

Abstract
Autofluorescence is a problem that interferes with immunofluorescent staining and complicates data analysis. Throughout the mouse embryo, red blood cells naturally fluoresce across multiple wavelengths, spanning the emission and excitation spectra of many commonly used fluorescent reporters, including antibodies, dyes, stains, probes, and transgenic proteins — making it difficult to distinguish assay fluorescence from endogenous fluorescence. Several tissue treatment methods have been developed to bypass this issue with varying degrees of success. Sudan Black B dye has been commonly used to quench autofluorescence, but can also introduce background fluorescence. Here we present a protocol for an alternative called TrueBlack Lipofuscin Autofluorescence Quencher (Biotium). The protocol described in this unit demonstrates how TrueBlack efficiently quenches red blood cell autofluorescence across red and green wavelengths in fixed embryonic tissue without interfering with immunofluorescent signal intensity or introducing background staining. We also identified optimal incubation, concentration, and multiple usage conditions for routine immunofluorescence microscopy.
Keywords: Autofluorescence, Red Blood Cell, Fluorescent Microscopy, Embryonic Tissue, Immunocytochemistry
INTRODUCTION
Advances in fluorescent microscopy have dramatically enhanced our ability to visualize the location of specific proteins and structures in tissue samples using fluorescent reporters such as labeled antibodies, probes, dyes, stains, and transgenic proteins. Although many new techniques in tissue processing and fluorophore visualization have been developed, autofluorescence remains a common problem plaguing experimental and clinical studies that rely on fixed mammalian tissue for research and diagnostic purposes. Autofluorescence occurs when internal tissue components or external processing cause the tissue to naturally fluoresce across a broad excitation and emission spectra (Schnell et al., 1999). This is problematic for immunofluorescent assays because it can mask or interfere with the experimental fluorescent signal of a specifically labeled protein. This in turn significantly effects tissue examination and analysis (Baschong et al., 2001; Romijn et al., 1999). For example, autofluorescence can hinder one’s ability to determine protein co— localization. Thus, for proper tissue analysis it is imperative to develop methods to quench autofluorescence to isolate the experimental signal of fluorescent reporters, without reducing its intensity, or altering the structural integrity of the tissue.
There are both exogenous and endogenous factors that can contribute to tissue autofluorescence. The major exogenous factors come from tissue preparation and include fixation and subsequent treatment prior to cutting. During aldehyde fixation, amines combine with aldehydes (glutaraldehyde>paraformaldehyde>formaldehyde) to form complexes known as Schiff bases which are fluorescent (Willingham, 1983). In addition, the material one choses to embed the tissue in, such as paraffin, may chemically alter molecules in the tissue to induce autofluorescence (Baschong et al., 2001; Davis et al., 2014). Many of the exogenous factors can be altered to reduce autofluorescence. Chemicals such as sodium borohydride (Clancy and Cauller, 1998) have been used to reduce autofluorescence induced by aldehyde fixation, however such chemicals tend to be caustic and may reduce the signal intensity of fluorescent antibodies as well as physically damage the tissue. Unfortunately, endogenous factors are more problematic than exogenous factors.
Extracellular matrix components such as collagen and elastin, flavins, red blood cells and lipofuscin are endogenous factors that can naturally fluoresce in mammalian tissue (Banerjee et al., 1999; Schnell et al., 1999; Viegas et al., 2007). Lipofuscin is a yellow/brownish, granular, iron— negative lipid pigment that accumulates in lysosomes as cells age. Lipofuscin is highly fluorescent across multiple wavelengths, and is found in the majority of fixed mammalian tissues, including neurons (Erben et al., 2016; Sun et al., 2011). Additionally, some tissues naturally induce more autofluorescence than others. This is particularly true for many abdominal cavity organs such as liver, kidneys, and pancreas, and areas of the brain (Clancy and Cauller, 1998; Erben et al., 2016; Sun et al., 2011; Viegas et al., 2007). These areas are filled with red blood cells, which contain fluorescent hemoglobin (Alpert et al., 1980; Hirsch et al., 1980). Hemoglobin effectively absorbs light at wavelengths below 600 nm and impedes imaging of experimental fluorescent reporters in deep tissues of mammals (Stamatas et al., 2003). The development of experimental fluorescent reporters that excite effectively in the optical window at 600 nm and above is therefore highly desirable, but for most procedures, the experimental fluorescent reporters currently used span red blood cell fluorescence wavelengths. Red blood cell autofluorescence is a major problem for researchers performing developmental work in embryonic tissue, with red blood cells often still nucleated and not contained in well— formed vessels.
Researchers are able to bypass the problem of red blood cell autofluorescence in adult animals by perfusing (i.e. flushing out) red blood cells from the tissue of interest. However, perfusion is not always possible for those who rely on embryonic tissues. Digital processing is an alternative that can be used to artificially remove background signal, but it may be user subjective and requires an accurate assessment of how much intensity to subtract, which is prone to false positives. Although imaging software can be set to detract or minimize autofluorescence of red blood cells (and other molecules), it is extremely difficult when the cells of interest are proximal to autofluorescent areas and/or similarly shaped to red blood cells. One of the most widely used methods to quench autofluorescence is treatment of tissue with Sudan Black B dye. Sudan Black B is a lipid soluble dye that binds to lipofuscin granules to reduce their fluorescence. Sudan Black B has been used with varying degrees of success to prevent autofluorescence, however it increases background staining and fluorescence at high red wavelengths. (Baschong et al., 2001; Romijn et al., 1999; Schnell et al., 1999). Other dyes have also been examined, including Pontamine sky blue and Trypan Blue, however these dyes also fluoresce at red wavelengths, and thus are not suitable for double fluorescent labeling (Cowen et al., 1985; Srivastava et al., 2011).
This unit presents an alternative to Sudan Black B, TrueBlack Lipofuscin Autofluorescence Quencher (Biotium), and a working protocol for use on non— perfused embryonic mouse tissue using formaldehyde fixation. In the protocol presented, TrueBlack reduces tissue autofluorescence, including that of red blood cells, across both red and green wavelengths. In addition, we show that TrueBlack does not mask the intensity of fluorescent antibody signal, and can be reused, which is ideal for laboratories that routinely perform immunofluorescent staining.
BASIC PROTOCOL REDUCTION OF AUTOFLUORESCENCE IN FIXED EMBRYONIC TISSUE USING TRUEBLACK
Reduction of tissue autofluorescence is imperative for accurate analysis of fluorescent immunostained tissue. Early mouse embryos contain red blood cells that naturally fluoresce, and fixation processes can augment the autofluorescence of embryonic tissue. While Sudan Black B has been traditionally used to reduce these issues, Sudan Black B introduces background fluorescence that can be problematic and does not efficiently reduce red blood cell autofluorescence. Therefore, another lipophilic dye, TrueBlack Lipofuscin Autofluorescence Quencher (Biotium), was tested on embryonic mouse tissue. For the protocol described in this unit, embryos from NIH Swiss mice were collected from time— mated pregnant dams at E12.5, however, this protocol has been tested on E9.5— 14.5 fixed embryos with similar results. All procedures using live animals were reviewed and approved by the National Institute of Neurological Disorders and Stroke (NINDS) Animal Care and Use Committee (ACUC) and performed in accordance with NIH guidelines for the use and care of laboratory animals. In order to optimize the use of the TrueBlack, we have tested additional parameters in this unit which include concentration, incubation duration, reusability, and mounting media. Sections were subsequently imaged using an upright epifluorescent microscope with 20X objective, and analyzed using ImageJ software (ImageJ, RRID:SCR_003070).
Materials
Mouse embryos (collected in accordance with our ACUC approved Animal Safety Protocol (ASP))
Phosphate— Buffered Saline (PBS), pH 7.4 ± 0.2 Forceps and small scissors
Tissue culture dishes
12— well plate
4% (v/v) Formaldehyde in
PBS 30% (w/v) Sucrose in
PBS
Tissue— Plus O.C.T. Compound (Thermo Fisher Scientific Cat# 4585)
Foil boxes (12 × 12 × 10 mm)
Charged Superfrost Microslides
Cryostat
- Immunostaining:
- Citrate Buffer (see recipe)
- 10% (v/v) Normal Horse Serum (NHS) + 0.3% (v/v) Triton X— 100 (Tx— 100)
- Primary Antibodies:
- Rabbit anti— Peripherin (1:3000) (Millipore Cat# AB1530
- RRID:AB_90725) Rabbit anti— Pax6 (1:400) (Millipore Cat# AB5409 RRID:AB_2315065)
- Goat anti— Sox21 (1:100) (R and D Systems Cat# AF3538
- RRID:AB_2195947) Guinea pig anti— DCX (1:3500) (Millipore Cat# AB5910 RRID:AB_2230227) Rabbit anti— GnRH— 1 (SW— 1, 1:15,000) (Wray et al., 1988)
- Secondary antibodies:
- Alexa Fluor 488 Donkey anti— Rabbit IgG (1:1000) (Thermo Fisher Scientific Cat# A— 21206 also A21206 RRID:AB_2535792)
- Alexa Fluor 555 Donkey anti— Rabbit IgG (1:1000) (Thermo Fisher Scientific Cat# A— 31572 also A31572 RRID:AB_162543)
- Alexa Fluor 488 Donkey anti— Goat IgG (1:1000) (Thermo Fisher Scientific Cat# A— 11055 also A11055 RRID:AB_2534102)
- Alexa Fluor 488 Affinity Pure Donkey anti— Guinea pig IgG (1:1000) (Jackson ImmunoResearch Labs Cat# 706— 545— 148 RRID:AB_2340472)
70% (v/v) Ethanol (EtOH)
20X TrueBlack Lipofuscin Autofluorescence Quencher in DMF (Biotium Cat# 23007) Staining box
Vectashield Antifade Mounting Medium with DAPI (Vectashield + DAPI) (Vector Laboratories Cat# H— 1200 RRID:AB_2336790)
Glass coverslips
Protocol Steps and Annotations
Tissue collection and preparation
Terminate a time pregnant mouse (at desired embryonic age) using an approved CO2 chamber followed by cervical dislocation as approved by NIH/IACUC and your institution.
Wet the abdomen with 70% EtOH, and use forceps to lift the skin over the abdomen and cut the upper and lower layers of skin from above the vaginal opening towards the chest.
Lift out the embryo— filled uterus and detach the uterus from the body cavity with small scissors. Transfer the uterus to a tissue culture dish filled with cold PBS (4°C) and rinse 2x in cold PBS.
Transfer uterus to a clean dish and remove embryos from the uterus by cutting through the muscular side of the uterus with small scissors, pulling off the amniotic sacks (with embryos inside), and removing embryos from their amniotic sacks with forceps.
-
Place embryos into a 12— well plate filled with PBS. Rinse embryos 3x in PBS, then fix each whole embryo in 1 ml 4% formaldehyde for 1 hour at room temperature with gentle shaking, followed by two quick rinses in PBS.
Older embryos will require longer fixation times (may incubate overnight at 4°C with gentle shaking).
Submerge embryos in 2 ml 30% (w/v) sucrose/PBS overnight at 4°C for cryoprotection. Embryos will initially float in 30% sucrose, but will sink to the bottom of the well after sufficient incubation. Ensure that embryos have sunk to the bottom of the well before proceeding. Older embryos may take an additional 1–2 days to sink.
-
Quickly rinse embryos 2x in Tissue— Plus O.C.T. compound, then embed individual embryos in foil boxes filled with O.C.T. compound.
Foil boxes are made using squares of aluminum foil and appropriate size template such as cuvette.
Freeze embryos on dry ice and store at −80°C until ready to cut.
Cut embryonic sections (10μm – 14μm) using a cryostat, and collect sections on charged super— frost slides, approximately 8–12 sections per slide.
Store cut sections at −80°C until processing.
Immunostaining
For this protocol, we let sections sit without shaking during PBS washes, blocking, and antibody incubations. However, gentle shaking may be added if desired.
Thaw sections at room temperature, then fix in 4% formaldehyde/PBS for 10 minutes.
Rinse sections 3×8min in PBS to remove formaldehyde.
For sections on which immunofluorescence is not needed: proceed to TrueBlack treatment (see below), then mount and coverslip with VectaShield + DAPI.
- For sections on which immunofluorescence will be performed: ± citrate antigen retrieval (if needed) prior to staining.
-
Citrate antigen retrieval: Boil samples in Citrate Buffer for 5 minutes, then cool for 20 minutes in Citrate Buffer.We take slides in plastic Coplin jar and submerge in a plastic beaker filled with 1000 ml Citrate Buffer, and microwave them for 17 minutes— it takes 12 minutes to reach a boil, and our samples boil for 5 minutes. We recommend testing a boiling apparatus prior to use.
- Rinse 3×8min in PBS.
- Block in 10% NHS + 0.3% Tx— 100 for 1 hour at room temperature, then rinse 6×8min in PBS.
- Incubate in primary antibody 1–2 nights at 4°C.
- Rinse 6×10min in PBS.
- Incubate in fluorescent secondary antibody for 1 hour at room temperature (or overnight at 4°C), then rinse 6×8min in PBS.
- For single labeling: Proceed to step i.
- For double labeling:
- Fix briefly in 4% formaldehyde for 5 min.
- Rinse 6×8min in PBS.
- Incubate in second primary antibody at least 2 nights at 4°C.
- Rinse 6×10min in PBS.
- Incubate in fluorescent secondary antibody for 1 hour at room temperature (or overnight at 4°C), then rinse 6×8min in PBS.
- Treat with TrueBlack as described below.
- Mount and coverslip with VectaShield + DAPI.
-
TrueBlack Treatment
Prepare 1X TrueBlack solution by diluting 20X TrueBlack stock solution in 70% EtOH, shielded from light.
Following immunostaining, remove sections from PBS and wipe excess PBS off of slide using a tissue wipe.
-
Cover sections with small volume of TrueBlack solution for 3 minutes
We place slides upside down in staining box with 400ul of TrueBlack.
Rinse sections 3X quickly in PBS until PBS is clear.
REAGENTS AND SOLUTIONS
Citrate Buffer
0.1M Citric Acid (Stock Solution 1)
19.2 g Citric Acid
Bring volume up to 1000 ml with distilled water
Solution can be stored at room temperature for 3 months, or longer at 4°C
0.1M Sodium Citrate (Stock Solution 2) 29.4g Sodium Citrate Dihydride
Bring volume up to 1000 ml with distilled water
Solution can be stored at room temperature for 3 months, or longer at 4°C
Working Citrate
Buffer 9ml Stock
Solution 1 41ml Stock
Solution 2
Bring volume up to 1000 ml with distilled water
COMMENTARY
Anticipated Results
TrueBlack versus Sudan Black B
To compare the quenching efficiency of dyes, fixed E12.5 mouse tissue (non— perfused) was treated with either TrueBlack or Sudan Black B, and analyzed with epifluorescent microscopy (Figure 1). Comparable atlas sections were treated with 400ul of either 1X TrueBlack for 3 minutes or 0.1% Sudan Black in 70% EtOH for 30 minutes. Normal (untreated) embryonic tissue is filled with red blood cells that fluoresce across both red and green wavelengths (Figure 1A, arrows). However, this autofluorescence was greatly reduced following treatment with both TrueBlack and Sudan Black B dye quenchers. While both dyes adequately reduced red blood cell fluorescence, Sudan Black B increased background tissue fluorescence (Figure 1A, asterisks). The manufacturers of TrueBlack suggest an incubation time of 30 seconds, however we have found that a 3— minute incubation is optimal for blockage across both wavelengths. Because some antibodies require antigen retrieval, TrueBlack was also tested on tissue following citrate antigen retrieval (Figure 1B), which consists of boiling samples in Citrate Buffer. While the boiling process enhanced the intensity of fluorescence compared to normal tissue, one can see that treatment of this tissue with TrueBlack greatly reduced, but did not abolish, the red blood cell fluorescence.
Figure 1. TrueBlack reduces red blood cell autofluorescence across green and red wavelengths.
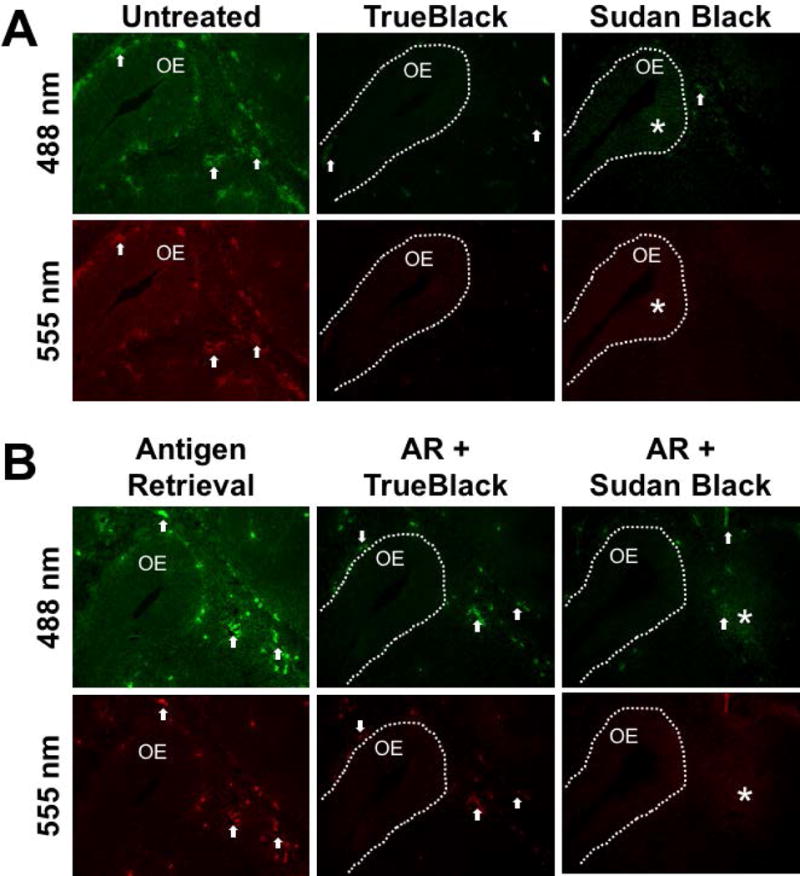
Untreated (A) and citrate antigen retrieval— treated (B) E12.5 fixed tissue sections were incubated in 1X TrueBlack for 3 minutes or 0.1% Sudan Black B for 30 minutes and the olfactory epithelium (OE) was imaged at green (488 nm) and red (555 nm) wavelengths. Arrows indicate autofluorescent red blood cells, which were reduced in tissue treated with TrueBlack and Sudan Black B, however Sudan Black B treatment also introduced background fluorescence, marked by asterisks.
In a similar manner, immunostained tissue sections were also treated with either TrueBlack or Sudan Black B (Figure 2). Sections were stained for GnRH— 1 to mark GnRH neurons, which are localized in areas that naturally contain aggregates of red blood cells (brain and nasal regions). Again it is clear that both TrueBlack and Sudan Black B efficiently reduced red blood cell autofluorescence and did not interfere with the signal intensity of GnRH fluorescent antibody. However, immunostained tissue treated with Sudan Black B contained background fluorescence. Together this shows that TrueBlack is an effective alternative to Sudan Black B to reduce red blood cell autofluorescence in fixed embryonic tissue.
Figure 2. TrueBlack reduces red blood cell fluorescence in immunostained tissue.
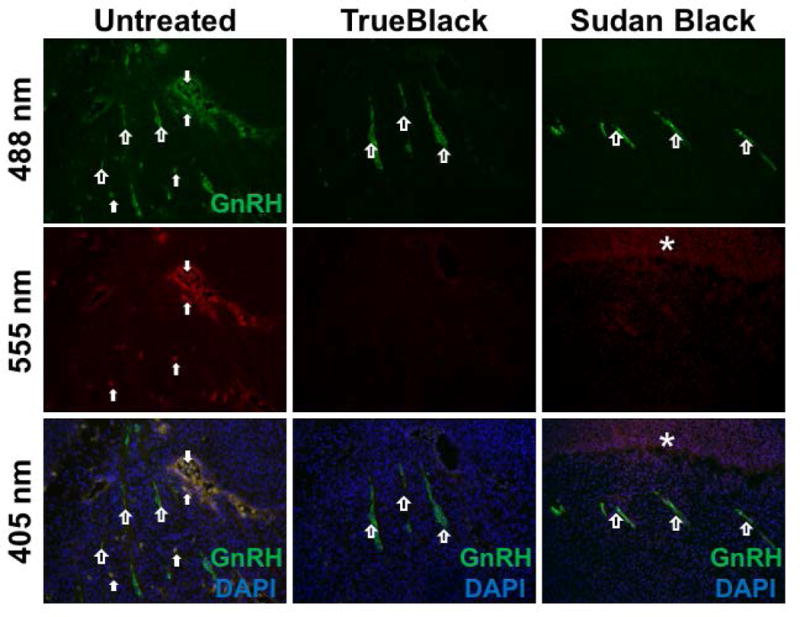
E12.5 fixed tissue sections were immunostained for GnRH and treated with 1X TrueBlack for 3 minutes or 0.1% Sudan Black for 30 minutes. Nasal tracks with stained GnRH neurons (green, open arrows) were imaged at green (488 nm), red (555 nm), and blue (405 nm) wavelengths. Untreated tissue contained many red blood cells (solid arrows) that fluoresced across all three wavelengths. Treatment with TrueBlack and Sudan Black B reduced autofluorescence, but Sudan Black B enhanced background fluorescence (asterisk).
TrueBlack Treatment for Double Immunofluorescence
With single immunofluorescence, red blood cells can be revealed by analyzing tissue across multiple wavelengths since only the red blood cells will fluoresce at wavelengths outside that of the labeled antibody. However, when double or triple immunofluorescence is desired, it can become difficult to differentiate fluorescent red blood cells from cells that are experimentally co— labeled by the fluorescent antibodies. Because of this, TrueBlack was assessed for autofluorescence quenching in double labeled tissue.
TrueBlack was examined in tissue subjected to antigen retrieval and double— labeled for expression of nuclear transcription factors Sox21 (labeled green) and Pax6 (labeled red) in the developing forebrain (Figure 3). While the two are expressed in unique regions, they are also co— localized in areas of the brain. Yet, because red blood cells fluoresce across both red and green wavelengths, they mimic cells with co— localized Sox21 and Pax6 expression, which can cause ambiguity in analysis. Treatment with TrueBlack abrogated red blood cell autofluorescence in this tissue, allowing for a clear view of co— localization. This was also tested on tissue without antigen retrieval with identical results.
Figure 3. TrueBlack quenches autofluorescence in double— labeled tissue.
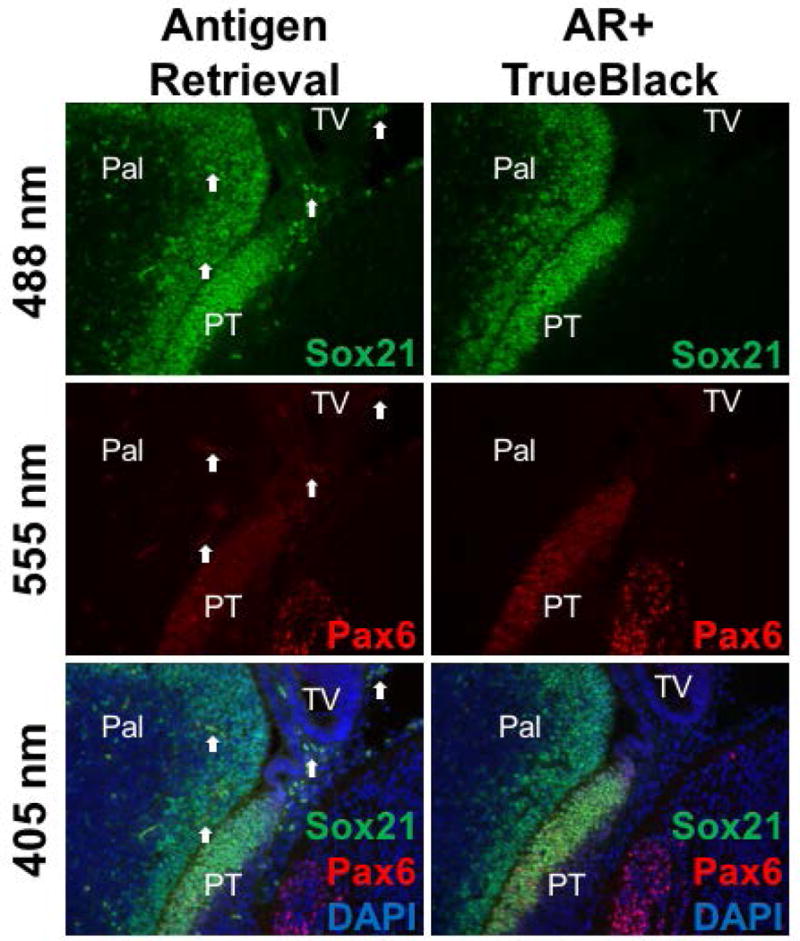
E12.5 fixed tissue sections were treated for citrate antigen retrieval, stained for Sox21 (green) and Pax6 (red) immunofluorescence, and treated with 1X TrueBlack for 3 minutes. Images of the forebrain (Pal= pallidum, PT= prethalamic eminence, TV= telencephalic vesicle) showed red blood cells (arrows) fluorescing across green (488 nm), red (555 nm), and blue (405 nm) wavelengths, which can imitate immunofluorescent signal co— localization. TrueBlack treatment eliminated this red blood cell autofluorescence.
Critical Parameters and Troubleshooting
Incubation Time and Concentration Considerations
We noticed that while TrueBlack treatment abolished autofluorescence on the red wavelength, there was still residual red blood cell fluorescence in green in unstained tissue. To determine the ideal concentration of TrueBlack and incubation time that are efficient to reduce autofluorescence on both red and green wavelengths, unstained sections were incubated with either 1X, 2X, or 3X TrueBlack for 3, 10, or 30 minutes (Figure 4). We found that increasing the concentration 1X–3X did not change the ability of TrueBlack to reduce red blood cell fluorescence within the sections of the gut, which naturally contain many fluorescent red blood cells. Additionally, increasing the incubation time from 3 minutes to 10 or 30 minutes did not enhance the ability of TrueBlack to quench autofluorescence. Thus, 3 minutes of 1X TrueBlack was sufficient to reduce autofluorescence and increasing either concentration or incubation time did not yield better results. The same was true in immunostained tissue (Figure 5), where Peripherin— labeled sections of the gut (A) and trigeminal ganglion (B) both show that a 3— minute incubation of 1X TrueBlack is adequate to reduce red blood cell fluorescence and increasing the incubation time or concentration does not change quenching of autofluorescence.
Figure 4. 1X TrueBlack with short incubation time is sufficient.
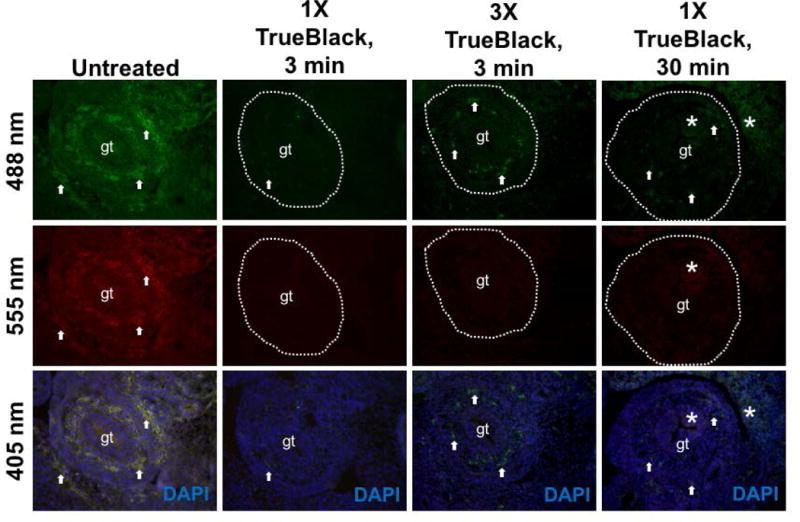
E12.5 fixed tissue sections were treated with different concentrations of TrueBlack (1X–3X) for 3 or 30 min, and sections of the gut (gt) were imaged at green (488 nm), red (555 nm), and blue (405 nm) wavelengths. Arrows indicate autofluorescent red blood cells, which are reduced but not abolished in unstained TrueBlack treated tissue. Increased concentration did not result in reduced autofluorescence, and longer incubation time introduced background fluorescence (asterisks).
Figure 5. 1X TrueBlack with short incubation time is sufficient for immunofluorescence.
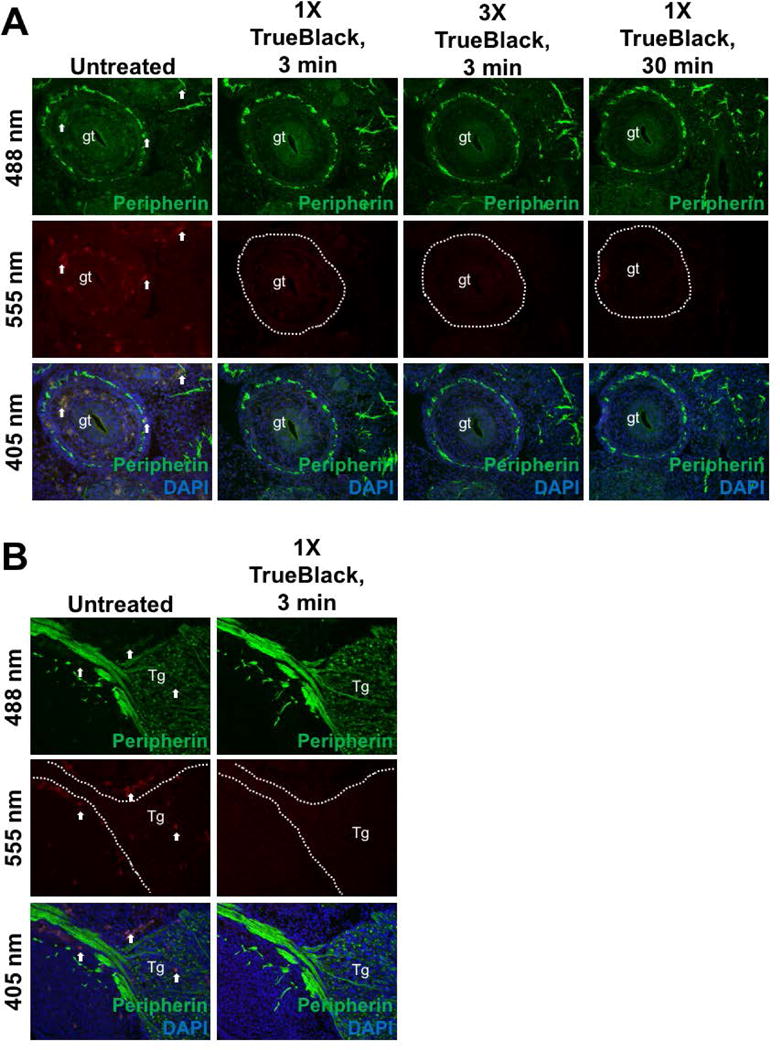
E12.5 fixed tissue sections were immunostained for Peripherin, then treated with different concentrations of TrueBlack (1X–3X) for 3 or 30 min. Sections of the gut (gt) (A) and trigeminal ganglion (Tg) (B) were imaged. Arrows indicate autofluorescent red blood cells, which were drastically diminshed in immunofluorescent TrueBlack treated tissue. Increased concentration or longer incubation time did not result in reduced autofluorescence.
In this protocol, we analyzed E12.5 embryonic sections with a thickness of 10μm, with 8–10 sections per slide, and found that a 3— minute incubation was sufficient to reduce red blood cell autofluorescence at both red and green wavelengths. As embryos age, they drastically increase in size and complexity, and therefore the size and number of sections on the slide as well as desired thickness may also change. As such, it will be important to consider optimizing TrueBlack incubation time for specific tissue conditions (size, thickness, and number of sections on each treated slide). It is possible that older embryos, or thicker sections, may require longer incubation times (up to 5 minutes) to sufficiently reduce red blood cell autofluorescence at both red and green wavelengths. We suggest testing a range of incubation times based on the age and thickness of your tissue.
Reusability
For laboratories that routinely perform immunofluorescence, reagents may be used multiple times for economic and environmental reasons. TrueBlack was tested to determine if it could be reused on stained and unstained tissue (Figure 6). In unstained tissue, fresh TrueBlack had the best results in reducing red blood cell autofluorescence (Figure 6A). When reused up to four times, TrueBlack reduced but did not eliminate red blood cell fluorescence (arrows), and an 8th use resulted in background fluorescence and reduced ability to quench red blood cell autofluorescence. A similar result was found in immunostained tissue (Figure 6B) — up to four uses of TrueBlack blocked autofluorescence without inducing background fluorescence. Therefore, we suggest that TrueBlack can be reused at least four times and no more than eight times without losing its activity. For older or thicker cut tissue, this may need to be adjusted.
Figure 6. TrueBlack may be reused for blocking autofluorescence.
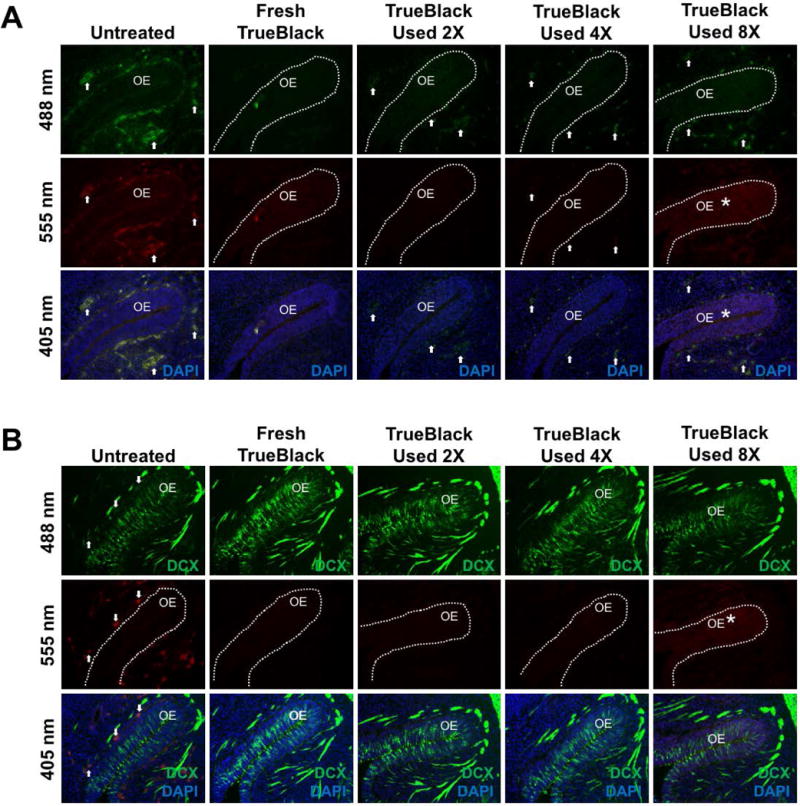
Unstained (A) and DCX— immunostained (B) E12.5 fixed tissue sections were treated with either fresh or reused TrueBlack as indicated for 3 minutes, and olfactory epithelia (OE) were imaged. Arrows indicate autofluorescent red blood cells, which are present in untreated tissue, but greatly reduced by treatment with fresh TrueBlack. With more reuse, TrueBlack gradually lost its ability to decrease red blood cell autofluorescence, and by the 8th use induced background fluorescence (asterisk).
Mounting Media
Following TrueBlack treatment, sections should be mounted and coverslipped using any standard aqueous— based fluorescence mounting media, as TrueBlack is not compatible with organic— based mountants. For this protocol, sections were coverslipped using Vectashield +DAPI. Other mounting media, such as EverBrite, may also be suitable for use. Although EverBrite was not tested with this protocol, the manufacturers suggest that it can be used with TrueBlack and will preserve the tissue without fading. Other mounting media were tested, including Vectashield HardSet and Fluoro— Gel. While Vectashield HardSet worked well for our use, Fluoro— Gel was inappropriate for use with TrueBlack. When mounted with Fluoro— Gel, the dye came off of the tissue and autofluorescence was no longer quenched. Therefore, one should test the compatibility of the mounting media used in one’s lab, if different from those tested here.
TrueBlack Pre— treatment
The manufacturers suggest that pre— treatment with TrueBlack prior to immunofluorescent staining is ideal because it does not affect the signal of fluorescent antibodies and nuclear stains, which could be a concern with post— treatment. We have compared pre— treated and post— treated tissues, and saw no difference in reduction of autofluorescence or change in fluorescent intensities of our antibodies or DAPI stains. We prefer post— treatment over pre— treatment because we found that the TrueBlack dye gradually comes off of the tissue and contaminates our blocking solutions and antibodies during longer incubations. Since we generally reuse these solutions, post— treatment is optimal for our use. If pre— treatment is preferred, one must not use buffers containing detergents, which can remove TrueBlack from the tissue. Thus, if detergent treatment is required, we suggest post— treatment as described in this protocol.
Time Considerations
TrueBlack treatment will add up to 5 additional minutes to the length of any standard immunocytochemistry staining protocol (see Incubation Time and Concentration Considerations above). The following time estimations are based on a single— labeling experiment using E12.5 embryos that have been cut into 10μm sections. These times will differ based on the age of the embryo and thickness of the sections, and may be extended an additional 3–5 days for double or triple labeling assays.
Total time (Tissue collection, preparation, immunostaining, and TrueBlack treatment): ≈ 40 hrs, or 4 days.
Tissue Collection and Preparation ≈ 19 hrs, or 2 days
Embryo collection ≈ 20 min
Embryo fixation and sucrose incubation ≈ 16 hrs (+ up to 3 days for older embryos)
Tissue embedding ≈ 1 hr
Tissue cutting ≈ 1 hr, depending on the size of embryo and thickness of sections
Immunocytochemistry ≈ 21 hrs, or 2 days
Antigen Retreival (with washes) ≈ 1 hr
Blocking, washes, and primary antibody incubation ≈ 17 hrs
Washes and secondary antibody incubation ≈ 3 hrs
TrueBlack Staining (with washes) and coverslipping ≈ 10 min.
Significance Statement.
Tissue autofluorescence occurs as components, such as red blood cells, naturally fluoresce across multiple wavelengths, and this can be enhanced by the fixation process. Auto— fluorescence is problematic because it can mask or interfere with the fluorescence of experimental reporters such as antibodies, probes, and transgenic proteins, making it difficult to distinguish assay fluorescence. Therefore, one must reduce autofluorescence to accurately analyze fluorescent reporters. Treatment with Sudan Black B has been used to reduce autofluorescence, however this also introduces background fluorescence that can affect analysis. Thus, another lipophilic dye, TrueBlack Lipofuscin Autofluorescence Quencher, was tested on fixed embryonic mouse tissue. This protocol shows that TrueBlack effectively reduces red blood cell autofluorescence without altering reporter fluorescence or introducing background fluorescence.
Acknowledgments
We thank Mr. H. Saadi (CDNS, NINDS) for sectioning of embryos. This work was supported by the Intramural research program of NINDS, NIH, Bethesda, MD, USA (NS002824--- 26).
Contributor Information
Niteace C. Whittington, Cellular and Developmental Neurobiology Section, NIH/NINDS, 35 Convent Drive, Bldg. 35 Rm 3A1010 Bethesda, Maryland 20892 (301), 496--- 8336
Susan Wray, Cellular and Developmental Neurobiology Section, NIH/NINDS, 35 Convent Drive, Bldg. 35 Rm 3A1012 Bethesda, Maryland 20892, (301) 496--- 6646.
LITERATURE CITED
- Alpert B, Jameson DM, Weber G. Tryptophan Emission From Human Hemoglobin and Its Isolated Subunits. Photochem Photobiol. 1980;31:1–4. doi: 10.1111/j.1751-1097.1980.tb03674.x. [DOI] [PubMed] [Google Scholar]
- Banerjee B, Miedema B, Chandrasekhar H. Role of basement membrane collagen and elastin in the autofluorescence spectra of the colon. J Investig Med. 1999;47:326–32. [PubMed] [Google Scholar]
- Baschong W, Suetterlin R, Laeng RH. Control of Autofluorescence of Archival Formaldehyde— fixed, Paraffin— embedded Tissue in Confocal Laser Scanning Microscopy (CLSM) Jounal Histochem Cytochem. 2001;49:1565–1572. doi: 10.1177/002215540104901210. [DOI] [PubMed] [Google Scholar]
- Clancy B, Cauller LJ. Reduction of background autofluorescence in brain sections following immersion in sodium borohydride. J Neurosci Methods. 1998;83:97–102. doi: 10.1016/s0165-0270(98)00066-1. [DOI] [PubMed] [Google Scholar]
- Cowen T, Haven AJ, Burnstock G. Pontamine sky blue: a counterstain for background autofluorescence in fluorescence and immunofluorescence histochemistry. Histochemistry. 1985;82:205–8. doi: 10.1007/BF00501396. [DOI] [PubMed] [Google Scholar]
- Davis AS, Richter A, Becker S, Moyer JE, Sandouk A, Taubenberger JK. Characterizing and Diminishing Autofluorescence in Formalin— fixed Paraffin— embedded Human Respiratory Tissue. J Histochem Cytochem. 2014;62:405–423. doi: 10.1369/0022155414531549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erben T, Ossig R, Naim HY, Schnekenburger J. What to do with high autofluorescence background in pancreatic tissues – an efficient Sudan black B quenching method for specific immunofluorescence labelling. Histopathology. 2016;69:406–422. doi: 10.1111/his.12935. [DOI] [PubMed] [Google Scholar]
- Hirsch RE, Zukin RS, Nagel RL. Intrinsic Fluorescence Emission of Intact Oxy Hemoglobins. Biochem Biophys Res Commun. 1980;93:432–439. doi: 10.1016/0006-291x(80)91096-7. [DOI] [PubMed] [Google Scholar]
- Romijn HJ, Uum JFM, Van Breedijk I, Emmering J, Radu I, Pool CW. Double Immunolabeling of Neuropeptides in the Human Hypothalamus as Analyzed by Confocal Laser Scanning Fluorescence Microscopy. J Histochem Cytochem. 1999;47:229–235. doi: 10.1177/002215549904700211. [DOI] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of Lipofuscin— like Autofluorescence in Fluorescently Labeled Tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Srivastava GK, Reinoso R, Singh AK, Fernandez-Bueno I, Hileeto D, Martino M, Garcia-gutierrez MT, Pigazo JM, Fernández N, Corell A, et al. Trypan Blue staining method for quenching the auto fl uorescence of RPE cells for improving protein expression analysis. Exp Eye Res. 2011;93:956–962. doi: 10.1016/j.exer.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Stamatas GN, Balas CJ, Kollias N. Hyperspectral Image Acquisition and Analysis of Skin. Proc SPIE, Spectr Imaging Instrumentation, Appl Anal II. 2003;4959:77–82. [Google Scholar]
- Sun Y, Yu H, Zheng D, Cao Q, Wang Y, Harris D, Wang Y. Sudan Black B Reduces Autofluorescence in Murine Renal Tissue. Arch Pathol Lab Med. 2011;135:1335–1342. doi: 10.5858/arpa.2010-0549-OA. [DOI] [PubMed] [Google Scholar]
- Viegas MS, Martins TC, Seco F, do Carmo A. An improved and cost— effective methodology for the reduction of autofluorescence in direct immunofluorescence studies on formalin— fixed paraffin— embedded tissues. Eur J Histochem. 2007;51:59–66. [PubMed] [Google Scholar]
- Willingham MC. An Alternative Fixation-Processing Method for Preembedding Ultrastructural Immunocytochemistry of Cytoplasmic Antigens: The GBS (Glutaraldehyde— Borohydride— Saponin) Procedure. Jounal Histochem Cytochem. 1983;31:791–798. doi: 10.1177/31.6.6404984. [DOI] [PubMed] [Google Scholar]
- Wray S, Gahwiler BH, Gainer H. Slice cultures of LHRH neurons in the presence and absence of brainstem and pituitary. Peptides. 1988;9:1151–1175. doi: 10.1016/0196-9781(88)90103-9. [DOI] [PubMed] [Google Scholar]