


Abstract
Although spatial and temporal elements of immune activation mediate the intensity of the immune response, few tools exist to directly examine these effects. To elucidate the spatiotemporal aspects of innate immune responses, we designed an optogenetic pattern recognition receptor that activates in response to blue light. We demonstrate direct receptor activation, leading to spatial and temporal control of downstream signaling pathways in a variety of relevant cell types. We combined our platform with Bi-molecular Fluorescence Complementation (BiFC), resulting in selective fluorescent labeling of cells in which receptor activation has occurred.
Graphical Abstract
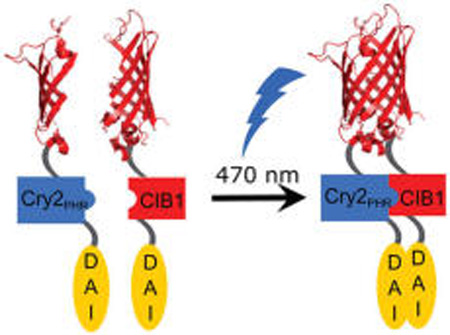
There is a common element in both preventing disease through vaccination and the chronic inflammation of autoimmune diseases— activation of the innate immune system by Pattern Recognition Receptors (PRRs).1 These recognition events are position- and time-dependent, leading to robust protection from disease in some cases and autoimmune disorders in others. The spatiotemporal sequence of activation and its cellular context determines the type and magnitude of the immune response.2,3 Despite the importance of understanding the pattern of receptor signaling, few spatial or temporal techniques exist to control PRR activation. In the field of neurobiology, light with its exquisite spatiotemporal precision has helped determine how complex networks of cells enact unique signaling mechanisms.4 Recently, we reported the use of photoactivated Toll-like receptor agonists to control temporal activation of innate immune receptors.5–7 However, spatial control using such technologies is limited by the available synthetic tools and the challenge of receptor activity and diverse location of immune cells within an organism. Here, we show a genetically encoded, photoactivated PRR that allows light-mediated control of innate immune signaling. The engineered receptor is a fusion of the lightactivated cryptochrome system, CRY2/CIB1, and the cytosolic pattern recognition receptor DAI (DNA-dependent activator of IFN-regulatory factors). This genetic fusion renders the DAI receptor inducible via dimerization by blue light. Genetic encoding allows the receptor to be expressed selectively within a designated cell type and to originate the signal only from selected cells. We show that the receptor activates cellular markers of innate immunity within 2 h of blue light exposure. Additionally, the system reports which cells have been activated using dimerization via a split-fluorescence reporter assay. The light-activated receptor was inserted into multiple cell types including RAW macrophages and HEK293. A genetically encoded, light-activated innate immune receptor will enable unprecedented control and analysis of complex innate immune interactions potentially including wound healing and paracrine signaling effects.
In designing a photoactivated receptor, we sought to apply a general design strategy that would work for many different innate immune receptors. As most PRRs use dimerization to control signaling, light-induced dimerization would yield a general method to gain light-mediated control of innate immune receptors.2,7,8 We chose the DAI receptor as a proof-of-concept effector module due to its convenient properties for protein engineering as well as a precedent for genetic manipulation and controlled dimerization resulting in controlled activation.9 DAI is a cytosolic PRR that homodimerizes upon binding dsDNA, activating the NF-κB and IRF3 pathways.9 Engineered DAI activated IFN pathways in response to chemical dimerization by a rapamycin derivative, making this receptor a promising candidate for light-induced dimerization.9 CRY2 (cryptochrome 2) and its binding partner CIB1 (cryptochrome interacting basic helix–loop–helix 1) are a robust blue light-induced dimerization system used in many mammalian cells.10–12 To minimize the increased molecular weight, we used the CRY2PHR subconstruct, which is sufficient to induce dimerization. Lacking nuclear import capability, it would maintain the CRY2-DAI fusion protein in the desired cytoplasmic compartment.10 The C-terminus of DAI is used for downstream signaling, and we hypothesized that an N-terminal fusion would not inhibit DAI activity.9,13 We therefore generated a photoinducible DAI system via coexpression of DAI variants as C-terminal fusions to either CRY2PHR or CIB1 (Figure 1).
Figure 1.
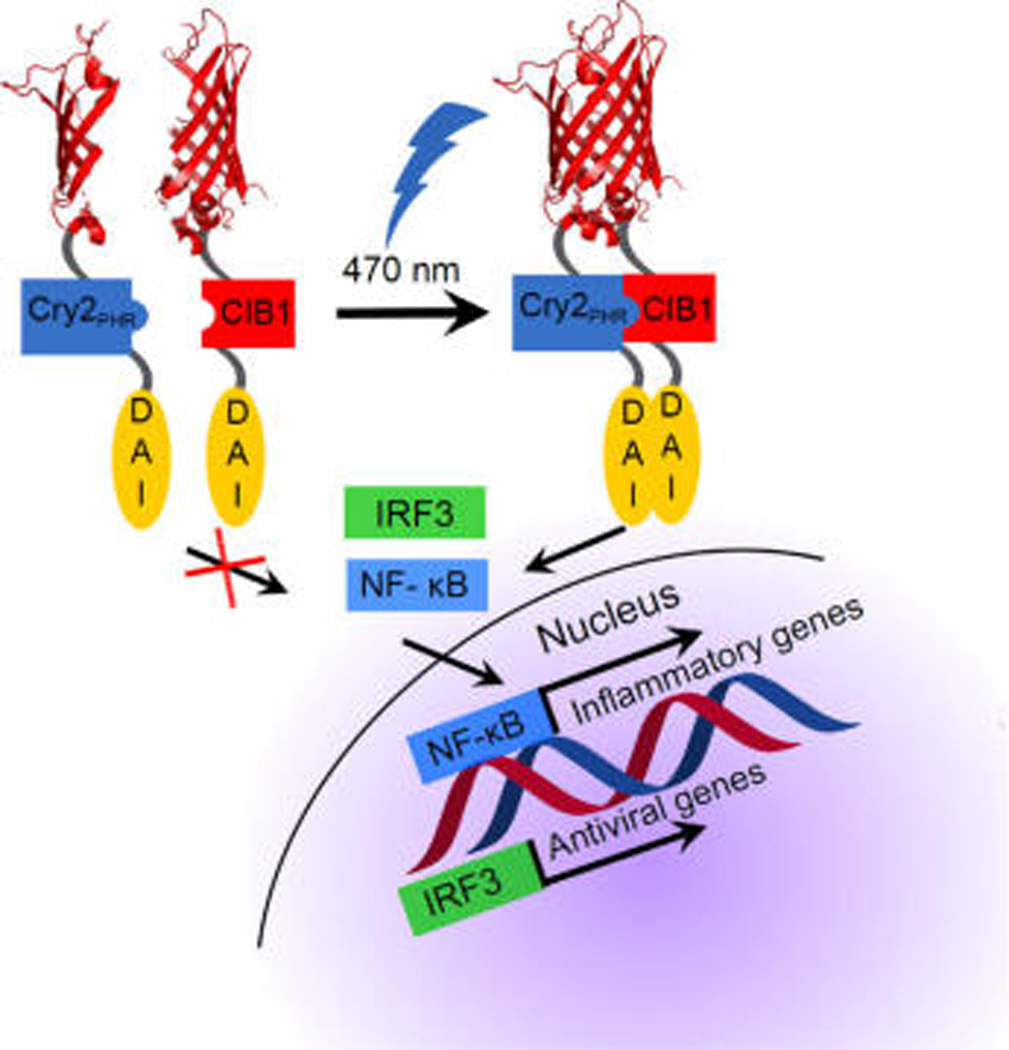
Schematic of photo-DAI activation. To achieve photoactivation, the CRY2/CIB1 domains are fused to one of two split domains of mCherry and identical copies of the DNA-dependent activator of IFN-regulatory factors (DAI) receptor. Activation with blue light (470 nm) induces dimerization of CRY2/CIB1, DAI, and subsequent fluorescence from the coupled mCherry. The photo-DAI enacts the transcription of interferons activating the innate immune system.
After completing the design of the light-activated DAI, we tested light-induced dimerization. We transfected HEK-Blue TLR4 cells with photo-DAI constructs (Figure 1). Cells were subjected to 470–490 nm light for 2 h (1.5 W/m2) and activation was measured using a colorimetric assay detecting NF-κB activity. NF-κB is an important transcription factor in immune activation and results in an upregulation of cytokines, chemokines, and cell surface receptors—alerting the immune system of potential pathogen invasion.19 Cells containing photo-DAI showed higher NF-κB activation than untransfected cells (Figure 2, Supporting Information (SI) Figure 1). Constructs fused to split-mCherry (Photo-DAI), a common label for BiFC,14 showed a larger fold-change between illuminated and dark states, demonstrating light-mediated control of activation.
Figure 2.
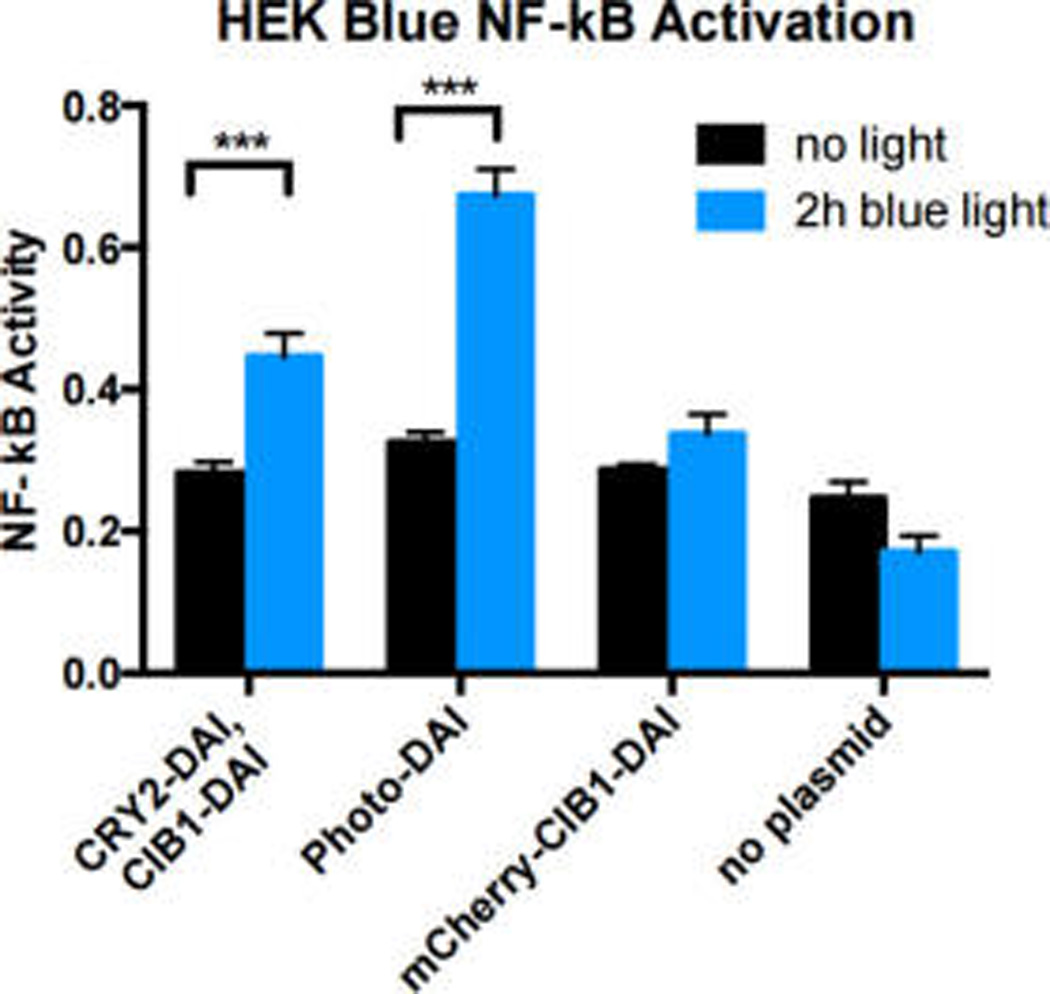
Photo-DAI activity in HEK Blue cells. Black bars: cells incubated in the dark. Blue bars: Cells exposed to light for 2 h. Error bars denote standard deviation. n = 3 replicates. ***p < 0.001.
To validate that dimerization was occurring in response to blue light and that the BiFC would mark immune activation, we examined the fluorescence from the photo-DAI system using flow cytometry and confocal microscopy (Figure 3, SI Figure 2). After 2 h of either blue light exposure or exposure to the native DAI agonist, 100 ng/mL Poly(dA:dT), cells began to show a shift in red fluorescence (Figure 3a). After 4 h of exposure to either blue light or native agonist, cells began to show a dramatic shift in fluorescence (Figure 3b). In addition to activation and fluorescence of mCherry, we observed a light-mediated temporal element to the dimerization of the receptor (Figure 3c). Blue light stimulation for 2 h elicited red fluorescence in 25% of transfected cells. Within 4 h of stimulation, 84% of cells expressing photo-DAI displayed mCherry fluorescence. In contrast, the native ligand for DAI, 100 ng/mL Poly(dA:dT), stimulated only 42% of transfected cells with two distinct populations of cells remaining after 4 h of agonist exposure (Figure 3a–c). In addition, untransfected cells exposed to blue light for 2 or 4 h demonstrated no significant change in fluorescence (Figure 3d).
Figure 3.
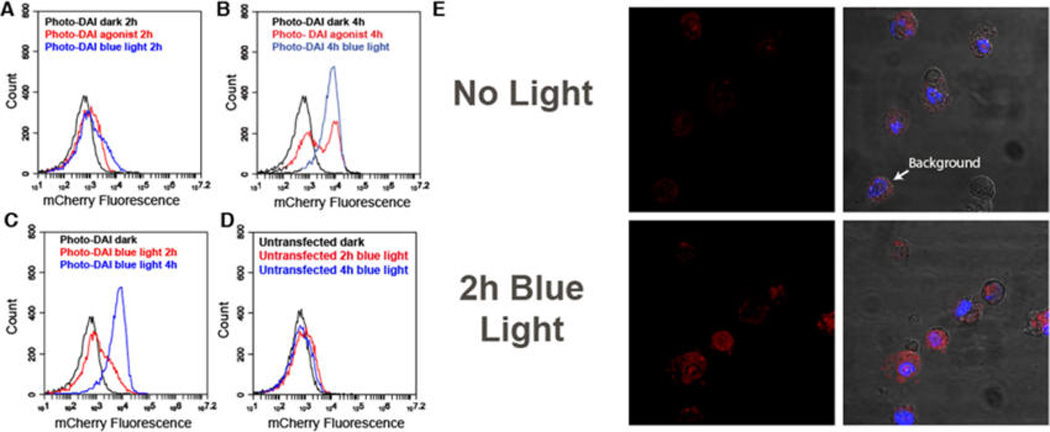
Fluorescent reporting of dimerization from cells expressing photo-DAI. (A) Flow cytometry of HEK cells expressing photo-DAI after 2 h of exposure to either blue light or the native DAI agonist (100 ng/mL Poly(dA:dT)). Cells incubated for 2 h in the dark (black), with Poly(dA:dT) (red), or with blue light (blue). (B) Cells expressing photo-DAI exposed to either blue light or Poly(dA:dT) for 4 h. Cells incubated for 4 h in the dark (black), with Poly(dA:dT) (red), or with blue light (blue). (C) Cells transfected with photo-DAI exposed to blue light for 0 h, 2 h, or 4 h. Cells incubated in the dark 4 h (black) and cells exposed to 2 h blue light (red) or 4 h (blue). (D) Untransfected cells exposed to blue light for 0 h, 2 h, or 4 h. Cells incubated in the dark 4 h (black) and cells exposed to blue light 2 h (red) or 4 h (blue). (E) Microscope images of HEK cells expressing photo-DAI Left: mCherry channel. Right: merge channel of mCherry and DAPI. Top: incubated in the dark. Bottom: 2 h blue light exposure.
To determine whether activated cells could be tracked, we imaged cells transfected with photo-DAI after 2 h of blue light exposure. Cells incubated in the dark demonstrated a low level of mCherry fluorescence, while cells exposed to blue light for 2 h elicited robust fluorescence (Figure 3e).
For reporting on the broad set of different immune cells, we tested if this receptor would function in other cell types. Beyond the initial HEK cells, we also examined the ability of photo-DAI to activate IRF3 signaling in RAW macrophages (Figure 4). IRF3 is an important transcription factor activated in response to viruses and upregulates cytokines and chemokines involved in an antiviral immune response.18 Accordingly, RAW cells were transduced with either the full photo-DAI system or the CRY2PHR or CIB1 half of the photo-DAI system using a lentiviral vector. Transduced cells were irradiated with blue light for 2 or 4 h, alongside a control plate that remained in the dark. The activity of cells expressing photo-DAI corresponded to the exposure time of blue light. Cells expressing mCherry-CRY2-DAI also showed a modest light-mediated response due to the propensity for light-activated CRY2 to self-oligomerize,10 although the activation was less than that of the full photo-DAI system—indicating that both dimerization partners are necessary for maximum DAI activity.
Figure 4.
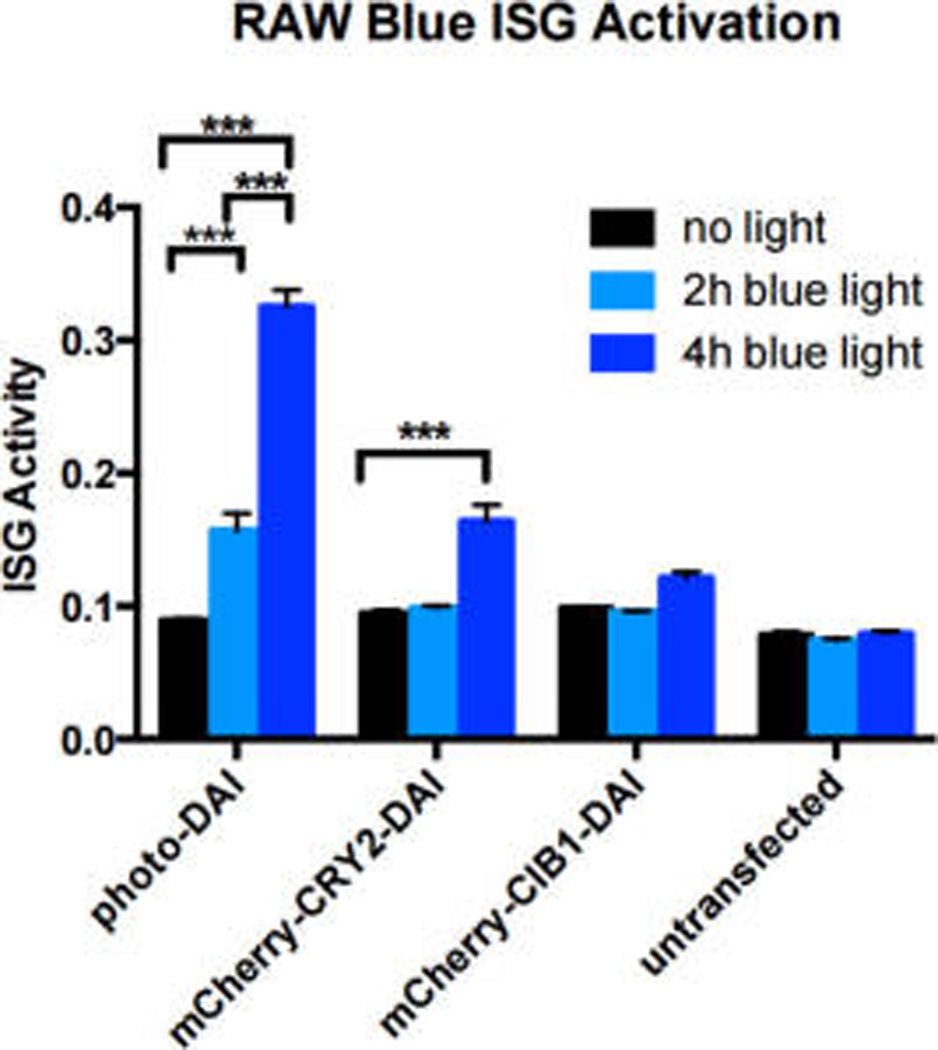
Biological activity of RAW Blue ISG cells transduced with photo-DAI system. Black bars: cells incubated in the dark. Light blue bars: cells exposed to blue light for 2 h. Blue bars: cells exposed to light for 4 h. Error bars denote standard deviation, n = 3 replicates. ***p < 0.001.
We have created an optogenetic DAI receptor via genetic fusion to a protein pair capable of undergoing photoinduced dimerization. Our engineered receptor activates NF-κB and IRF3 nuclear translocation in response to blue light stimulation. By fusing a split fluorescent reporter to this complex, cells containing the dimerized receptor can be identified. This method can be widely applied to a variety of innate immune receptors and their downstream pathways. This method could be applied in vivo to examine the effects of spatial and temporal induction of inflammation, providing useful information for understanding its effects on vaccination, diabetes, arthritis, and development.
EXPERIMENTAL SECTION
Materials and Methods
Plasmid Construction
Linearized DNA was obtained by a standard Q5 HotStart PCR protocol and the following PCR conditions: initial denaturation (98 °C, 30 s), 27 cycles (98 °C, 10 s; appropriate Tm, 30 s; 72 °C 30 s kb−1), and final extension (72 °C, 2 min) in a BioRad C1000 thermocycler. Linearized DNA fragments (1:1 molar ratios of 100 ng vector/insert) were added to 10 µL of Gibson Master Mix (NEB) and incubated for 1 h at 50 °C and transformed into chemically competent Top10 E. coli (Life Technologies). All plasmids were sequenced, Retrogen (San Diego, CA).
Plasmid Isolation
All plasmids were transformed into Mach1 T1R or Top10 chemically competent E. coli (Life Technologies). A single colony was placed in 3 mL of 2xYT media with appropriate antibiotic and allowed to grow overnight in a 37 °C shaking incubator. Plasmids were miniprepped using a QIAprep Spin Miniprep Kit (Qiagen) according to the manufacturer’s protocol. Plasmid was eluted using 30 µL of HyClone purified endotoxin- free distilled water (GE healthcare). Concentration was determined using NanoDrop2000 (Thermo Scientific).
HEK-Blue TLR4 Assay
Cells were grown in a six-well plate to 60% confluency and transfected using Lipofectamine 3000 (Life Technologies). Cells were incubated for 3 days to ensure that the cytoplasmic plasmid was destroyed and would not interfere with the assay. Cells were passaged and plated in a 96 well plate at 2.5 × 105 cells per well in 200 µL of DMEM supplemented with 10% (v/v) HIFBS. Cells were exposed to blue light for 2 h using a UVP Chromato-Vue UV transilluminator (115 V 60 Hz 1.8 Amp) and UVP UV/Blue converter plate (1.2 W/m2) and incubated at 37 °C and 5% CO2 for 20 h. After 20 h, 20 µL of the cell supernatant was placed in 180 µL of freshly prepared QuantiBlue (Invivogen) solution and incubated at 37 °C and 5% CO2 for up to 6 h. The plate was analyzed every hour using a Multiskan FC plate reader (Thermo Scientific), and absorbance was measured at 620 nm.
RAW Macrophage Plasmid Transduction
HEK293T cells were plated in a 10 cm plate at 1 × 106 cells and grown for 2 days to 90% confluency. Cells were split 1:3 in 10 cm plates and grown for 24 h to 70% confluency. Plasmid was obtained as previously described. Cells were transfected with transfer plasmid containing the gene of interest, packing plasmids, and envelope plasmid at a 5:2:2:1 ratio, respectively, via Lipofectamine 3000 transfection (pMDLg/pRRE, Addgene:12251; pRSV-Rev, Addgene:12253; pMD2.G, Addgene:12259). Cells were washed after 15 h and the medium was replaced with DMEM supplemented with 20 mM HEPES, 4 mM sodium butyrate, and 10% (v/v) FBS. Viral supernatant was collected after 48 h and replaced with growth media. A second batch of viral supernatant was collected 48 h later. Viral stocks were pooled and centrifuged at 500g to get rid of cellular debris. The virus was concentrated using a Lenti-X Concentrator (Clontech) and resuspended at 1:100 original volume. The virus was immediately titrated at 1 µL, 10 µL, and 100 µL, and the remainder stored at −80 °C. Raw macrophages were spinfected in a tabletop centrifuge for 90 min at 30 °C and 2500 rpm and incubated at 37 °C and 5% CO2 for 2 days.
RAW-Blue ISG Assay
Cells were grown in a six-well plate to 60% confluency and transduced at a MOI of 1. Cells were incubated for 3 days to ensure cytoplasmic plasmid was destroyed and would not interfere with the assay. Infected cells were selected by sorting for mTurqoise (FACS AriaFusion). Cells were passaged and plated in a 96 well plate at 2.5 × 105 cells per well in 200 µL of DMEM supplemented with 10% (v/v) HIFBS. Cells were exposed to blue light for 2 h using a UVP Chromato-Vue UV transilluminator (115 V 60 Hz 1.8 Amp) and UVP UV/Blue converter plate (1.2 W/m2) and incubated at 37 °C and 5% CO2 for 20 h. After 20 h, 20 µL of the cell supernatant was placed in 180 µL of freshly prepared QuantiBlue (Invivogen) solution and incubated at 37 °C and 5% CO2 for up to 6 h. The plate was analyzed every hour using a Multiskan FC plate reader (Thermo Scientific), and absorbance was measured at 620 nm.
Flow Cytometry of Transduced RAW Macrophages
Cells plated for continued growth were passaged and resuspended in DMEM supplemented with 10% FBS. A total of 1.5 µL (3 × 105 cells) was placed in 1.5 mL microcentrifuge tubes and washed twice with PBS supplemented with 5% (v/v) FBS and placed in a final volume of 300 µL of PBS supplemented with 5% (v/v) FBS. Cells were analyzed using an Accuri C6 flow cytometer. Gating parameters were selected for size of a single cell.
Supplementary Material
Acknowledgments
The authors would like to thank the Optical Biology Core at University of California, Irvine, for assistance in image acquisition. We thank D. Trono for the gift of the third generation lentiviral plasmids (pMDLg/pRRE, Addgene:12251; pRSV-Rev, Addgene:12253; pMD2.G, Addgene:12259),15 C. Tucker for the gift of CIB1 and CRY2PHR genes (Addgene 28245 and 28244),16 and M. Davidson for the gift of mTurquoise (Addgene 54521).17
Funding
The authors acknowledge the financial support provided by National Institutes of Health (1U01Al124286-01 and 1DP2Al112194-01, GM099594), A.E.-K. thanks the Pew Scholars Program and the Cottrell Scholars Program for generous support. B.M. thanks the NSF GRFP (DGE-1321846). This work was supported, in part, by a grant from the Alfred P. Sloan foundation.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Additional cell assay data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
REFERENCES
- 1.Mogensen T. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009;22(2):240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 3.Xue Q, Lu Y, Eisele M, Sulistijo E, Khan N, Fan R, Miller-Jensen K. Analysis of single-cell cytokine secretion reveals a role for paracrine signaling in coordinating macrophage responses to TLR4 stimulation. Sci. Signaling. 2015;8(381):ra59. doi: 10.1126/scisignal.aaa2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lima S, Miesenböck G. Remote control of behavior through genetically targeted photostimulation of neurons. Cell. 2005;121:141–152. doi: 10.1016/j.cell.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Mancini R, Stutts L, Moore T, Esser-Kahn A. Controlling the origins of inflammation with a photoactive lipopeptide immunopotentiator. Angew. Chem., Int. Ed. 2015;54:5962–5965. doi: 10.1002/anie.201500416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stutts L, Esser-Kahn AP. Directing the Immune System with Chemical Compounds. ChemBioChem. 2015;16(20):1744–1748. [Google Scholar]
- 7.Ryu K, Stutts L, Tom J, Mancini R, Esser-Kahn A. Stimulation of Innate Immune Cells by Light-Activated TLR7/8 Agonists. J. Am. Chem. Soc. 2014;136:10823–10825. doi: 10.1021/ja412314j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mancini R, Tom J, Esser-Kahn A. A Light-Controlled TLR4 Agonist and Selectable Activation of Cell Subpopulations. Angew. Chem., Int. Ed. 2014;53:189–192. doi: 10.1002/cbic.201500164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takaoka A, Wang Z, Choi M, Yanai H, Negishi H, Ban T, Taniguchi T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy M, Hughes R, Peteya L, Schwartz J, Ehlers M, Tucker C. Rapid blue-light–mediated induction of protein interactions in living cells. Nat. Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konermann S, Brigham M, Trevino A, Hsu P, Heidenreich M, Cong L, Platt R, Scott D, Church G, Zhang F. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polstein L, Gersbach C. A light-inducible CRISPR/Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015;11:198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rebsamen M, Heinz L, Meylan E, Michallet M, Schroder K, Hofmann K, Vazquez J, Benedict C, Tschopp J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 2009;10(8):916–922. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan J, Cui Z, Wei H, Zhang Z, Zhou Y, Wang Y, Zhang X. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem. Biophys. Res. Commun. 2008;367(1):47–53. doi: 10.1016/j.bbrc.2007.12.101. [DOI] [PubMed] [Google Scholar]
- 15.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72(11):8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ai H, Olenych S, Wong P, Davidson M, Campbell R. Hue-shifted monomeric variants of Clavulariacyan fluorescent protein: identification of the molecular determinants of color and applications in fluorescence imaging. BMC Biol. 2008;6(13):1–13. doi: 10.1186/1741-7007-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow J, Franz K, Kagan J. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology. 2015;479–480:104–109. doi: 10.1016/j.virol.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunol. Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.