

Abstract
Despite containing an α-amino acid, the versatile cofactor S-adenosylmethionine (SAM) is not a known building block for non-ribosomal peptide synthetase (NRPS) assembly lines. Here we report an unusual NRPS module from colibactin biosynthesis that uses SAM for amide bond formation and subsequent cyclopropanation. Our findings showcase a new use for SAM and reveal a novel biosynthetic route to a functional group that likely mediates colibactin’s genotoxicity.
S-adenosylmethionine (SAM) is an important and versatile cofactor. It serves as a source of electrophilic methyl, aminopropyl, and 5′-deoxyadenosyl groups for SN2-type nucleophilic substitutions.1 Transfer of these groups to metabolites and macromolecules is critical for regulation of gene expression, polyamine biosynthesis, and many other biological processes.2 Radical SAM enzymes, the largest known enzyme superfamily, use SAM as a source of carbon-based radicals, enabling a variety of challenging transformations.3
SAM is also a non-proteinogenic α-amino acid. While non-proteinogenic amino acids are important building blocks for non-ribosomal peptide synthetase (NRPS) assembly lines,4 SAM was not previously known to be used in this manner. Here, we report that an NRPS module from colibactin biosynthesis uses SAM as a substrate for amide bond formation and subsequent generation of a cyclopropane. Our study expands the chemistry known for this important cofactor and identifies a novel pathway for cyclopropane biosynthesis, a functional group that is implicated in colibactin’s biological activity.
Colibactin is a genotoxin of unknown structure produced by human gut commensal Escherichia coli strains that contain a 54-kilobase nonribosomal peptide-polyketide biosynthetic gene cluster (the pks island; Supplementary Results, Supplementary Fig. 1).6 E. coli strains harboring the pks island (pks+) induce DNA double strand breaks in HeLa cells,6 promote the development of colitis-associated colorectal cancer in animal models,7 and are found with higher frequency in colon tissues of inflammatory bowel disease and colorectal cancer patients.7 In efforts to identify the active genotoxin, we and others have isolated non-genotoxic, pks-dependent metabolites from pks+ E. coli mutants.8–14 Notably, a subset of these molecules (Fig. 1a and Supplementary Fig. 1) contain cyclopropanes, a structural motif found in DNA-alkylating agents (e.g. duocarmycins15 and illudins16). Recent studies using synthetic model substrates suggest that the cyclopropane group enables DNA alkylation.17 The pks enzymes required for generating cyclopropane-containing molecules have been identified by gene inactivation studies.10, 12 Because deletion of the pks gene clbH abolishes the production of both 1 and a putative cyclopropane-containing metabolite (2),10,12 it has been proposed that this NRPS is involved in cyclopropane biosynthesis.
Figure 1. SAM is activated as a non-ribosomal peptide synthetase (NRPS) building block and loaded onto the phosphopantetheinyl arm of ClbH.
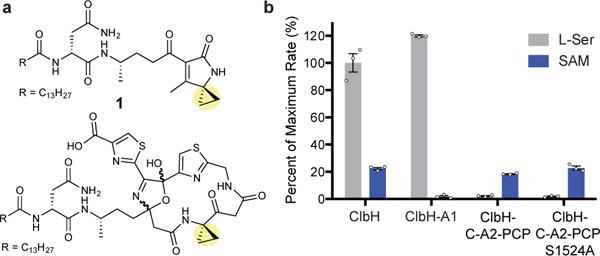
(a) Pks-dependent, cyclopropane-containing metabolites isolated from pks+ E. coli mutant strains. (b) ATP-[32P]-PPi exchange assay for the substrate specificity of full-length ClbH, ClbH-A1, ClbH-C-A2-PCP, and ClbH-C-A2-PCP-S1524A. C, condensation; A, adenylation; PCP, peptidyl carrier protein. Results of scintillation counting were averaged and scaled relative to the activation of L-Ser by full-length ClbH (data points are shown). Error bars represent the standard error of the mean (s.e.m.) of three replicates.
In addition to condensation (C), adenylation (A), and peptidyl carrier protein (PCP) domains (C-A2-PCP), ClbH contains a second N-terminal A domain (A1) (Supplementary Fig. 2). It has been proposed based on feeding studies that ClbH uses the L-methionine (L-Met)-derived cyclopropane-containing amino acid, 1-aminocyclopropane-1-carboxylic acid (ACC) as a building block,10–12, 18 and that ACC is generated either prior to activation by the NRPS or after loading of L-Met onto the ClbH PCP domain (Supplementary Fig. 2).10, 11 However, in vitro characterization of excised ClbH A domains does not support this hypothesis. While ClbH-A1 activated and loaded L-serine (L-Ser) onto the free-standing acyl carrier protein (ACP) ClbE, generating a precursor to the PKS extender unit aminomalonyl-S-ACP,13, 18 neither A domain activated ACC or L-Met.18
We envisioned a distinct route for ACC formation: synthesis from SAM, a possibility without precedent but consistent with the results of L-Met feeding experiments.10, 11 In plants, SAM is directly converted to ACC by the enzyme ACC synthase.1 E. coli lack this enzyme, leading us to hypothesize that ClbH might use SAM as a building block for cyclopropane formation on the NRPS assembly line. Since bioinformatic analyses suggested ClbH-A2 may not use a proteinogenic amino acid,10 we proposed that ClbH-A2 might activate SAM (Supplementary Fig. 2).
To test this hypothesis, we cloned, overexpressed, and purified full-length ClbH, truncated ClbH constructs (ClbH-A1, ClbH-C-A2-PCP), and a ClbH-C-A2-PCP with a mutant PCP domain (S1524A) that cannot be phosphopantetheinylated (Supplementary Fig. 3). Using an ATP-[32P]-PPi exchange assay, we found that full-length ClbH activated L-Ser and SAM (Fig. 1b), but not L-Met, ACC, or the SAM degradation products L-homocysteine or adenosine (Supplementary Fig. 4). ClbH-C-A2-PCP and ClbH-C-A2-PCP-S1524A activated SAM but not L-Ser (Fig. 1b), suggesting that SAM is the substrate of ClbH-A2. The isolated ClbH-A2 domain does not activate SAM in vitro,18 suggesting that the addition of the C and PCP domains to make a tri-domain construct is critical for activity.
We next examined whether SAM could be loaded onto ClbH. Both a radiometric loading assay and gel autoradiography with [carboxyl-14C]-SAM showed charging of the ClbH PCP domain with SAM in the presence of ATP and the promiscuous phosphopantetheinyl (ppant) transferase Sfp19 (Supplementary Figs. 5 and 6). We further confirmed SAM is covalently tethered to the ppant arm of the PCP domain using protein mass spectrometry (MS).20 When SAM was incubated with holo-ClbH and ATP for 1 hour, multiple charge states of the digested SAM-ClbH peptide were observed in MS1 (Supplementary Fig. 7). Identification of the SAM-loaded peptide was also supported by the detection of peptide backbone cleavage and ppant ejection ions in MS2 (Supplementary Fig. 10). Taken together, the in vitro characterization of ClbH shows that SAM is activated by ClbH-A2 and then loaded onto its PCP domain. However, no ACC-ClbH thioester was detected by protein MS when SAM-ClbH was incubated overnight (Supplementary Fig. 11), suggesting that additional enzymes are needed for cyclopropane formation.
To identify these enzymes, we attempted to reconstitute the biosynthesis of putative cyclopropane-containing metabolite 2 (m/z 567.3752) (Fig. 2a).11, 12 Previous gene inactivation studies had indicated that clbN, clbB, clbC, and clbH are required for generating 2 in vivo.12 We cultured an E. coli DH10B mutant strain expressing the ppant transferase ClbA, ClbN, ClbB, ClbC, and either ClbH or ClbH-C-A2-PCP. Although LC/MS analysis of pellet extracts revealed the production of a compound with m/z 567.3752 in both strains, its retention time did not match that of a synthetic standard of 2 (Supplementary Note). Rather, this metabolite corresponded to an isomeric γ-lactone (3) (Fig. 2a, Supplementary Figs. 12–14, and Supplementary Note). These observations indicate that the structure of this metabolite was previously misassigned12 and indicate that enzymes beyond ClbN, ClbB, ClbC, and ClbH are likely involved in cyclopropane formation.
Figure 2. ClbH-mediated SAM elongation precedes ClbI-catalyzed cyclopropanation.
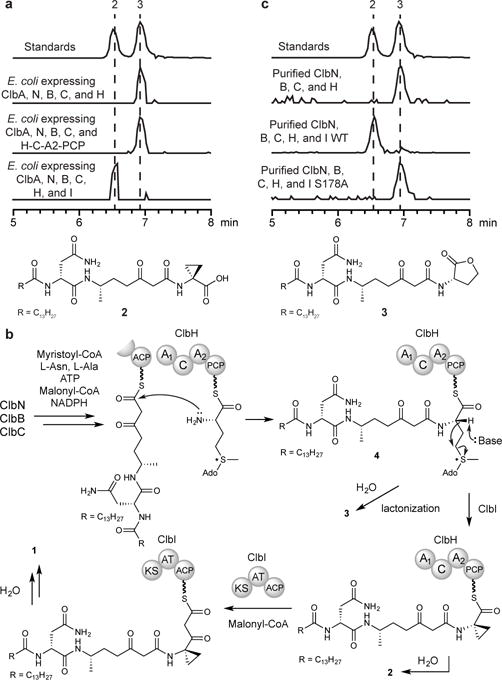
(a) LC/MS analysis of pellet extracts of E. coli DH10B mutant strains expressing ClbA, ClbN, ClbB, ClbC, and either (i) ClbH, (ii) ClbH-C-A2-PCP, or (iii) ClbH and ClbI. (b) Biosynthetic hypothesis for the formation of SAM-containing assembly line intermediate 4 and cyclopropane-containing metabolites 2 and 1. (c) LC/MS analysis of in vitro assays of purified enzymes. These assays have been replicated three times in the laboratory. (a,c) Extracted ion chromatograms of m/z 567.3752.
To deduce the minimal set of pks enzymes required for cyclopropane construction, we analyzed metabolites produced by an E. coli strain expressing ClbA, ClbN, ClbB, ClbC, ClbH, and ClbI, a PKS enzyme necessary for the biosynthesis of 1.10, 12 Instead of γ-lactone 3, we detected both 1 and 2 in these extracts, indicating that ClbI is essential for cyclopropane formation (Fig. 2a and Supplementary Fig. 15). Metabolite 2 may derive from non-enzymatic hydrolysis of the corresponding assembly line-bound intermediate, and related off-loaded products have been observed previously in vivo.11, 12
To decipher the enzymatic chemistry involved in cyclopropane formation, we fed L-[U-13C, 15N]-Met to an E. coli DH10B mutant strain expressing ClbA, ClbN, ClbB, ClbC, and ClbH. This yielded a new mass feature (m/z 572.3857), indicating incorporation of the aminobutyryl group of SAM into 3 (Supplementary Fig. 16). Guided by known chemical transformations of SAM,21 we reasoned that both 2 and 3 could be derived from intermediate 4, which would be generated if elongation with SAM occurs prior to cyclopropane formation (Fig. 2b). From 4, enolate formation, cyclopropanation, and thioester hydrolysis would provide 2. Alternatively, thioester hydrolysis and non-enzymatic alkylation of the resulting carboxylic acid 5 (m/z 864.4648, Supplementary Fig. 17) would generate 3. Consistent with previous reports,10, 11 feeding of L-[U-13C, 15N]-Met to an E. coli DH10B mutant strain expressing ClbA, ClbN, ClbB, ClbC, ClbH, and ClbI yielded an additional mass feature (m/z 552.3959), demonstrating that the aminobutyryl group of SAM is also incorporated into 1 (Supplementary Fig. 16).
To explore whether 2 and 3 are derived from intermediate 4, we sought to generate this species in vitro. A reaction containing Sfp, CoA, ClbN, ClbB, ClbC, ClbH, myristoyl-CoA, malonyl-CoA, L-Asn, L-Ala, NADPH, ATP, and SAM was quenched with methanol to precipitate enzymes, and the pellet was then treated with KOH to cleave covalently tethered intermediates from thiolation domains. Consistent with in vivo data, LC/MS analysis did not reveal 2 in the supernatant or pellet extract. The base-sensitive compounds 3 and 5 (m/z 864.4648) were detected only in supernatant (Fig. 2c and Supplementary Fig. 18). A new metabolite 6 (m/z 615.3786) detected in the pellet extract is likely a base-promoted degradation product of 5 (Supplementary Fig. 19). 3, 5, and 6 were not detected in a control reaction lacking ClbH (Supplementary Fig. 20). Assays with [methyl-2H3]-SAM yielded two additional ions 3 Da higher in mass (m/z 867.4836 and m/z 618.3974), confirming that 5 and 6 are derived from this cofactor (Supplementary Fig. 21). We were unable to detect 2 or ACC, strongly suggesting that SAM is loaded onto ClbH and undergoes amide bond formation with an upstream intermediate to yield 4 prior to cyclopropane formation.
When we added ClbI to the assay described above, 1 was detected in the supernatant and 2 was observed in both the supernatant and the pellet extracts, indicating that this PKS is required for cyclopropane formation in vitro (Fig. 2c and Supplementary Fig. 22). We were unable to detect the malonate elongation product of 2 and the corresponding decarboxylated product in vivo or in vitro, potentially because the Knoevenagel condensation leading to 1 is rapid. Although proposed to perform a canonical elongation in the biosynthesis of 1,11, 12 the ClbI KS domain contains a serine (S178) in place of the active site cysteine typically required for this reaction (Supplementary Fig. 23).5 This substitution is characteristic of starter KS domains, which decarboxylate ACP domain-tethered malonyl extender units.22, 23 Mutating S178 to alanine abolished the production of 2 in an in vitro assay (Fig. 2c and Supplementary Fig. 24), suggesting that this residue participates in cyclopropane formation. S178 could act as a general base to promote cyclization of the ClbH-tethered intermediate 4 or function as a nucleophile, covalently tethering an intermediate to ClbI. Despite multiple attempts, we have been unable to excise the ClbI KS domain to examine its activity in vitro. It is also possible that another KS domain (e.g. ClbB-KS, ClbC-KS) may catalyze elongation to generate 1. Further studies will be needed to identify the precise timing of cyclopropane formation on the assembly line and define the role of S178 in this process, information that could ultimately enable application of this enzymatic machinery in combinatorial biosynthesis.
Utilization of SAM by NRPS assembly lines requires an adenylation domain to protect the reactive methyl thioadenosine group. Bioinformatic analyses provided initial insights into how this chemical challenge may be accomplished. A multiple sequence alignment indicates that ClbH-A2 lacks the conserved aspartate residue in core motif A4 that typically interacts with the α-amino group of amino acid substrates24 (Supplementary Fig. 25), suggesting a different arrangement of the ClbH active site. Homology modeling suggests that ClbH-A2 harbors an expanded specificity pocket in comparison to the structurally characterized A domain PheA24 (Supplementary Fig. 26). In particular, substitution of Trp239 in PheA with glycine in ClbH-A2 could help to accommodate a larger substrate. Overall, these analyses imply that the ClbH-A2 active site has evolved to handle SAM. As ClbH-A2 has modest sequence identity to structurally characterized A domains (33–35%), further structural and mechanistic characterization will be required to elucidate the details of SAM recognition and utilization. Interestingly, homologs of ClbH-A2 and ClbI are only found together in pks islands (Supplementary Table 6) and are not present in other gene clusters known to produce cyclopropane-containing natural products.
In summary, this work has identified the first use of SAM as a building block for assembly line enzymes and a new strategy for cyclopropane biosynthesis that requires an unusual direct activation of SAM’s carboxylic acid.1 This logic is distinct from other PKS and NRPS assembly lines that generate cyclopropane-containing natural products. In coronatine, kutzneride 2, and curacin A biosynthesis, cyclopropanes are derived from chlorinated intermediates through intramolecular alkylation of a thioester enolate.1, 25 Cyclopropane formation in other pathways has been proposed to involve SAM-dependent alkylation (jawsamycin25), Favorskii-like rearrangement (ambruticin25), or intramolecular nucleophilic ring-opening of a lactone (hormaomycin1, 4). Finally, in clarifying the origin of a functional group that is likely important for colibactin’s genotoxicity,17 we have revealed potential targets for inhibition of colibactin biosynthesis. More broadly, this work highlights SAM-utilizing biosynthetic machineries available for assembly line engineering.
Online methods
General materials and methods
Oligonucleotide primers were synthesized by Integrated DNA Technologies or Sigma-Aldrich. Recombinant plasmid DNA was purified with a QIAprep® Spin Miniprep Kit from Qiagen. Gel extraction of DNA fragments and restriction endonuclease clean up were performed using an Illustra GFX PCR DNA and Gel Band Purification Kit from GE Healthcare. DNA sequencing was performed by Beckman Coulter Genomics and Eton Bioscience. Optical densities of E. coli cultures were determined with a DU 730 Life Sciences UV/Vis spectrophotometer (Beckman Coulter) by measuring absorbance at 600 nm. All chemicals and solvents were obtained from Sigma-Aldrich except where noted.
During protein purification, proteolysis was inhibited by adding Pierce™ Protease Inhibitor tablets (EDTA free; Thermo Scientific) to lysis buffer. Affinity chromatography and SDS-PAGE analysis were conducted using HisPur™ Nickel-nitrilotriacetic acid-agarose (Ni-NTA) resin (Thermo) and 10% or 4–15% Mini-PROTEAN TGX Precast SDS-PAGE gels (Bio-Rad) with 2× Laemmli sample buffer (Bio-Rad). Protein concentrations were determined by measuring absorbance at 280 nm with a NanoDrop 2000 spectrophotometer (Thermo).
Cloning of pks proteins
The cloning, overexpression, and purification of ClbN, ClbC, ClbI, and Sfp were reported previously.13, 19, 26 The genes clbB and clbH were PCR amplified from E. coli CFT073 genomic DNA (purchased from the American Type Culture Collection [ATCC]) using the primers shown in Supplementary Table 1. A typical PCR reaction (50 μL) contained 25 μL of Phusion High-Fidelity PCR Master Mix (New England Biolabs), 2 ng of DNA template, and 500 pmoles of each primer. Thermocycling was carried out in a MyCycler gradient cycler (Bio-Rad) using the parameters outlined in Supplementary Table 2.
PCR reactions were analyzed by agarose gel electrophoresis with ethidium bromide staining, pooled, and purified. Amplified fragments were digested with the appropriate restriction enzymes (New England Biolabs) for 2.5 h at 37 °C. A typical digest contained 1 μL of water, 3 μL of NEB Buffer (10×), 3 μL of BSA (10×, if needed), 20 μL of PCR product, and 1.5 μL of each restriction enzyme (20,000 U/μL). Restriction digests were purified directly using agarose gel electrophoresis. Gel fragments were further purified using the Illustra GFX kit. The digests were ligated into linearized expression vectors using T4 DNA ligase (New England Biolabs). ClbB and clbH were ligated into the pET-28a vector to encode an N-terminal His6-tagged construct.
Ligations were incubated at 16 °C for ~16 h and they contained 3 μL of water, 1 μL of T4 Ligase Buffer (10×), 1 μL of digested vector, 3 μL of digested insert DNA, and 2 μL of T4 DNA Ligase (400 U/μL). 5 μL of each ligation was used to transform a single tube of chemically competent E. coli TOP10 cells (Invitrogen). The identities of the resulting constructs were confirmed by sequencing of purified plasmid DNA. These constructs were transformed into chemically competent E. coli BAP1 cells (ClbB) or BL21 (DE3) Tuner cells (ClbH; Invitrogen) and stored at −80 °C as frozen 1:1 LB/glycerol stocks.
ClbH-C-A2-PCP was PCR amplified from pET28a-clbH plasmid, digested, and ligated into the pET-28a or pET-29b vector to encode an N-terminal or C-terminal His6-tagged construct. The constructs were transformed into E. coli BL21 cells (Invitrogen).
Site-directed mutagenesis of pET29b-clbH-C-A2-PCP and pET29b-clbI was performed using 25 μL of Phusion High-Fidelity PCR Master Mix (New England Biolabs), 50 ng of template, and 500 pmoles of each primer in a total volume of 50 μL. Thermocycling was carried out in a MyCycler gradient cycler (Bio-Rad) using the parameters outlined in Supplementary Table 2. The primers used were listed in Supplementary Table 1. Digestion of the template in each PCR reaction was performed by the addition of 1 μL of DpnI (20,000 U/μL, New England Biolabs) and incubation at 37 °C for 1 h, followed by the addition of another 1 μL of DpnI and incubation at 37 °C for 1 h. 2 μL of each digestion reaction were used to transform 50 μL chemically competent E. coli TOP10 cells (Invitrogen). The success of mutagenesis was confirmed by sequencing of purified plasmid DNA. These sequenced constructs were then transformed into chemically competent E. coli BL21 (DE3) cells (Invitrogen) and stored at −80 °C as frozen LB/glycerol stocks.
Overexpression and purification of pks proteins (Supplementary Fig. 3)
A 25 mL starter culture of BL21 E. coli was inoculated from frozen stock except for ClbB (from plated colonies on LB agar plates containing 50 μg/mL kanamycin) and grown overnight at 37 °C in LB medium supplemented with 50 μg/mL kanamycin. Overnight cultures were diluted 1:100 into 2 L of LB medium containing 50 μg/mL kanamycin. Cultures were incubated at 37 °C with shaking at 175 rpm, moved to 15 °C at OD600 = 0.2–0.3, induced with 25 μΜ (ClbB) or 100 μΜ IPTG (ClbH, ClbH-C-A2-PCP, ClbH-C-A2-PCP S1524A & ClbI S178A) at OD600 = 0.5–0.6, and incubated at 15 °C while shaking for ~ 16 h.
Cells from 2 L of culture were harvested by centrifugation (6,500 rpm × 15 min) and re-suspended in 40 mL of lysis buffer (50 mM Tris-HCl pH 8.3, 250 mM NaCl, 10 mM MgCl2, protease inhibitor [ClbB & ClbH], 10% v/v glycerol). The cells were lysed by passage through a cell disruptor (Avestin EmulsiFlex-C3) twice at 7,500 psi, and the lysate was clarified by centrifugation at 4 °C (13,000 rpm × 30 min). The supernatant was incubated with 2 mL (ClbH & ClbI-S178A) or 3 mL (ClbB, ClbH-C-A2-PCP & ClbH-C-A2-PCP S1524A) of Ni-NTA resin equilibrated with elution buffer (50 mM Tris-HCl pH 8.3, 500 mM NaCl, 10 mM MgCl2, 10% v/v glycerol or 10% w/v sucrose) containing 1 mM (ClbB) or 10 mM (all others) imidazole on a nutator for 2 h at 4 °C. The mixture was centrifuged (3,500 rpm × 10 min) and the unbound fraction discarded. The Ni-NTA resin was re-suspended in 1.5 mL of elution buffer containing 1 or 10 mM imidazole, loaded onto a glass column, and washed with ~15 mL of elution buffer containing 25 mM imidazole. Protein was eluted from the column using a stepwise imidazole gradient in elution buffer (50 mM, 75 mM, 100 mM, 125 mM, 150 mM, 200 mM), collecting 2 mL fractions. The resin was rinsed with elution buffer containing 250 mM imidazole until all resin-bound protein was eluted. SDS–PAGE analysis (4–15% Tris-HCl gel) was employed to confirm the presence and purity of protein in fractions. Fractions containing the desired protein were combined and dialyzed twice against 2 L of storage buffer (50 mM Tris-HCl pH 8.3, 200 mM NaCl, 10 mM MgCl2, 10% v/v glycerol). Solutions containing protein were concentrated with a Spin-X concentrator (Corning) to 0.8–1.0 mL and centrifuged at 4 °C (13,200 rpm × 10 min) to remove particulate. All proteins were further purified by gel filtration chromatography on a GE HighLoad™ 26/600 Superdex 200 pg column. This procedure yielded 0.5–2 mg/L of purified protein. All proteins used in the current study are shown in Supplementary Fig. 3.
ATP-[32P]PPi exchange assay of full-length ClbH, ClbH-A1, ClbH-C-A2-PCP, and ClbH-C-A2-PCP S1524A (Supplementary Fig. 4)
Assays (100 μL) contained 50 mM Tris-HCl (pH 8.3), 200 mM NaCl, 10 mM MgCl2, 5 mM dithiothreitol, 5 mM ATP, 1 mM amino acid, and 4 mM Na4PPi/[32P]PPi (2–6 × 105 cpm/mL; Perkin Elmer). Reactions were initiated by the addition of enzyme (1 μM) and incubated at room temperature for 30 min. Reactions were quenched by the addition of 200 μL of charcoal suspension (16 g/L activated charcoal, 100 mM Na4PPi, 3.5% [v/v] HClO4). The samples were centrifuged (13,000 rpm × 3 min), and the supernatant was removed. The charcoal pellet was washed twice with 200 μL of wash buffer (100 mM Na4PPi, 3.5% [v/v] HClO4). The pellet was re-suspended in 300 μL of wash buffer and added to 10 mL of scintillation fluid (Ultima Gold, Perkin Elmer). Radioactivity was measured on a Beckman LS 6000SC scintillation counter.
PCP-domain loading assay of full-length ClbH, ClbH-C-A2-PCP, and ClbH-C-A2-PCP S1524A (Supplementary Fig. 5)
Assays for reconstitution of SAM-ClbH formation (50 μL) contained 50 mM Tris-HCl (pH 8.3), 200 mM NaCl, 10 mM MgCl2, 1 mM TCEP, 250 μM CoA, 3 μM ClbH, ClbH-C-A2-PCP or ClbH-C-A2-PCP S1524A, 0.6 μM Sfp, 3 mM ATP, and 25 μM [carboxyl-14C]-SAM (55 mCi/mmol, American Radiolabeled Chemicals). ClbH, ClbH-C-A2-PCP or ClbH-C-A2-PCP S1524A was incubated with Sfp and CoA at 22 °C for 1.5 h for phosphopantetheinylation. The complete reaction was initiated by the addition of [carboxyl-14C]-SAM and ATP. The assay mixtures were incubated at 30 °C and quenched after 1 h by the addition of 100 μL BSA (1 mg/mL) and 500 μL trichloroacetic acid (10% m/v). The samples were centrifuged (10,000 rpm × 8 min), and the supernatant was removed. The protein pellet was washed twice with 250 μL trichloroacetic acid (10% m/v), re-suspended in 100 μL formic acid, and added to 10 mL of scintillation fluid (Ultima Gold, Perkin Elmer). Radioactivity was measured on a Beckman LS 6000SC scintillation counter.
14C gel autoradiography of SAM loading on ClbH, ClbH-C-A2-PCP, and ClbH-C-A2-PCP S1524A (Supplementary Fig. 6)
Assays for reconstitution of SAM-ClbH formation (50 μL) contained 50 mM Tris-HCl (pH 8.3), 200 mM NaCl, 10 mM MgCl2, 1 mM TCEP, 250 μM CoA, 3 μM ClbH, ClbH-C-A2-PCP or ClbH-C-A2-PCP S1524A, 0.6 μM Sfp, 3 mM ATP, and 25 μM [carboxyl-14C]-SAM (55 mCi/mmol, American Radiolabeled Chemicals). ClbH, ClbH-C-A2-PCP or ClbH-C-A2-PCP S1524A was incubated with Sfp and CoA at 22 °C for 1.5 h for phosphopantetheinylation. The complete reaction was initiated by the addition of [carboxyl-14C]-SAM and ATP, and incubated at 30 °C. At 45 min, 20 μL aliquots of the reaction mixtures were quenched by addition to an equal volume of 2× Laemmli loading buffer that did not contain a reducing agent. Quenched reactions were not boiled. Samples were analyzed on a 10% SDS-PAGE gel. The [14C]-labeled protein standard was purchased from Perkin Elmer ([Methyl-14C] Methylated, Protein Molecular Weight Markers, 1 μCi, catalog number = NEC811001UC) and used as directed. The gel was dried on a Labconco Benchtop Gel Dryer (80 °C for 2 h and then heating off for 1 h; on vacuum throughout 3 h), and were then exposed on a BAS-IP MS 2025E Multipurpose Standard storage phosphor screen (20 × 25 cm, GE Healthcare) for 2 days. Phosphor images were captured on a GE (Amersham) Typhoon Trio Imager (Storage Phosphor mode, best sensitivity, resolution 200 μm). Since data were stored in the .gel file format in square-root scale rather than a linear scale, the raw phosphor images were opened in ImageJ and linearized using the Linearize GelData plugin (available from National Institutes of Health at http://rsb.info.nih.gov/ij/plugins/linearize-gel-data.html; accessed October 18, 2016; scale factor = 1/21025). After linearization, the maximum intensity in the display was adjusted to the most intense band (maximum intensity was set in Adjust → Brightness and Contrast).
Protein mass spectrometry analysis of SAM-ClbH thioester (Supplementary Figs. 7–11)
Assays for reconstitution of SAM-ClbH formation (50 μL) contained 50 mM Tris-HCl (pH 8.3), 200 mM NaCl, 10 mM MgCl2, 1 mM TCEP, 250 μM CoA, 3 μM ClbH, 0.6 μM Sfp, 3 mM ATP, and 25 μM SAM. ClbH was incubated with Sfp and CoA at 22 °C for 1.5 h for phosphopantetheinylation. The complete reaction was initiated by the addition of SAM and ATP, and incubated at 22 °C. At 1 h or overnight (12 h), the reaction was centrifuged (13,200 rpm × 10 min). The supernatant was frozen in liquid N2, shipped from Harvard University to Northwestern University on dry ice, and stored at −80 °C until LC/MS analysis.
Samples were thawed at room temperature, and 0.5 μg of trypsin (Promega) was added to each sample. After incubating at 37 °C for 20 minutes, reactions were quenched with 1 μL formic acid and diluted with 10 μL of 0.2% v/v formic acid. 5 μL of the diluted mixture was injected onto a Phenomenex Jupiter C18(2) column, 1 mm × 250 mm at a flow rate of 150 μL/min using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B. The following gradient was applied: 0–35 min, 2–50% B.
MS data were collected on a Q-Exactive with the following parameters: sheath gas flow rate: 25 L/min, spray voltage: 5.1 kV, capillary temperature: 250 °C, and S-lens RF level: 50.0. MS1 data were collected from m/z 500 to 2000 at 35,000 resolution, MS2 data were collected at 17,500 resolution with an Automatic Gain Control (AGC) target of 1e5 charges, a maximum inject time of 1s, and an isolation window of 2 m/z. Normalized collisional energy (NCE) was set to 25.
An inclusion list of the theoretical m/z values for the 2, 3, 4 and 5+ charge states of the ClbH tryptic active site peptide for the apo, holo, SAM-loaded and ACC-loaded, which was never observed, was used to seed MS2 events. Several ATP and coenzyme A ions were excluded from MS2.
Thermo .raw files were converted to .mgf with COMPASS27 with the following parameters: assumed precursor charge state: 2 to 6; and the following peak filtering options were selected: clean precursor and enable ETD pre-processing. These .mgf were searched against a database of Clb proteins with Mascot28 with the following parameters: trypsin with allowance for 1 missed cleavage; peptide charge, 2 through 4+; MS1 peptide tolerance, 20 ppm; phosphopantetheinyl-serine as a variable modification; an error tolerant search was performed for MS2 with a 30 mili-mass units (mmu) tolerance for fragment ions. These parameters returned both the apo and holo peptides confidently. From there, loaded peptide variants were manually found in raw files.
Chemdraw was used to predict masses for tryptic peptides and ejected pantetheine (pant) ions seen in MS2. Xcalibur was used to view and interact with raw files. Figures were generated using Xcalibur and Inkscape. The following files are uploaded to MassIVE (massive.ucsd.edu/MSV000080499): .raw for apo and holo and 1 h and overnight (4 files) and the corresponding .mgf for the apo and holo files used for searching.
Generation of E. coli DH10B BACpks knockout strains using λ Red recombinase-mediated gene disruption
The target genes clbDEFGHIJKLM (clbDtoM) and ClbOPQ (clbOtoQ) were disrupted by the PCR-targeting λ Red recombinase-mediated gene disruption system.29 The disruption cassette for clbDtoM included the aac(3)IV gene flanked by 39 bp arms homologous to clbD and clbM. The disruption cassette for clbOtoQ included the Kan gene flanked by 39 bp arms homologous to clbO and clbQ. The disruption cassettes were generated by PCR amplification using primers shown in Supplementary Table 3. PCR reactions (20 μL) contained 10 μL of Q5 High Fidelity 2× Master Mix (New England Biolabs), 1 ng of DNA template (plasmid pIJ773 for clbDtoM or plasmid pKD4 for clbOtoQ), and 500 pmoles of primers. Thermocycling was carried out in a MyCycler gradient cycler (Bio-Rad) using the following condition: denaturation for 1 min at 98 °C, followed by 35 cycles of 10 sec at 98 °C, 30 sec at 72 °C for ClbDtoM or at 66 °C for ClbOtoQ, 1 min at 72 °C, and a final extension of 5 min at 72 °C. The disruption cassettes were purified using an Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare).
To generate a BACpksΔclbDtoM mutant, the purified disruption cassette for clbDtoM was electroporated into E. coli BW25113 harboring the λ Red recombinase expression plasmid pkD46 and BACpks. Mutants were selected on LB plates containing 50 μg/mL apramycin. To generate an E. coli mutant strain expressing ClbA, ClbB, ClbC, ClbN, ClbO, ClbP, and ClbQ, E. coli DH10B were electroporated with mutant BACpksΔclbDtoM and selected on LB plates containing 50 μg/mL apramycin and 50 μg/mL kanamycin.
To further generate a BACpksΔclbDtoMΔclbOtoQ mutant, the purified disruption cassette for clbOtoQ was electroporated into E. coli BW25113 harboring pKD46 and BACpksΔclbDtoM. Mutants were selected on LB plates containing 50 μg/mL apramycin and 50 μg/mL kanamycin. The mutant BACs were isolated from these colonies using ZR BAC DNA Miniprep Kit (Zymo research), and gene disruption was verified by PCR amplification of knockout regions. To generate an E. coli mutant strain expressing ClbA, ClbB, ClbC, and ClbN, E. coli DH10B were electroporated with mutant BACpksΔclbDtoMΔclbOtoQ and selected on LB plates containing 50 μg/mL apramycin and 50 μg/mL kanamycin.
Design and introduction of clbH, clbH-C-A2-PCP, and clbH-clbI overexpression plasmids into E. coli DH10B BACpks knockout strains
Linearized pTrcHisA vector was PCR amplified from pTrcHisA vector (Invitrogen) using primers shown in Supplementary Table 4. Full-length clbH, truncated clbH (clbH-C-A2-PCP), and two adjacent genes clbH and clbI (clbH-clbI) were PCR amplified from E. coli CFT073 genomic DNA (ATCC) using primers shown in Supplementary Table 4. PCR reactions (20 μL) contained 10 μL Q5 High-Fidelity 2× Master Mix (New England Biolabs), 1 ng of DNA template, and 500 pmoles of each primer. Thermocycling was carried out in a MyCycler gradient cycler (Bio-rad) using the following condition: denaturation for 1 min at 98 °C, followed by 35 cycles of 10 sec at 98 °C, 30 sec at 72 °C, 3 min for clbH (2 min for clbH-C-A2-PCP and 4 min for clbH-clbI) at 72 °C, and a final extension of 5 min at 72 °C.
Gibson assembly reactions (10 μL) contained 100 ng linearized pTrcHisA vector, 3-fold of molar excess purified PCR products of clbH, clbH-C-A2-PCP, or clbH-clbI, and 5 μL of 2× Gibson Assembly Master Mix (New England Biolabs). The mixtures were incubated at 50 °C for 15 min and used to transform 50 μL of chemically competent E. coli TOP10 cells (Invitrogen). The identity of the assembled plasmids was confirmed by sequencing.
The pTrcHisA-clbH, -clbH-C-A2-PCP, and -clbH-clbI plasmids were electroporated into electrocompetent E. coli DH10B BACpksΔclbDtoMΔclbOtoQ and stored at −80 °C as frozen LB/glycerol stocks.
Metabolite analyses of E. coli DH10B BACpks knockout strain harboring overexpression plasmids (Figure 2a, Supplementary Figs. 13–16)
A starter culture of E. coli DH10B BACpksΔclbDtoMΔclbOtoQ (5 mL) harboring an appropriate overexpression plasmid was inoculated from frozen cell stock and grown overnight at 37 °C in LB medium supplemented with 50 μg/mL apramycin, 50 μg/mL kanamycin, and 100 μg/mL ampicillin. The starter culture was used to inoculate 50 mL of fresh LB medium containing 50 μg/mL apramycin, 50 μg/mL kanamycin, and 100 μg/mL ampicillin with a normalized number of cells, such that an OD600 of 1 of the overnight culture gave a 1:100 volume of inoculum. In some experiments, the culture was supplemented with 1 mg/mL [13C5,15N]-L-methionine (Cambridge Isotope Laboratories) at the time of inoculation. The culture was incubated at 37 °C with 200 rpm shaking for 24 h, transferred into 50 mL Falcon tubes, and centrifuged (4,000 rpm × 10 min at 4 °C). The cell pellets were flash frozen in liquid nitrogen and lyophilized overnight. The resulting dried biomass was then extracted with MeOH (750 μL) by vortexing. Samples were centrifuged (13,000 rpm × 20 min at 4 °C), and 300 μL of the supernatant was analyzed by LC/MS. The m/z values reported correspond to monoisotopic peaks.
LC/MS and LC/MS/MS analyses were carried out on an Agilent 1290 Infinity UHPLC system (Agilent Technologies) coupled to a maXis impact UHR time-of-flight mass spectrometer system (Bruker Daltonics Inc) equipped with an electrospray ionization (ESI) source. Data were acquired with Bruker Daltonics HyStar software version 3.2 for UHPLC and Compass OtofControl software version 3.4 for mass spectrometry, and processed with Bruker Compass DataAnalysis software version 4.2.
For the UHPLC system, 8 μL sample were injected onto the UHPLC including a G4220A binary pump with a built-in vacuum degasser and a thermostatted G4226A high performance autosampler. An XTerra MS C18 analytical column (2.1 × 150 mm, 3.5 μm) from Waters Corporation was used at the flow rate of 0.3 mL/min using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B. The column temperature was maintained at room temperature. The following gradient was applied: 0–3 min, 5–60% B; 3–11 min, 60–68% B; 11–11.1min, 68–100% B; 11.1–13.1 min, 100% B isocratic; 13.1–13.2 min, 100–5%B; 13.2–19.2 min, 5% B isocratic.
For the MS system for LC/MS and LC/MS/MS, the ESI mass spectra data were recorded on a positive ionization mode with a mass range of m/z 50 to 1200 under the auto MS/MS mode monitoring the detection of the targets and performing MS/MS on them if detected: calibration mode, HPC; spectra rate, 1.00 Hz; capillary voltage, 3800 V; nebulizer pressure, 25.0 psi; drying gas (N2) flow, 9.3 L/min; source temperature, 220 °C. A mass window of ± 0.005 Da was used to extract the ion of [M+H]+ for all targets. Targets were considered detected when the mass accuracy was less than 5 ppm and there was a match between the observed and the theoretical isotopic patterns and a match of retention time between the samples and standards.
In vitro reconstitution of 2 and 3 with purified ClbN, ClbB, ClbC, ClbH, and ClbI (Supplementary Figs. 18–22, 24)
The assay for reconstituting 3 (50 μL) contained 50 mM Tris-HCl (pH 8.3), 200 mM NaCl, 10 mM MgCl2, 1 mM TCEP, 1.7% v/v DMSO, 125 μM CoA, 500 μM myristoyl-CoA, 500 μM malonyl-CoA, 2 mM NADPH (Enzo Life Sciences), 5 μM ClbN, 5 μM ClbB, 5 μM ClbC, 10 μM ClbH, 3 μM Sfp, 0.3 mM L-Asn, 0.3 mM L-Ala, 0.3 mM unlabeled SAM or [methyl-2H3]-SAM (CDN Isotopes, catalog No. D-4093), and 2.3 mM ATP. A control reaction where ClbH was omitted was also analyzed.
The assay for reconstituting 2 and 1 (50 μL) contained 50 mM Tris-HCl (pH 8.3), 200 mM NaCl, 10 mM MgCl2, 1 mM TCEP, 1.7% v/v DMSO, 125 μM CoA, 500 μM myristoyl-CoA, 500 μM malonyl-CoA, 2 mM NADPH, 5 μM ClbN, 5 μM ClbB, 5 μM ClbC, 5 μM ClbH, 10 μM ClbI, 3 μM Sfp, 0.3 mM L-Asn, 0.3 mM L-Ala, 0.3 mM unlabeled SAM, and 2.3 mM ATP. ClbN, ClbB, ClbC, ClbH, and ClbI were incubated with Sfp and CoA at 22 °C for 1.5 h for phosphopantetheinylation. The complete reaction was initiated by the addition of myristoyl-CoA, malonyl-CoA, NADPH, L-Asn, Ala, SAM, and ATP. To identify key residues of ClbI, this assay was repeated with 10 μM ClbI S178A instead of 10 μM ClbI.
The assay mixtures were incubated at 22 °C, quenched after 16 h by the addition of ice cold 250 μL methanol, incubated on ice for 30 min, and centrifuged at 4 °C (13,200 rpm × 15 min). The supernatant was saved for LC/MS and LC/MS/MS analysis. The protein pellet was washed twice with 250 μL ice cold methanol and dried under a stream of N2 for 1 min. Products bound to PCP- and ACP-domains were hydrolyzed by the addition of 0.1 M KOH (40 μL) followed by heating at 70 °C for 20 min. The samples were cooled on ice, and 0.1 M HCl (80 μL) was added to the solutions. Finally, methanol (120 μL) was added to the samples, which were then incubated at −20 °C overnight to precipitate protein. The base-treated pellet samples were centrifuged at 4 °C (13,200 rpm × 15 min), and the liquid phase was analyzed by LC/MS and LC/MS/MS.
In the supernatant samples 1, 2, and 3 were identified using the same method as for the metabolite analyses of E. coli DH10B BACpks knockout strain harboring overexpression plasmids. In the supernatant and base-treated pellet samples, 5 and 6 were identified on the same instrument using a Thermo Hypersil GOLD aq Polar Endcapped C18 analytical column (3 × 50 mm, 3 μm). The injection volume was 15 μL and the flow rate was 0.4 mL/min using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B. The following gradient was applied: 0–2 min, 2% B; 2–12 min, 2–100% B; 12–17 min, 100% B isocratic; 17–17.5 min, 100–2% B; 17.5–25 min, 2% B isocratic. The m/z values reported correspond to monoisotopic peaks.
Multiple sequence alignments of ClbI-KS (Supplementary Fig. 23) and ClbH-A2 (Supplementary Table 5, Supplementary Fig. 25)
Amino acid sequences of eleven ketosynthases and six adenylation domains from various PKS and hybrid PKS/NRPS enzymatic machineries were obtained from the NCBI public database, and domain boundaries were determined using the Maryland PKS/NRPS Analysis Web Server.30 Multiple sequence alignments were generated using Clustal Omega31 and visualized in JalView.
Searches for gene clusters that harbor both ClbH-A2 and ClbI homologs (Supplementary Table 6)
Homologs of ClbH-A2 (predicted sequence from Maryland PKS/NRPS Analysis Web Server30) and ClbI were identified using Protein BLAST. The domain architectures of the detected homologs were predicted using Maryland PKS/NRPS Analysis Web Server.30 The genomic context of the detected homologs was examined by analyzing the nucleotide sequences of gene clusters using antiSMASH.32
Generation of homology model of ClbH-A2 using HHPred (Supplementary Fig. 26)
The ClbH A2 domain was used as the query to search for structural homologs using HHpred.33 The highest hit was the phenylalanine-activating adenylation domain PheA of gramicidin synthetase A (GrsA).34 A homology model of the ClbH A2 domain was generated by Modeller33 using the crystal structure of PheA as a template and the resulting PDB file was aligned with the PheA PDB file (1AMU) using PyMOL version 1.8 (Schrödinger, LLC).
Supplementary Material
Acknowledgments
We thank C. Brotherton and A. Sieg for help with cloning; J. May (Kahne Lab, Harvard University, Cambridge, MA) for help with radiometric assays; M. McCallum for help with bioinformatic analyses; and P. Boudreau, C. Chittim, D. Kenny, H. Nakamura, and S. Peck for helpful discussions. L. Zha, Y. Jiang, M. Wilson, and E. Balskus acknowledge financial support from National Cancer Institute (1R01CA208834-01), the Damon Runyon-Rachleff Innovation Award, and the Packard Fellowship for Science and Engineering. M. Henke and N. Kelleher were supported by Northwestern University and the National Institutes of Health (GM 067725 & AT 009143). M. Wilson is supported by an American Cancer Society-New England Division Postdoctoral Fellowship (PF-16-122-01-CDD).
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Author Contributions
L.Z. cloned, overexpressed, and purified colibactin biosynthetic enzymes and completed all enzymatic assays. Y.J. generated mutants used in the study and prepared culture extracts. M.T.H. analyzed enzymatic assays by protein mass spectrometry. M.R.W. prepared and characterized synthetic standards. J.X.W. helped to develop the analysis method for small-molecule LC/MS/MS. L.Z., M.T.H, N.L.K., and E.P.B. prepared and revised the manuscript.
Data availability
Protein mass spectrometry data supporting the findings of this study are available in MassIVE (ftp://massive.ucsd.edu/MSV000080499). All other data generated or analyzed during this study are available within the published article and its Supplementary Information files.
References
- 1.Thibodeaux CJ, Chang WC, Liu HW. Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang S. Histone methyltransferases, diet nutrients and tumour suppressors. Nat Rev Cancer. 2002;2:469–476. doi: 10.1038/nrc819. [DOI] [PubMed] [Google Scholar]
- 3.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-adenosylmethionine enzymes. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh CT, O’Brien RV, Khosla C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew Chem Int Ed. 2013;52:7098–7124. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 6.Nougayrède JP, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 7.Arthur JC, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vizcaino MI, Engel P, Trautman E, Crawford JM. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J Am Chem Soc. 2014;136:9244–9247. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brotherton CA, Wilson M, Byrd G, Balskus EP. Isolation of a metabolite from the pks island provides insights into colibactin biosynthesis and activity. Org Lett. 2015;17:1545–1548. doi: 10.1021/acs.orglett.5b00432. [DOI] [PubMed] [Google Scholar]
- 10.Bian XY, Plaza A, Zhang YM, Muller R. Two more pieces of the colibactin genotoxin puzzle from Escherichia coli show incorporation of an unusual 1-aminocyclopropanecarboxylic acid moiety. Chem Sci. 2015;6:3154–3160. doi: 10.1039/c5sc00101c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vizcaino MI, Crawford JM. The colibactin warhead crosslinks DNA. Nat Chem. 2015;7:411–417. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li ZR, et al. Critical Intermediates reveal new biosynthetic events in the enigmatic colibactin pathway. ChemBioChem. 2015;16:1715–1719. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]
- 13.Zha L, Wilson MR, Brotherton CA, Balskus EP. Characterization of polyketide synthase machinery from the pks island facilitates isolation of a candidate precolibactin. ACS Chem Biol. 2016;11:1287–1295. doi: 10.1021/acschembio.6b00014. [DOI] [PubMed] [Google Scholar]
- 14.Li ZR, et al. Divergent biosynthesis yields a cytotoxic aminomalonate-containing precolibactin. Nat Chem Biol. 2016;12:773–775. doi: 10.1038/nchembio.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh N, Sheldrake HM, Searcey M, Pors K. Chemical and biological explorations of the family of CC-1065 and the duocarmycin natural products. Curr Top Med Chem. 2009;9:1494–1524. doi: 10.2174/156802609789909812. [DOI] [PubMed] [Google Scholar]
- 16.Tanasova M, Sturla SJ. Chemistry and biology of acylfulvenes: sesquiterpene-derived antitumor agents. Chem Rev. 2012;112:3578–3610. doi: 10.1021/cr2001367. [DOI] [PubMed] [Google Scholar]
- 17.Healy AR, Nikolayevskiy H, Patel JR, Crawford JM, Herzon SB. A mechanistic model for colibactin-induced genotoxicity. J Am Chem Soc. 2016;138:15563–15570. doi: 10.1021/jacs.6b10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brachmann AO, et al. Colibactin biosynthesis and biological activity depend on the rare aminomalonyl polyketide precursor. Chem Commun. 2015;51:13138–13141. doi: 10.1039/c5cc02718g. [DOI] [PubMed] [Google Scholar]
- 19.Yin J, Lin AJ, Golan DE, Walsh CT. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 20.Dorrestein PC, et al. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontecave M, Atta M, Mulliez E. S-adenosylmethionine: nothing goes to waste. Trends Biochem Sci. 2004;29:243–249. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Convergence of isoprene and polyketide biosynthetic machinery: isoprenyl-S-carrier proteins in the pksX pathway of Bacillus subtilis. Proc Natl Acad Sci USA. 2006;103:8977–8982. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simunovic V, Muller R. Mutational analysis of the myxovirescin biosynthetic gene cluster reveals novel insights into the functional elaboration of polyketide backbones. ChemBioChem. 2007;8:1273–1280. doi: 10.1002/cbic.200700153. [DOI] [PubMed] [Google Scholar]
- 24.Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 25.Sundaram S, Hertweck C. On-line enzymatic tailoring of polyketides and peptides in thiotemplate systems. Curr Opin Chem Biol. 2016;31:82–94. doi: 10.1016/j.cbpa.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Brotherton CA, Balskus EP. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J Am Chem Soc. 2013;135:3359–3362. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]
- 27.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachmann BO, Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Method Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 31.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber T, et al. AntiSMASH 3.0 — a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alva V, Nam SZ, Soding J, Lupas AN. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016;44:W410–415. doi: 10.1093/nar/gkw348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conti E, Stachelhaus T, Marahiel MA, Brick P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.