0 | >4 | PR | Int | High | |
| KT-12 | Rhabdoid | 33.5 | <0.001 | 2.6 | >4 | PD2 | Int | Int | |
| KT-10 | Wilms | > EP | <0.001 | > 3.5 | 0.7 | CR | High | High | R290H |
| KT-11 | Wilms | 32.1 | <0.001 | 2.4 | >4 | PD2 | Int | Int | |
| EW5 | Ewing | 9.4 | <0.001 | 1.4 | >4 | PD1 | Low | Low | R337C |
| CHLA258 | Ewing | 40.0 | <0.001 | 4.1 | >4 | CR | Int | High | |
| ES4 | Ewing | 39.4 | <0.001 | 3.3 | >4 | PD2 | Int | Int | |
| ES6 | Ewing | 26.9 | <0.001 | 1.9 | >4 | PD2 | Low | Int | |
| Rh10 | Alveolar RMS | 21.2 | 0.012 | 1.6 | >4 | PD2 | Low | Int | |
| Rh30 | Alveolar RMS | 31.1 | <0.001 | 2.0 | >4 | PD2 | Int | Int | |
| Rh30R | Alveolar RMS | 34.7 | <0.001 | 2.5 | >4 | PD2 | Int | Int | R273C & Y205C |
| Rh65 | Alveolar RMS | > EP | <0.001 | > 3.5 | 3.5 | SD | Int | Int | |
| Rh18 | Embryonal RMS | > EP | <0.001 | > 2.7 | 0.0 | MCR | High | High | |
| Rh36 | Embryonal RMS | 26.8 | <0.001 | 2.1 | >4 | PD2 | Int | Int | |
| BT-50 | Medulloblastoma | > EP | 0.211 | . | 0.5 | CR | NE | High | |
| BT-36 | Ependymoma | > EP | 1.000 | . | 0.0 | MCR | NE | High | |
| BT-41 | Ependymoma | > EP | 1.000 | . | 0.4 | MCR | NE | High | |
| GBM2 | Glioblastoma | 17.1 | 0.117 | 1.4 | >4 | PD1 | Low | Low | R273C |
| BT-39 | Glioblastoma | 14.6 | <0.001 | 2.7 | >4 | PD2 | Int | Int | |
| D645 | Glioblastoma | 9.4 | 0.956 | 1.0 | >4 | PD1 | Low | Low | Y205C |
| D456 | Glioblastoma | 9.4 | 0.763 | 1.0 | >4 | PD1 | Low | Low | |
| NB-SD | Neuroblastoma | 13.0 | 0.032 | 1.2 | >4 | PD1 | Low | Low | C176F |
| NB-EBc1 | Neuroblastoma | 6.5 | <0.001 | 1.6 | >4 | PD2 | Low | Int | |
| CHLA-79 | Neuroblastoma | 11.1 | 0.032 | 1.2 | >4 | PD1 | Low | Low | |
| NB-1643 | Neuroblastoma | 11.7 | <0.001 | 1.9 | >4 | PD2 | Low | Int | |
| OS-1 | Osteosarcoma | 20.3 | <0.001 | 1.3 | >4 | PD1 | Low | Low | Low Expression |
| OS-2 | Osteosarcoma | 18.4 | 0.003 | 1.1 | >4 | PD1 | Low | Low | Low Expression |
| OS-17 | Osteosarcoma | 23.6 | <0.001 | 1.2 | >4 | PD1 | Low | Low | Low Expression |
| OS-9 | Osteosarcoma | 21.7 | 0.006 | 1.3 | >4 | PD1 | Low | Low | Low Expression |
| OS-33 | Osteosarcoma | 17.4 | <0.001 | 1.1 | >4 | PD1 | Low | Low | Low Expression |
| OS-31 | Osteosarcoma | 18.0 | 0.003 | 1.3 | >4 | PD1 | Low | Low | Low Expression |
| ALL-2 | ALL B-precursor | 33.9 | <0.001 | 4.1 | >25 | PD2 | Int | Int | |
| ALL-4 | ALL B-precursor | 32.5 | <0.001 | 5.4 | >25 | PR | Int | High | |
| ALL-7 | ALL B-precursor | 33.1 | <0.001 | 5.0 | >25 | PR | Int | High | |
| ALL-8 | ALL T-cell | 41.2 | <0.001 | 6.4 | >25 | CR | Int | High | |
| ALL-17 | ALL B-precursor | 27.8 | 0.107 | 4.4 | >25 | PR | Low | High | |
| ALL-19 | ALL B-precursor | 27.0 | 0.107 | 6.6 | >25 | PR | Low | High | |
| ALL-31 | ALL T-cell | 39.2 | 0.002 | 6.6 | >25 | PR | Int | High | |
| MLL-7 | MLL ALL | 41.0 | <0.001 | 8.9 | >25 | CR | Int | High | |
| RS4;11 | MLL ALL | 20.3 | <0.001 | 3.1 | >4 | PR | Int | High | |
| MV4;11 | MLL AML | > EP | <0.001 | > 3.7 | 0.0 | MCR | High | High |
Tumor Volume T/C value: Relative tumor volumes (RTV) for control (C) and treatment (T) mice were calculated at day 21 or when all mice in the control and treated groups still had measurable tumor volumes (if less than 21 days). The T/C value is the mean RTV for the treatment group divided by the mean RTV for the control group. High activity = T/C ≤ 0.15; Intermediate activity = T/C ≤ 0.45 but > 0.15; and Low activity = T/C > 0.45.
EFS T/C values = the ratio of the median time to event of the treatment group and the median time to event of the respective control group. High activity requires: a) an EFS T/C > 2; b) a significant difference in EFS distributions, and c) a net reduction in median tumor volume for animals in the treated group at the end of treatment as compared to at treatment initiation. Intermediate activity = criteria a) and b) above, but not having a net reduction in median tumor volume for treated animals at the end of the study. Low activity = EFS T/C < 2.
Objective response measures are described in detail in the Supplemental Response Definitions. PD1 = progressive disease with EFS T/C ≤ 1.5, and PD2 = progressive disease with EFS T/C > 1.5.
MK-8242 induced statistically significant differences in event-free survival (EFS) distribution compared to control in 27 of 31 (87%) of the solid tumor xenografts evaluable for this measure and in 8 of 10 (80%) of the evaluable ALL xenografts (Table II). The two ependymoma xenografts were not evaluable for this measure because of their slow growth rate. Note that RS4;11 and MV4;11 are included with the ALL xenografts for describing efficacy, but that they were tested as subcutaneous models at 125 mg/kg rather than as systemic models at 75 mg/kg as were the other ALL models. The TP53 wild-type xenografts showed a similar proportion of models with a significant difference in EFS distribution as did the TP53 defective xenografts (17 of 19, 89% versus 10 of 12, 83%, respectively) [23].
For those xenografts with a statistically significant difference in EFS distribution between treated and control groups, the EFS T/C activity measure additionally requires an EFS T/C value of > 2.0 for intermediate activity and indicates a substantial agent effect in slowing tumor progression. High activity further requires a reduction in final tumor volume compared to the starting tumor volume. Among TP53 wild-type xenografts, MK-8242 induced tumor growth inhibition meeting criteria for intermediate or high EFS T/C activity in 10 of 17 (59%) solid tumor xenografts evaluable for this measure. Among TP53 defective xenografts, only 2 of 12 (17%) xenografts showed intermediate or high activity. Intermediate/high activity for the EFS T/C metric occurred most frequently in the rhabdomyosarcoma panel (5 of 6), Wilms tumor panel (2 of 2) and Ewing sarcoma panel (2 of 4). No neuroblastoma (n=4) or osteosarcoma (n=6) xenografts met criteria for intermediate/high EFS T/C activity. For the ALL panel, 8 of 10 xenografts met criteria for intermediate/high activity.
An objective response [partial response (PR), complete response (CR), or maintained CR (MCR)] was observed in 7 of 33 (21.2%) evaluable solid tumor xenografts. Objective responses were observed in the following tumor panels: rhabdoid tumor (1 of 3); Wilms tumor (1 of 2); Ewing sarcoma (1 of 4), rhabdomyosarcoma (1 of 6), medulloblastoma (1 of 2), and ependymoma (2 of 2). Of note, the KT-10 Wilms tumor has a TP53 missense mutation (Arg290His), yet was still responsive to treatment. Responses for the non-GBM brain tumors are presented in Figure 2. The MDM2-amplified rhabdomyosarcoma xenograft Rh18 showed a MCR response to MK-8242, while the MDM2-amplified neuroblastoma xenograft NB-1691 showed only tumor growth delay to MK-8242. (NB-1691 was not evaluable by PPTP criteria as 3 of 10 animals experienced toxicity, but among the remaining 7 animals there were no objective responses.) For the ALL panel, 3 of 10 xenografts achieved CR or MCR with 6 additional xenografts showing PRs, Figure 3. The objective response results for both solid tumor and leukemia models are represented in Figure 4 using a ‘COMPARE’ format, based on the objective response scoring criteria centered around the midpoint score of 0 that represents stable disease. In the figure, xenografts with PD2 are indicated by a score of −3, and xenografts with regression (PR or CR) are indicated by bars to the right of the midpoint line. Red bars indicate xenografts with significant differences in EFS distribution between the treated and control groups. Figure 4 also shows the objective response activity in a “heat map” format.
Figure 2.
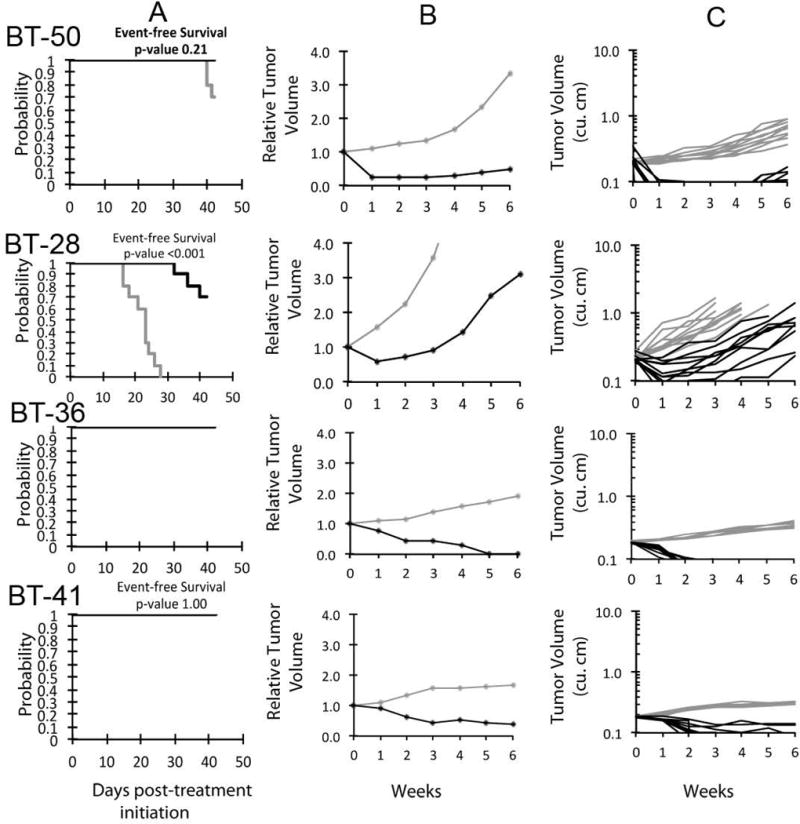
MK-8242 in vivo objective response activity for non-glioblastoma brain tumor models. Medulloblastomas (BT-28 and BT-50) and two ependymoma lines (BT-36, BT-41). A. Kaplan-Meier curves for EFS, B. median relative tumor volume graphs, and C. individual tumor volume graphs are shown for selected lines. Control (gray lines); Treated (black lines), statistical significance (p values) of the difference between treated and control groups are included.
Figure 3.
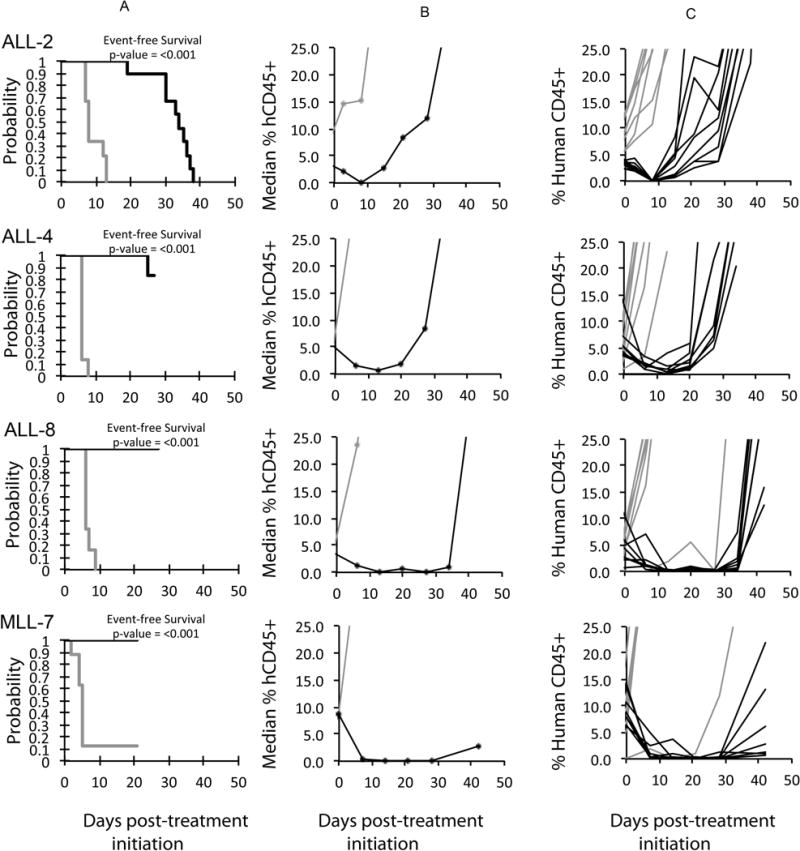
MK-8242 in vivo objective response activity for ALL models. ALL-2 (B-precursor), ALL-4 (B-precursor; BCR-ABL), ALL-8 (T-cell ALL) and MLL-7 (Infant, Precursor B-ALL). A. Kaplan-Meier curves showing the EFS, B. median leukemia engraftment as detected in peripheral blood, and C. individual leukemia engraftment (right). Controls (gray lines); Treated (black lines), statistical significance (p values) of the difference between treated and control groups are included.
Figure 4.
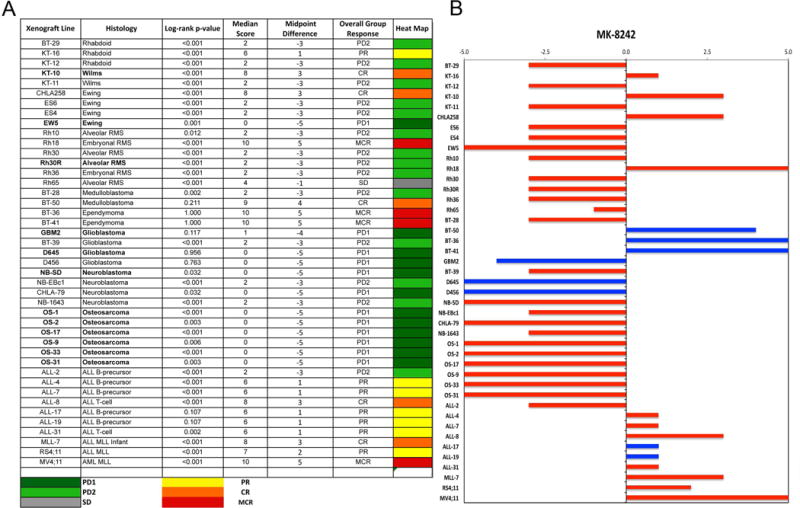
A. The colored heat map depicts group response scores. A high level of activity is indicated by a score of 6 or more, intermediate activity by a score of ≥2 but <6, and low activity by a score of <2. Lines with bolded font have mutant TP53 or very low TP53 expression (osteosarcomas).
B. Representation of tumor sensitivity based on the difference of individual tumor lines from the midpoint response (stable disease). Bars to the right of the median represent lines that are more sensitive, and to the left are tumor models that are less sensitive. Red bars indicate lines with a significant difference in EFS distribution between treatment and control groups, while blue bars indicate lines for which the EFS distributions were not significantly different.
Pharmacokinetic studies
A pharmacokinetic evaluation was performed using non-tumor-bearing SCID mice employing the whole blood micro-sampling method for rodents supplied by Merck. Three mice were administered 125 mg/kg MK-8242 by oral gavage, and samples collected at 2, 4, 6, 8 and 24 hours. Drug levels were determined by Merck & Co., Inc. and are shown in Supplemental Table II and in Supplemental Figure 2. Cmax was approximately 40 μM, and by 24 hr drug levels were below the limit of quantitation (0.05 μM). The AUC was approximately 200 μM·h.
Pharmacodynamic studies
Changes in expression of MDM2, TP53 and TP53 regulated genes (p21, PUMA) were examined after a single administration of MK-8242. Tumor samples were derived from non-responsive EW-5 (TP53 mutant) as well as tumors with wild-type TP53 having modest (PD2) to maintained complete responses, Supplemental Figure 3. As anticipated, MK-8242 induced neither stabilization of MDM2, or induction of TP53 or TP53-responsive genes in the EW-5 tumor. In contrast, there was robust induction of MDM2, TP53 and p21 in Rh18, BT-36, MV4;11 and in BT-50, all of which were highly responsive to treatment. Of interest is that MK-8242 induced MDM2, p21 and to a lesser extent, TP53 in BT-28 xenografts that were only modest responders to treatment (PD2). Only in Rh18 xenografts was there a consistent and maintained elevation of PUMA.
DISCUSSION
The PPTP in vitro and in vivo results for MK-8242, in combination with prior PPTP results for the MDM2 inhibitor RG7112, document a consistent response pattern among the PPTP cell lines and xenografts to MDM2 inhibition. For the in vitro panel, there is the expected pattern of sensitivity to MK-8242 for cell lines with wild-type TP53, while there is resistance for cell lines with mutant TP53 [29]. The degree of cytotoxic response was consistent between MK-8242 and RG7112, with BT-12 and CHLA-9 showing Relative In/Out% values far from −100% to both agents and with other sensitive cell lines showing values approaching −100% to both agents. The median rIC50 for MK-8242 for TP53 wild-type cell lines (0.07 μM) was approximately 6-fold lower than the rIC50 observed for RG7112 for the same cell lines (0.44 μM).
There were also commonalities in the response of the in vivo tumor panels to MK-8242 compared to RG7112. These commonalities were observed despite the different schedules employed: RG7112 was administered daily for 14 days, and MK-8242 was administered for 5 days followed by a second 5-day course at Day 15. The 5-day intermittent schedule for MK-8242 was selected as it represents a more plausible clinical schedule given the myelosuppression induced by MDM2 inhibitors. Among the best responders to RG7112 were KT-10, CHLA-258, Rh65 and BT-50. These were also among the best responders to MK-8242: KT-10 (CR, EFS T/C >3.5), CHLA-258 (CR, EFS T/C > 4.1), Rh65 (SD, EFS T/C >3.5) and BT-50 (CR, EFS T/C not evaluable). The two ependymoma xenografts (BT-36, BT-41) that had MCRs to MK-8242 were not tested for sensitivity to RG7112. A major discrepancy between RG7112 and MK-8242 was for Rh18, which had a PD1 response to RG7712 and a MCR response to MK-8242. Of note, Rh18 was also unresponsive to RG7388, a second generation agent, although it was highly synergistic when combined with radiation treatment in this model [30]. Rh18 has amplification and overexpression of MDM2 [31]. Also of interest, recent exome sequencing results indicate that KT-10 tumors have a TP53 missense mutation (R290H), however the functional significance of this mutation is not clear [32]. The osteosarcoma panel showed no response to either RG7112 or MK-8242. Each of the osteosarcoma xenografts has very low TP53 gene expression (Supplemental Figure 1), which may be the result of intronic translocations previously described for osteosarcoma that block TP53 gene transcription [33].
The response of the leukemia xenografts was similar between RG7112 and MK-8242. For both agents, the most responsive lines were two with MLL gene rearrangement: MLL-7 and MV4;11, with each achieving CR or MCR responses. A third line with MLL gene rearrangement, RS4;11, was less responsive to both RG7112 (PD2, EFS T/C 1.9) and MK-8242 (PR, EFS T/C 3.1). Richmond, et al., documented recently the consistent high-level activity of RG7112 against an extended panel of MLL ALL xenografts [34]. Most of the non-MLL ALL xenografts showed partial responses to MK-8242, indicating the absence of complete remission and a moderate level of anti-leukemia activity.
The clinical experience of MDM2 inhibitors for liposarcomas provides important lessons for pediatric oncologists to consider when developing this class of agents for childhood cancers. Most cases of dedifferentiated and well-differentiated liposarcoma show MDM2 amplification, so this diagnosis should represent a best case scenario for clinical development of MDM2 inhibitors [17]. Similar to the activity of MDM2 inhibitors observed against wildtype pediatric cancer cell lines, the MDM2 inhibitor Nutlin-3a inhibited growth and induced apoptosis in MDM2-amplified liposarcoma cell lines [35]. Additionally, MDM2 inhibitors commonly induce complete regression of the MDM2-amplified ostesoarcoma line SJSA-1 [36–39]. Despite this impressive in vitro activity against liposarcoma cell lines and in vivo activity against an MDM2-amplified xenograft, MDM2 inhibitors have shown few objective responses for patients with MDM2-amplified liposarcoma, with response rates in the 5% to 10% range [18,21,22]. Additionally, treatment with the MDM2 inhibitor SAR405838 appeared to rapidly select for TP53 mutations as evidenced by the appearance of TP53 mutations in circulating cell-free DNA following initiation of treatment with SAR405838 [40].
Comparison of the the MK-8242 drug exposure in SCID mice to that observed in humans is important in interpreting the PPTP data for MK-8242. The pharmacokinetic analysis using whole blood specimens reported here indicates a systemic exposure of approximately 200 μM·h for SCID mice at the 125 mg/kg dose used for testing. This systemic exposure exceeds that observed in adults with solid tumors for MK-8242 at its MTD (400 mg) (plasma AUC0–12hr of 16.7 μM*hr) [22]. Factoring in the blood-to-plasma ratio (~0.6 in both mouse and human) and the 1.5- to 3-fold higher unbound fraction in humans versus mice reduces the relevant difference between systemic exposures. That said, the comparison of the drug exposure tolerated in adults to the drug exposure observed in mice suggests that the PPTP preclinical in vivo studies are unlikely to be under-predicting MK-8242 clinical activity. Further pharmacokinetic analysis for MK-8242 and for other MDM2 inhibitors that have entered clinical evaluation will be important to better understand the relationship between drug levels of MDM2 inhibitors tolerated by mice compared to those tolerated by humans.
Barone et al. reviewed the preclinical rationale for MDM2 inhibitors for pediatric solid tumors, pointing out that most childhood cancers have wild-type TP53 and that preclinical testing has shown activity for MDM2 inhibitors across a range of pediatric solid tumors, including neuroblastoma, osteosarcoma, retinoblastoma, rhabdomyosarcoma, and Ewing sarcoma [41]. Most of the published preclinical data for pediatric cancers is for in vitro testing (aside from the MDM2-amplified osteosarcoma line SJSA-1 that is commonly used for in vivo proof of principle experiments for MDM2 inhibitors). Our in vitro results also show consistent activity across a range of histologies for cell lines with wild-type TP53. Similarly, the pharmacodynamic testing that we present is consistent with both the proposed mechanism of action of MK-8242 and with published data for other MDM2 inhibitors against pediatric preclinical models [41]. MK-8242 induced TP53 and the TP53-regulated gene p21, in all tumor models with wild-type TP53, but not in EW-5 xenografts that are mutant for TP53.
Looking to the future for MDM2 inhibitors for childhood cancers, our results offer two cautionary notes regarding potential pediatric clinical evaluations of MDM2 inhibitors. One is that the consistent in vitro activity observed for MDM2 inhibitors against pediatric cell lines with wild-type TP53 is not mirrored by consistent, robust in vivo activity. While tumor regression is observed for solid tumor xenografts, this represents a minority of TP53 wild-type models tested. Additionally, pharmacokinetic analysis shows that MK-8242 drug exposure in SCID mice appears to exceed that observed in adult phase 1 trials. A final point for consideration when evaluating pediatric clinical development of MDM2 inhibitors is the delayed thrombocytopenia that appears to be an on-target pharmacodynamic effect for this class of agents [20,41].
Supplementary Material
Supplemental Figure 1. TP53 and MDM2 gene expression data for PPTP cell lines and xenografts.
Supplemental Figure 2. Whole blood levels of MK-8242 in three individual mice following oral administration of 125 mg/kg MK-8242. The broken line indicates the average drug level.
Supplemental Figure 3. Pharmacodynamic changes induced by MK-8242.
Supplemental Table I. Efficacy of MK-8242 against PPTP Xenograft Models
Supplemental Table II. Pharmacokinetic Testing Results for MK-8242
Acknowledgments
This work was supported by NO1-CM-42216, CA21765, and CA108786 from the National Cancer Institute and used MK-8242 supplied the Merck & Co., Inc, Kenilworth, NJ. In addition to the authors this paper represents work contributed by the following: Sherry Ansher, Joshua Courtright, Kathryn Evans, Edward Favours, Henry S. Friedman, Danuta Gasinski, Nicholas Pettit, Jennifer Richmond, Melissa Sammons, Joe Zeidner, Jianrong Wu, Ellen Zhang, and Jian Zhang. Children’s Cancer Institute Australia for Medical Research is affiliated with the University of New South Wales and the Sydney Children’s Hospitals Network.
Abbreviations
- PPTP
Pediatric Preclinical Testing Program
- EFS T/C
Ratio of Event Free Survival Treated/Control
- Cmax
Maximum plasma concentration
- (NOD)/scid−/−
Non-Obese diabetic/severe combined immune deficient
Footnotes
Conflict of interest statement: The authors consider that there are no actual or perceived conflicts of interest.
References
- 1.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ. p53, the Cellular Gatekeeper for Growth and Division. Cell. 1997;88(3):323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 3.Royds JA, Iacopetta B. p53 and disease: when the guardian angel fails. Cell Death Differ. 2006;13(6):1017–1026. doi: 10.1038/sj.cdd.4401913. [DOI] [PubMed] [Google Scholar]
- 4.Pollack IF, Hamilton RL, Finkelstein SD, Campbell JW, Martinez AJ, Sherwin RN, Bozik ME, Gollin SM. The Relationship between TP53 Mutations and Overexpression of p53 and Prognosis in Malignant Gliomas of Childhood. Cancer Research. 1997;57(2):304–309. [PubMed] [Google Scholar]
- 5.Tweddle DA, Pearson AD, Haber M, Norris MD, Xue C, Flemming C, Lunec J. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 2003;197(1–2):93–98. doi: 10.1016/s0304-3835(03)00088-0. [DOI] [PubMed] [Google Scholar]
- 6.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358(6381):80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 7.Overholtzer M, Rao PH, Favis R, Lu X-Y, Elowitz MB, Barany F, Ladanyi M, Gorlick R, Levine AJ. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proceedings of the National Academy of Sciences. 2003;100(20):11547–11552. doi: 10.1073/pnas.1934852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pishas KI, Al-Ejeh F, Zinonos I, Kumar R, Evdokiou A, Brown MP, Callen DF, Neilsen PM. Nutlin-3a Is a Potential Therapeutic for Ewing Sarcoma. Clinical Cancer Research. 2011;17(3):494–504. doi: 10.1158/1078-0432.CCR-10-1587. [DOI] [PubMed] [Google Scholar]
- 9.Malkin D, Sexsmith E, Yeger H, Williams BRG, Coppes MJ. Mutations of the p53 Tumor Suppressor Gene Occur Infrequently in Wilms’ Tumor. Cancer Research. 1994;54(8):2077–2079. [PubMed] [Google Scholar]
- 10.Marks D, Kurz B, Link M, Ng E, Shuster J, Lauer S, Carroll D, Brodsky I, Haines D. Altered expression of p53 and mdm-2 proteins at diagnosis is associated with early treatment failure in childhood acute lymphoblastic leukemia. Journal of Clinical Oncology. 1997;15(3):1158–1162. doi: 10.1200/JCO.1997.15.3.1158. [DOI] [PubMed] [Google Scholar]
- 11.Wada M, Bartram C, Nakamura H, Hachiya M, Chen D, Borenstein J, Miller C, Ludwig L, Hansen-Hagge T, Ludwig W. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82(10):3163–3169. [PubMed] [Google Scholar]
- 12.Kusafuka T, Fukuzawa M, Oue T, Komoto Y, Yoneda A, Okada A. Mutation analysis of p53 gene in childhood malignant solid tumors. Journal of Pediatric Surgery. 1997;32(8):1175–1180. doi: 10.1016/s0022-3468(97)90677-1. [DOI] [PubMed] [Google Scholar]
- 13.Van Maerken T, Vandesompele J, Rihani A, De Paepe A, Speleman F. Escape from p53-mediated tumor surveillance in neuroblastoma: switching off the p14ARF-MDM2-p53 axis. Cell Death Differ. 2009;16(12):1563–1572. doi: 10.1038/cdd.2009.138. [DOI] [PubMed] [Google Scholar]
- 14.Van Maerken T, Rihani A, Dreidax D, De Clercq S, Yigit N, Marine J-C, Westermann F, De Paepe A, Vandesompele J, Speleman F. Functional Analysis of the p53 Pathway in Neuroblastoma Cells Using the Small-Molecule MDM2 Antagonist Nutlin-3. Molecular Cancer Therapeutics. 2011;10(6):983–993. doi: 10.1158/1535-7163.MCT-10-1090. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. Journal of medicinal chemistry. 2015;58(3):1038–1052. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (New York, NY. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 17.Abbas Manji G, Singer S, Koff A, Schwartz GK. Application of molecular biology to individualize therapy for patients with liposarcoma. American Society of Clinical Oncology educational book / ASCO American Society of Clinical Oncology Meeting. 2015;35:213–218. doi: 10.14694/EdBook_AM.2015.35.213. [DOI] [PubMed] [Google Scholar]
- 18.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, Nichols GL, Bui BN. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The Lancet Oncology. 2012;13(11):1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 19.Kurzrock R, Blay JY, Nguyen BB, Wagner AJ, Maki RG, Schwartz GK, Patnaik A, Gore L, Wu L, Vassilev LT, Ding M, Geho D, Zhi J, Middleton S, Nichols GL. A phase I study of MDM2 antagonist RG7112 in patients (pts) with relapsed/refractory solid tumors. J Clin Oncol. 2012;30(suppl) abstr e13600. [Google Scholar]
- 20.Iancu-Rubin C, Mosoyan G, Glenn K, Gordon RE, Nichols GL, Hoffman R. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Experimental hematology. 2014;42(2):137–145. doi: 10.1016/j.exphem.2013.11.012. e135. [DOI] [PubMed] [Google Scholar]
- 21.de Weger V, Lolkema MP, Dickson M, Le Cesne A, Wagner A, Merqui-Roelvink M, Varga A, Tap W, Schwartz G, Demetri G, Zheng W, Tuffal G, Mace S, Miao H, Schellens JHM, de Jonge M. A first-in-human (FIH) safety and pharmacological study of SAR405838, a novel HDM2 antagonist, in patients with solid malignancies. European Journal of Cancer. 2014;50:121–122. [Google Scholar]
- 22.Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch HA, Fancourt C, Levonas A, Lam R, Meister A, Kemp RK, Knox C, Rose S, Hong DS. A phase I trial of the human double minute 2 (HDM2) inhibitor MK-8242 in patients (pts) with advanced solid tumors. J Clin Oncol. 2015;33(suppl) doi: 10.1200/JCO.2016.70.7117. abstr 10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carol H, Reynolds CP, Kang MH, Keir ST, Maris JM, Gorlick R, Kolb EA, Billups CA, Geier B, Kurmasheva RT, Houghton PJ, Smith MA, Lock RB. Initial testing of the MDM2 inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatric blood & cancer. 2013;60(4):633–641. doi: 10.1002/pbc.24235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian HM, Levonas A, Fancourt C, Lam R, Meister A, Jones ME, Kemp RK, Knox C, Rose S, Patel PS, Tibes R. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML) J Clin Oncol. 2015;33(suppl) abstr 7070. [Google Scholar]
- 25.Frgala T, Kalous O, Proffitt RT, Reynolds CP. A fluorescence microplate cytotoxicity assay with a 4-log dynamic range that identifies synergistic drug combinations. Mol Cancer Ther. 2007;6(3):886–897. doi: 10.1158/1535-7163.MCT-04-0331. [DOI] [PubMed] [Google Scholar]
- 26.Kang MH, Smith MA, Morton CL, Keshelava N, Houghton PJ, Reynolds CP. National Cancer Institute Pediatric Preclinical Testing Program: Model description for in vitro cytotoxicity testing. Pediatric blood & cancer. 2011;56(2):239–249. doi: 10.1002/pbc.22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R, Carol H, Tajbakhsh M, Reynolds CP, Maris JM, Courtright J, Keir ST, Friedman HS, Stopford C, Zeidner J, Wu J, et al. The Pediatric Preclinical Testing Program: description of models and early testing results. Pediatric blood & cancer. 2007;49(7):928–940. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- 28.Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, Ramirez CD, Rice AM, Haber M, Norris MD, MacKenzie KL, Lock RB. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103(10):3905–3914. doi: 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 29.Saiki AY, Caenepeel S, Cosgrove E, Su C, Boedigheimer M, Oliner JD. Identifying the determinants of response to MDM2 inhibition. Oncotarget. 2015;6(10):7701–7712. doi: 10.18632/oncotarget.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phelps D, Bondra K, Seum S, Chronowski C, Leasure J, Kurmasheva RT, Middleton S, Wang D, Mo X, Houghton PJ. Inhibition of MDM2 by RG7388 confers hypersensitivity to X-radiation in xenograft models of childhood sarcoma. Pediatric blood & cancer. 2015 doi: 10.1002/pbc.25465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenzie PP, McPake CR, Ashford AA, Vanin EF, Harris LC. MDM2 does not influence p53-mediated sensitivity to DNA-damaging drugs. Mol Cancer Ther. 2002;1(12):1097–1104. [PubMed] [Google Scholar]
- 32.Soussi T, Kato S, Levy PP, Ishioka C. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. Human mutation. 2005;25(1):6–17. doi: 10.1002/humu.20114. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, Parker M, Rusch M, Nagahawatte P, Wu J, Mao S, Boggs K, Mulder H, Yergeau D, Lu C, Ding L, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell reports. 2014;7(1):104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richmond J, Carol H, Evans K, High L, Mendomo A, Robbins A, Meyer C, Venn NC, Marschalek R, Henderson M, Sutton R, Kurmasheva RT, Kees UR, Houghton PJ, Smith MA, Lock RB. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(6):1395–1405. doi: 10.1158/1078-0432.CCR-14-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singer S, Socci ND, Ambrosini G, Sambol E, Decarolis P, Wu Y, O’Connor R, Maki R, Viale A, Sander C, Schwartz GK, Antonescu CR. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 2007;67(14):6626–6636. doi: 10.1158/0008-5472.CAN-07-0584. [DOI] [PubMed] [Google Scholar]
- 36.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, To KH, Linn M, Podlaski F, Wovkulich P, Vu B, Vassilev LT. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73(8):2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, Stuckey JA, Meagher JL, Bai L, Liu L, Hoffman-Luca CG, Lu J, Shangary S, Yu S, Bernard D, Aguilar A, Dos-Santos O, Besret L, Guerif S, Pannier P, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74(20):5855–5865. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalez-Lopez De Turiso F, Houze JB, Huang X, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. Journal of medicinal chemistry. 2014;57(4):1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 40.Watters JW, Dickson MA, Schwartz GK, Le Cesne A, Bahleda R, Wagner AJ, Choy E, De Jonge MJ, Light M, Jung J, Lee JS, Mace S. TP53 mutations emerge in circulating cell-free DNA obtained from patients undergoing treatment with the HDM2 antagonist SAR405838. J Clin Oncol. 2015;33(suppl) abstr 2515. [Google Scholar]
- 41.Barone G, Tweddle DA, Shohet JM, Chesler L, Moreno L, Pearson AD, Van Maerken T. MDM2-p53 interaction in paediatric solid tumours: preclinical rationale, biomarkers and resistance. Current drug targets. 2014;15(1):114–123. doi: 10.2174/13894501113149990194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. TP53 and MDM2 gene expression data for PPTP cell lines and xenografts.
Supplemental Figure 2. Whole blood levels of MK-8242 in three individual mice following oral administration of 125 mg/kg MK-8242. The broken line indicates the average drug level.
Supplemental Figure 3. Pharmacodynamic changes induced by MK-8242.
Supplemental Table I. Efficacy of MK-8242 against PPTP Xenograft Models
Supplemental Table II. Pharmacokinetic Testing Results for MK-8242