

Abstract
Genome rearrangements underlie different human diseases including many cancers. Determining the rates at which genome rearrangements arise and isolating unique, independent genome rearrangements is critical to understanding the genes and pathways that prevent or promote genome rearrangements. Here we describe quantitative S. cerevisiae genetic assays for measuring the rates of accumulating genome rearrangements including deletions, translocations and broken chromosomes healed by de novo telomere addition that result in deletion of two counter-selectable genes, CAN1 and URA3, placed in non-essential regions of the S. cerevisiae genome. The assays also allow for the isolation of individual genome rearrangements for structural studies, and a method for analyzing genome rearrangements by next-generation DNA sequencing is provided.
Keywords: Genome instability, deletion, monocentric translocation, dicentric translocation, de novo telomere addition, genetics, GCR rates, whole-genome sequencing
1 Introduction
Maintaining the stability of the genome during cell division is critical for cell survival and normal cell growth. Understanding the pathways and mechanisms by which cells prevent genome rearrangements (herein called Gross Chromosomal Rearrangements or GCRs) has been greatly facilitated by the development of simple genetic methods for measuring the rate at which GCRs occur in different wild-type and mutant S. cerevisiae strains [1–3]. Such methods have made it possible to identify genes in which defects cause increased or decreased GCR rates, perform epistasis analysis of these mutations and isolate independent GCRs for structural analysis (for examples see [4, 5]). These types of studies have provided a growing picture of the cellular pathways that act to prevent GCRs as well as the pathways that produce GCRs.
The key observation that led to the development of a broad series of assays for measuring the rate of accumulation of GCRs was that the CAN1-containing portion of the left arm of chromosome V from the telomere to PCM1 was nonessential [6]. This allowed the development of the first GCR assay (referred to as the “classical” GCR assay) in which the URA3 gene was inserted into HXT13 between CAN1 and the telomere (Figure 1; [2]). During normal mitotic growth of haploid cells containing this genetically marked chromosome, progeny arise that are resistant to both canavanine (Can) and 5-fluoroorotic acid (5-FOA). These double-drug-resistant progeny contain a rearranged chromosome V in which URA3 and CAN1 have been deleted by the formation of GCRs including: 1) interstitial deletions, 2) terminal deletions of chromosome V L that are healed by the addition of a new telomere, and 3) terminal deletions of chromosome V L healed by fusion to a fragment of another chromosome, generating a translocation, which can either be stable monocentric translocations or dicentric translocations that are unstable and undergo additional rounds of rearrangement (Figure 1) [2, 6–8]. By measuring the rate of occurrence of Carr 5-FOAr progeny, GCR rates can be determined and by determining the structure of the GCRs present in individual progeny, the rates of occurrence of specific types of GCRs can be determined.
Figure 1. GCR assays.
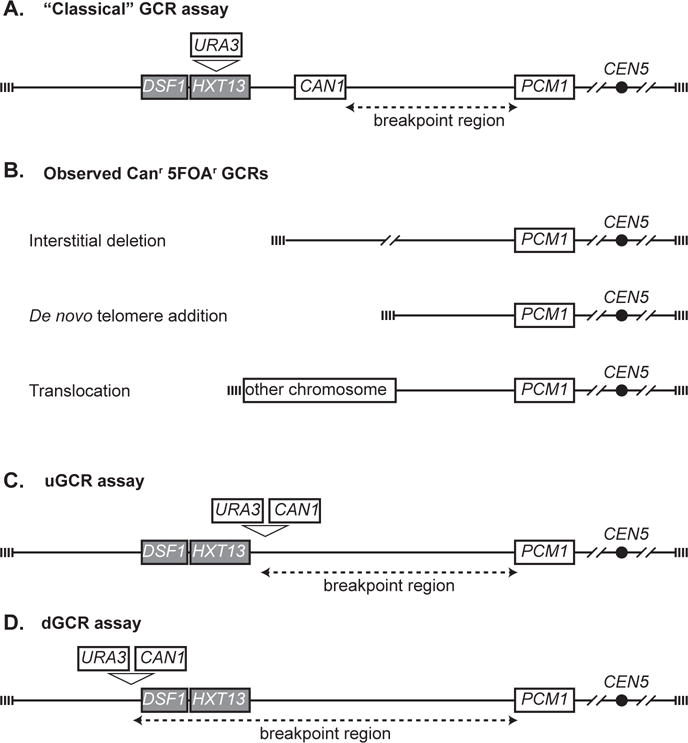
A. The “classical” GCR assay is constructed by inserting URA3 into HXT13. The breakpoint region (dashed line) is between the most telomeric essential gene, PCM1, and the most centromeric marker gene, CAN1. B. A variety of GCR products have been observed, including interstitial deletions, terminal deletions healed by de novo telomere additions, and translocations. The types of translocations are dictated by the orientation, presence or absence of a centromere and source of the sequence joined to the broken chromosome V. C. The unique sequence or uGCR assay contains a CAN1 URA3 cassette centromeric to the DSF1-HXT13 segmental duplication (grey). D. The duplication-mediated or dGCR assay contains a CAN1 URA3 cassette telomeric to the DSF1-HXT13 segmental duplication (grey).
To increase the utility of GCR assays, CAN1 and URA3 were inserted into a cassette. This made it possible to move the two counter-selectable genes to different chromosomal locations in haploid strains containing mutations at the CAN1 and URA3 chromosomal loci to evaluate the effect of chromosomal context on GCR rates. In two such assays (Figure 1), the CAN1 URA3 cassette was placed either centromeric (the yel068c::CAN1/URA3 or uGCR assay) or telomeric (the yel072w::CAN 1/URA3 or dGCR assay) to the HXT13-DSF1 segmental duplication region on chromosome V L [3]. These assays revealed strong influences of having a sequence in the breakpoint region with divergent homology with other regions of the genome on GCR rates, on the pathways that suppress GCRs and the mechanisms by which GCRs are formed [3]. In addition to these GCR assays, a number of other haploid strain-based GCR assays utilizing counter-selectable markers in non-essential regions of the genome have been described (for examples see [9, 10]). In addition, GCR assays in which amplification of genetic markers is selected for in haploid strains have been described, as have a limited number diploid strain-based GCR assays (for examples see [11–13]).
GCR structures can be analyzed using a variety of methods such as PCR-based mapping and amplification of breakpoint junctions, multiplex ligation-dependent probe amplification (MLPA), pulsed-field gel electrophoresis (PFGE) combined with Southern blotting, and microarray-based comparative genome hybridization (aCGH), and usually, determining the complete structure of an individual GCR requires multiple methods and approaches [2, 3, 7, 14, 15]. A method that has been recently adapted for analysis of GCR structures is next-generation whole-genome sequencing (NGS) (Figure 3; [16, 17]). Analysis of GCRs using paired-end sequencing, which provides sequence from both ends of genomic DNA fragments, is particularly useful for the analysis of GCRs and should be the method used when performing NGS on GCR-containing strains. Compared to prior methods, NGS is less labor-intensive and more rapidly provides genome-wide data on copy number, point mutations and genome rearrangements. Furthermore, the small size of the S. cerevisiae genome allows multiple strains to be sequenced simultaneously (multiplexed), greatly reducing the cost of analysis.
Figure 3. Examples of GCR structures deciphered through analysis of NGS data.
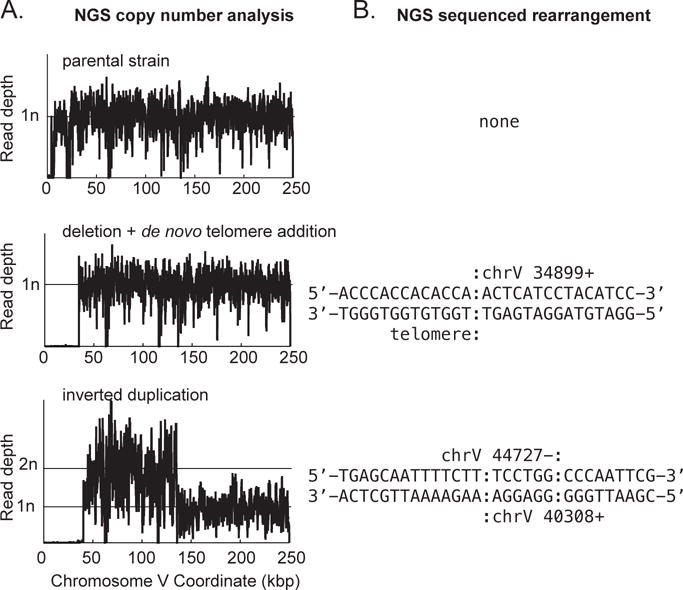
A. Copy number analysis for the left arm of chromosome V is plotted for a parental strain (top), a de novo telomere addition GCR (middle), and a GCR involving an inverted duplication (bottom). Note that both rearranged chromosomes have deleted the region containing the CAN1 URA3 cassette (around coordinate 25,700 in this GCR assay strain). The inverted duplication has additionally duplicated the region from around coordinate 44,700 to the Ty-related sequence YELWdelta6 (around coordinate 138,300). B. Sequence of the novel junctions involved in the observed GCR. A de novo telomere (sequence prior to the colon) healed the terminal deletion at chromosome V 34,899 for the strain displayed in the middle of the panel. For the strain displayed at the bottom of the panel, non-concordant read pairs in the NGS data revealed the presence of an inverted duplication, which was also confirmed by the increase in copy number (panel A) and through the sequence of the reads that are associated with the novel junction and hence did not map to the reference sequence. For this inverted duplication, there is a six base identity (sequence between the colons) at the junction between the source and target of the inversion. The inversion was not a perfect hairpin, however, as ~4,000 bp separate the two fused sequences.
In this article, we describe the use of GCR assays based on selection against the presence of URA3 and CAN1 to both measure GCR rates and isolate progeny containing unique GCRs for structural analysis. We also describe a method for preparing 600 bp multiplexed libraries from S. cerevisiae strains for paired-end whole-genome sequencing using an Illumina HiSeq 2500 for characterizing GCR structures (see Note 1). The general approach described is applicable to many other types of GCR assays.
2 Materials
2.1 Determining GCR rates
YPD (Yeast extract Peptone Dextrose) liquid medium: For 1 L of medium, add 10 g Bacto-yeast extract and 20 g Bacto-peptone to 950 ml deionized water, and sterilize by autoclaving at 121°C for 20 min. Cool and add 50 ml of a 40% (w/v) sterile glucose (D-dextrose) solution.
YPD plates: For 1 L of medium (40 plates), add 10 g Bacto-yeast extract, 20 g Bacto-peptone, and 21 g of Bacto-agar to 950 ml deionized water in a 2 L flask, place a magnetic stir bar in the flask and sterilize by autoclaving at 121 °C for 20 min. Cool to 60°C and add 50 ml of a 40% (w/v) sterile glucose (D-dextrose) solution. Mix well by stirring and pour 25 ml per plate in 100×15 mm round petri dishes. Allow the plates to dry for 1–2 days at room temperature.
40% (w/v) glucose solution: Dissolve 400 g glucose (D-dextrose) in 600 ml warm deionized water with continuous stirring, adjust the volume to 1000 ml, and sterilize by autoclaving at 121°C for 20 min.
Can 5-FOA plates: Add 23–25 g Bacto-agar to 750 ml deionized water in a 2 L flask and place a magnetic stir bar in the flask. Sterilize the agar mixture by autoclaving at 121°C for 20 min and then stir on a magnetic stir plate to allow the mixture to cool to ~65°C (see Note 2). While the agar mixture cools, prepare the dropout mix as follows. Dissolve 60 mg L-canavanine sulfate (Can) and 1 g 5-fluoroorotic acid (5-FOA) in 200 ml warm deionized water. Add 2 g of dropout powder minus arginine (US Biological, D9518), 6.7 g of yeast nitrogen base without amino acids (with ammonium sulfate), and 50 ml of 40% (w/v) sterile glucose solution, and stir well to dissolve. The dropout solution can be filter sterilized, although this is generally not necessary because the combination of Can and 5-FOA is capable of killing most potential contaminants. Keep the dropout mix at 55–60°C until the autoclaved agar mixture has cooled sufficiently. Carefully transfer the dropout mix to the autoclaved agar mixture, avoiding the formation of air bubbles. Pour approximately 70 ml per plate in 150×15 mm round petri dishes or 25 ml per plate in 100×15 mm round petri dishes as needed. Allow the plates to dry for 1–2 days at room temperature.
Sterile deionized water.
Sterile plastic culture tubes (15 ml round-bottomed tubes, 50 ml conical tubes).
Sterile glass 250 ml culture flasks for larger cultures.
Sterile toothpicks to streak out, patch, and inoculate colonies.
Surgical scalpel.
Sterile microcentrifuge tubes – 1.5 ml.
Sterile 4 mm glass beads.
Benchtop centrifuge.
Centrifuge with swinging bucket rotor.
Incubator set to 30°C.
Shaker set to 30°C.
Sterile velvets.
Replica-plating block.
2.2 For preparing libraries for NGS to analyze GCR structures
YPD liquid medium.
YPD plates.
40% (w/v) sterile glucose solution.
Sterile deionized water.
Gentra Puregene Yeast/Bact. kit (Qiagen) or preferred genomic DNA extraction kit
4 mg/ml RNase A (Qiagen or equivalent).
Covaris microTUBEs: microTUBE-15AFA beads screw-cap vials and M220 holder XTU insert microTUBE 15 μl. For 50 μl sample volumes, use microTUBE-50AFA fiber screw-cap vials and M220 holder XTU insert microTUBE 50 μl.
Covaris Focused Ultrasonicator M220.
FlashGel DNA Cassette 1.2%, 12+1, single tier (Lonza).
FlashGel Loading Dye 5 × 1 ml, 5× concentrate (Lonza)
FlashGel DNA Marker 100 bp–4 kb (Lonza).
FlashGel Dock System (Lonza).
End-It DNA end-repair kit (Epicentre Technologies).
Klenow DNA polymerase (3′–5′ exo−) (5U/μl) (New England Biolabs (NEB) or equivalent).
dATP.
Quick ligation kit (NEB or equivalent).
Library amplification readymix (KAPA Biosystems).
TruSeq PCR-Free LT DNA sample prep kit set A (Illumina).
MinElute PCR purification kit (Qiagen or equivalent).
MinElute gel extraction kit (Qiagen or equivalent).
Certified low-range ultra agarose (Bio-Rad or equivalent).
50× TAE Buffer.
10× Orange Loading Buffer (NEB or equivalent).
100 bp DNA ladder (Bioline or similar).
SYBR Safe DNA gel stain.
Benchtop microcentrifuge.
Microcentrifuge tubes − 0.5 ml (clear, thin-walled; Thermo Fisher Scientific or equivalent) and 1.5 ml.
Centrifuge with swinging bucket rotor.
Sterile toothpicks to streak out and inoculate strains.
Sterile culture tubes (15 ml round-bottomed tubes, 50 ml conical tubes).
Razor blades and forceps.
Nanodrop 2000 UV-Vis spectrophotometer.
Dark Reader transilluminator (Iso BioExpress) or other UV transilluminator.
Electrophoresis power supply, slab gel trays and electrophoresis tank.
Thermal cycler.
Qubit fluorometer.
Qubit dsDNA HS assay kit.
3 Methods
3.1 Determining GCR rates
Streak out the S. cerevisiae strains of interest containing the desired GCR assay (see Note 3) on YPD plates (or appropriate selective medium if plasmid selection is required), and incubate the plates for 2–3 days at 30°C to obtain well-separated single colonies. Streak out at least 2 independent biological isolates for each strain.
Using a sterile surgical scalpel (see Note 4), excise and inoculate at least 7 colonies from each biological isolate into individual sterile tubes or flasks containing an appropriate volume (see Note 5) of YPD liquid medium (or appropriate selective medium, if plasmid selection is required). Ensure that the entire colony is used for inoculation. Grow the cultures to saturation (16–18 hours; see Note 6) in a 30°C shaker with vigorous shaking (250–300 rpm).
To obtain the viable cell count, plate an appropriate dilution of each culture on YPD (or appropriate selective medium) plates as follows. For each culture, prepare 5 microcentrifuge tubes containing 90 μl sterile deionized water. Transfer 10 μl of the saturated culture into the 1st tube and vortex vigorously. Then transfer 10 μl from the 1st tube into the 2nd tube, and so on, until the 5th tube, vortexing vigorously before each serial dilution, to obtain a 10-fold dilution series. Transfer the entire 0.1 ml of the 10−5 dilution to a YPD plate and spread well using sterile glass beads.
To select for cells that contain a GCR, plate an appropriate volume (see Note 7) of each culture on Can 5-FOA plates. Use 100×15 mm and 150×15 mm petri dishes to plate up to 1 ml (108 cells) and 10 ml (109 cells) of the culture, respectively. Use multiple plates if larger culture volumes must be plated; for instance, while plating RDKY3615 (the classical GCR assay), which requires ~50 ml cultures, use five 150×15 mm Can 5-FOA plates per culture, i.e., 10 ml culture (109 cells) per plate as follows. After plating the appropriate dilution on YPD plates (step 3), centrifuge the remaining culture at 3000 rpm for 10 min at room temperature and discard the supernatant. For up to 1 ml of culture, resuspend the cells in 0.1 ml of sterile deionized water and plate on a 100×15 mm Can 5-FOA plate. For larger culture volumes, resuspend the cells in 0.2 ml of sterile deionized water per 10 ml of original culture, and plate 0.2 ml per 150×15 mm Can 5-FOA plate.
Incubate the YPD and Can 5-FOA plates at 30°C for 2–3 days and 3–5 days, respectively.
Count and record the number of colonies on each YPD plate (see Note 8) and the corresponding Can 5-FOA plates (see Note 9).
Calculate the GCR rate using the Lea-Coulson method of the median [18]. The spreadsheet illustrated in Figure 2 provides a method to calculate the median and 95% confidence interval of the median based on the total volume of the cultures and the count of the colonies on the non-selective and selective plates. A crucial step in the calculation is the use of the number of mutants per culture, r, to estimate m, which is the most likely number of mutations per culture. The non-linear relationship between m and r requires, in each spreadsheet, the CalcM() macro, which calculates the Lea-Coulson median estimator using the Newton-Raphson method (Figure 2).
Figure 2. Spreadsheet for calculating the median GCR rate of a series of cultures and 95% confidence intervals for the median GCR rate.
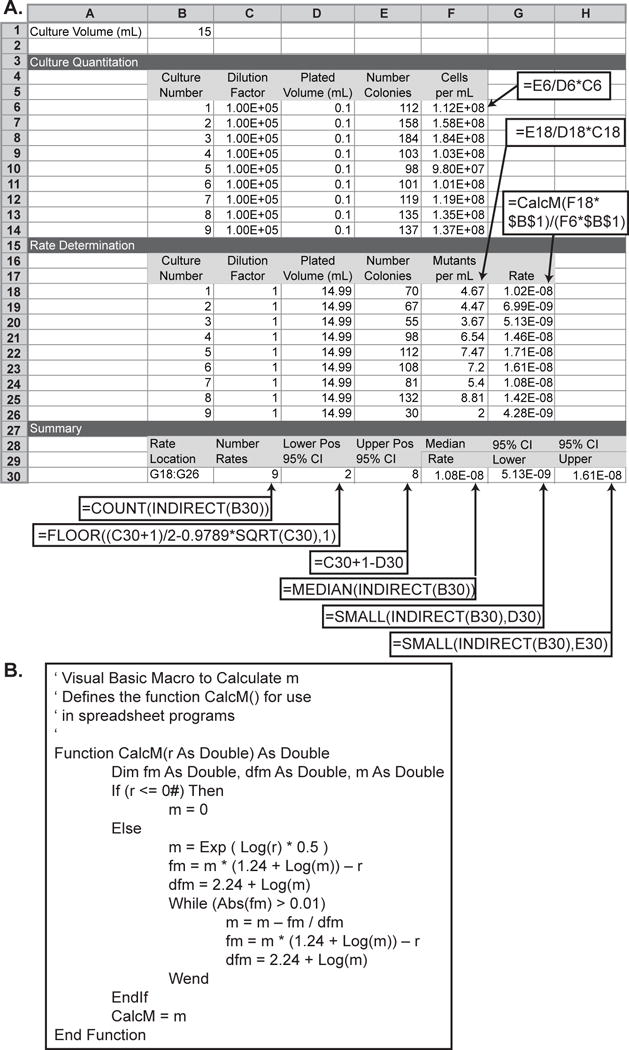
A. The spreadsheet is displayed with both sample data and the underlying formulas. Callout boxes indicate cells that should contain formulas; the required formulas are indicated the callout boxes. For cells F6 to F14, F18 to F26, and G18 to G26, the row specifiers in the formula should be incremented in each row (for example F6 should have the formula =E6/D6*C6 and F7 should have the formula =E7/D7*C7). Most modern spreadsheets have fill functions that will automatically increment these values. B. In order to calculate the rate in cells G18 to G26, the CalcM() Visual Basic macro must be defined for the spreadsheet. CalcM() initializes the test value for m to the square root of r and uses the iterative Newton-Raphson method to solve the transcendental equation r/m − ln(m) − 1.24 = 0. This macro functions in both Microsoft Excel and in LibreOffice Calc.
3.2 Preparation of S. cerevisiae genomic DNA libraries for multiplexed paired-end NGS
Select strains for analysis by NGS. Multiplexed libraries containing DNA from up to 12 strains can be sequenced on an Illumina HiSeq 2500 to obtain sufficient read depth (>20-fold) to reliably identify rearrangements, point mutations and copy number changes. GCR-containing strains can be obtained from Canr 5FOAr colonies on plates from fluctuation tests or from patches replica plated onto Can 5-FOA medium when strains have sufficiently high GCR rates (see Note 7). It is critical, however, to ensure that all GCR-containing strains analyzed are independently isolated so that multiple descendants of a single GCR-containing cell are not analyzed. Hence, only one colony per culture used in fluctuation analysis or only one colony per patch should be analyzed. When sequencing genomic DNA from GCR-containing strains, it is preferable to also sequence genomic DNA from any parental strains from which the GCR-containing strains were derived so that pre-existing rearrangements and point mutations can be identified. A typical sample of multiplexed libraries from a GCR experiment will contain libraries from 1 parental strain and 11 independently isolated GCR-containing strains; however, the method can be scaled up or down in regard to the number of strains analyzed so long as sufficient coverage for each genome can be achieved.
Streak selected GCR-containing strains onto Can 5-FOA plates for single colonies to purify the isolate. Incubate the freshly streaked Can 5-FOA plates for 3–5 days at 30°C. Inoculate a single colony for each GCR-containing strain into 5 ml of YPD liquid and grow in a 30°C shaker for 12–16 hr or until saturation is reached. In parallel, inoculate the parent strain(s) from which the GCR-containing isolates were derived. To generate a glycerol stock for each overnight culture of a GCR-containing strain, take 0.5 ml of the culture, mix with 0.5 ml of 40% glycerol and freeze at −80°C.
Prepare genomic DNA from the overnight cultures as follows (see Note 10). Harvest the cells from 1.5 ml of each culture by centrifugation at 14,000 rpm for 30 sec–1 min in a benchtop microcentrifuge. Carefully remove the supernatant by pipetting and discard. Suspend the cells in 1 ml of sterile deionized water, harvest the cells again by centrifugation and discard the supernatant. The genomic DNA purification method described here is based on the Gentra PureGene Yeast/Bact. kit with some modifications. Resuspend the cell pellet in 300 μl Cell Suspension Solution and pipet to mix well. Add 1.5 μl Lytic Enzyme Solution, mix well and incubate the cells for 1 hr at 37°C. Pellet the cells by centrifugation at 14,000 rpm for 1 min and carefully remove the supernatant by pipetting and discard. Resuspend the cells in 300 μl Cell Lysis solution and pipet to mix well. Add 100 μl Protein Precipitation Solution and vortex at high speed for 20–30 sec. Centrifuge the samples at 14,000 rpm for 3 min to pellet the proteins. Transfer the supernatant to a fresh microcentrifuge tube, add 1.5 μl of 4 mg/ml RNAse A to the supernatant, mix well and incubate at 37°C for 1 hr. Then add 300 μl isopropanol, mix well, and centrifuge the samples at 14,000 rpm for 1 min to precipitate the DNA (the pellet may not always be visible). Discard the supernatant carefully by pouring. Wash the pellet with 300 μl 70% ethanol, and centrifuge the samples at 14,000 rpm for 1 min. Remove the ethanol carefully and air-dry the pellet for 5 min. Add 50–100 μl DNA Hydration Solution and dissolve the DNA by incubating the tubes at 65°C for 1 hr. Measure the DNA concentration using a Nanodrop spectrophotometer (see Note 11). If needed, the genomic DNA samples can be stored at −20°C overnight or longer.
Prepare sonicated DNA samples as follows. Reserve 1 μl of each genomic DNA sample as a “pre-sonicated” control. Sonicate 1–5 μg of the remainder of the genomic DNA to fragments with an average size of 600 bp using the Covaris M220 or preferred sonicator (see Note 12). To verify fragment size range, load 1 μl of the pre-sonicated and sonicated DNA samples on a 1.2% FlashGel (use 2 μl Flash Gel Dye + 2 μl sterile deionized water + 1 μl DNA) with 2 μl Flash Gel Ladder, and run the gel at 200 V for 5 min. Sonication should result in a smear of fragments ranging from 200–800 bp. The fragment size range can be adjusted by changing the sonication time. If needed, the sonicated DNA samples can be stored at −20°C overnight or longer.
- Prepare blunt-ended 5′-phosphorylated DNA fragments using T4 DNA polymerase and T4 polynucleotide kinase. If using the End-It DNA End-Repair kit, treat up to 5 μg DNA in a 50 μl reaction prepared as follows:
- 1–34 μl DNA (see Note 13)
- 5 μl 10× End-Repair buffer
- 5 μl 2.5 mM dNTP mix
- 5 μl 10 mM ATP
- 1 μl End-Repair enzyme mix
- Sterile water (for a final volume of 50 μl)
- Adenylate the 3′ ends of the DNA fragments using Klenow DNA polymerase as follows:
- 32 μl end-repaired DNA
- 5 μl 10× NEBuffer 2
- 10 μl 1mM dATP
- 3 μl Klenow DNA polymerase (exo−, 5U/μl)
- Ligate Illumina adapters to the 5′ and 3′ ends of the A-tailed DNA fragments using Quick DNA ligase. Each adapter mix contains both a universal adapter and an indexed adaptor that tags the fragments so that multiplexed sequences can be assigned to specific samples after sequencing. Ensure that a different indexed adapter mix is used for each sample within one multiplexed library. Set up ligation reactions as follows:
- 15 μl A-tailed DNA
- 20 μl 2× DNA ligase buffer
- 2 μl 1:10 diluted Illumina indexed adapter mix
- 3 μl Quick DNA ligase
Purify 500–700 bp ligated samples by gel electrophoresis and gel extraction as follows. Prepare 200 ml of 2% high-resolution agarose in 1× TAE (diluted from 50× TAE in sterile deionized water), add 20 μl Sybr Safe, and cast 2 gels (100 ml each) using 15-well combs. Load 10 μl HyperLadder 100 bp in the first lane of the gel. Add 5 μl Orange gel loading dye to each DNA sample from the previous step and load the first 6 samples on the first gel, leaving a gap of one lane after each sample to prevent cross-contamination. Load 10 μl HyperLadder 100 bp on the final lane of the gel. Flanking the samples with markers facilitates excision of the desired fragment sizes. Similarly, load the next set of 6 samples and 100 bp ladder on the second gel. Run the gels at 120 V for 60 min (6–10 V/cm). View the fragments using a Dark Reader transilluminator. At this stage, it is normal to observe very weak or no DNA signal using a transilluminator. Using a fresh razor blade, excise a piece of the gel containing fragments ranging from 500 to 700 bp from each sample lane. Use a fresh blade for each sample or clean the blade thoroughly by washing in water and ethanol between samples to avoid cross contamination. Transfer the gel piece to a microcentrifuge tube using a forceps, cleaning the forceps thoroughly between samples. Purify the DNA from the excised gel pieces using the Qiagen MinElute gel extraction kit, eluting in 11.5 μl elution buffer. The purified samples can be stored at −20°C overnight or longer.
-
Enrich adapter-ligated DNA fragments by PCR amplification (see Note 14) using the following reaction mix:
- 11.5 μl DNA from the previous step
- 12.5 μl 2× KAPA master mix
- 1 μl 25 μM PCR primer mix
Use the following PCR conditions:- Denaturation: 98°C for 45 seconds
- 18 cycles (see Note 15): denaturation (98°C for 15 seconds), annealing (65°C for 30 seconds), and extension (72°C for 30 seconds)
- Final extension: 72°C for 1 min
- Hold: 4°C as long as desired
Purify the final libraries by running the samples on 2% agarose gels as described in Step 3.2.8. Excise 500–700 bp fragments as described above, and purify the fragments from the gel pieces using the Qiagen MinElute gel extraction kit, eluting the DNA in 15 μl elution buffer. The samples can be stored at −20°C overnight or longer.
Measure the DNA concentrations using an assay that is highly specific to double-stranded DNA, such as the Qubit dsDNA HS assay kit, which uses a double-stranded DNA-specific fluorescent dye. Standard spectrophotometric measurements based on the absorbance at 260 nm are often influenced by the presence of RNA and other contaminants in the sample and hence are not suitable for obtaining accurate DNA concentrations for NGS libraries. To prepare samples for the Qubit fluorometer, add 2 μl of each sample to 198 μl of Qubit working solution (prepared by diluting the Qubit dsDNA HS reagent 1:200 in the Qubit dsDNA HS buffer) in thin-walled, clear 0.5 ml microcentrifuge tubes. To prepare the standards, add 190 μl of Qubit working solution to 10 μl of each Qubit standard and mix well. Calibrate the Qubit fluorometer before measuring the sample concentrations. Calculate the molar concentration (nM) of each library based on the Qubit reading (ng/μl) assuming that the average fragment length is 600 bp, and hence the average molecular weight is 600 bp × 660 g/mol/bp = 396,000 g/mol. Dilute all samples to the same concentration (5 or 10 nM but no less then 2 nM). Pool 2 μl of each sample for multiplexing and mix well. Use 5 μl of the pooled libraries for paired-end sequencing using the Illumina HiSeq 2500.
Sequencing of prepared NGS libraries is now a regularly available service at both commercial companies as well as sequencing facilities at universities and research institutes. The analyses performed and details such as the file formats of the data returned are very facility-dependent. This protocol assumes that the data returned will be unmapped reads, separated by sample/index, in either a FASTQ or BAM format (which can be converted to a FASTQ format by samtools [19]).
Analysis of the sequencing data first requires mapping the sequencing reads to the reference sequence of the S. cerevisiae genome (http://www.yeastgenome.org) using programs such as Bowtie2 [20]. When mapping paired-end data, many read-mapping programs filter out reads that are not concordant with the reference genome, which eliminates reads that span novel junctions present in GCRs. To preserve these reads, provide the paired-end data as two sets of “unpaired” data to the mapping program (for example using the “−U” flag to Bowtie2) and add back the read identifier using programs such as graft in the Pyrus suite ([16], http://www.sourceforge.net/p/pyrus-seq). The Pyrus suite of programs can then be used to sort the mapped reads and perform copy number analysis, sequence variant identification, and identification and sequencing of genome rearrangements by identification of anomalous paired-end alignments. Many other programs for identifying structural variants that use a variety of strategies have been developed and can also be used to perform these types of analyses [21]. The combination of copy number analysis and identification of rearrangements targeting single copy sequences in the genome can provide a great deal of insight into the structure of individual GCRs (Figure 3), particularly if the junction sequence can be identified using programs such as comice in the Pyrus suite [16]. Many GCRs, however, are mediated by non-allelic homologous recombination between repetitive elements (such as Ty elements) that are larger than 600 bp. These rearrangements cannot be spanned by the NGS libraries described here and will not be identifiable by anomalous paired-end alignment analysis. In these cases, copy number analysis can provide hints as to the structures of these rearrangements, but proof of these structures requires the use of complementary techniques such as PCR and PFGE [7, 9].
Acknowledgments
This work was supported by NIH grant GM26017 and the Ludwig Institute for Cancer Research.
Footnotes
The method for generating NGS libraries was modified from the Illumina library preparation guide (Illumina). It should be noted that there are continual improvements in the methods and reagents for NGS library generation that should be evaluated from time to time.
Do not allow the autoclaved agar mixture to cool too much because it will rapidly cool once the dropout mix is added and can solidify before it is poured.
Strains containing three widely used haploid strain-based assays include RDKY3615 (classical GCR assay; MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2Δ1 ade8 hxt13::URA3), RDKY6677 (uGCR assay; MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2Δ1 ade8 yel068c::CAN1-URA3 iYEL072W::hph) and RDKY6678 (dGCR assay; MATa ura3-52 leu2Δ1 trp1 Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2Δ1 ade8 yel072w::CAN1-URA3 iYEL072W::hph) [2, 3]. Mutations of interest can be introduced into these strains by methods such as PCR-based gene disruption using the following selectable markers: HIS3, TRP1, G418-resistance (e.g., kanMX4), and nourseothricin-resistance (e.g., natNT2). Alternatively, the classical GCR assay can be introduced into any CAN1 ura3 strain of interest by replacing HXT13 with URA3 (hxt13::URA3) [2]. The uGCR or dGCR assay can be introduced into any can1 ura3 strain of interest by inserting the desired GCR assay cassette (yel068c::CAN1-URA3 for uGCR and yel072w::CAN1-URA3 for dGCR), which can be amplified from the plasmids pRDK1379 and pRDK1378, respectively [3], at the appropriate chromosomal site. To facilitate systematic GCR studies using the BY4741 MATa yeast deletion collection, MATalpha query strains that contain the appropriate markers as well as the uGCR or dGCR assay have been constructed [22]: RDKY8625 (uGCR; MATalpha hom3-10 ura3Δ0 leu2Δ0 trp1Δ63 his3Δ200 lyp1::TRP1 cyh2-Q38K iYFR016::PMFA1-LEU2 can1::PLEU2-NAT yel068c::CAN 1-URA3) and RDKY7635 (dGCR; MATalpha hom3-10 ura3Δ0 leu2Δ0 trp1Δ63 his3Δ200 lyp1::TRP1 cyh2-Q38K iYFR016::PMFA1-LEU2 can1::PLEU2-NAT yel072w::CAN1-URA3). Additional mutations can be introduced into these query strains by PCR-based gene disruption using the HIS3 and hygromycin-resistance (e.g., hphNT1) markers.
Sterilize a scalpel/surgical blade by dipping in ethanol and flaming. Before excising colonies, ensure that the blade is sufficiently cooled by pressing into a region of agar on the edge of the plate, away from any colonies.
The culture volume depends on the assay used to monitor the formation of GCRs. The wild-type rates for the classical, uGCR, and dGCR assays are 3.5 × 10−10, 2.27 × 10−9, and 1.97 × 10−8, respectively [2, 3]. For strains with estimated GCR rates that are similar to or greater than wild-type, use 50 ml, 10 ml, and 4 ml cultures, respectively, for these 3 assays. For mutant strains that have estimated GCR rates that are lower than wild-type, larger culture volumes can be used if very few or no colonies are observed in patch tests (see Note 7) on Can 5-FOA plates. To allow sufficient aeration, the cells mus be cultured in tubes or flasks with a capacity of at least 4–5 times the desired culture volume.
For strains with a severe growth defect, use longer incubation times and/or larger culture volumes.
The volume of culture plated depends on the estimated GCR rate, which is influenced by the sequences within the breakpoint region (i.e., the GCR assay used) and the mutations present in the strain of interest. Patch tests can be performed to make a preliminary assessment of the effect of any mutation of interest on the GCR rate before performing fluctuation tests. The dGCR assay is most amenable to this type of test because the rate of accumulating GCRs in the dGCR assay is sufficiently high. Patch tests can be performed as follows. The strains of interest (including a wild-type control) are streaked on YPD plates and incubated for 2–3 days at 30°C. Using sterile toothpicks, three independent colonies of each strain are spread into individual ~1 cm2 squares on YPD plates (including the wild-type strain as a control). The plates are incubated for 2–3 days at 30°C, and the patches are then replica-plated onto GCR plates using sterile velvets. The number of papillae in each patch is counted after 3–5 days of growth at 30°C. The wild-type dGCR strain generally yields 1–5 papillae per patch, and different mutations can cause either increased or decreased papillation. The number of papillae can be used to make an initial estimate of the volume of saturated culture to be plated on GCR plates for fluctuation tests as follows: 1–5 papillae, 0.75–1 ml; 6–15 papillae, 0.3–0.4 ml; ~16–150 papillae, 0.1–0.25 ml; papillae that are too many or too close together to count, 0.1 to 0.2 ml of a 1:10 dilution of the saturated culture. If plating these volumes yields too few or too many colonies on the Can 5-FOA plates, then increase or decrease the volume plated accordingly. It is important to note that the patch test is significantly influenced by the growth rate of the strain(s) of interest, and therefore patch tests are not reliable predictors of GCR rates for strains with growth defects. Additionally, certain mutations can have assay-specific effects, for instance by increasing the GCR rate in the uGCR but not the dGCR assay (as observed when genes like EXO1, RAD10, or RAD6 are deleted [3]). In such cases, performing patch tests using only the dGCR assay can lead to the potentially inaccurate conclusion that the mutation does not impact the GCR rate. Therefore, while patch tests are extremely useful in rapidly screening large numbers of mutations, it is crucial to perform fluctuation tests especially when the mutation of interest causes a growth defect or potentially has an assay-specific effect on the GCR rate.
S. cerevisiae strains yield on average 108 cells per ml of saturated culture in YPD; therefore 0.1 ml of the 10−5 dilution plated on YPD will yield approximately 100 colonies.
If all Can 5-FOA plates contain very few or no colonies, plate larger culture volumes.
The genomic DNA extraction method described here uses the Gentra Puregene Yeast/Bact. kit (Qiagen), but other genomic DNA extraction methods can be used at this step, with the modification that RNase treatment should be performed prior to DNA precipitation and resuspension.
If the DNA yield from 1.5 ml of the overnight culture is low, the culture volume used for the DNA isolation should be increased. For example, a 10 ml overnight culture can be split into multiple separate 1.5 ml scale DNA isolations. When multiple DNA isolations per culture are performed, the DNA can be combined in the final step by resuspending the DNA from all tubes with the same 50 μl of DNA Hydration Solution.
The Covaris microTUBE-15 and microTUBE-50 vials have a DNA input limit of 1 μg and 5 μg, respectively; therefore, use 1 or 2 μg genomic DNA in a final volume of volume of 15 to 20 (+/−) 1 μl and 50 (+/−) 3 μl, respectively, depending on the DNA concentration. Use the manufacturer settings to obtain 600 bp fragments in each case.
If the DNA concentration is low, use the entire 50 μl of sonicated DNA sample and scale up the end-repair reaction to 125 μl with 50 μl DNA, 12.5 μl each of the 10× End-repair buffer, 2.5 mM dNTP mix, and 10 mM ATP, 2.5 μl of End-It enzyme mix, and sterile water to a final volume of 125 μl.
There are alternative PCR-free methods to enrich adapter-ligated fragments, as described at www.illumina.com.
If insufficient amplification is observed, use real-time PCR to determine the optimal number of cycles.
References
- 1.Chan JE, Kolodner RD. A genetic and structural study of genome rearrangements mediated by high copy repeat Ty1 elements. PLoS Genet. 2011;7:e1002089. doi: 10.1371/journal.pgen.1002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 3.Putnam CD, Hayes TK, Kolodner RD. Specific pathways prevent duplication-mediated genome rearrangements. Nature. 2009;460:984–989. doi: 10.1038/nature08217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myung K, Chen C, Kolodner RD. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 5.Pennaneach V, Kolodner RD. Recombination and the Tel1 and Mec1 checkpoints differentially effect genome rearrangements driven by telomere dysfunction in yeast. Nat Genet. 2004;36:612–617. doi: 10.1038/ng1359. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Umezu K, Kolodner RD. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell. 1998;2:9–22. doi: 10.1016/s1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- 7.Pennaneach V, Kolodner RD. Stabilization of dicentric translocations through secondary rearrangements mediated by multiple mechanisms in S. cerevisiae. PLoS One. 2009;4:e6389. doi: 10.1371/journal.pone.0006389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putnam CD, Pennaneach V, Kolodner RD. Chromosome healing through terminal deletions generated by de novo telomere additions in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2004;101:13262–13267. doi: 10.1073/pnas.0405443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narayanan V, Mieczkowski PA, Kim HM, Petes TD, Lobachev KS. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006;125:1283–1296. doi: 10.1016/j.cell.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 10.Kanellis P, Gagliardi M, Banath JP, Szilard RK, Nakada S, Galicia S, et al. A screen for suppressors of gross chromosomal rearrangements identifies a conserved role for PLP in preventing DNA lesions. PLoS Genet. 2007;3:e134. doi: 10.1371/journal.pgen.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koszul R, Caburet S, Dujon B, Fischer G. Eucaryotic genome evolution through the spontaneous duplication of large chromosomal segments. EMBO J. 2004;23:234–243. doi: 10.1038/sj.emboj.7600024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Umezu K, Hiraoka M, Mori M, Maki H. Structural analysis of aberrant chromosomes that occur spontaneously in diploid Saccharomyces cerevisiae: retrotransposon Ty1 plays a crucial role in chromosomal rearrangements. Genetics. 2002;160:97–110. doi: 10.1093/genetics/160.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Saini N, Sheng Z, Lobachev KS. Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet. 2013;9:e1003979. doi: 10.1371/journal.pgen.1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- 15.Chan JE, Kolodner RD. Rapid analysis of Saccharomyces cerevisiae genome rearrangements by multiplex ligation-dependent probe amplification. PLoS Genet. 2012;8:e1002539. doi: 10.1371/journal.pgen.1002539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Putnam CD, Pallis K, Hayes TK, Kolodner RD. DNA repair pathway selection caused by defects in TEL1, SAE2, and de novo telomere addition generates specific chromosomal rearrangement signatures. PLoS Genet. 2014;10:e1004277. doi: 10.1371/journal.pgen.1004277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng SK, Yin Y, Petes TD, Symington LS. Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol Cell. 2015;60:500–508. doi: 10.1016/j.molcel.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. J Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guan P, Sung WK. Structural variation detection using next-generation sequencing data: A comparative technical review. Methods. 2016;102:36–49. doi: 10.1016/j.ymeth.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Putnam CD, Srivatsan A, Nene RV, Martinez SL, Clotfelter SP, Bell SN, et al. A genetic network that suppresses genome rearrangements in Saccharomyces cerevisiae and contains defects in cancers. Nat Commun. 2016;7:11256. doi: 10.1038/ncomms11256. [DOI] [PMC free article] [PubMed] [Google Scholar]