

Abstract
In this chapter, we provide a review of the challenges and advances in developing successful positron emission tomography (PET) imaging agents for three major types of aggregated amyloid proteins – amyloid-beta (Aβ), tau, and alpha-synuclein (α-syn). These three amyloids are involved in the pathogenesis of a variety of neurodegenerative diseases, referred to as proteinopathies or proteopathies, that include Alzheimer’s disease, Lewy body dementias, multiple system atrophy, and frontal temporal dementias among others. In the Introduction, we briefly discuss the history of amyloid in neurodegenerative diseases and describe why progress in developing effective imaging agents has been hampered by the failure of crystallography to provide definitive ligand-protein interactions for rational radioligand design efforts. Instead, the field has relied on largely serendipitous, trial and error methods to achieve useful and specific PET amyloid imaging tracers for Aβ, tau, and α-syn deposits. Because many of the proteopathies involve more than one amyloid protein, it is important to develop selective PET tracers for the different amyloids to help assess the relative contribution of each to total amyloid burden. We utilize Pittsburgh Compound B (PiB) to illustrate some of the critical steps in developing a potent and selective Aβ PET imaging agent. Other selective Aβ and tau PET imaging compounds have followed similar pathways in their developmental processes. Success for selective α-syn PET imaging agents has not been realized yet, but work is ongoing in multiple laboratories throughout the world. In the tau sections, we provide background regarding 3-repeat (3R) and 4-repeat (4R) tau proteins and how they can affect the binding of tau radioligands in different tauopathies. We review the ongoing efforts to assess the properties of tau ligands, which are useful in 3R, 4R or combined 3R/4R tauopathies. Finally, we describe in the α-syn sections recent attempts to develop selective tracers to image α-synucleinopathies.
Introduction
The human genome codes for approximately 20,000 proteins (1). This chapter focuses on imaging three proteins in the brain that are prone to aggregation - amyloid-beta (Aβ), tau, and alpha-synuclein (α-syn). These proteins are associated with a variety of neurodegenerative diseases including Alzheimer’s disease (AD), cerebral amyloid angiopathy (CAA), Pick’s disease (PiD), corticobasal degeneration (CBD), primary supranuclear palsy (PSP), chronic traumatic encephalopathy (CTE), Parkinson’s disease (PD), dementia with Lewy bodies (DLB), Parkinson’s disease with dementia (PDD), and multiple system atrophy (MSA) (Figure 1) (2–5). Aggregated proteins not discussed in this chapter include huntingtin found in Huntington’s disease, prions found in Creutzfeldt-Jakob disease (sCJD, fCJD, vCJD), Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, Kuru, and Alpers syndrome, and proteins comprising a variety of systemic amyloidoses (6–10).
Figure 1.
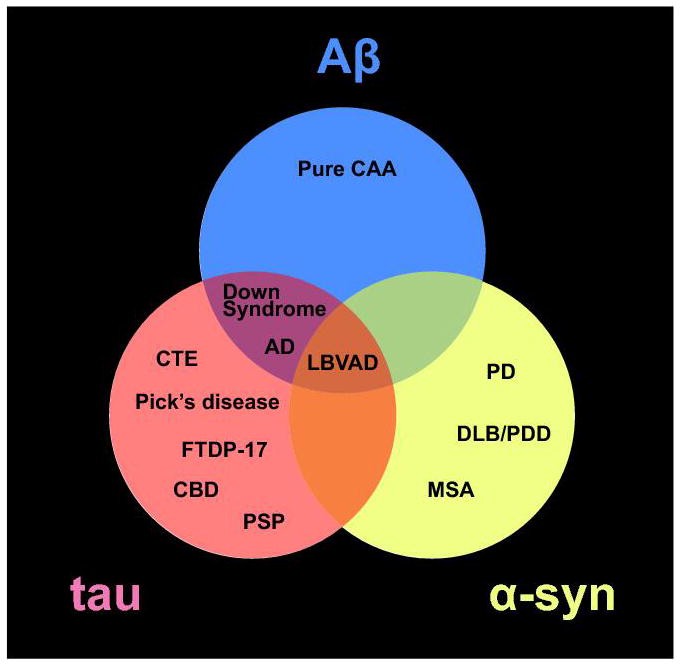
Venn diagram of the three aggregated amyloid proteins discussed in the chapter and their associated neurodegenerative diseases. Abbreviations: cerebral amyloid angiopathy (CAA); Alzheimer’s disease (AD); AD Parkinson’s disease (PD); dementia with Lewy bodies (DLB); Parkinson’s disease with dementia (PDD); multiple system atrophy (MSA); Lewy body variant Alzheimers’s disease (LBVAD); chronic traumatic encephalopathy (CTE); frontal temporal dementia with parkinsonism-17 (FTDP-17); corticobasal degeneration (CBD); and progressive supranuclear palsy (PSP).
Under normal physiological conditions, protein production, incorporation, degradation, and clearance are in homeostatic equilibrium. A variety of diseases can perturb these equilibria and impair cellular functions (11–13). Amyloids comprise a group of proteins of varying cellular origin, size, concentration, and function, and they are characterized by their propensity to self-aggregate into oligomeric species that progressively form fibrillar beta-pleated sheet structures (14) (Figure 2). These extended beta-sheet structures are the distinguishing feature of amyloids (15). “Amyloid” derives from the Greek “ámulon” meaning starch and was first used by the German botanist Matthias Schleiden to describe starch-like plant extracts that he identified using a crude staining method employing iodine and sulphuric acid (16). In the medical literature, the term amyloid was first used by German pathologist Rudolph Virchow to describe abnormal hyaline cerebral inclusions, which he termed corpora amylacea, that are often associated with astrocytes in the aging brain. Virchow noted that these deposits were stained by the same crude iodine-based method used by Schleiden, leading him to conclude that they were cellulose or starch-like in composition (17), when in fact they are composed of polysaccharides and variable amounts of protein (18). Friedreich and Kekule correctly identified the protein content in amyloids four years later (19). Today, the term amyloid denotes extracellular protein aggregates that are congophilic (avidly stained by the dye Congo red, see below) and exhibit birefringence when viewed with polarized light (10). Tau and α-syn were considered “intracellular amyloid” or “amyloid-like” proteins for many years because of their intracellular locations, but the Nomenclature Committee of the International Society of Amyloidosis (ISA) recommended reclassification of tau and α-syn as full-fledged amyloids in 2016, as they can also form extracellular amyloid deposits upon cell death (10). Thus, the ISA currently lists 36 different amyloid proteins, among which are Aβ, tau, α-syn, huntingtin, prions, and many systemic amyloids.
Figure 2.
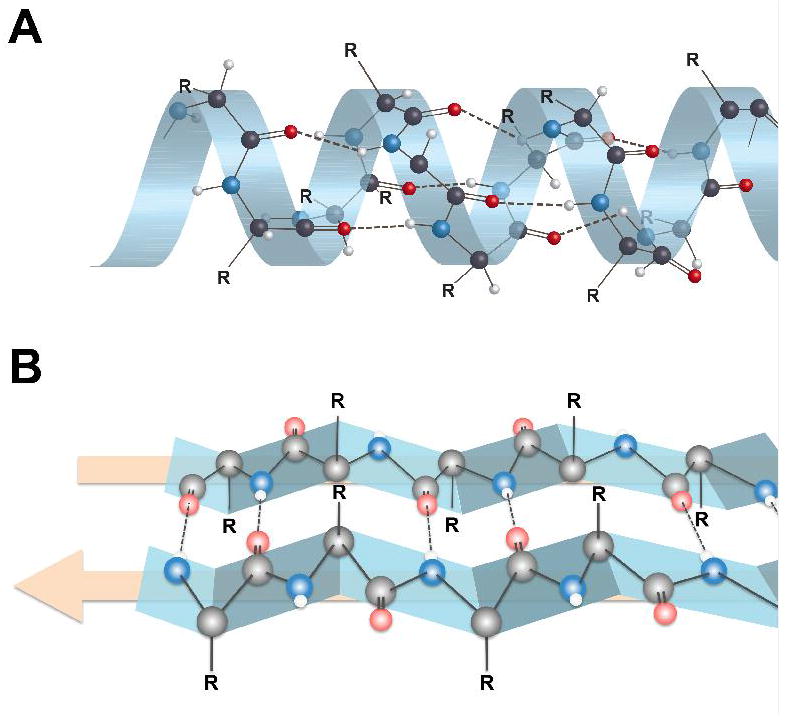
Secondary structures of proteins showing alpha-helix (A) and beta-pleated sheet (B) configurations. Hydrogen bonding between an amide nitrogen (blue) and an amide oxygen (red) in beta-pleated sheet structures takes place at the interfaces between two protein strands (edge-on-edge interactions), which is very different than the intramolecular hydrogen bonding of alpha-helix proteins. Note that the R groups on the α-carbons are either above the beta-sheet or below the beta-sheet. This presents sites for potentially different binding interactions with ligands on the two sides of the beta-sheet. Figure 4 displays the R group orientations from a different perspective.
Amyloid aggregations found in human tissues have proven difficult to fully characterize at the structural level because they do not form regular crystal lattices readily amenable to conventional crystallography techniques (20). The lack of detailed structural data has complicated the rational design of ligands intended to interact selectively and with high affinities with different amyloids, as the specific amino acids most involved in the ligand-protein interactions cannot be readily determined and modeled. For more than a century, investigators have noted empirically the selective interactions of different compounds with amyloids in synthetic model systems and postmortem human tissues and used these compounds to detect and quantify amyloids. Using the Bielschowsky silver staining method, Alois Alzheimer observed Aβ plaques and tau-containing neurofibrillary tangles (NFTs) in a postmortem brain in 1907 (21). More recently, neuropathologists have utilized small-molecule dyes such as Congo red, thioflavin-S and thioflavin-T to microscopically visualize amyloid deposits in tissue sections from autopsy brains (Figure 3). Further advances in the field have been made studying synthetic amyloid fibrils with solid state NMR structural assignment techniques (22–26). A limitation of these NMR methods is their reliance on synthetic amyloids that may not be truly representative of post-translationally modified native amyloids. Post-translational modifications can affect protein aggregation propensity and secondary and tertiary structures (27–29). In vitro seeding methods using native human amyloids to initiate synthetic fibril formation may overcome some of the inherent limitations of synthetic fibrils (30–35), but seeded synthetic fibrils do not incorporate extensive post-translational modifications. While synthetic amyloid fibrils have proven useful for some purposes, such as convenient model systems for 3D structural studies and high throughput screening assays requiring large amounts of screening substrate, the native amyloids found in the human neurodegenerative diseases are the final arbiters to definitively and quantitatively assess various ligand-amyloid interactions.
Figure 3.

Fluorescent images of consecutive paraffin sections from the frontal cortex of an Alzheimer’s disease brain stained with: (A) the pan-amyloid dye X-34 (100 μM), which binds to both Aβ-containing plaques and cerebral amyloid angiopathy (CAA) and tau-containing neurofibrillary tangles (NFTs); and (B) the Aβ-selective dye 6-CN-PiB (10 μM), which is a highly fluorescent, close structural analogue of PiB with identical binding properties. Note that X-34 stains Aβ plaques and CAA as well as NFTs while 6-CN-PiB stains only Aβ-containing plaques and CAA. Panels C and D are high magnification images of the area boxed in A and B, respectively. Bar = 50 μm in C.
Radioligands for Amyloids
Successful positron emission tomography (PET) brain amyloid imaging radioligands should possess many of the properties of successful PET neuroreceptor radioligands (36, 37). Some of the important requisite properties are listed in Table 1 and are well established in the field of PET radiopharmaceutical development. The radioligand should bind selectively, reversibly, and with high affinity to the amyloid targets in the brain. Target selectivity is an important criterion, although the degree of selectivity required depends on many factors such as the relative affinities of the radioligand to target and non-target sites, as well as the brain distribution and the relative concentrations of the target and non-target sites. It is possible for high-capacity, low affinity (high Bmax and Kd) interactions to obscure low-capacity, high-affinity (low Bmax and Kd) interactions at early times post-injection, so the relative concentrations, affinities, and brain distributions of the target and non-target binding sites should be considered. An equilibrium dissociation binding constant (Kd) in the range of 1 nM reflects relatively slow and reversible off-rates coupled with relatively high on-rates (Kd = koff/kon), and is needed for use with short-lived PET radionuclides such as 11C (t1/2 = 20.3 min) and 18F (t1/2 = 109.8 min). A low nanomolar Kd value helps ensure that the radioligand-amyloid target complex remains bound sufficiently long for non-specific binding to clear, permitting specific binding signal to be more readily determined. For effective blood-brain barrier (BBB) penetration via passive diffusion, candidate agents should be relatively small (<700 Da) and moderately lipophilic with an octanol-water partition coefficient logP value in the range of 1–3 at pH 7.4 (logD7.4). Standardized uptake values (SUV) in the brain greater than 1.0 within a few minutes of intravenous injection are a hallmark of nearly all successful PET neuroreceptor radioligands, and represent a reasonable goal for brain amyloid imaging agents. While large molecules, antibodies, and nanobodies can penetrate the brain, attainment of an SUV value greater than 1.0 a few minutes after injection has proven to be disqualifying for large species labeled with short-lived radionuclides. The clearance of non-bound radioligand from brain tissues should be relatively rapid so that specifically bound radioligand in the brain can be accurately quantified; most successful neuroreceptor radioligands demonstrate brain non-specific clearance half-times of 30 min or less. It is also desirable that the radiotracer not produce radiometabolites that cross the BBB and that radiometabolites are not produced in brain tissues, thereby confounding pharmacokinetic assessments, as PET imaging follows the radionuclide distribution without regard to its chemical composition.
Table 1.
Some Amyloid PET Radioligand Properties
|
From a non-invasive imaging perspective, aggregated amyloids are a logical target for radioligand development. Pathologists have employed small-molecule dyes, such as Congo red, thioflavin-S, and thioflavin–T, to selectively detect and visualize the density of extended beta-pleated sheet species in tissues for many years (38–41). One approach would be to develop radioligands that bind selectively to the beta-sheet structures of amyloids in a manner analogous to well-known fluorescent dyes (Figure 4). These dyes bind less avidly to monomeric and oligomeric amyloid species than to the extended beta-sheet structures of aggregated amyloids found in amyloid fibrils and larger deposits such as Aβ plaques, NFTs, and Lewy bodies. Aggregation concentrates amyloid protein molecules into macroscopic extended beta-sheet fibrils which deposit in the brain with regional concentrations in the high nanomolar to low micromolar range in advanced AD cases, making non-invasive tomographic detection feasible. Small-molecule dyes initially used to detect the beta-sheet structure of amyloids in postmortem tissues were found to bind non-selectively to different amyloids. Hence, Congo red, chrysamine-G, thioflavin-S, thioflavin-T are pan-amyloid dyes that avidly bind beta-sheets of different amyloids with widely diverse amino contents. However, radioligands possessing binding selectivity for one type of amyloid could be important in many neurodegenerative disease imaging applications, as variable amounts of more than one amyloid are frequently present. Prime examples (Figure 1) are AD, with high levels of Aβ plaques and NFTs, and Lewy body variant of AD (LBVAD), with high levels of Lewy bodies (α-syn), Aβ plaques, and NFTs. Other neurodegenerative diseases such as PD, DLB, PDD and some non-AD tauopathies contain varying amounts of α-syn, tau, and Aβ aggregates. Non-selective amyloid agents cannot readily distinguish the different amyloids present in these diseases, which might prove to be important for disease detection, disease staging, and assessing responses to different anti-amyloid therapies. 18F-FDDNP was the first pan-amyloid PET imaging agent reported in human studies, and it has been utilized for Aβ, tau, huntingtin, and prion PET imaging applications (42–46). Approximately 20 years ago, the Pittsburgh group developed fluorescent pan-amyloid agents: X-34, which has been used to image amyloids in vitro in tissue sections (Fig 3) (47–49), and methoxy-X04, which has been used widely for in vivo multi-photon amyloid imaging studies in transgenic mice (50–55). Until the discovery of the first selective amyloid imaging agent for Aβ, Pittsburgh Compound B (PiB) (56, 57), it was not clear if successful development of selective small-molecule amyloid imaging agents was feasible. It is instructive to outline some of the key steps in the development of PiB (58), as many of these principles have been applied subsequently to the development of other selective amyloid imaging agents.
Figure 4.
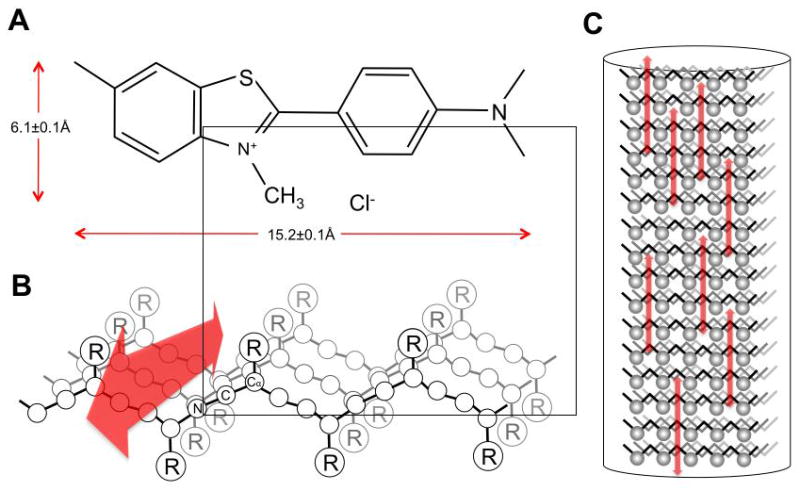
Hypothetical binding orientation of thioflavin-T (A) to a beta-sheet structure (B) resulting from interactions with R groups oriented above the plane of the fibril’s long axis. PanelC shows multiple thiofavin-T molecules binding to the fibril. (adapted from (14))
Selective Aβ Imaging Agents
The synthesis and evaluation by the Pittsburgh group of several hundred Congo red and chrysamine-G derivatives led to the “X” series of pan-amyloid imaging agents that displayed nanomolar binding affinities (Kd values) for aggregated Aβ, tau, α-syn, and prions. However, many of these compounds were charged species at physiological pH and did not achieve high brain penetration levels (>1 SUV) a few minutes after intravenous injection (59, 60). The Pittsburgh group changed direction and began the examination of thioflavin-T (Th-T) derivatives (Figure 5). Removal of the methyl group (and the positive charge) from the nitrogen in the benzothiazole ring led to a series of neutral, lipophilic compounds (termed benzothiazole anilines or BTAs) that readily entered rodent brain (Figure 6) (61, 62). Fortunately, these compounds exhibited much greater affinity for synthetic Aβ fibrils than did the parent compound, Th-T (Figure 7). Subsequent derivatization of the 6-position of the benzothiazole ring and variation of the degree of methylation of the aniline nitrogen provided a series of BTA compounds with low nanomolar affinities for Aβ fibrils (Table 2). Importantly, these BTA compounds readily entered and rapidly cleared from normal rodent brain, indicating the potential to provide radioligands with low non-specific binding in vivo (Table 3). One compound stood out from the others with a very rapid normal brain clearance half-time of 6 min. That compound was the 6-hydroxy, monomethylated aniline derivative that became known as PiB. For a history and discussion of Pittsburgh Compound A (PiA), interested readers are referred to a recent review (58). Subsequent binding assays demonstrated that PiB bound with very low affinity to aggregated tau, with a relative binding ratio of tau-to-Aβ (Kitau/KiAβ) greater than 100-fold (63). Histopathology studies confirmed that PiB predominantly labeled Aβ in plaques and CAA, but very weakly labeled tau in classic NFTs (64, 65). Additional binding analyses demonstrated that PiB bound to α-syn-containing Lewy bodies with very low affinity as well (66, 67). It is interesting to note that PiB bound with high affinity to synthetic α-syn fibrils, but not to Lewy body inclusions of α-syn in human brain (66, 67). As mentioned in the Introduction, radioligand binding to synthetic amyloid fibrils may differ from binding to endogenous amyloid deposits in human brain. Another example of binding differences was noted with PiB binding to synthetic Aβ fibrils vs. Aβ plaques in human brain homogenates (56, 68). PiB bound to Aβ1–40 and Aβ1–42 synthetic fibrils (the two most prevalent forms of Aβ) and to human plaques with similarly high affinity (Kd ~2 nM), but the Bmax value was about 500-fold higher for Aβ plaques than for synthetic Aβ fibrils. Many Aβ transgenic mice models displayed a similarly low Bmax value for PiB binding as well (68), highlighting the need to critically evaluate the fidelity of the amyloid model system relative to amyloid found in the authentic human disease.
Figure 5.
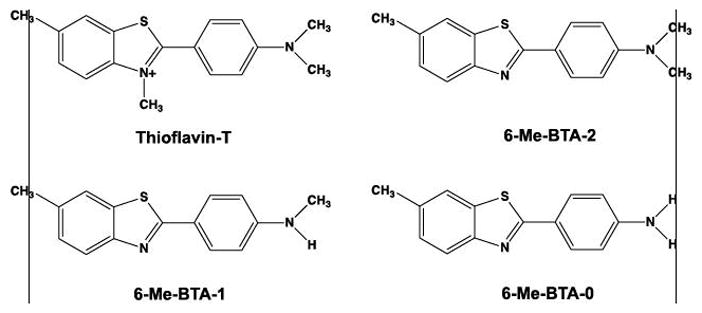
Structure of cationic thioflavin-T and three neutral thioflavin-T analogues resulting from the removal of the quaternary methyl group on the benzothiazole nitrogen. The resulting compounds were termed benzothiazole anilines or BTAs and possessed different degrees of N-methylation (denoted by the trailing number).
Figure 6.
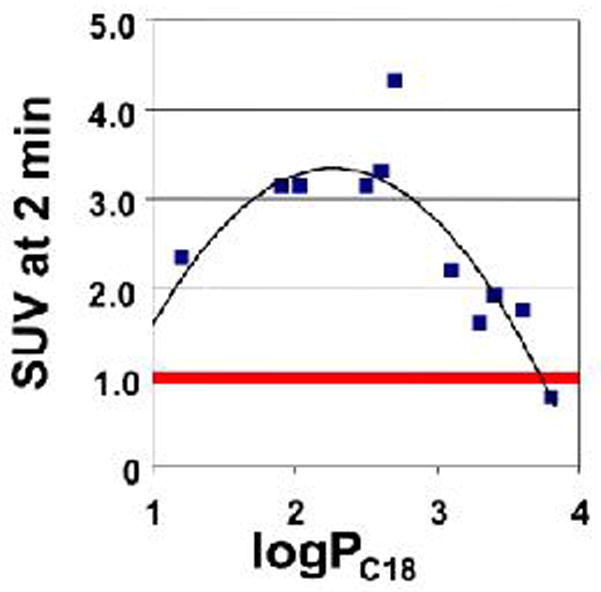
Relationship of normal mouse whole brain uptake and logPC18 (logarithm of the octanol-water partition coefficient estimated by relative HPLC retention on a lipophilic C18 column) of different 11C-labeled-BTA compounds at 2 min following intravenous tail-vein injection. Note that the brain uptake SUV of most BTAs exceeded 1.0. (adapted from (56)).
Figure 7.
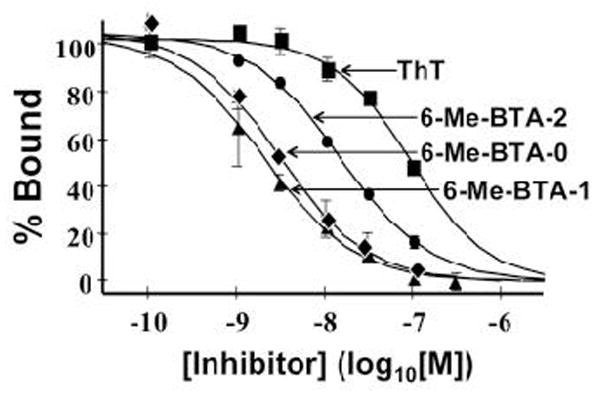
Competition binding plot of three neutral BTA analogues shown in Fig. 5 and thioflavin-T (Th-T) for Aβ1–40 fibrils with [3H]BTA-1. All three BTA analogues demonstrated much higher affinity for Aβ1–40 fibrils than thioflavin-T.
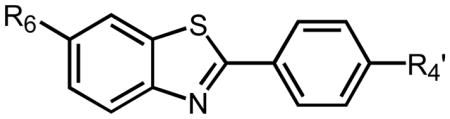
Table 2.
Binding affinities (Ki values in nM) of different BTAs for Aβ1–40 fibrils in competition with [3H]BTA-1.
| ||||
|---|---|---|---|---|
|
| ||||
| Ki (nM) | ||||
|
| ||||
| R4′ | ||||
| NH2 | NHCH3 | N(CH3)2 | ||
|
| ||||
| R6 | CH3 | 9.5 | 10 | 64 |
| H | 37 | 10 | 4.0 | |
| HO | 46 | 4.3 | 4.4 | |
| CH3O | 7.0 | 4.9 | 1.9 | |
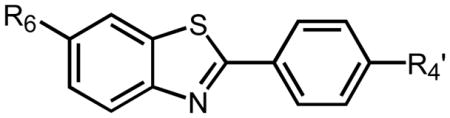
Table 3.
Clearance of different 11C-labeled BTAs from mouse brain (ratio of 2 min brain concentration to 30 min brain concentration). The 6-OH-BTA-1 compound (PiB) demonstrated the highest clearance ratio of 11 indicating a clearance half-time of ~6 min.
| ||||
|---|---|---|---|---|
|
| ||||
| Ratio of 2′-to-30′ mouse brain uptake | ||||
|
| ||||
| R4′ | ||||
| NH2 | NHCH3 | N(CH3)2 | ||
|
| ||||
| R6 | CH3 | - | 2.7 | 0.5 |
| H | - | 7.6 | 2.5 | |
| HO | - | 11 | 3.0 | |
| CH3O | 3.8 | 3.2 | 1.1 | |
The peripheral radiometabolites of PiB found in animal and human blood were highly polar and did not readily cross the BBB (56, 69). However in rat brain, PiB was rapidly metabolized to polar species (i.e., 6-O-sulfate and others) that built up over time and complicated pharmacokinetic analyses (70, 71). Fortunately, the intracerebral metabolism of PiB appeared to be unique to rats, as mouse, non-human primate, and human brain did not produce radiometabolites of PiB (70). These findings reinforce the need to carefully assess and evaluate potentially confounding inter-species differences in all aspects of radiopharmaceutical development.
The pharmacokinetics of PiB in human subjects have been thoroughly evaluated over the past 15 years (57, 69, 72–74). In general, the in vivo kinetics of PiB are relatively rapid and amenable to accurate quantitation within the constraints of the 20.3 min half-life of 11C. PiB late-time regional brain SUV ratios (SUVRs) relative to cerebellar grey matter (which is typically devoid of fibrillar Aβ) are known to be biased (5–25%) relative to arterial input function-derived specific binding outcomes (73, 75, 76). Yet SUVRs have proven useful and practical by eliminating long scanning times and the need to sample and process arterial blood for input function assessments. An example of SUVR PiB images in an elderly cognitively normal subject and an AD subject acquired 40–60 min post injection is shown in Figure 8. While this example demonstrates large differences in specific signal between an Aβ-negative control and an Aβ-positive AD subject, the most promising and useful application of Aβ imaging is in subjects who are not demented but are at risk for developing AD by virtue of being Aβ-positive (77–88). These subjects can be followed longitudinally and assessed using PiB (or another Aβ imaging agent) to determine whether they go on to develop AD, and, importantly, whether different therapeutic interventions can prevent or delay the onset of AD in non-demented Aβ-positive subjects (89–92).
Figure 8.

Pittsburgh Compound B (PiB) standardized uptake value ratio (SUVR) images relative to cerebellar grey matter in an Aβ-negative (Aβ−) cognitively normal elderly subject (top row) and an Aβ-positive (Aβ+) Alzheimer’s disease (AD) subject (bottom row) determined 40–60 min post injection. Retention of PiB in the cognitively normal subject is primarily a result of non-specific binding in white matter while the AD subject shows extensive tracer retention in frontal cortex, posterior cingulate, and parietal regions.
While PiB continues to be very useful in research settings in close proximity to a production cyclotron, its clinical utility is limited by the short half-life of the 11C radiolabel. As a result, 18F-labeled Aβ imaging agents have been developed and make use of commercial 18F-FDG cyclotron production and distribution networks. The structures of three agents approved for clinical studies in the US, Europe, and Asia are shown in Figure 9. All three compounds are highly selective for binding aggregated Aβ in the brains of human subjects (93–96). As with PiB, these 18F-labeled Aβ imaging agents are used to detect the presence of Aβ deposits in brain for diagnostic purposes and in conjunction with drug therapy clinical trial studies (97–100). Appropriate use criteria for these agents have recently been established (101–105), and Procedure Standards and Practice Guidelines have been recommended by the Society of Nuclear Medicine and Molecular Imaging and the European Association of Nuclear Medicine (106).
Figure 9.
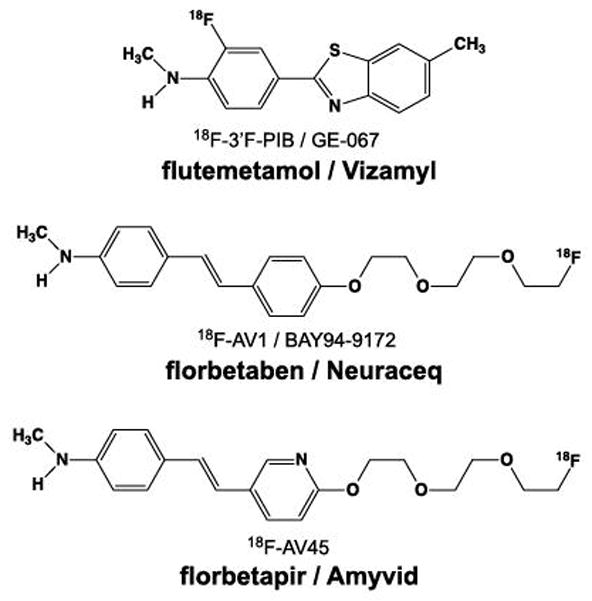
Structures of three 18F-labeled Aβ imaging agents approved for clinical scanning in the US, Europe, and Asia. Vizamyl is an 18F-labeled derivative of PiB, and Amyvid and Neuraceq differ in structure by one atom in the central ring (N or C).
Tau Protein in Disease
Although neurofibrillary pathology was first recognized and associated with senile dementia more than one hundred years ago (21, 107), knowledge of the origin and composition of what is recognized as one of the pathologic hallmarks of AD would not be elucidated for many decades. Electron microscopy studies in the 1960s revealed that the dense, fibrous argyrophilic inclusions termed NFTs, frequently observed in specific populations of neurons from AD brain tissue, were comprised mainly of paired helical filaments (PHFs) of a then unknown composition (108). It would later be demonstrated that the major constituent of PHFs is a hyperphosphorylated and insoluble form of the microtubule-associated protein (MAP) tau (109–111). Microtubules are filamentous intracellular structures assembled from heterodimers of α- and β-tubulin protein that are a major constituent of the eukaryotic cytoskeleton. The dominant functional role of physiologic MAPs is to interact with tubulin to promote the assembly and stability of microtubules (112, 113). Abnormal hyperphosphorylation of positively-charged tau promotes its aggregation into insoluble PHFs and ultimately NFTs, although the mechanisms that drive this process are not fully described. Nevertheless, the relationship of pathologic tau deposits to the progressive cognitive symptoms of dementia is well established in AD (114–121). More generally, pathological aggregations of tau protein are a defining characteristic of a broader class of neurodegenerative diseases, termed tauopathies, which also include PSP and CBD. Additionally, some types of frontotemporal lobar degeneration (FTLD) are considered primary tauopathies, notably PiD and some rare hereditary forms (Table 4) (28, 122, 123).
Table 4.
Classification of Brain Proteopathies
| Disease | Phenotype | Origin | Major Proteopathy | Prominent Histologic Findings |
|---|---|---|---|---|
|
| ||||
| Primary tauopathies | ||||
| Pick’s disease | FTD | SP | 3R tau as TF | Pick bodies, ramified astrocytes |
| Progressive supranuclear palsy (PSP) | FTD, MD | SP | 4R tau as SF | Tufted astrocytes, globose tangles |
| Corticobasal degeneration (CBD) | FTD, MD | SP | 4R tau as SF | Astrocytic plaques |
| Argyrophilic grain disease | DEM | SP | 4R tau as SF | Oligodendroglial coiled bodies, limbic argyrophilic grains |
| FTDP-17 | FTD, MD | IN | 3R,4R tau as PHF, SF, TF | Highly variable depending on specific mutation: NFT, Pick bodies, glial inclusions are common |
|
| ||||
| Secondary tauopathies | ||||
| Alzheimer’s disease | DEM | SP, IN | 3R,4R tau as PHF, Aβ as NP | Cortical NFT and NP |
| Down syndrome | DEM | IN | 3R,4R tau as PHF, Aβ as NP | Dense cortical NFT and NP |
| Chronic traumatic encephalopathy | DEM | ACQ | 3R,4R tau as PHF | NFT in cortical convexities |
|
| ||||
| Synucleinopathies | ||||
| Parkinson’s disease | MD | SP, IN | α-synuclein | Cortical and brainstem Lewy bodies and neurites |
| Dementia with Lewy bodies | DEM, MD | SP, IN | α-synuclein | Cortical and brainstem Lewy bodies and neurites |
| Multiple system atrophy | MD | SP | α-synuclein | Glial cytoplasmic inclusions (Papp-Lantos bodies) |
Abbreviations: 3R: three repeat; 4R: four repeat; ACQ: acquired; DEM: dementia; FTD: frontotemporal dementia; IN: inherited; MD: movement disorder; NFT: neurofibrilliary tangles; NP: neuritic plaques; PHF: paired helical filaments; SF: straight filaments; SP: sporadic; TF: twisted filaments
Tau is encoded by the MAPT gene, located on chromosome 17 at position 17q21.31 and spans 133.9 kb with 16 exons. Rare inherited mutations in the domain of MAPT are known to give rise to hereditary forms of FTLD (e.g., FTDP-17). Developmentally regulated alternative RNA splicing of exons 2, 3, and 10 gives rise to six different tau isoform proteins expressed in the adult human brain that range between 352 to 441 amino acids in length (Figure 10). These six isoforms are differentiated by either 3 or 4 repeat (3R, 4R) regions of a highly conserved, positively-charged microtubule (MT) binding domain in the carboxy-terminus of the protein and the insertion of zero, one, or two 29-amino acid inserts (0N, 1N, 2N) in the amino terminus (124–126). In the adult human brain, all six isoforms are expressed with roughly equivalent amounts of 3R and 4R species. Tau isoforms with two amino inserts (2N) comprise only ~10% of total tau (123, 127). Interestingly, in the fetal brain only the shortest isoform (0N3R) is expressed, indicating developmentally regulated expression of tau isoforms (124, 128, 129). It is important to note that the 3R and 4R regions of the hyperphosphorylated tau molecule aggregate to form the extended beta-sheet structure of tau amyloids (Figure 2), and, because the amino acid content of these regions can vary significantly, one might expect differing binding properties of radioligands to different aggregated 3R and 4R isoforms (Figure 4). Hence, the important features of the 3R and 4R tauopathies are discussed in some detail in this review to highlight the potential complexities of radioligand-tau fibril interactions.
Figure 10.

Diagrammatic representation of the six isoforms of tau varying in length from 352 to 441 amino acids. Three isoforms contain 3-repeats (3R) and three isoforms contain 4-repeats (4R) in the critical microtubule binding region that is prone to aggregation and subsequent beta-sheet formation.
Post-translational modifications of tau are numerous and further diversify tau species at a molecular level. Phosphorylation of tau is a post-translational modification and is of particular relevance to the disease process (28, 122, 130). The tau protein contains numerous phosphate acceptor sites associated with serine, threonine, and tyrosine residues, with as many as 85 present in the largest (2N4R) tau isoform. Phosphorylation of tau at a subset of preferred residues appears to be a highly regulated process that provides a dynamic mechanism for regulating its microtubule-stabilizing activity (131, 132). Phosphorylation of tau is also developmentally regulated, with a high degree of phosphorylation present in infancy and decreasing thereafter (133, 134). Hyperphosphorylated tau is a characteristic of pathologic tau aggregates, which in AD contains several-fold more phosphate per mole of protein than soluble cytosolic tau (135). The conversion from physiologic to pathologic species of hyperphosphorylated tau likely involves the dysregulation of tau protein kinases and phosphatases (122, 136). Hyperphosphorylation of tau adversely affects its binding to microtubules, leading to increased concentrations of unbound tau, and hyperphosphorylation helps to neutralize positive-charges, thereby promoting aggregation. Non-fibrillar tau accumulation in cells, called pre-tangles, is detectible in early disease phases, while continued accumulation of tau leads to conformational changes that promote beta-sheet formation and ultimately self-aggregation of tau into larger filaments (131, 137). While NFTs are a common form of pathologic tau fibrils observed in AD brain, a diverse morphology of neurofibrillary pathology is observed, including neuropil threads in dendritic processes of neurons, straight filaments (SF), twisted ribbons (TR), and dystrophic neurites associated with classic Aβ plaques (138–140). All six tau isoforms occur in PHFs and are present in a hyperphosphorylated state (127, 141). In AD, neurofibrillary pathology consists of a roughly equal balance of 3R and 4R species (141), whereas in other tauopathies the isoform composition can be skewed such that either 3R isoforms (e.g., PiD) or 4R (e.g., PSP and CBD) isoforms dominate the insoluble tau fraction (142–146). In AD and other mixed 3R/4R tauopathies (e.g., Down syndrome, CTE, and FTDP-17), NFTs consist mainly of PHFs, although SF are often a minor constituent (147). Conversely, in 3R or 4R dominant tauopathies, neurofibrilliary pathology consists mainly of SF, suggesting that the presence of both 3R and 4R species is required for PHF assembly. Tauopathies may also be distinguished by their cellular distribution, which can be predominantly neuronal (e.g., AD and PiD), predominantly glial (e.g., age-related tau astrogliopathy (ARTAG)), or mixed neuronal/glial (e.g., PSP and CBD) (148). It is not known how and why these diverse tau morphologies arise, or whether conversion from one tau morphology to another is possible. It is also noteworthy that some evidence implicates soluble aggregates of oligomeric tau as an additional, if not a primary, substrate for tau-mediated neurotoxicity (149–151).
In AD, tau pathology evolves following a fairly predictable and well-defined pattern of regional involvement that is typically described in terms of pathological stages such as those defined by Braak (152). The earliest pathologic tau deposits are observed in the transentorhinal region of the medial temporal lobe, followed by the entorhinal region as a result of trans-synaptic or other processes (153–155). It is noteworthy that in these early stages (Braak I/II), tau positive neurons in the medial temporal lobe are a common pathologic finding in elderly subjects who are cognitively normal or exhibit only mild symptoms of cognitive impairment. Such cases may indicate an age-related process, termed primary age-related tauopathy (PART), characterized by mild to moderate NFT pathology in the medial temporal lobe (156). In intermediate Braak stages (III/IV), tau pathology becomes more pronounced in transentorhinal and entorhinal regions and infringes upon the hippocampus, while in most severe Braak stages (V/VI) NFTs are found in neocortical association areas. Paralleling the development of more extensive and pronounced neocortical pathology, progressive cognitive abnormalities assume a more characteristic presentation that is indicative of AD.
Non-AD Tauopathies
Tauopathies are a class of more than 20 neurodegenerative diseases and disorders characterized by the presence of abnormal tau-positive neuropathology, albeit of variable morphology and distribution (148). Hyperphosphorylation, somatodentric sequestration, fibrillization, and aggregation of insoluble tau in neurons and glial cells appear to be common factors leading to the development of disease-specific pathologies. Tauopathies are subclassified as either primary or secondary, depending on the degree to which tau pathology occurs independently of other proteopathies (e.g., Aβ, α-syn, TDP-43). FTLD is a collection of non-AD neurodegenerative disorders affecting primarily the frontal and temporal lobes (157). FTLD can be further divided into three histological subtypes based upon the predominant proteopathy: FTLD-tau, FTLD-TDP, and FTLD-FUS (158). The FTLD-tau subtype includes most primary tauopathies, such as PSP, CBD, and PiD. These neurodegenerative diseases have in common the presence of abnormal neuronal and/or glial tau inclusions, although they are clinically and pathologically heterogeneous (159). Secondary tauopathies include AD, Down syndrome, and dementia with Lewy bodies (DLB), and are characterized by tau pathology in association with other proteopathies (e.g., Aβ and α-syn).
Need for Both Aβ and Tau Imaging Agents
The development of PET Aβ imaging agents was strongly motivated by the body of empirical evidence supporting the amyloid cascade hypothesis as an early and perhaps initiating event in the pathologic cascade of AD (160), and furthermore for its potential utility as a surrogate marker of brain Aβ burden for assessing the efficacy of experimental anti-Aβ therapeutics. Similarly, the close association between the stage of cerebral tau pathology and the severity of clinical dementia symptoms suggests that tau imaging biomarkers could be useful indices of disease progression in AD and primary tauopathies, aid challenging differential diagnoses, and support the development of novel therapeutic approaches targeting tau deposits. Although histological analyses of post-mortem tissues remain the standard for recognizing Aβ and tau pathology in AD, they do not permit longitudinal progression follow-up, and are therefore of more limited value for determining causality and evaluating treatment efficacy.
Much evidence has been gathered in support of the amyloid cascade hypothesis, most importantly the observation that rare dominantly-inherited mutations in the genes encoding amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2) lead to increased Aβ production and result in an early-onset form of AD that is phenotypically indistinguishable from the common sporadic form (161–164). However, evidence from clinical pathological studies suggests that deposition of Aβ in its insoluble, fibrillar form may be a necessary but insufficient phase in the development of sporadic AD. Several key observations support this view. First, post-mortem studies show a poor association between brain Aβ load and the severity of cognitive symptoms in AD, which have been shown to be more tightly associated with NFT pathology (114, 116, 118–120). Secondly, cross-sectional and longitudinal studies using PET Aβ imaging agents suggest that a plateau in Aβ load is reached in early AD, in spite of progressive cognitive decline (165, 166). Thirdly, significant Aβ deposits are frequently observed in cognitively normal elderly (77, 79, 83, 85, 87, 167–170), including those of very advanced age (82), and often in the absence of other abnormal biomarkers of neurodegeneration. Finally, primary tauopathies are sometimes associated with cognitive symptoms that resemble those that are characteristic of AD (148). In pure tauopathies, such abnormalities develop in the absence of any other notable pathologic feature, suggesting that tau-mediated processes alone have the capacity to promote neurodegeneration (171, 172). Although the associations between Aβ and tau are not completely understood in AD, the emerging picture is one in which Aβ and tau likely assume synergistic roles in the pathophysiology of AD. In this view, Aβ deposition is the initiating pathological event that triggers or facilitates increased tau aggregation and ultimately NFT formation (79, 173–177). Complicating this view somewhat are recent reports indicating that, in a subset of cognitively normal elderly subjects, detectible abnormalities in biomarkers of neurodegeneration (e.g., hippocampal atrophy) antecede abnormalities in Aβ biomarkers. Jack and colleagues suggest that this observation may indicate a parallel disease trajectory in AD that is initiated by non-Aβ processes (178). Another explanation is that this neurodegeneration-first trajectory arises due to distinct co-morbid processes associated with aging and AD. Indeed, medial temporal lobe atrophy is a gross pathologic feature that is closely associated with, but not unique to, AD. Tau pathology restricted to the medial temporal lobe in brains lacking any significant Aβ deposits is a frequent autopsy finding in non-demented elderly (PART) and a possible cause of medial temporal lobe atrophy (156). It is not known with certainty whether or not PART represents a prodromal phase of a parallel AD disease trajectory or merely one that occurs frequently in the elderly, approximately 20–25% of whom will develop detectible Aβ deposits by age 65 (179). In any case, tau imaging agents, in concert with other biomarkers of neurodegeneration and clinical assessments, will almost certainly lead to a greater understanding of the evolution of tau pathology in AD and primary tauopathies.
Development of Tau Imaging Agents
In addition to satisfying the general criteria for a PET neuroimaging agent, the development of a successful tau agent faces additional challenges. Unlike extracellular Aβ plaques and cell surface receptors, tau aggregates are mainly intraneuronal, and thus a tau imaging agent must penetrate the cell membrane or be otherwise transported into the cell in order to bind to tau pathology. Passively delivered CNS tracers have crossed the BBB in order to access the brain parenchyma, and the neuronal cell membrane likely presents no more impedance to passage than presented by the BBB. The different isoforms, conformational states, and ultrastructural forms (e.g., PHF or SF) of tau pathology across the spectrum of tauopathies present a complex environment for imaging agent development. The relatively large size of tau compared to Aβ (~400 amino acids vs. ~40) provides a greater number of potential binding sites and opportunities for polymorphisms to arise that are associated with tau morphology and pathogenicity. Indeed, several known MAPT polymorphisms have been associated with increased risk for sporadic tauopathies such as PSP (180) and CBD (181, 182). Numerous post-translational modifications to tau, which include but are not limited to phosphorylation, yield an even greater diversity of molecular states. Both tau and Aβ aggregates form β-sheet conformations, presenting a complication for tau agent discovery, as it can be reasonably expected that candidate molecules will exhibit a considerable degree of cross-reactivity for both tau and Aβ aggregates. A high level of tau selectivity is desirable for AD imaging applications due to the relative abundance of Aβ compared to tau in many cortical regions, particularly in more advanced disease where there is considerable overlap of the pathologies. Importantly, selectivity must be determined at ligand concentrations representative of the application. A typical PET neuroimaging study at routinely achievable radiotracer specific activities introduces a total ligand mass (labeled+unlabeled) of a few micrograms, yielding low nanomolar concentrations in the adult brain. High-affinity binding interactions dominate at these concentrations. At higher ligand concentrations, lower-affinity binding sites may also contribute to the specific signal. It is noteworthy that PiB and its close structural analogues bind to tau at micromolar concentrations but not at nanomolar concentrations (63).
Competitive in vitro assays are performed using synthetic tau fibrils, human tissue sections, or tissue homogenates to screen candidate ligands for binding characteristics such as affinity and selectivity. Synthetic tau PHFs are readily available, although a drawback of synthetic tau fibrils is that they are typically composed of a single 3R or 4R isoform that lacks the diversity of post-translational modifications characteristic of native PHF-tau. Binding experiments performed with synthetic PHF-tau cannot be expected to fully recapitulate the binding to native pathology (183). Brain tissue homogenates are less readily available, but do not suffer from this limitation, although there may be considerable variability in the pathology within the brain regions and also between donors.
Tau imaging in Alzheimer’s Disease
Until recently, it was possible to visualize tau pathology only using histologic dyes or immunohistochemical methods with post-mortem tissue. Biochemical measurements of CSF concentrations of total tau and phospho-tau may indicate the presence of cerebral tau pathology but not the extent and location of its distribution in brain. Clearly, a technique able to visualize and quantitatively assess tau burden in the living brain would be an important advancement, mirroring earlier developments in targeted Aβ probes. Despite the many obstacles to the successful development of a selective tau imaging agent (e.g., its intracellular location, lower brain concentration relative to Aβ in AD, presence of six isoforms, differing conformational states, and post-translational modifications), the field has recently realized considerable success. Several classes of compounds have been identified as potential radioligands for imaging tau pathology in AD (Figure 11). Lead candidates for further characterization and development were identified from preclinical evaluations assessing criteria such as binding affinity (often to synthetic PHFs), selectivity for PHFs over other amyloids (e.g., Aβ and α-syn), co-localization of autoradiographic signal with NFT pathology, lipophilicity, and brain uptake and clearance parameters. Several of these lead agents have advanced to the stage of clinical investigations in human subjects to assess their suitability for assessing tau burden in AD and other tauopathies. Parallel efforts to refine the tools and techniques for tau imaging are ongoing, with new tau agents approaching the threshold of human subject experimentation (184, 185). This review will focus on the development of tau imaging agents that have advanced to human research studies published in the scientific literature.
Figure 11.
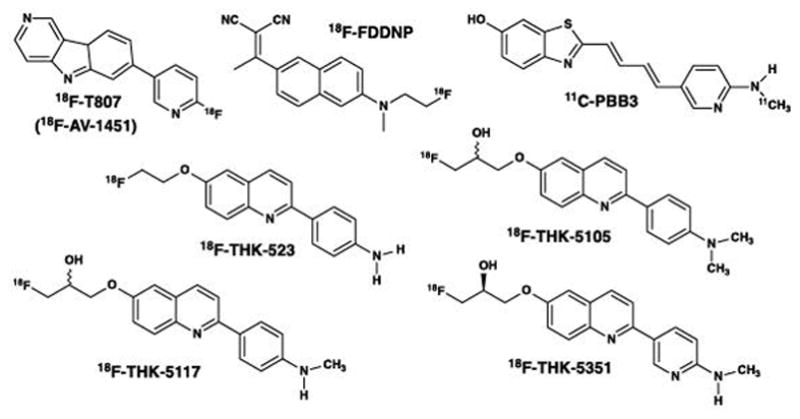
Structures of tau PET imaging tracers reported in human research studies in the scientific literature. 18F-FDDNP is a pan-amyloid imaging agent, while the other compounds are relatively selective for tau over other amyloids. THK-5351 has been shown recently to bind with high affinity to MAO-B. The relative binding affinities of all tau PET imaging tracers for 3R and 4R tauopathies remain to be clearly defined.
Developed initially as an Aβ imaging agent, 18F-FDDNP was the first non-invasive imaging agent to label fibrillar tau deposits in vivo (42), as suggested by fluorescence confocal microscopy studies showing detectable labeling of FDDNP to NFTs (186). In vivo 18F-FDDNP imaging studies showed patterns of retention that reflected the cortical distribution of Aβ, except for the medial temporal lobe where the retention of Aβ imaging agents such as PiB is relatively low (42). Based on correlations between in vivo 18F-FDDNP imaging and post-mortem histology, evidence suggests that in vivo 18F-FDDNP binding reflects contributions from both Aβ and NFTs (187). As it is not possible to differentiate the contributions of the respective pathologies to the in vivo signal, 18F-FDDNP is of limited utility as a tau imaging agent in AD where there is often significant overlap in the brain distribution of Aβ and NFTs in more advanced stages. In pure tauopathies where mixed binding to Aβ and tau is not as great a concern, 18F-FDDNP may be appropriate. Indeed, reports of its use in PSP suggest possible utility for imaging tau deposits (45, 46), although its specific binding signal is quite low.
From a screen of a large number of candidate small-molecules, a series of quinoline derivatives with high affinity and good selectivity for tau were identified with the potential to be exploited as tau imaging agents (188). The first member of this class to be described was 18F-THK-523 (Figure 11), which showed excellent affinity for synthetic tau fibrils (Kd ~1.7 nM) and approximately 10-fold binding selectivity for tau relative to Aβ fibrils. Autoradiograpic and fluorescence microscopy studies of THK-523 in AD hippocampal sections demonstrated robust labeling of NFTs with no detectible binding fibrillar Aβ, even at high concentrations (100 μM)(189). Despite these promising preclinical findings, human 18F-THK-523 studies revealed high white matter retention that complicated visual interpretation of images and severely confounded quantification of grey matter signal (190). For this reason, development of 18F-THK-523 was halted in favor of other lead agents identified from this class.
Like 18F-THK-523, the 2-arylquinoline derivatives 18F-THK-5105 and 18F-THK-5117 (Figure 11) were identified based upon in vitro binding to synthetic tau fibrils, autoradiography on AD brain tissue, and mouse brain uptake studies (191, 192). Initial human studies using these agents demonstrated small but significant increases in radiotracer retention in AD subjects relative to controls in regions where NFT pathology is expected (e.g., medial temporal lobe) (193, 194). 18F-THK-5117 was also used to illustrate the evolution of tau pathology in longitudinal studies of AD subjects, which showed significantly increased annual increases in 18F-THK-5117 binding relative to healthy controls. A potential confound for their use was the observation of significant non-specific white matter retention in all subjects with the potential to contribute spill-in to adjacent NFT-rich grey matter structures. Significant off-target binding was observed in the putamen and brain stem (193, 194). These agents were soon eclipsed by another 2-arylquinoline derivative, THK-5351, the S-enantiomer of a pyridine derivative of THK-5117 (195). 18F-THK-5351 was shown to have improved signal-to-background characteristics and low white matter retention compared to 18F-THK-5105 and 18F-THK-5117, which the authors attributed to its optical purity and lower lipophilicity. Like other tau radiotracers, significant off-target binding in the basal ganglia was observed (196). While investigating the source of the off-target binding of 18F-THK-5351 in basal ganglia, it was discovered that the in vivo binding of 18F-THK-5351 was vulnerable to pretreatment with selegiline, a potent inhibitor of monoamine oxidase B (MAO-B). Pretreatment with a single oral dose of selegiline (10 mg) resulted in 30–50% reductions in 18F-THK-5351 retention across the cortical and subcortical regions examined (197). Although it appears that 18F-THK-5351 binds to both PHFs and MAO-B with high affinity, the overlapping brain distribution of NFT pathology and MAO-B expression presents a serious confound complicating the interpretation of 18F-THK-5351 images. A further complication is the observation of significant increases in brain MAO-B availability (~9% per decade) with increasing age (198), superimposing an age effect upon neurodegenerative disease processes.
Xia and colleagues described a candidate tau radiotracer from a series of benzimidazoles showing high affinity for tau and good binding selectivity for tau versus Aβ (25:1) (183). Designated initially as 18F-T807 (later renamed 18F-AV-1451 and flortaucipir F18), autoradiographic studies reported that 18F-AV-1451 signal is strongly associated with mature NFT pathology comprised of PHF-tau in AD brain (199, 200). These and other properties indicated 18F-AV-1451 to be a potentially useful tau imaging agent to advance to human studies.
Initial 18F-AV-1451 proof-of-concept human imaging studies were reported in AD, mild cognitive impairment (MCI), and healthy control subjects (201). Late-scan (80–100 min) SUVRs were employed relating the radioactivity concentrations in target regions to the cerebellum, which is characterized by low pathologic tau burden. 18F-AV-1451 SUVR values were highest in the medial temporal, lateral temporal, and parietal lobes of the AD and MCI subjects relative to controls, consistent with more advanced Braak stages. Importantly, SUVR values did not approach a constant value after 100 min of acquisition in AD and MCI subjects, suggesting that SUVR outcomes could be influenced by the measurement interval. Although promising, additional studies were warranted to further characterize 18F-AV-1451 as a robust and reliable index of cerebral aggregated tau burden.
The early findings of Chien et al. (201) would soon be confirmed and extended by larger 18F-AV-1451 studies involving AD, MCI, and cognitively normal elderly subjects (202–205). These studies in general showed a correlation between 18F-AV-1451 specific signal and disease severity, as well as concordance between patterns of 18F-AV-1451 retention and the expected brain distribution of NFT pathology in AD described by Braak. Further 18F-AV-1451 studies demonstrated the pattern of 18F-AV-1451 retention to differ in a predictable way across a spectrum of clinical AD phenotypes (e.g., amnestic variant, visual variant, and language variant), showing increases in clinically affected regions and also strong inverse associations with indices of cerebral glucose metabolism (206).
An unexpected finding in these larger studies were areas of markedly increased 18F-AV-1451 retention in brain regions not associated with significant NFT burden in AD, often involving the choroid plexus, basal ganglia, and midbrain (Figure 12). These off-target signals are frequently observed in elderly subjects regardless of diagnostic status and are generally unremarkable or altogether absent in younger subjects, suggesting that the phenomenon is related to normal aging processes and not dementia. Although the source of off-target binding is unknown, histological and autoradiographic studies suggest a possible association with tau in choroid epithelial cells (207) and neuromelanin in some brain regions such as the substantia nigra (199, 200). However, the absence of neuromelanin-containing cells in the basal ganglia and the presence of significant off-target binding of 18F-AV-1451 suggest that the source of off-target binding is potentially multi-factorial. Off-target binding is a particular concern for in vivo assessments of tau burden in some brain regions with relevance to dementia such as the hippocampus, which has the potential to be contaminated by spill-in from signal from the adjacent choroid plexus. It is important to carefully assess region-of-interest definition strategies to minimize the potential confound of off-target binding on assessment of tau burden, particularly in medial temporal lobe structures.
Figure 12.

[18F]AV-1451 standardized uptake value ratio (SUVR) images in a tau-negative (tau-) cognitively normal elderly subject (top row) and a tau-positive (tau+) Alzheimer’s disease subject (bottom row) determined 80–100 min post injection. These are the same subjects shown in Fig. 8. Retention of [18F]AV-1451 in the cognitively normal subject shows elevated retention in basal ganglia (indicated by arrow) that is representative of off-target binding in elderly subjects. The AD subject also exhibits [18F]AV-1451 off-target binding in basal ganglia, but also high levels of radiotracer retention in the lateral temporal lobes.
PBB3 (Figure 11) is a highly fluorescent compound that was identified as a lead candidate tau radiotracer based on preclinical tissue studies and also in PS19 transgenic mice bearing a specific FTDP-17 MAPT mutation that promotes fibrillar tau pathology in the brainstem (208). Confocal fluorescence microscopy studies reported selective labeling of tau pathology in tissue sections of AD brain, PiD (a 3R tauopathy), and PS19 mice, whereas real-time two-photon microscopy in transgenic mice demonstrated in vivo labeling of intraneuronal tau inclusions by PBB3 in transgenic mouse brain. In vitro and ex vivo autoradiography using 11C-PBB3 in AD and transgenic mouse brain tissue showed increased autoradiographic signal in a manner consistent with the distribution of tau pathology. In vivo microPET 11C-PBB3 imaging in PS19 mice also showed increased retention in the brainstem where histology indicated abundant fibrillar tau pathology (208). However in both humans and mice, 11C-PBB3 was observed to rapidly decompose to a radiolabeled metabolite, with less than 10% unmetabolized in human plasma after 3 min. The major radiometabolite was identified to be a sulfated conjugate of 11C-PBB3 mediated by a sulfotransferase enzyme (209). Analysis of mouse brain tissue revealed that the labeled metabolite entered brain and accounted for approximately 40% of brain radioactivity at 5 min after injection (210). Exploratory in vivo 11C-PBB3 studies in human brain showed modest increases in signal retention in the medial temporal lobe of AD subjects that contrasted sharply with comparative PiB scans indicating Aβ load (208). Interestingly, 11C-PBB3 exhibited minimal white-matter retention, and the pattern of off-target binding in basal ganglia and brain stem characteristic of 18F-AV-1451 was not observed.
A larger human study in healthy elderly control (HC), MCI, and AD subjects employing 11C-PBB3 and PiB as complementary indices of tau and Aβ burden showed significant positive correlations in a composite cortical region, suggesting widely overlapping brain distributions of Aβ and tau pathologies (177). Indeed, widespread cortical tau pathology is a feature of late-stage (Braak V/VI) AD (204). The study also found tau burden to be positively correlated with age in PiB-negative HC subjects. These associations were driven primarily by elevated 11C-PBB3 retention in the medial temporal lobe, consistent with the pathological description of PART. Voxel-based analyses showed retention of 11C-PBB3 to be tightly associated with MR-based assessments of brain atrophy in the medial temporal lobe of AD spectrum subjects (AD and PiB-positive MCI), whereas PiB retention was not (177). Despite the shortcomings of low specific signal and a radiometabolite that enters brain, these studies demonstrated that 11C-PBB3 was able to recapitulate the most important features of tau pathology in AD and normal aging.
This review focuses on published peer-reviewed reports of tau imaging agents in human AD PET imaging studies (Figure 11). A number of second generation tau PET imaging agents are under active development, and recent reviews summarize the status of promising new agents that are beginning to enter human research studies such as 18F-RO69558948, 18F-MK-6240, 18F-GTP1, and 18F-N-methyl-lansoprazole (184, 185).
PET imaging in Non-AD Tauopathies
Although the current tau imaging agents were developed in the context of detecting tau deposits in AD, continued investigation of tau imaging agents as putative tools for indexing tau burden across the spectrum of tauopathies is ongoing. As described earlier, primary tauopathies differ from AD in terms of the tau isoform composition, tau fibril morphology, and the brain distribution of tau pathology. Given the heterogeneous nature of tau pathology in different tauopathies, it is reasonable to expect some heterogeneity in the binding properties of prospective tau agents to these diverse pathologies. For instance, although 18F-AV-1451 PET appears to be a useful index of tau burden in AD, where the hallmark pathology are NFTs comprised of a mixture of 3R and 4R tau isoforms arranged in PHFs, an early report comparing in vivo 18F-AV-1451 PET images and post-mortem histology from cases of suspected non-AD tauopathies suggested that AV-1451 binding characteristics may be dependent upon the dominant tau isoform, conformational state, or specific post-translational modifications (211). Examined in this report were two pathologically confirmed PSP cases and a familial FTLD-17 case (MAPT P301L), all of which were characterized by a preponderance of 4R tau species. In vivo 18F-AV-1451 imaging showed the highest retention in basal ganglia and brainstem in all three subjects. Lower 18F-AV-1451 signal was observed in some cortical areas and also cerebellar dentate nuclei in PSP subjects. Autoradiographic imaging of tissue sections using 18F-AV-1451 consistently showed no detectible autoradiographic signal in cortical and subcortical regions shown by immunohistochemistry to contain characteristic and abundant tau inclusions in these cases. Interestingly, no autoradiographic signal was detected in basal ganglia, despite this being a consistently prominent feature in the in vivo 18F-AV-1451 images attributed to off-target binding. These findings are in stark contrast to autoradiographic imaging of AD brain tissue sections using AV-1451, which showed a robust signal in NFT-rich regions (199, 212). Furthermore, in vivo 18F-AV-1451 indices of tau load showed no significant associations with either in vitro measures of [3H]AV-1451 binding site density (pmol/g tissue) or tau burden determined by PHF-1 immunostaining. A broader autoradiographic survey of 18F-AV-1451 specific signal in primary tauopathies provides additional support for the view that 18F-AV-1451 performs much better as a marker for AD tau pathology than for non-AD tau pathology (199, 211, 212), although some preliminary evidence suggests 18F-AV-1451 may be capable of discriminating tau pathology in specific FTLD-17 MAPT mutations (213). The phenomenon of specificity for NFTs does not appear to be unique to AV-1451. THK-523 readily labeled NFTs and neuropil threads in AD tissue specimens, but failed to label tau pathology in non-AD tauopathies (214).
The concept that the binding of prospective tau imaging agents may depend on the specific tau conformational states and strain variations was illustrated by a recent work (215). Using autoradiography and fluorescence microscopy, the authors demonstrated that while both PBB3 and AV-1451 strongly labeled NFTs and ghost tangles in AD, PBB3 also strongly labeled neuropil threads and PHF-positive plaque-associated neurites whereas AV-1451 did not. In 4R tauopathies such as PSP, CBD, and FTLD-17 (N279K mutation), differential labeling of tau pathology was even more distinct, where PBB3 consistently showed robust labeling of neuronal and glial tau inclusions in contrast to comparatively weak labeling by AV-1451. These observations were supported by radioligand binding studies in AD and PSP brain, which showed vastly more binding sites for 11C-PBB3 compared to 18F-AV-1451, and, moreover, that the binding sites for these radioligands were distinct (215).
More recent investigations of non-AD tauopathies involving larger subject cohorts have shown mixed results. Several published reports in PSP have failed to demonstrate a clinically useful 18F-AV-1451 specific signal in regions characterized by abundant tau inclusions (216–218) or a correlation between 18F-AV-1451 outcomes and measures of clinical disease severity (216). This conclusion is not universal, however, with at least one 18F-AV-1451 study concluding that the radiotracer exhibited a characteristic pattern of retention in PSP, consistent with the expected distribution of tau pathology, that was distinguishable from controls and AD (219). Studies in PSP and CBD have also been reported using other putative tau agents, namely 18F-THK-5351 (220, 221) and 18F-FDDNP (46), which suggest potential utility in these and other non-AD tauopathies. A complication in part is that tau pathology in PSP is often most remarkable in the basal ganglia and midbrain, a pathological distribution that overlaps with regions in which high off-target binding of 18F-AV-1451 has been described (222) and also for which MAO-B expression is maximal (197, 198). A definitive statement regarding the utility of these agents and other agents under development in non-AD tauopathies will likely require correlational studies between imaging and histological measures of tau burden and very much remains a work in progress at this time.
α-Synucleinopathies
Alpha-synuclein (α-syn) is an abundant 140 amino acid protein found in human brain and comprises ~1% of the total protein content in brain cytosol (223). The α-syn protein was first identified as the non-amyloid-β component (NAC) of Aβ plaques in postmortem brain tissues of AD subjects (224). The exact functions of α-syn are unknown, but it is believed to play a role in maintaining synaptic vesicles in presynaptic terminals. The protein has also been suggested to be involved in regulating the release of the dopamine in controlling voluntary and involuntary movements. The α-syn protein is concentrated in the presynaptic terminal regions of neurons and is found to a lesser extent in glial cells (225). α-Syn has three distinct structural domains (Figure 13): the N-terminal region (residues 1 to 60) containing four imperfect 11 amino acid repeats including the sequence KTKEGV, which is important in α-helix formation; the central hydrophobic region (residues 61 to 95) containing the NAC region including residues 71–82, which are highly involved in beta-sheet formation and generate insoluble aggregates that comprise Lewy bodies (LBs) and Lewy neurites (LNs) (226); and finally, the C-terminal region (residues 96 to 140), which is highly acidic and proline rich. LBs are found in the cell bodies of neurons located mainly in the brain stem and subcortical regions in early disease stages and spread to neocortical regions later (227), and are composed of a dense core of α-syn aggregates (228). LNs are abnormal neuronal processes (axons or dendrites) that contain α-syn aggregates with a more diffuse α-syn morphology than LBs. LBs and LNs are the primary pathological features of the Lewy body diseases (LBD) (229, 230). In addition, aggregated α-syn is also a major constituent of oligodendrocyte glial cytoplasmic inclusions (GCIs) found in MSA (231). Together, LBD and MSA comprise the neurological disorders termed α-synucleinopathies (Figure 1). LBD is a classification which includes PD, PDD, and DLB subjects. While PD is primarily a movement disorder characterized by extrapyramidal symptoms (EPS), PDD and DLB also involve severe cognitive deficits (dementia) and visual hallucinations (232). There are no neuropathological features that clearly distinguish PDD and DLB. Instead, classification distinction is made on clinical grounds: in PDD, EPS precedes dementia onset by 12 months or more; and in DLB, EPS occurs in close time proximity to or after dementia onset (232).
Figure 13.

Diagrammatic representation of the 140 amino acid α-synuclein molecule. The amphipathic repeat region contains both hydrophilic and hydrophobic sub-regions, and the NAC (non-amyloid-β component) is involved in beta-sheet formation. Some of the important post-translational modification amino acid sites are shown.
The aggregated proteins distinguishing some of the clinical groups in Figure 1 may not be as distinct as indicated in the Venn diagram. Different aggregated proteins can co-exist in brain, with or without clinical manifestations associated with the individual pathologies (230). For example, LBs and LNs are found in ~40% of AD patients who come to autopsy (233), Aβ pathology is found in over 80% of α-synucleinopathies (LBD and MSA), and Braak stage III or greater (relatively frequent NFTs and neuropil threads containing aggregated tau) is found in more than 50% of α-synucleinopathies (234). In LBD, Aβ deposits are present in ~85% of cases with dementia (234, 235) and tau pathology is found in ~30% of LBD cases in high Braak stages (V/VI) (234). Subjects with combined DLB and AD pathologies can be neuropathologically diagnosed as mixed dementia AD/DLB (or Lewy body variant AD (LBVAD) in Figure 1). Hence in LBD, the frequent existence of mixed pathologies makes the development and application of selective α-syn, Aβ, and tau radiotracers necessary to help identify overlapping pathologies in vivo.
Several pathogenic roles have been proposed for oligomeric and aggregated α-syn including involvement in mitochondrial and proteasomal dysfunctions and abnormal vesicular trafficking within presynaptic dopaminergic neurons that adversely affects dopamine release (236). While dopamine-replacement therapy is effective in treating the motor symptoms of PD, efficacious therapies to halt disease progression in LBD and MSA are not available. Early stage patients with minimal dopaminergic cell loss might benefit the most from treatments that reverse, halt, or slow down disease progression (237). Braak PD staging indicates that significant dopaminergic loss occurs at stage III or later, which is well after the appearance of LBs and LNs (238, 239). Sensitive and specific α-syn imaging might detect pre-symptomatic PD earlier than current dopaminergic or vesicular (VMAT2) imaging approaches, as dopamine cell loss appears to be downstream of α-syn deposition. Early imaging of α-syn load will be important in disease-modifying LBD and MSA therapeutic trials. Anti-synuclein clinical trials are currently underway and are aimed at slowing down or reversing neurodegenerative damage caused by oligomeric or fibrillar α-syn species (240, 241). As the AD therapy community has learned, it will be important to accurately diagnose and identify α-syn patients for early treatment. The use of a PET radiotracer to image concentrations of insoluble α-syn deposits at treatment entry, during treatment, and in follow-up studies will be critical in α-syn clinical trials (242).
PET Imaging Agents for α–Synucleinopathies
PET radiotracers for imaging aggregated α-syn in LBs, LNs, and GCIs should meet the criteria listed in Table 1, and several important features of these targets should be noted. First, the amount of insoluble α-syn protein in LBD and MSA brain is 10-fold or more lower than the amount of Aβ in AD brain, and is in the range of 50–200 nM in brainstem and subcortical regions in advanced cases (243). Like NFTs, LBs are intraneuronal and GCIs are intraglial, and the tracer must readily pass through the BBB as well as the cell membrane to access the binding target. Like NFTs, post-translational modification of α-syn is prevalent (244). The most common post-translational modification of α-syn is phosphorylation, which occurs mainly at serine residues S129 and S87 and at tyrosine residues Y125, Y133 and Y135. In DLB brains, approximately 90% of insoluble α-syn is phosphorylated at S129 compared with only 4% in soluble cytosolic α-syn (245). The second most common post-translational modification is ubiquitination, which is the attachment of ubiquitin to α-syn at lysine residues (mainly K6, K10 and K12). Another common post-translational modification is nitration, which is the attachment of a nitro group to α-syn at tyrosine residues (mainly Y39, Y125, Y133 and Y136). While all of the post-translational modifications could affect radioligand-target binding interactions, those in the hydrophobic central region (residues 61–95) that form the amyloid beta-sheet structure likely would be most affected. Finally as described previously, α-syn is often co-localized with other amyloids, and high binding specificity of the radioligand for α-syn is critical for imaging assessments of regional brain α-syn pathology.
α-Syn PET Radioligands
BF227 (Table 5) was among the first PET radioligands evaluated as a potential α-syn imaging agent (246). BF227 was known to have high affinity for aggregated Aβ and low affinity for NFTs (247), and its in vitro binding properties with α-syn fibrils and human brain tissues from AD, DLB with Aβ, pure DLB (no Aβ), and age-matched control subjects were evaluated (246). 18F-BF227 bound with high affinity to synthetic α-syn fibrils and DLB (Aβ+), but resulted in no detectable specific binding to pure DLB (α-syn+ and Aβ−) and control brain (α-syn- and Aβ−) tissues (Table 5). Staining of α-syn-containing Lewy bodies within PD substantia nigra tissues with highly fluorescent BF227 at 100 μM demonstrated that BF227 bound to LBs in substantia nigra at high concentrations. Despite its non-selective binding with high affinity to Aβ and low affinity to α-syn in LBs, the authors suggested that BF227 warranted further evaluation as a PD diagnostic marker. To overcome potential interpretation issues with mixed LBD pathologies, the Tohoku University group evaluated BF227 in MSA (a relatively pure α-synucleinopathy) (248). They found that 100 μM BF227 stained GCIs in vitro in the pontine base of MSA, and also found that high specific activity 11C-BF227 accumulated in GCI-rich brain regions (subcortical white matter, putamen, posterior and anterior cingulate cortices, globus pallidus, primary motor cortex, and substantia nigra) in higher concentrations in MSA patients than in normal control subjects.
Table 5.
Structures and binding properties of three classes of α-syn radioligands.
| Compound | Ki or Kd (nM) α-syn fibrils | Ki or Kd (nM) Aβ fibrils | Ki or Kd (nM) tau fibrils | Ki or Kd (nM) α-syn positive human brain | |
|---|---|---|---|---|---|
|
| |||||
|
18F-BF227 (246) |
9.6 | 1.3 | - | Pure DLB (no Aβ) resulted in no binding detected | |
|
| |||||
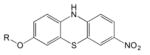 Phenothiazines (249–251) |
SIL5 R= 11CH3 |
66 | 110 | 136 | 83 (PD brain) |
| SIL26 R=CH2CH218F |
16 | 103 | 125 | 34 (PD brain) | |
|
| |||||
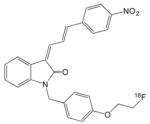 18F-WC58a (252) |
8.9 | 271 | 50 | Too lipophilic to assay with human brain tissues | |
The Washington University group reported a series of phenothiazine derivatives (Table 5) that bound with moderate affinities and selectivity to α-syn fibrils and PD brain (249–251). The binding affinities of SIL5 and SIL26 for α-syn in PD brain likely are not sufficiently strong to merit imaging studies in human LBD and MSA patients. More recently, the Washington University group examined a series of 3-(benzylidene)indolin-2-one derivatives (Table 5) of which 18F-WC-58a is a lead candidate (252). The affinity and selectivity of 18F-WC-58a for synthetic α-syn fibrils over Aβ and tau fibrils was promising, but the compound proved to be too lipophilic (logP=4.18) to conduct meaningful human PD brain tissue homogenate binding assays and cleared very slowly from non-human primate brain in vivo in microPET imaging studies (253).
Attainment of a high affinity, selective PET radioligand to image α-syn in vivo in human subjects remains elusive. Groups throughout the world continue to work in this research area, and accounts of the evaluation of potential lead compounds likely will result in useful α-syn PET imaging agents in the future.
Future Directions in PET Tracers for Imaging Proteinopathies
The development of selective PET radioligands for fibrillar Aβ has been realized with three FDA-approved agents – Amyvid, Neuraceq, and Vizamyl. Efforts in this area to develop Aβ agents more sensitive to lower levels of fibrillar Aβ found in diffuse plaques are ongoing (254). The successful development of PET radioligands to image Aβ oligomers in brain is hampered by their low concentration (<1 nM) and the relatively high concentration of fibrillar Aβ which complicate the development of a selective agent for oligomeric Aβ. In the tau area, the development of PET tracers selective for tau has recently advanced with several promising agents and other agents in the later stages of development. The binding properties of all of the tau radioligands for the different tau isoforms remain to be fully characterized, and it is important to sort out their abilities to image different 3R and 4R tauopathies. The development of selective and potent PET α-syn radiotracers requires new lead compounds for structural modification and subsequent evaluation, and work in this area is ongoing. A need exists to develop selective PET radioligands for other proteopathies not discussed in this chapter, such as TDP-43, prions, and huntingtin, and much work remains to achieve useful tracers for these aggregated protein targets.
Figure 14.
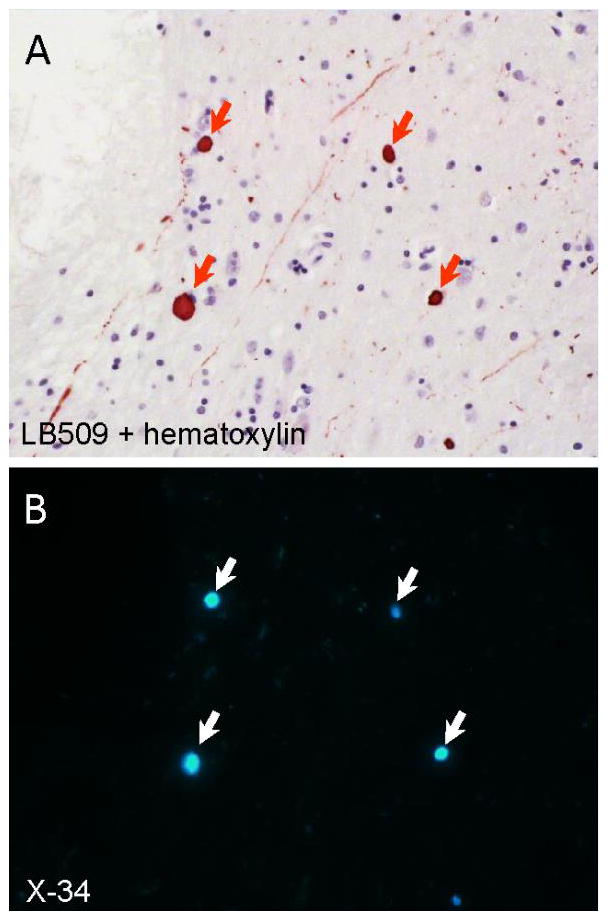
Co-localization of α-synuclein immunoreactivity and X-34 fluorescence in the amygdala from a case of dementia with Lewy bodies. A 10 μm thin paraffin section was first processed using anti-α-synuclein antibody LB509 immunohistochemistry with hematoxylin counterstain to visualize cell bodies (panel A). The section was cleared of chromogen using potassium permanganate, overstained with the pan-amyloid dye X-34 (100 μM), and re-imaged (panel B). Arrows point to Lewy bodies double-labeled with LB509 antibody and X-34.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ezkurdia I, Juan D, Rodriguez JM, et al. Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum Mol Genet. 2014;23(22):5866–78. doi: 10.1093/hmg/ddu309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar V, Sami N, Kashav T, et al. Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur J Med Chem. 2016;124:1105–1120. doi: 10.1016/j.ejmech.2016.07.054. [DOI] [PubMed] [Google Scholar]
- 3.Stroo E, Koopman M, Nollen EA, et al. Cellular Regulation of Amyloid Formation in Aging and Disease. Front Neurosci. 2017;11:64. doi: 10.3389/fnins.2017.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong YC, Krainc D. alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourdenx M, Koulakiotis NS, Sanoudou D, et al. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol. 2015 doi: 10.1016/j.pneurobio.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Saa P, Harris DA, Cervenakova L. Mechanisms of prion-induced neurodegeneration. Expert Rev Mol Med. 2016;18:e5. doi: 10.1017/erm.2016.8. [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–23. doi: 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Will RG, Ironside JW. Sporadic and Infectious Human Prion Diseases. Cold Spring Harb Perspect Med. 2017;7(1) doi: 10.1101/cshperspect.a024364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641–54. doi: 10.1016/S0140-6736(15)01274-X. [DOI] [PubMed] [Google Scholar]
- 10.Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016;23(4):209–213. doi: 10.1080/13506129.2016.1257986. [DOI] [PubMed] [Google Scholar]
- 11.Walker LC, LeVine H., 3rd Corruption and spread of pathogenic proteins in neurodegenerative diseases. J Biol Chem. 2012;287(40):33109–15. doi: 10.1074/jbc.R112.399378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patterson BW, Elbert DL, Mawuenyega KG, et al. Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann Neurol. 2015;78(3):439–53. doi: 10.1002/ana.24454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013;126(4):461–77. doi: 10.1007/s00401-013-1182-x. [DOI] [PubMed] [Google Scholar]
- 14.Krebs MR, Bromley EH, Donald AM. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J Struct Biol. 2005;149(1):30–7. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Pulawski W, Ghoshdastider U, Andrisano V, et al. Ubiquitous amyloids. Appl Biochem Biotechnol. 2012;166(7):1626–43. doi: 10.1007/s12010-012-9549-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schleiden MJ, Leipzig FABV. Grundzüge der wissenschaftlichen Botanik: nebst einer methodologischen Einleitung als Anleitung zum Studium der Pflanze. T. Leipzig: Verlag von Wilhelm Engelmann; 1842. pp. 1–2. [Google Scholar]
- 17.Cohen AS. General introduction and a brief history of the amyloid fibril. In: Marrink J, Van Rijswijk MH, editors. Amyloidosis. Nijhoff: Dordrecht; 1986. pp. 3–19. [Google Scholar]
- 18.Rohn TT. Corpora Amylacea in Neurodegenerative Diseases: Cause or Effect? Int J Neurol Neurother. 2015;2(3) doi: 10.23937/2378-3001/2/2/1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedreich N, Kekulé A. Zur Amyloidfrage. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin. 1859;16(1):50–65. [Google Scholar]
- 20.Makin OS, Serpell LC. Structures for amyloid fibrils. FEBS J. 2005;272(23):5950–61. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 21.Alzheimer A, Stelzmann RA, Schnitzlein HN, et al. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8(6):429–31. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 22.Tycko R. Alzheimer’s disease: Structure of aggregates revealed. Nature. 2016;537(7621):492–493. doi: 10.1038/nature19470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daebel V, Chinnathambi S, Biernat J, et al. beta-Sheet core of tau paired helical filaments revealed by solid-state NMR. J Am Chem Soc. 2012;134(34):13982–9. doi: 10.1021/ja305470p. [DOI] [PubMed] [Google Scholar]
- 24.Peterson DW, Zhou H, Dahlquist FW, et al. A soluble oligomer of tau associated with fiber formation analyzed by NMR. Biochemistry. 2008;47(28):7393–404. doi: 10.1021/bi702466a. [DOI] [PubMed] [Google Scholar]
- 25.Eisenberg DS, Sawaya MR. Implications for Alzheimer’s disease of an atomic resolution structure of amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2016;113(34):9398–400. doi: 10.1073/pnas.1610806113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez JA, Ivanova MI, Sawaya MR, et al. Structure of the toxic core of alpha-synuclein from invisible crystals. Nature. 2015;525(7570):486–90. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feligioni M, Marcelli S, Iannuzzi F, et al. The involvement of post-translational modifications in Alzheimer’s disease. Curr Alzheimer Res. 2017 doi: 10.2174/1567205014666170505095109. [DOI] [PubMed] [Google Scholar]
- 28.Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665–704. doi: 10.1007/s00401-017-1707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vicente Miranda H, Szego EM, Oliveira LM, et al. Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain. 2017 doi: 10.1093/brain/awx056. [DOI] [PubMed] [Google Scholar]
- 30.Scherpelz KP, Lu JX, Tycko R, et al. Preparation of Amyloid Fibrils Seeded from Brain and Meninges. Methods Mol Biol. 2016;1345:299–312. doi: 10.1007/978-1-4939-2978-8_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peelaerts W, Bousset L, Van der Perren A, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522(7556):340–4. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 32.Duyckaerts C. Neurodegenerative lesions: seeding and spreading. Rev Neurol (Paris) 2013;169(10):825–33. doi: 10.1016/j.neurol.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 33.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70(4):532–40. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao Y, Ma B, McElheny D, et al. Abeta(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol. 2015;22(6):499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuttle MD, Comellas G, Nieuwkoop AJ, et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23(5):409–15. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laruelle M, Slifstein M, Huang Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol Imaging Biol. 2003;5(6):363–75. doi: 10.1016/j.mibio.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Pike VW. Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Curr Med Chem. 2016;23(18):1818–69. doi: 10.2174/0929867323666160418114826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennhold H. Specific staining of amyloid by Congo red. Muenchen Med Wochenschr. 1922;69:1537–8. [Google Scholar]
- 39.Divry P. Etude histochimique des plaques seniles. J Belge Neurol Psychiat. 1927;27:643–657. [Google Scholar]
- 40.Vassar PS, Culling CF. Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol. 1959;68:487–98. [PubMed] [Google Scholar]
- 41.Elghetany MT, Saleem A. Methods for staining amyloid in tissues: a review. Stain Technol. 1988;63(4):201–12. doi: 10.3109/10520298809107185. [DOI] [PubMed] [Google Scholar]
- 42.Shoghi-Jadid K, Small GW, Agdeppa ED, et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10(1):24–35. [PubMed] [Google Scholar]
- 43.Shin J, Kepe V, Barrio JR, et al. The merits of FDDNP-PET imaging in Alzheimer’s disease. J Alzheimers Dis. 2011;26(Suppl 3):135–45. doi: 10.3233/JAD-2011-0008. [DOI] [PubMed] [Google Scholar]
- 44.Kepe V, Ghetti B, Farlow MR, et al. PET of brain prion protein amyloid in Gerstmann-Straussler-Scheinker disease. Brain Pathol. 2010;20(2):419–30. doi: 10.1111/j.1750-3639.2009.00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrio JR, Small GW, Wong KP, et al. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci U S A. 2015;112(16):E2039–47. doi: 10.1073/pnas.1409952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kepe V, Bordelon Y, Boxer A, et al. PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J Alzheimers Dis. 2013;36(1):145–53. doi: 10.3233/JAD-130032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Styren SD, Hamilton RL, Styren GC, et al. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem. 2000;48(9):1223–32. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 48.Link CD, Johnson CJ, Fonte V, et al. Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol Aging. 2001;22(2):217–26. doi: 10.1016/s0197-4580(00)00237-2. [DOI] [PubMed] [Google Scholar]
- 49.Ikonomovic MD, Abrahamson EE, Isanski BA, et al. X-34 labeling of abnormal protein aggregates during the progression of Alzheimer’s disease. Methods Enzymol. 2006;412:123–44. doi: 10.1016/S0076-6879(06)12009-1. [DOI] [PubMed] [Google Scholar]
- 50.Klunk WE, Bacskai BJ, Mathis CA, et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61(9):797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- 51.Sadowski M, Pankiewicz J, Scholtzova H, et al. Targeting prion amyloid deposits in vivo. J Neuropathol Exp Neurol. 2004;63(7):775–84. doi: 10.1093/jnen/63.7.775. [DOI] [PubMed] [Google Scholar]
- 52.Brendza RP, Bacskai BJ, Cirrito JR, et al. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115(2):428–33. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, et al. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J Neurosci. 2008;28(52):14156–64. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burgold S, Bittner T, Dorostkar MM, et al. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 2011;121(3):327–35. doi: 10.1007/s00401-010-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bertoncini CW, Celej MS. Small molecule fluorescent probes for the detection of amyloid self-assembly in vitro and in vivo. Curr Protein Pept Sci. 2011;12(3):205–20. doi: 10.2174/138920311795860151. [DOI] [PubMed] [Google Scholar]
- 56.Mathis CA, Wang Y, Holt DP, et al. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46(13):2740–54. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 57.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 58.Klunk WE, Mathis CA. Whatever happened to Pittsburgh Compound-A? Alzheimer Dis Assoc Disord. 2008;22(3):198–203. doi: 10.1097/WAD.0b013e318188c0c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mathis CA, Wang Y, Klunk WE. Imaging beta-amyloid plaques and neurofibrillary tangles in the aging human brain. Curr Pharm Des. 2004;10(13):1469–92. doi: 10.2174/1381612043384772. [DOI] [PubMed] [Google Scholar]
- 60.Mathis CA, Mason NS, Lopresti BJ, et al. Development of positron emission tomography beta-amyloid plaque imaging agents. Semin Nucl Med. 2012;42(6):423–32. doi: 10.1053/j.semnuclmed.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathis CA, Bacskai BJ, Kajdasz ST, et al. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett. 2002;12(3):295–8. doi: 10.1016/s0960-894x(01)00734-x. [DOI] [PubMed] [Google Scholar]
- 62.Klunk WE, Wang Y, Huang GF, et al. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001;69(13):1471–84. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- 63.Klunk WE, Wang Y, Huang GF, et al. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23(6):2086–92. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt 6):1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lockhart A, Lamb JR, Osredkar T, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130(Pt 10):2607–15. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 66.Fodero-Tavoletti MT, Smith DP, McLean CA, et al. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J Neurosci. 2007;27(39):10365–71. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye L, Velasco A, Fraser G, et al. In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J Neurochem. 2008;105(4):1428–37. doi: 10.1111/j.1471-4159.2008.05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klunk WE, Lopresti BJ, Ikonomovic MD, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25(46):10598–606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25(11):1528–47. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 70.Mathis CA, Holt D, Wang YM, et al. Species-dependent formation and identification of the brain metabolites of the amyloid imaging agent [C-11]PiB. Neurobiol Aging. 2004;25(Suppl 2):S277–S278. [Google Scholar]
- 71.Cole GB, Keum G, Liu J, et al. Specific estrogen sulfotransferase (SULT1E1) substrates and molecular imaging probe candidates. Proc Natl Acad Sci U S A. 2010;107(14):6222–7. doi: 10.1073/pnas.0914904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McNamee RL, Yee SH, Price JC, et al. Consideration of optimal time window for Pittsburgh compound B PET summed uptake measurements. J Nucl Med. 2009;50(3):348–55. doi: 10.2967/jnumed.108.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lopresti BJ, Klunk WE, Mathis CA, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46(12):1959–72. [PubMed] [Google Scholar]
- 74.Tolboom N, Yaqub M, Boellaard R, et al. Test-retest variability of quantitative [11C]PIB studies in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36(10):1629–38. doi: 10.1007/s00259-009-1129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Berckel BN, Ossenkoppele R, Tolboom N, et al. Longitudinal amyloid imaging using 11C-PiB: methodologic considerations. J Nucl Med. 2013;54(9):1570–6. doi: 10.2967/jnumed.112.113654. [DOI] [PubMed] [Google Scholar]
- 76.Yaqub M, Tolboom N, Boellaard R, et al. Simplified parametric methods for [11C]PIB studies. Neuroimage. 2008;42(1):76–86. doi: 10.1016/j.neuroimage.2008.04.251. [DOI] [PubMed] [Google Scholar]
- 77.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65(11):1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cohen AD, Mowrey W, Weissfeld LA, et al. Classification of amyloid-positivity in controls: comparison of visual read and quantitative approaches. Neuroimage. 2013;71:207–15. doi: 10.1016/j.neuroimage.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Knopman DS, Jack CR, Jr, Lundt ES, et al. Evolution of neurodegeneration-imaging biomarkers from clinically normal to dementia in the Alzheimer disease spectrum. Neurobiol Aging. 2016;46:32–42. doi: 10.1016/j.neurobiolaging.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lao PJ, Betthauser TJ, Hillmer AT, et al. The effects of normal aging on amyloid-beta deposition in nondemented adults with Down syndrome as imaged by carbon 11-labeled Pittsburgh compound B. Alzheimers Dement. 2016;12(4):380–90. doi: 10.1016/j.jalz.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lim HK, Nebes R, Snitz B, et al. Regional amyloid burden and intrinsic connectivity networks in cognitively normal elderly subjects. Brain. 2014;137(Pt 12):3327–38. doi: 10.1093/brain/awu271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mathis CA, Kuller LH, Klunk WE, et al. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol. 2013;73(6):751–61. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 84.Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130(Pt 11):2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 85.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68(20):1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 86.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–92. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924–38. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chetelat G, La Joie R, Villain N, et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. Neuroimage Clin. 2013;2:356–65. doi: 10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN-TU trial. Rev Neurol (Paris) 2013;169(10):737–43. doi: 10.1016/j.neurol.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hsu D, Marshall GA. Primary and Secondary Prevention Trials in Alzheimer Disease: Looking Back, Moving Forward. Curr Alzheimer Res. 2017;14(4):426–440. doi: 10.2174/1567205013666160930112125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26(Suppl 3):321–9. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305(3):275–83. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wolk DA, I, Grachev D, Buckley C, et al. Association between in vivo fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging and in vivo cerebral cortical histopathology. Arch Neurol. 2011;68(11):1398–403. doi: 10.1001/archneurol.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rinne JO, Wong DF, Wolk DA, et al. [(18)F]Flutemetamol PET imaging and cortical biopsy histopathology for fibrillar amyloid beta detection in living subjects with normal pressure hydrocephalus: pooled analysis of four studies. Acta Neuropathol. 2012;124(6):833–45. doi: 10.1007/s00401-012-1051-z. [DOI] [PubMed] [Google Scholar]
- 96.Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 2015;11(8):964–74. doi: 10.1016/j.jalz.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 97.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–72. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 98.Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–6. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 99.Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016;12(2):110–20. doi: 10.1016/j.jalz.2015.06.1893. [DOI] [PubMed] [Google Scholar]
- 100.Panza F, Solfrizzi V, Imbimbo BP, et al. Efficacy and safety studies of gantenerumab in patients with Alzheimer’s disease. Expert Rev Neurother. 2014;14(9):973–86. doi: 10.1586/14737175.2014.945522. [DOI] [PubMed] [Google Scholar]
- 101.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. J Nucl Med. 2013;54(3):476–90. doi: 10.2967/jnumed.113.120618. [DOI] [PubMed] [Google Scholar]
- 102.Johnson KA, Minoshima S, Bohnen NI, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. J Nucl Med. 2013;54(7):1011–3. doi: 10.2967/jnumed.113.127068. [DOI] [PubMed] [Google Scholar]
- 103.Arbizu J, Garcia-Ribas G, Carrio I, et al. Recommendations for the use of PET imaging biomarkers in the diagnosis of neurodegenerative conditions associated with dementia: SEMNIM and SEN consensus. Rev Esp Med Nucl Imagen Mol. 2015;34(5):303–13. doi: 10.1016/j.remn.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 104.Grundman M, Johnson KA, Lu M, et al. Effect of Amyloid Imaging on the Diagnosis and Management of Patients with Cognitive Decline: Impact of Appropriate Use Criteria. Dement Geriatr Cogn Disord. 2016;41(1–2):80–92. doi: 10.1159/000441139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Laforce R, Rosa-Neto P, Soucy JP, et al. Canadian Consensus Guidelines on Use of Amyloid Imaging in Canada: Update and Future Directions from the Specialized Task Force on Amyloid imaging in Canada. Can J Neurol Sci. 2016;43(4):503–12. doi: 10.1017/cjn.2015.401. [DOI] [PubMed] [Google Scholar]
- 106.Minoshima S, Drzezga AE, Barthel H, et al. SNMMI Procedure Standard/EANM Practice Guideline for Amyloid PET Imaging of the Brain 1.0. J Nucl Med. 2016;57(8):1316–22. doi: 10.2967/jnumed.116.174615. [DOI] [PubMed] [Google Scholar]
- 107.Bielschowsky M, Brodmann K. Zur feineren histologie und histopathologie der grosshirnrinde. J Psychol Neurol. 1905;5:173–199. [Google Scholar]
- 108.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–3. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 109.Wischik CM, Novak M, Thogersen HC, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85(12):4506–10. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goedert M, Wischik CM, Crowther RA, et al. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85(11):4051–5. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Terry RD. The Fine Structure of Neurofibrillary Tangles in Alzheimer’s Disease. J Neuropathol Exp Neurol. 1963;22:629–42. doi: 10.1097/00005072-196310000-00005. [DOI] [PubMed] [Google Scholar]
- 112.Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev. 1995;75(4):835–64. doi: 10.1152/physrev.1995.75.4.835. [DOI] [PubMed] [Google Scholar]
- 113.Mandelkow E, Mandelkow EM. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7(1):72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 114.Arriagada PV, Growdon JH, Hedley-Whyte ET, et al. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–9. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 115.Bancher C, Jellinger K, Lassmann H, et al. Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur Arch Psychiatry Clin Neurosci. 1996;246(3):137–46. doi: 10.1007/BF02189115. [DOI] [PubMed] [Google Scholar]
- 116.Berg L, McKeel DW, Jr, et al. Clinicopathologic studies in cognitively healthy aging and alzheimer disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein e genotype. Archives of Neurology. 1998;55(3):326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 117.Bierer LM, Hof PR, Purohit DP, et al. Neocortical neurofibrillary tangles correlate with dementia severity in alzheimer's disease. Archives of Neurology. 1995;52(1):81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 118.Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 119.Nagy Z, Esiri MM, Jobst KA, et al. Relative roles of plaques and tangles in the dementia of Alzheimer’s disease: correlations using three sets of neuropathological criteria. Dementia. 1995;6(1):21–31. doi: 10.1159/000106918. [DOI] [PubMed] [Google Scholar]
- 120.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. Journal of Neuropathology & Experimental Neurology. 2012;71(5):362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xia C, Makaretz SJ, Caso C, et al. Association of In Vivo [18F]AV-1451 Tau PET Imaging Results With Cortical Atrophy and Symptoms in Typical and Atypical Alzheimer Disease. JAMA Neurol. 2017;74(4):427–436. doi: 10.1001/jamaneurol.2016.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016;126(Pt 3):238–292. doi: 10.1016/j.brainresbull.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 123.Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21(10):428–33. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- 124.Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–26. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 125.Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739(2–3):179–97. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 126.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 127.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9(13):4225–30. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31(43):10626–33. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 129.Jovanov-Milosevic N, Petrovic D, Sedmak G, et al. Human fetal tau protein isoform: possibilities for Alzheimer’s disease treatment. Int J Biochem Cell Biol. 2012;44(8):1290–4. doi: 10.1016/j.biocel.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fontaine SN, Sabbagh JJ, Baker J, et al. Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci. 2015;72(10):1863–79. doi: 10.1007/s00018-015-1839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lippens G, Landrieu I, Smet C, et al. NMR Meets Tau: Insights into Its Function and Pathology. Biomolecules. 2016;6(2) doi: 10.3390/biom6020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Simic G, Babic Leko M, Wray S, et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules. 2016;6(1):6. doi: 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bramblett GT, Goedert M, Jakes R, et al. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10(6):1089–99. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 134.Goedert M, Jakes R, Crowther RA, et al. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci U S A. 1993;90(11):5066–70. doi: 10.1073/pnas.90.11.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Köpke E, Tung YC, Shaikh S, et al. Microtubule-associated protein tau: Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. Journal of Biological Chemistry. 1993;268(32):24374–24384. [PubMed] [Google Scholar]
- 136.Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kuret J, Chirita CN, Congdon EE, et al. Pathways of tau fibrillization. Biochim Biophys Acta. 2005;1739(2–3):167–78. doi: 10.1016/j.bbadis.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 138.Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88(6):2288–92. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Goedert M, Jakes R, Spillantini MG, et al. Tau protein in Alzheimer’s disease. Biochem Soc Trans. 1995;23(1):80–5. doi: 10.1042/bst0230080. [DOI] [PubMed] [Google Scholar]
- 140.Iqbal K, del Alonso AC, Chen S, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739(2–3):198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 141.Goedert M, Spillantini MG, Cairns NJ, et al. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8(1):159–68. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 142.Delacourte A, Sergeant N, Wattez A, et al. Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann Neurol. 1998;43(2):193–204. doi: 10.1002/ana.410430209. [DOI] [PubMed] [Google Scholar]
- 143.Arai T, Ikeda K, Akiyama H, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 2001;101(2):167–73. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- 144.Umeda Y, Taniguchi S, Arima K, et al. Alterations in human tau transcripts correlate with those of neurofilament in sporadic tauopathies. Neurosci Lett. 2004;359(3):151–4. doi: 10.1016/j.neulet.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 145.Yoshida M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology. 2006;26(5):457–70. doi: 10.1111/j.1440-1789.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- 146.Chambers CB, Lee JM, Troncoso JC, et al. Overexpression of four-repeat tau mRNA isoforms in progressive supranuclear palsy but not in Alzheimer’s disease. Ann Neurol. 1999;46(3):325–32. doi: 10.1002/1531-8249(199909)46:3<325::aid-ana8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 147.Perry G, Mulvihill P, Manetto V, et al. Immunocytochemical properties of Alzheimer straight filaments. J Neurosci. 1987;7(11):3736–8. doi: 10.1523/JNEUROSCI.07-11-03736.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kovacs GG. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int J Mol Sci. 2016;17(2) doi: 10.3390/ijms17020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ward SM, Himmelstein DS, Lancia JK, et al. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. 2012;40(4):667–71. doi: 10.1042/BST20120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gerson JE, Castillo-Carranza DL, Kayed R. Advances in therapeutics for neurodegenerative tauopathies: moving toward the specific targeting of the most toxic tau species. ACS Chem Neurosci. 2014;5(9):752–69. doi: 10.1021/cn500143n. [DOI] [PubMed] [Google Scholar]
- 151.Shafiei SS, Guerrero-Muñoz MJ, Castillo-Carranza DL. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Frontiers in Aging Neuroscience. 2017;9(83) doi: 10.3389/fnagi.2017.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271–8. doi: 10.1016/0197-4580(95)00021-6. discussion 278–84. [DOI] [PubMed] [Google Scholar]
- 153.Liu L, Drouet V, Wu JW, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Holmes BB, Diamond MI. Cellular mechanisms of protein aggregate propagation. Curr Opin Neurol. 2012;25(6):721–6. doi: 10.1097/WCO.0b013e32835a3ee0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pooler AM, Polydoro M, Wegmann S, et al. Propagation of tau pathology in Alzheimer’s disease: identification of novel therapeutic targets. Alzheimers Res Ther. 2013;5(5):49. doi: 10.1186/alzrt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755–66. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Olney NT, Spina S, Miller BL. Frontotemporal Dementia. Neurol Clin. 2017;35(2):339–374. doi: 10.1016/j.ncl.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mann DMA, Snowden JS. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017 doi: 10.1111/bpa.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dickson DW, Kouri N, Murray ME, et al. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau) J Mol Neurosci. 2011;45(3):384–9. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 161.Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J Neurol. 2006;253(2):139–58. doi: 10.1007/s00415-005-0019-5. [DOI] [PubMed] [Google Scholar]
- 162.Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–70. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Finckh U, Alberici A, Antoniazzi M, et al. Variable expression of familial Alzheimer disease associated with presenilin 2 mutation M239I. Neurology. 2000;54(10):2006–8. doi: 10.1212/wnl.54.10.2006. [DOI] [PubMed] [Google Scholar]
- 164.Bird TD, Lampe TH, Nemens EJ, et al. Familial Alzheimer’s disease in American descendants of the Volga Germans: probable genetic founder effect. Ann Neurol. 1988;23(1):25–31. doi: 10.1002/ana.410230106. [DOI] [PubMed] [Google Scholar]
- 165.Jack CR, Jr, Wiste HJ, Lesnick TG, et al. Brain beta-amyloid load approaches a plateau. Neurology. 2013;80(10):890–6. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 167.Jack CR, Jr, Wiste HJ, Weigand SD, et al. Age-specific population frequencies of cerebral beta-amyloidosis and neurodegeneration among people with normal cognitive function aged 50–89 years: a cross-sectional study. Lancet Neurol. 2014;13(10):997–1005. doi: 10.1016/S1474-4422(14)70194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17(2):101–18. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 169.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 170.Mufson EJ, Malek-Ahmadi M, Perez SE, et al. Braak staging, plaque pathology, and APOE status in elderly persons without cognitive impairment. Neurobiol Aging. 2016;37:147–53. doi: 10.1016/j.neurobiolaging.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ghetti B, Oblak AL, Boeve BF, et al. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41(1):24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ludolph AC, Kassubek J, Landwehrmeyer BG, et al. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur J Neurol. 2009;16(3):297–309. doi: 10.1111/j.1468-1331.2008.02513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Insel PS, Mattsson N, Mackin RS, et al. Accelerating rates of cognitive decline and imaging markers associated with beta-amyloid pathology. Neurology. 2016;86(20):1887–96. doi: 10.1212/WNL.0000000000002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jack CR, Jr, Therneau TM, Wiste HJ, et al. Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. Lancet Neurol. 2016;15(1):56–64. doi: 10.1016/S1474-4422(15)00323-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jack CR, Jr, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80(6):1347–58. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tosun D, Landau S, Aisen PS, et al. Association between tau deposition and antecedent amyloid-beta accumulation rates in normal and early symptomatic individuals. Brain. 2017 doi: 10.1093/brain/awx046. [DOI] [PubMed] [Google Scholar]
- 177.Shimada H, Kitamura S, Shinotoh H, et al. Association between Abeta and tau accumulations and their influence on clinical features in aging and Alzheimer’s disease spectrum brains: A [11C]PBB3-PET study. Alzheimers Dement (Amst) 2017;6:11–20. doi: 10.1016/j.dadm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jack CR, Jr, Wiste HJ, Weigand SD, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology. 2013;81(20):1732–40. doi: 10.1212/01.wnl.0000435556.21319.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine. 2016;1(6):53–57. doi: 10.29245/2572.942x/2016/6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Conrad C, Andreadis A, Trojanowski JQ, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997;41(2):277–81. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- 181.Di Maria E, Tabaton M, Vigo T, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47(3):374–7. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 182.Houlden H, Baker M, Morris HR, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56(12):1702–6. doi: 10.1212/wnl.56.12.1702. [DOI] [PubMed] [Google Scholar]
- 183.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013;9(6):666–76. doi: 10.1016/j.jalz.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 184.Villemagne VL, Dore V, Bourgeat P, et al. Abeta-amyloid and Tau Imaging in Dementia. Semin Nucl Med. 2017;47(1):75–88. doi: 10.1053/j.semnuclmed.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 185.Hall B, Mak E, Cervenka S, et al. In vivo tau PET imaging in dementia: Pathophysiology, radiotracer quantification, and a systematic review of clinical findings. Ageing Res Rev. 2017;36:50–63. doi: 10.1016/j.arr.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 186.Agdeppa ED, Kepe V, Liu J, et al. 2-Dialkylamino-6-acylmalononitrile substituted naphthalenes (DDNP analogs): novel diagnostic and therapeutic tools in Alzheimer’s disease. Mol Imaging Biol. 2003;5(6):404–17. doi: 10.1016/j.mibio.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 187.Smid LM, Kepe V, Vinters HV, et al. Postmortem 3-D brain hemisphere cortical tau and amyloid-beta pathology mapping and quantification as a validation method of neuropathology imaging. J Alzheimers Dis. 2013;36(2):261–74. doi: 10.3233/JAD-122434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Okamura N, Suemoto T, Furumoto S, et al. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J Neurosci. 2005;25(47):10857–62. doi: 10.1523/JNEUROSCI.1738-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fodero-Tavoletti MT, Okamura N, Furumoto S, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain. 2011;134(Pt 4):1089–100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 190.Villemagne VL, Furumoto S, Fodero-Tavoletti MT, et al. In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2014;41(5):816–26. doi: 10.1007/s00259-013-2681-7. [DOI] [PubMed] [Google Scholar]
- 191.Lemoine L, Saint-Aubert L, Marutle A, et al. Visualization of regional tau deposits using (3)H-THK5117 in Alzheimer brain tissue. Acta Neuropathol Commun. 2015;3:40. doi: 10.1186/s40478-015-0220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Okamura N, Furumoto S, Harada R, et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med. 2013;54(8):1420–7. doi: 10.2967/jnumed.112.117341. [DOI] [PubMed] [Google Scholar]
- 193.Harada R, Okamura N, Furumoto S, et al. [(18)F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2015;42(7):1052–61. doi: 10.1007/s00259-015-3035-4. [DOI] [PubMed] [Google Scholar]
- 194.Okamura N, Furumoto S, Fodero-Tavoletti MT, et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain. 2014;137(Pt 6):1762–71. doi: 10.1093/brain/awu064. [DOI] [PubMed] [Google Scholar]
- 195.Tago T, Furumoto S, Okamura N, et al. Structure-Activity Relationship of 2- Arylquinolines as PET Imaging Tracers for Tau Pathology in Alzheimer Disease. J Nucl Med. 2016;57(4):608–14. doi: 10.2967/jnumed.115.166652. [DOI] [PubMed] [Google Scholar]
- 196.Harada R, Okamura N, Furumoto S, et al. 18F-THK5351: A Novel PET Radiotracer for Imaging Neurofibrillary Pathology in Alzheimer Disease. J Nucl Med. 2016;57(2):208–14. doi: 10.2967/jnumed.115.164848. [DOI] [PubMed] [Google Scholar]
- 197.Ng KP, Pascoal TA, Mathotaarachchi S, et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res Ther. 2017;9(1):25. doi: 10.1186/s13195-017-0253-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fowler JS, Volkow ND, Wang GJ, et al. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging. 1997;18(4):431–5. doi: 10.1016/s0197-4580(97)00037-7. [DOI] [PubMed] [Google Scholar]
- 199.Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun. 2016;4(1):58. doi: 10.1186/s40478-016-0315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78(5):787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34(2):457–68. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 202.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110–9. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain. 2016;139(Pt 5):1539–50. doi: 10.1093/brain/aww023. [DOI] [PubMed] [Google Scholar]
- 204.Scholl M, Lockhart SN, Schonhaut DR, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89(5):971–82. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol. 2016;80(2):247–58. doi: 10.1002/ana.24711. [DOI] [PubMed] [Google Scholar]
- 206.Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain. 2016;139(Pt 5):1551–67. doi: 10.1093/brain/aww027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ikonomovic MD, Abrahamson EE, Price JC, et al. [F-18]AV-1451 positron emission tomography retention in choroid plexus: More than “off-target” binding. Ann Neurol. 2016;80(2):307–8. doi: 10.1002/ana.24706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79(6):1094–108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hashimoto H, Kawamura K, Takei M, et al. Identification of a major radiometabolite of [11C]PBB3. Nucl Med Biol. 2015;42(12):905–10. doi: 10.1016/j.nucmedbio.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 210.Hashimoto H, Kawamura K, Igarashi N, et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med. 2014;55(9):1532–8. doi: 10.2967/jnumed.114.139550. [DOI] [PubMed] [Google Scholar]
- 211.Marquie M, Normandin MD, Meltzer AC, et al. Pathological correlations of [F-18]-AV-1451 imaging in non-alzheimer tauopathies. Ann Neurol. 2017;81(1):117–128. doi: 10.1002/ana.24844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sander K, Lashley T, Gami P, et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement. 2016;12(11):1116–1124. doi: 10.1016/j.jalz.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 213.Spina S, Schonhaut DR, Boeve BF, et al. Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology. 2017;88(8):758–766. doi: 10.1212/WNL.0000000000003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Fodero-Tavoletti MT, Furumoto S, Taylor L, et al. Assessing THK523 selectivity for tau deposits in Alzheimer’s disease and non-Alzheimer’s disease tauopathies. Alzheimers Res Ther. 2014;6(1):11. doi: 10.1186/alzrt240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ono M, Sahara N, Kumata K, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. 2017;140(3):764–780. doi: 10.1093/brain/aww339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cho H, Choi JY, Hwang MS, et al. Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord. 2017;32(1):134–140. doi: 10.1002/mds.26844. [DOI] [PubMed] [Google Scholar]
- 217.Coakeley S, Cho SS, Koshimori Y, et al. Positron emission tomography imaging of tau pathology in progressive supranuclear palsy. J Cereb Blood Flow Metab. 2016:271678x16683695. doi: 10.1177/0271678X16683695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Smith R, Schain M, Nilsson C, et al. Increased basal ganglia binding of 18 F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord. 2017;32(1):108–114. doi: 10.1002/mds.26813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Whitwell JL, V, Lowe J, Tosakulwong N, et al. [18 F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord. 2017;32(1):124–133. doi: 10.1002/mds.26834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ishiki A, Harada R, Okamura N, et al. Tau imaging with [18 F]THK-5351 in progressive supranuclear palsy. Eur J Neurol. 2017;24(1):130–136. doi: 10.1111/ene.13164. [DOI] [PubMed] [Google Scholar]
- 221.Kikuchi A, Okamura N, Hasegawa T, et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology. 2016;87(22):2309–2316. doi: 10.1212/WNL.0000000000003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Passamonti L, Vazquez Rodriguez P, Hong YT, et al. 18F-AV-1451 positron emission tomography in Alzheimer’s disease and progressive supranuclear palsy. Brain. 2017;140(3):781–791. doi: 10.1093/brain/aww340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther. 2014;6(5):73. doi: 10.1186/s13195-014-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ueda K, Fukushima H, Masliah E, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(23):11282–6. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Giasson BI, I, Murray V, Trojanowski JQ, et al. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276(4):2380–6. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 227.Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 228.Spillantini MG, Crowther RA, Jakes R, et al. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95(11):6469–73. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Villar-Pique A, Lopes da Fonseca T, Outeiro TF. Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J Neurochem. 2016;139(Suppl 1):240–255. doi: 10.1111/jnc.13249. [DOI] [PubMed] [Google Scholar]
- 230.Spires-Jones TL, Attems J, Thal DR. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017 doi: 10.1007/s00401-017-1709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Spillantini MG. Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy are alpha-synucleinopathies. Parkinsonism Relat Disord. 1999;5(4):157–62. doi: 10.1016/s1353-8020(99)00031-0. [DOI] [PubMed] [Google Scholar]
- 232.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–24. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 233.Uchikado H, Lin WL, DeLucia MW, et al. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65(7):685–97. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16(1):55–65. doi: 10.1016/S1474-4422(16)30291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Jellinger KA, Attems J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006;112(3):253–60. doi: 10.1007/s00401-006-0088-2. [DOI] [PubMed] [Google Scholar]
- 236.Ottolini D, Cali T, Szabo I, et al. Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications. Biol Chem. 2017;398(1):77–100. doi: 10.1515/hsz-2016-0201. [DOI] [PubMed] [Google Scholar]
- 237.Dehay B, Bourdenx M, Gorry P, et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14(8):855–66. doi: 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–7. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 239.Hawkes CH, Del Tredici K, Braak H. A timeline for Parkinson’s disease. Parkinsonism Relat Disord. 2010;16(2):79–84. doi: 10.1016/j.parkreldis.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 240.Lawand NB, Saade NE, El-Agnaf OM, et al. Targeting alpha-synuclein as a therapeutic strategy for Parkinson’s disease. Expert Opin Ther Targets. 2015;19(10):1351–60. doi: 10.1517/14728222.2015.1062877. [DOI] [PubMed] [Google Scholar]
- 241.Schenk DB, Koller M, Ness DK, et al. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32(2):211–218. doi: 10.1002/mds.26878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Eberling JL, Dave KD, Frasier MA. alpha-synuclein imaging: a critical need for Parkinson’s disease research. J Parkinsons Dis. 2013;3(4):565–7. doi: 10.3233/JPD-130247. [DOI] [PubMed] [Google Scholar]
- 243.Deramecourt V, Bombois S, Maurage CA, et al. Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2006;65(3):278–88. doi: 10.1097/01.jnen.0000205145.54457.ea. [DOI] [PubMed] [Google Scholar]
- 244.Barrett PJ, Timothy Greenamyre J. Post-translational modification of alpha-synuclein in Parkinson’s disease. Brain Res. 2015;1628(Pt B):247–53. doi: 10.1016/j.brainres.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 245.Anderson JP, Walker DE, Goldstein JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739–52. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 246.Fodero-Tavoletti MT, Mulligan RS, Okamura N, et al. In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur J Pharmacol. 2009;617(1–3):54–8. doi: 10.1016/j.ejphar.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 247.Kudo Y, Okamura N, Furumoto S, et al. 2-(2-[2-Dimethylaminothiazol-5-yl]ethenyl)-6- (2-[fluoro]ethoxy)benzoxazole: a novel PET agent for in vivo detection of dense amyloid plaques in Alzheimer’s disease patients. J Nucl Med. 2007;48(4):553–61. doi: 10.2967/jnumed.106.037556. [DOI] [PubMed] [Google Scholar]
- 248.Kikuchi A, Takeda A, Okamura N, et al. In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain. 2010;133(Pt 6):1772–8. doi: 10.1093/brain/awq091. [DOI] [PubMed] [Google Scholar]
- 249.Yu L, Cui J, Padakanti PK, et al. Synthesis and in vitro evaluation of alpha-synuclein ligands. Bioorg Med Chem. 2012;20(15):4625–34. doi: 10.1016/j.bmc.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Bagchi DP, Yu L, Perlmutter JS, et al. Binding of the radioligand SIL23 to alpha-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One. 2013;8(2):e55031. doi: 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zhang X, Jin H, Padakanti PK, et al. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging alpha-Synuclein. Appl Sci (Basel) 2014;4(1):66–78. doi: 10.3390/app4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chu W, Zhou D, Gaba V, et al. Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for alpha-Synuclein Fibrils. J Med Chem. 2015;58(15):6002–17. doi: 10.1021/acs.jmedchem.5b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Mach RH, Chu W, Zhou D, et al. A PET radiotracer for imaging alpha synuclein aggregates in Parkinson’s disease. J Labelled Comp Radiopharm. 2015;58(Suppl 1):S42. [Google Scholar]
- 254.Sundaram GS, Dhavale DD, Prior JL, et al. Fluselenamyl: A Novel Benzoselenazole Derivative for PET Detection of Amyloid Plaques (Abeta) in Alzheimer’s Disease. Sci Rep. 2016;6:35636. doi: 10.1038/srep35636. [DOI] [PMC free article] [PubMed] [Google Scholar]