

Abstract
Arthritis represents a family of complex joint pathologies responsible for the majority of musculoskeletal conditions. Nearly all diseases within this family, including osteoarthritis, rheumatoid arthritis, and juvenile idiopathic arthritis, are chronic conditions with few or no disease-modifying therapeutics available. Advances in genome engineering technology, most recently with CRISPR-Cas9, have revolutionized our ability to interrogate and validate genetic and epigenetic elements associated with chronic diseases such as arthritis. These technologies, together with cell reprogramming methods, including the use of induced pluripotent stem cells, provide a platform for human disease modeling. We summarize new evidence from genome-wide association studies and genomics that substantiates a genetic basis for arthritis pathogenesis. We also review the potential contributions of genome engineering in the development of new arthritis therapeutics.
Keywords: inflammation, GWAS, iPSC, autoimmune, drug screening, genome engineering
Arthritis – A Family of Complex Diseases of the Joints
Arthritis represents a family of the most prevalent musculoskeletal diseases, collectively forming the greatest source of disability in the United States, and affecting over 54 million Americans. (https://www.cdc.gov/arthritis/data_statistics/national-statistics.html) In particular, osteoarthritis (OA), rheumatoid arthritis (RA), juvenile idiopathic arthritis (JIA), and psoriatic arthritis are the most common arthritides, characterized by pain and loss of joint function due to inflammation, bone erosion and remodeling, and the progressive degeneration of articular cartilage and other joint tissues.[1–4] Traditionally, disparate disease paradigms have been applied to RA and OA: RA has been defined as an autoimmune systemic condition, while OA has been described as a mechanically-driven, wear-and-tear arthropathy.[5,6] However, growing evidence indicates that OA consists of a large family of diseases with a similar endpoint, but potentially different pathological mechanisms; in many cases, these may involve dysregulated systemic metabolic or inflammatory cascades.[7,8] For example, multiplex immunoassays have detected inflammatory cytokines in OA patient synovial fluid, supporting the notion that OA might also arise from long-term low level chronic inflammation, either driven by resident cell types or by systemic metabolic syndromes (e.g., associated with diabetes or obesity), rather than by flares of high inflammation, typically seen in rheumatic conditions[7,9].
Pharmacological recommendations from the American College of Rheumatology for rheumatic conditions include synthetic and biologic disease-modifying anti-rheumatic drugs (DMARDs).[10–12] Synthetic DMARDS, such as methotrexate and leflunomide, are lymphotoxic therapeutics used to treat RA and a range of systemic inflammatory conditions (e.g. Crohn’s disease, ulcerative colitis) through their immunosuppressive effects. Biologic DMARDs, such as tumor necrosis factor (TNF) inhibitors (e.g., adalimumab and etanercept) and interleukin (e.g., IL-1 and IL-6) antagonists have also been used to treat RA patients refractory to synthetic DMARDS, and more recently, have been used for patients presenting with moderate to severe RA.[10–12] Although these drugs are aimed at targeting specific inflammatory pathways and cell-types, and have shown great promise, they have also led to clinical remission in only approximately 50% of patients with RA or JIA, with highly variable outcomes between individual patients.[13] Furthermore, up to 20% of patients satisfying the American College of Rheumatology (ACR) criteria for remission continue to exhibit radiographic deterioration of the joint space due to sustained low-level inflammation.[14,15] Moreover, systemic delivery of these therapies can lead to the development of significant side-effects, such as autoimmune reactions (e.g. vasculitis, psoriasis, and lupus), or increased risk of opportunistic infections through immunosuppressive effects.[16–18]
By contrast, attempts to identify clinically effective disease-modifying osteoarthritis drugs (DMOADs) have been unsuccessful. The IL-1 receptor antagonist (IL-1Ra, i.e., anakinra), while effective for a subset of individuals with RA and post-traumatic arthritis (PTA), has not demonstrated efficacy in treating OA in the clinical setting.[19–22] Therefore, pharmacological treatment options for OA continue to be palliative, consisting of administration of non-steroidal anti-inflammatory drugs (NSAIDs) to reduce pain, glucocorticoids to decrease production of inflammatory cytokines, and hyaluronic acid injections to lubricate eroded joint(s).[23] However, recent studies beyond the scope of this review, suggest that even these treatments do not provide beneficial effects and may in fact lead to increased cartilage loss in OA patients.[24] Consequently, it is becoming increasingly clear that patients with unique disease etiologies will require distinct, personalized therapeutics.[2]
Indeed, the future of new arthritis therapies might need to involve combinatorial approaches that seek to restore metabolic balance systemically as well as in joint tissues exhibiting strong degradative responses to inflammatory cytokines. We envision therapeutics tailored to patients that consider both their genetic susceptibility (e.g. combinations of deleterious single nucleotide polymorphisms (SNPs)) as well as environmental risk factors (e.g. injury). [25,26] This might be accomplished only through a better and updated understanding of distinct pathomolecular mechanisms and alterations leading to various forms arthritis. Here, we review the growing body of evidence on the contribution of genetic variation to arthritis susceptibility. We also discuss recent advances that highlight our ability to precisely re-write the genome and epigenome to interrogate the role of genetic variation in arthritic diseases, as well as the application of tissue and genome engineering to human disease modeling platforms. Together, these fields are opening promising avenues toward tailored therapeutics through the interrogation and manipulation of genomic coding, and through the modulation of regulatory genetic elements that might confer arthritis susceptibility.[27,28]
Understanding the Genetic Determinants of Arthritis to Effectively Tailor Therapeutics
A variety of polygenic inheritance modalities have been implicated in the pathogenesis of RA, JIA, and OA, and development of novel therapies might be enhanced by achieving an improved understanding of specific genetic factors able to modulate varying disease states.[29,30] Until recently, genetic studies investigating the role of naturally occurring arthritis were limited to gene mapping in animal models of arthritis.[31] While these studies have improved our understanding of genetic variation in RA – particularly the role of variants at major histocompatibility complex (MHC) loci – their relevance to human disease has remained poorly defined. Based on these data, candidate association studies have further elucidated the effect of various mutations on arthritis pathogenesis in human patients. For example, the first study identifying the human OA candidate variant rs143383, a single nucleotide polymorphism (SNP) in the 5′ untranslated region(UTR) of the Growth/Differentiation 5 (GDF5) locus as a candidate allele, was based on the arthritic phenotype of Gdf5 loss of function in mice.[32] It has become the most well-characterized OA risk variant, thought to function as an expression quantitative trait locus (eQTL).[33–35] Moreover, patient-derived tissue and human cancer cell lines harboring the risk (T) allele, located at the 5′ UTR of GDF5 exhibit decreased expression of GDF5 compared to the protective (C) allele (Key Figure, Figure 1).[33,34] Homozygotes and heterozygotes for the risk allele are at an increased risk of developing both hip and knee OA.[33–36] Similarly, candidate association studies have substantiated the association of several SNPs with RA, such as variants found in tumor necrosis factor alpha-induced protein 3(TNFAIP3), transforming growth factor beta-1 (TGFB1), methylenetetrahydrofolate reductase (MTHFR), vitamin D receptor (VDR), protein tyrosine phosphatase, non-receptor 22 (PTPN22), and TNF receptor associated factor 1 (TRAF1).[37–40]
Key Figure, Figure 1. Functional analysis of candidate arthritis causal variants in a human iPSC arthritis modeling platform.
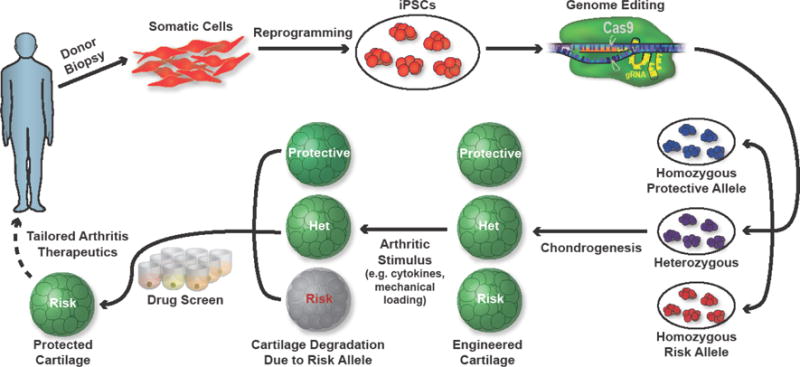
Shown, is a schematic of arthritis modeling by editing candidate variants in human iPSCs and assaying the changes in induced cartilage tissue after administering disease-associated stimuli (i.e. inflammatory cytokines, mechanical loading). Candidate disease-modifying therapeutics might then be assayed to potentially reverse deficiencies in cells harboring genetic variants.
Subsequently, highly powered genome wide association studies (GWAS) have begun to provide an unbiased scan of known genomic loci and variants associated with human disease states. Many loci identified by OA and RA GWAS have been shown to regulate known inflammatory genes, arthrodial developmental programs, and structural components of articular cartilage, as well as various genes with little or no known function in joint development. For example, arcoGen, the first GWAS undertaken for OA, identified several human loci associated with OA at genome-scale significance (p<1×108) from 7410 genomes.[41] One such locus harbors rs11177, a coding variant of nucleostemin (GNL3). Nucleostemin, a nucleolar GTPase, was initially discovered in human neuronal tissue and was recently found to be upregulated in synovial fluid from patients with knee osteoarthritis.[42] Since rs11177 is a coding variant which may affect nucleostemin function in the joint, it may be a causal variant. The majority of identified variants, however, are non-coding and likely influence gene regulation in cis or trans (Fig 2A). A trans-ethnic meta-analysis evaluating over 100,000 patients for RA-associated SNPs identified non-coding variants near the transcription factor ETV7, synaptogryin1 (SYNGR1), and surfactant protein D (SFTPD) as conferring the highest risk of RA development with odds ratios of 1.22–1.23.[43] To date, two JIA GWAS have been carried out which identified several variants, particularly in genes involved in immune cell activation and epigenetic modifiers.[44,45] These genes include IL-2 and its receptors, the histone demethylase JMJD1C, and variants in 3q13. Many of these variants, such as those found in OA and RA GWAS, are in eQTLs and may act in cis or trans to regulate other genes.[44,45] For instance, an HLA allele associated with RA (HLA-DRB1*04), has been shown to be protective in JIA patients by contrast, emphasizing the possibility that different pathogenic mechanisms can contribute to causing these diseases, and suggesting that these traits could be exploited to tailor different therapeutic options.[46]
Fig 2. Description of eQTLs and identification of candidate variants using CRISPR-Cas9 screening technology.
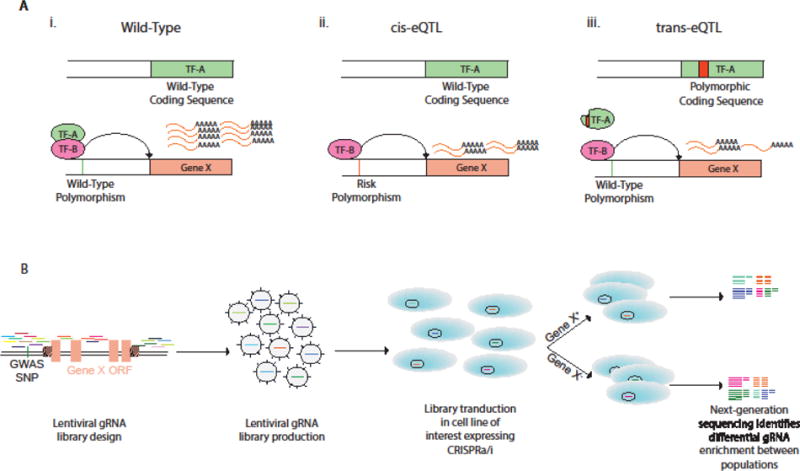
A) Illustration of cis- and trans-expression quantitative trait loci (eQTL). i) Transcription factor A and B (TF-A/TF-B) bind to enhancer elements upstream of Gene X to promote transcription. ii) A polymorphism in the promoter sequence of Gene X inhibits binding of TF-A, reducing transcription of Gene X (cis-eQTL). iii) A polymorphism in the coding region of TF-A prevents TF-A binding to Gene X enhancer element, attenuating expression (trans-eQTL). B) Non-coding CRISPR screen to identify enhancer elements of Gene ×. gRNAs are saturated in non-coding regions flanking and within the open reading frame (ORF). The cell line of interest is transduced with a pool of lentivirus encoding the DNA library. Cells are sorted by expression of Gene X or phenotype of interest and gRNAs within positive and negative populations are sequenced to determine enrichment of gRNAs. Enriched gRNAs bind to candidate enhancers. GWAS-SNPs within these regions can be edited for functional validation as illustrated in Key Figure, Figure 1.
While GWAS have identified loci associated with arthritis, validation and characterization of the causal variants at these loci will be a key challenge in the identification of therapeutic targets. Furthermore, understanding how various loci interact to modulate arthritis development will be important in furthering our understanding of arthritis susceptibility. With recent advances in gene editing and genome engineering technologies, the functional validation of genetic variation in regulatory elements and coding sequences of human cell types is now feasible. Moreover, the generation of cell lines harboring targeted editing susceptibility variants in an isogenic background can now be generated using specific gene editing tools.
The Genome/Epigenome-Engineering Toolbox
Early efforts to alter gene sequences in a site-specific manner, particularly in human cells, sought to exploit natural DNA-binding domains of transcription factors (TFs) fused to an endonuclease catalytic domain to create double-strand break(s) (DSB). After a DSB is formed, error-prone molecular repair machinery is recruited to mend the break, but after many rounds of cleavage and repair, this can result in the accumulation of random insertions or deletions in a process termed non-homologous end joining (NHEJ), the predominant repair mechanism in mammalian cells (Fig 3A).[47] Homology-directed repair (HDR), by contrast, can be used for genome editing by introduction of a donor template with homology to the target site along with the endonuclease (Fig 3B).[48] In addition, multiple DSBs can be used to delete large regions of the genome (Fig 3C). As DSBs can ultimately lead to gene knockout or site-directed mutagenesis, gene editing has revolutionized reverse genetics approaches, and their applications (see below). Primarily three classes of programmable nucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)-associated Cas9 nuclease, are used for genome engineering. Epigenome editing can be accomplished by either removal or mutation of the catalytic endonuclease domain, and fusion of the resulting DNA-binding protein to transcriptional activators, transcriptional repressors, or epigenetic modifiers (Fig 2B).[49] Activation of endogenous genes has been accomplished by fusing potent transcriptional activators such as VP64, a tetramer of the transactivation domain of the viral transcriptional factor VP16, to the deactivated endonuclease.[50] Moreover, epigenetic modifiers such as histone acetylase p300, Krüppel associated box (KRAB) domain, and DNA methyltransferase 3a (DNMT3a) have also been fused to modify chromatin marks on both histones and DNA that can in turn regulate gene expression.[51] The ability to recruit these domains to desired regions of the genome has allowed the identification of novel regulatory elements controlling gene expression.[52] A brief overview of the major editing platforms is described below.
Fig 3. Repair Mechanisms Following Targeted Double Strand Break by Site-Specific Nucleases.
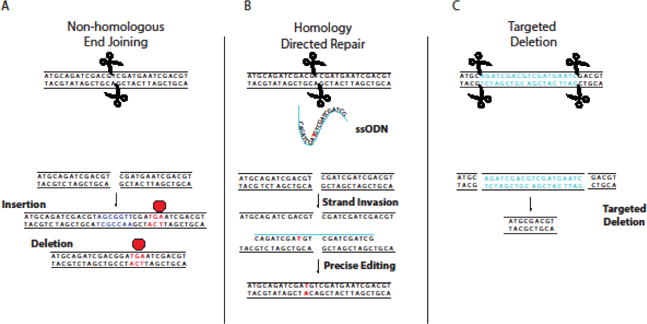
A) Non-homologous end joining (NHEJ) can result in repair of dsDNA with random insertions or deletions that may cause frameshift mutations, resulting in premature termination of translation. B) Introduction of a homologous donor sequence (ssODN shown) can facilitate precise editing through homologous recombination repair pathways. C) Use of two nucleases can create specific deletions, allowing the removal of entire regions of the genome.
Zinc Finger Nucleases (ZFNs)
Initial attempts to engineer site-specific programmable nucleases attempted to modulate the DNA-binding domains of transcription factors (TFs). Zinc finger proteins (ZFPs) are abundant TFs that recognize DNA in sets of three to four base pairs per modular zinc finger domain (Fig 4A). Novel modules were engineered for optimal binding to target sequences, and the modular nature of ZFNs allowed the fusion of recombinant chimeric ZFPs to a FokI endonuclease domain.[53,54] Thus, ZFNs could cleave DNA based on sequence specificity determined by the zinc finger modules used. The need to optimize specific combinations of modules for new sequences limited the applicability of this technology for most purposes, since creating new ZFNs could take weeks to months to engineer and optimize.
Fig 4. Common Genome Engineering Tools.
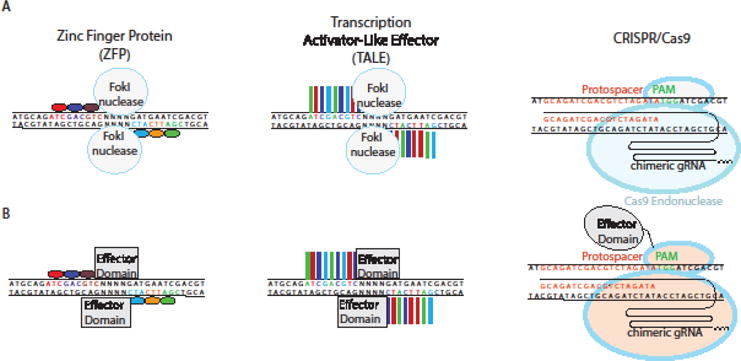
A) Zinc Finger Proteins (ZFPs) and Transcription Activator-Like Effector (TALE) modules are engineered to bind a desired sequence, and then fused to the FokI endonuclease to generate targeted double strand breaks. In the CRISPR system, a custom designed 20 base pair (bp) guide RNA (gRNA) recruits Cas9 endonuclease to induce double strand breaks. B) ZFPs and TALEs fused to transcriptional activators and epigenetic modifiers are shown. In the CRISPR system, these domains are fused to a deactivated Cas9 (dCas9)
As a precursor to epigenome editing, efforts to repurpose DNA-binding proteins for programmable transcriptional regulation were first pioneered with ZFPs.[55] VP64 or KRAB fusion to ZFPs allowed endogenous gene activation or repression, respectively, at multiple loci.[56] In an early attempt to site-specifically edit the epigenome, ZFPs were fused to histone methyltransferase domains to induce H3K9 methylation, demonstrating the ability of histone methylation to initiate a gene repression pathway in human cells.[57] Thus, ZFP engineering created tools for editing both the genome and epigenome.
Transcription Activator Like Effector Nucleases (TALENs)
Transcription Activator Like Effector (TALEs) are another class of programmable nucleases derived from transcription factors that were originally discovered in Xanthomonas.[58] Like ZFPs, TALEs are modular, but composed of repeats of thirty-four amino acids. Two residues within these repeats, known as the repeat variable diresidue (RVD), are responsible for specifying binding to a single base pair (Fig 4A).[59] Because each module recognizes one base pair, combinations of only 4 modules are needed for assembly of TALENs targeting new sequences.[60] Like ZFNs, TALE nucleases (TALENS) were developed by fusion to the FokI endonuclease catalytic domain.[60–62] Epigenome engineering became more tenable with the relative simplicity of TALE engineering. Similar to ZFPs, effector domain fusions to engineered TALEs included transcriptional activation and repression domains, DNA demethylases, and recombinases (Fig 4B).[63–66]
CRISPR-Cas9
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system is a recently developed gene editing tool, first discovered as belonging to the adaptive immune system in prokaryotes.[67]. Its discovery has revolutionized the field of genome/epigenome editing by eliminating the need for protein engineering to obtain site specificity, thereby increasing accessibility to the platform. Editing in mammalian cells using the Type II CRISPR system utilizes a CRISPR RNA (crRNA) and Cas9, an endonuclease that cleaves DNA at targeted sites complementary to the crRNA. A second trans-activating CRISPR RNA (tracrRNA) facilitates binding of the crRNA to Cas9, allowing the complex to scan the genome for complementarity between the crRNA and the target sequence (Fig 4A). Engineered CRISPR-Cas9 systems make use of a single chimeric guide RNA (sgRNA), in which the crRNA and the tracrRNA have been fused into a single RNA species.[68] (Fig 4A and B) Through Watson-Crick base pairing, the sgRNA directs Cas9 to its target in a sequence-specific manner (Fig 4A and B). The target sequence, however, must be followed by a protospacer adjacent motif (PAM), which for the commonly used S. pyogenes Cas9 is a 5′-NGG-3′ trinucleotide sequence. The CRISPR-Cas9 system has become the most accessible genome-editing tool due to the ease of sgRNA synthesis along with specificity and efficiency of targeted DSBs. Musculoskeletal applications of CRISPR technology are rapidly advancing; recent studies demonstrated the efficacy of CRISPR-Cas9 delivery in vivo to restore the dystrophin reading frame and treat a murine model of Duchenne muscular dystrophy (DMD).[69–71] However, applying gene editing tools to treat monogenic joint and skeletal diseases, such as thanatophoric dwarfism (FGFR3 mutation) or Stickler syndrome (COL2A1 mutation), will be challenging because these congenital conditions typically lead to irreversible pathology. Further studies assessing the ability of various viral vectors to effectively deliver CRISPR-Cas9 to the joint will be essential for the potential translation of this technology to the clinic. (Box 1)
Box 1. Therapeutic Applications of Gene Editing Technology.
While this review focuses on the use of genome engineering to identify personalized therapeutics, gene-editing technology is itself being tested as a therapy. Use of this technology has rapidly moved forward for clinical applications in which genetic modifications result in amelioration of disease phenotypes. For example, ZFN-mediated ex vivo editing of CCR5 in CD4+ T-lymphocytes is being considered as a putative strategy for HIV-1 infection treatment, which is currently in Phase II clinical trials, following results of Phase I trials demonstrating safety.[120] Furthermore, an emerging treatment strategy for β-hemoglobinopathies consists of the re-activation of γ-globin, necessary to produce fetal hemoglobin [121]. Specifically, CRISPR-mediated saturating mutagenesis has been used dissect cell-type specific enhancers of human BCL11a, a repressor of γ-globin; targeted mutation of the enhancer sequence resulted in both γ-globin upregulation in immortalized human erythroid progenitor cell lines and delayed switching of fetal hemoglobin to adult hemoglobin in mouse embryos without the consequences of a Bcl11a knockout phenotype, which are known to include impaired lymphogenesis and neurological deficiencies in mice.[121] These advances highlight the potential of ex vivo editing to develop cell-based therapies for HIV-1 infections and β-hemoglobinopathies such as sickle cell disease.
In vivo delivery of programmable nucleases is also being explored as a therapy in models of Duchenne Muscular Dystrophy (DMD)[69,70,122] and other monogenic diseases (via adeno-associated viral (AAV) vectors or nanoparticles for nuclease delivery). A recent study assessed the protective capacity of Nf-κb knockdown to prevent a mouse-model of PTA, conjugating siRNA to a short carrier peptide. Articular injection of siRNA-peptide nanoparticles penetrated deep within murine articular cartilage and reduced tissue degradation relative to controls.[123] Future exploration of intra-articular delivery of CRISPR-Cas9 may involve delivery of nanoparticles carrying sgRNA and Cas9 protein.[124]
In addition to fusion with KRAB and VP64, deactivated Cas9 (dCas9) has been fused to several other combinations of activators and epigenetic modifiers such as p300 and DNMT3a. p300 acts a histone acetyl transferase, activating gene expression by opening chromatin structure at regulatory regions.[72] Likewise, DNA modifications can be specifically altered with DNMT3a methyltransferase.[73] These gene activators and repressors might have therapeutic potential for arthritic joints by targeting epigenetic modifications in both inflammatory cells and resident cell types, although this remains largely unexplored.
Regenerative Medicine and Tissue Engineering with Genetically Engineered Cells
Regenerative medicine integrates solutions from materials science, cell biology, and gene therapy to provide cellular or tissue substitutes capable of restoring function to overcome degeneration caused by chronic diseases or injuries. Musculoskeletal regenerative medicine has made great progress due to advances in genetic engineering, which has provided promising avenues for engineering cartilage and bone substitutes with tissue mimetic properties.[74–79] Due to the ease-of-use and specificity of the CRISPR-Cas9 system, genome engineering and epigenome editing offer to extend the promise of personalized medicine.[67,80] Perhaps the most obvious approach would entail using nucleases to edit disease-causing mutations and subsequently transplanting cells with genetic corrections as therapies. While such an approach is certainly feasible for many types of diseases (e.g. hematologic disorders), the multifactorial nature of arthropathies suggests that a more nuanced strategy may be required. Offering much more promise than simply as a tool to correct genetic mutations, site-specific nucleases open new avenues toward cell-based therapies for musculoskeletal diseases by improving stem cell differentiation protocols and by enabling the rewiring of intrinsic cell signaling pathways.
Interest in generating cell-based therapies to combat the pro-inflammatory, arthritic environment has led to strategies that incorporate gene delivery vehicles driving expression of antagonists of IL-1 or TNF, or of anti-inflammatory cytokines such as IL-10.[81–85] While pioneering work toward this goal has involved constitutive ectopic expression of therapeutic transgenes, recent work has exploited the use of drug-inducible or synthetic cytokine-responsive promoters that allow for tuned expression of immunomodulatory factors.[86,87] Such strategies generally involve viral vectors for gene delivery, which can be impaired via epigenetic silencing or be subjected to the oncogenic consequences of insertional mutagenesis.[88,89] The CRISPR-Cas9 gene editing nuclease platform is attractive for applications where the use of viral vectors may not be permissive; indeed, nuclease-mediated targeted gene addition can enable epigenetically stable, robust transgene expression from safe-harbor loci.[90] Furthermore, building on advances in genome and epigenome engineering, there are ongoing efforts to develop fine control over cellular responses in an environmental context (e.g. injury/inflammation). For example, targeted gene modification of endogenous inflammation-responsive promoters might circumvent the need to ectopically express therapeutic transgenes and permit transient, context-dependent expression.
Our laboratories have recently illustrated the efficacy of using this approach to engineer cytokine-activated and feedback-controlled expression of biologic therapies in murine pluripotent stem cells.[91] These stem cells were used to engineer articular cartilage capable of inducing transient anti-inflammatory responses to the catabolic effects of IL-1 or TNF. Specifically, using CRISPR-Cas9 gene editing, targeted gene addition of IL1Ra or soluble TNF receptor (sTNFR) cDNA downstream of the Ccl2 promoter was used to produce “smart” induced pluripotent stem cells (iPSCs) [91]. These initiated a dynamic negative feedback loop upon stimulation with inflammatory cytokines; once iPSCs were differentiated into chondrocyte-like cells, their response to inflammatory stimuli was attenuated, as evidenced by the decreased expression of catabolic enzymes (Mmp-9/13) and the maintenance of glycosaminoglycan (GAG)-rich cartilage matrix [91].
By ensuring tight control of therapeutic production, manipulation of endogenous circuitry may serve effectively as a cell-based “vaccine” for the treatment of chronic diseases such as OA and RA. “Designer” cell approaches (as in the previous example) may overcome limitations and presumably modify the unwanted consequences that are associated with the delivery of large drug doses, or the constitutive overexpression of biologic therapies, given that these might lead to systemic pleiotropic effects. Moreover, by specifically targeting and cleaving predetermined DNA sequences, the CRISPR-Cas9 platform can be exploited to selectively delete genes from living cells. Such an approach could be used as an alternative to existing RNAi strategies to modify the cellular response to pro-inflammatory signals in an arthritic environment. Proof-of-principle for this approach has been demonstrated by engineering murine pluripotent stem cells (PSCs) with functional deletion of the interleukin-1 receptor I (Il1r1).[92] Knockout of Il1r1 resulted in iPSCs capable of producing articular cartilage resistant to IL-1α-mediated tissue degradation, measured from biochemical analysis of GAG content.[92] In other studies, epigenome editing – using dCas9-KRAB at loci encoding the cytokine receptors IL1R1 and TNFR1 – was used attenuate the catabolic response of human adipose stem cell (hASC)-derived chondrocyte-like cells to inflammatory environments.[93] Specifically, epigenetic silencing of IL1R1 and TNFR1 to their promoters (dCas9-KRAB targeting), mitigated the downstream activation of NF-κB, a transcription factor that initiates catabolic pathways; this protected cartilage matrix integrity in the presence of IL-1 or TNF.[93] Moreover, CRISPR-based epigenome editing approaches have also been used to activate anti-inflammatory cytokines such as IL1RN, and these may hold promise for engineering inflammation-resistant cartilage tissue.[66]
A major challenge that limits the use of PSCs as a source for regenerative medicine is the inability to efficiently and robustly drive differentiation of these cells towards a terminal fate of choice. To overcome this, investigators often employ multi-staged differentiation protocols that involve fluorescence-activated sorting of purified cells based on the upregulation of differentiation markers.[94–96] In many cases, cell surface antigens associated with desirable cell states are not known, or do not exist. In such cases, purified, iPSC-derived populations can be sorted using genetically-encoded fluorescent reporters that are activated upon cell fate induction. For instance, studies have demonstrated the use of collagen-specific promoters to purify chondro- and osteo-progenitor populations from multi-staged, iPSC differentiation protocols, for both human and mouse cells lines.[95,97] Indeed, the use of reporter lines will help optimize differentiation conditions for regenerative medicine therapeutic approaches, and presumably uncover surface markers that could facilitate the purification of differentiated cells. Additionally, epigenome editing using the CRISPR-Cas9 system has the potential for programming gene networks to deterministically direct cell lineage specification or transdifferentiation.[51,98,99] Therefore the CRISPR-Cas9 platform offers the possibility of repurposing PSCs towards clinical use, and potentially, for personalized regenerative therapy.
Disease Modeling Applications
In addition to the regenerative potential of engineered tissue, stem cell-derived cartilage and bone tissue can be used in model systems to test the consequences of various environmental and genetic perturbations. The ability to target any locus in the genome holds great potential for modeling orthopedic disease. As discussed above, the most common types of arthritis are not monogenic diseases and are not amenable to targeted disruption of a single gene. Furthermore, identifying DMOADs has been challenging as we are only beginning to understand the fundamental pathways of OA pathogenesis. Because a combination of environmental and genetic factors can predispose to developing such chronic diseases, functional interrogation of causative genetic elements – including coding/noncoding SNPs or variable number tandem repeats (VNTRs) – might provide a means by which to gain further insight into disease physiology and targeted drug development for personalized therapeutics (Key Figure, Figure 1).
A collection of GWAS and candidate association studies have identified hundreds of candidate SNPs as potentially causative of various forms of arthritis, as discussed above. To our knowledge, no studies published to date have rigorously interrogated GWAS-associated SNPs to implicate their causal role in OA and RA pathogenesis. This is due in part to the difficulty of arthritis disease modeling. Recent efforts directed towards arthritis disease modeling have shown that administration of inflammatory cytokines IL1 and TNF to engineered tissues derived from murine iPSCs can result in tissue degradation, which is consistent with the detrimental effects of these inflammatory cytokines in the joints of patients harboring various forms of arthritis.[95,100] Degradative changes in induced cartilage might thus be ameliorated through small molecule-mediated inhibition of catabolic enzymes (e.g. Mmp-13) or transcription factors (e.g. Nf-κB).[100] Such findings demonstrate the use of an in vitro arthritis disease modeling platform to identify candidate DMOADs. Future efforts should consider extending this approach to human iPSC-based OA modeling platforms, where human iPSC-derived cartilage tissue might be exposed to OA-relevant stimuli, such as inflammatory cytokines and/or mechanical loading (Key figure, Figure 1). Recent advances in chondrogenic differentiation of human iPSCs support the establishment of disease modeling platforms on which candidate DMOADs that potentially prevent or reverse tissue degradation are tested for clinical efficacy.[101–103] Subsequent disease modeling studies might ask how cell lines with edited genetic backgrounds are differentially affected by these degradative stimuli; drugs efficacious in specific backgrounds might then be identified. For example, interrogation of GDF5 rs143383 variants on OA susceptibility can be considered an attractive proof of concept test, given the known functional role of GDF5 in gene regulation, joint development, and OA pathogenesis.[32–34,104]
Thus, genome/epigenome editing screens can be used to annotate and elucidate the effect of uncharacterized variants identified from these GWAS. Because sgRNA sequences can be readily synthesized and cloned into appropriate vectors, genome-scale CRISPR libraries (similar to siRNA libraries) have been created to screen each of the ~25000 annotated genes in the genome for their effect on a phenotype of interest.[105–109] CRISPR-based activators and repressors are powerful tools allowing non-coding CRISPR activator/interference (CRISPRa/i) screens to be performed for context-independent elucidation of novel enhancer regions that might harbor GWAS hits (Fig 2B).[52,110–112] For example, a preliminary study that carried out a non-coding CRISPRa screen at the human IL2RA locus (previously associated with RA), identified an enhancer region containing rs61839660, a SNP implicated in several autoimmune conditions.[45,113] Generation of knock-in mice bearing the human risk variant resulted in decreased induction of IL2RA surface expression following antibody-mediated CD4+ T-cell activation, relative to controls [113]. Deletion of the enhancer in primary human CD4+ T-cells was reported to have a similar effect, inducing IL2RA upregulation following antibody-mediated activation [113]. In a similar manner, enhancers containing SNPs associated with arthritis might be identified using CRISPRa screens, and subsequently incorporated into human iPSCs via genome editing in order to assay phenotypic differences after differentiation. Large drug screens might then potentially reveal small molecules capable of rescuing the phenotype (Key Figure, Figure 1).
With the ability to model the effects of disease-causing variants by generating isogenic lines harboring targeted combinations of genetic modifications, gene editing technology holds immense potential to uncover the role of arthritis-associated variants. To date, few studies have sought to use this technology in disease modeling of joint physiology and pathology. Indeed, most studies investigating cartilage development in human cells have relied on comparisons between diseased and healthy patient-derived stem cells. For example, comparisons have been made among patient-derived iPSCs harboring mutations in FGFR3 (as well as controls) to understand the effect of FGFR3 signaling/activation during chondrogenesis; this study found that statin therapy could rescue in vitro chondrogenesis in FGFR3-mutant human iPSCs.[114]. Another study found that iPSCs from a patient with an NLRP3 mutation causing neonatal onset multisystem inflammatory disease (NOMID) presented enhanced chondrogenesis.[115] iPSCs derived from unique patients have thus been shown to have highly variable differentiation potential as a result of different genetic backgrounds.[116,117] As these studies use patient-derived iPSCs, they cannot control for inherent genetic variability among cell lines that might also alter chondrogenic potential. Consequently, the ability to use gene editing tools to create multiple isogenic cell lines from a single patient in which only the gene of interest is mutated, can control for genetic variation between different patients, and might enable the undertaking of genetic studies with greater fidelity.
Accordingly, the creation of isogenic lines to characterize protein function in cell types relevant to orthopedic diseases has been carried out in a few studies. In one example, gRNAs were designed to knockout hyaluronan synthase 2 (Has2) in a rat chondrosarcoma (RCS) line constitutively expressing Cas9 [118] [119]. Has2 is an enzyme that generates hyaluronan, and whose loss of function is an embryonic lethal in mice.[118] [119] By editing Has2 in the chondrosarcoma line, Has2 was shown to retain aggrecan (an important proteoglycan of articular cartilage) in the pericellular matrix of mature chondrocytes [118] [119]. Thus, genome engineering can be applied to tissue engineering platforms to both replicate knockout studies in animal models and further elucidate protein function when traditional in vivo knockout is cumbersome. Analysis of clonal lines with isogenic backgrounds will become increasingly important when evaluating the function of genetic variation in orthopedic biology.
Concluding Remarks
The various forms of arthritis constitute the majority of musculoskeletal diseases. Targeted regenerative medicine therapies for these conditions will require a more precise understanding of the various etiologies of disease (see Outstanding Questions and Box 2). Dissecting these mechanisms can now be performed using genome engineering techniques. The development of new cell or drug therapies has required testing patient-derived tissues, which are characterized by varying genetic backgrounds which can act as confounding factors in the analysis. However, recent approaches can exploit genome and epigenome editing to characterize specific genetic elements in an isogenic background, allowing the functional characterization of GWAS-identified SNPs and enhancer elements, as well as defining their role in cell differentiation and pathophysiology. Applying these approaches to arthritis disease models may greatly facilitate the continuing search for novel disease-modifying therapeutics. While research in other fields has extensively used these platforms for various genetic applications and synthetic biology, great opportunities lie that may revolutionize via gene editing tools, the fields of musculoskeletal biology and rheumatic diseases.
Outstanding Questions.
Of the loci identified in OA, RA, and JIA GWAS, which are causal for pathogenesis? What are the mechanisms by which they contribute to disease?
As many candidate loci for RA and JIA are near genes implicated in immune functions (MHC loci, genes encoding cytokine receptors, etc.), what appropriate model systems (i.e. engineering cartilage or lymphatic tissue) can be used to test the effects of these SNPs? One of the biggest challenges for validating the effects of causal variants will be to test them in the correct cell types in order to faithfully model the pathomechanisms responsible for disease phenotypes.
Delivery of gene editing tools in vivo has been shown to be feasible in a variety of cell types and organs using viral vectors. What are the best ways of delivering gene editing tools such as CRISPR-Cas9 to the joint, to either introduce new therapeutic genes or to delete ‘unwanted’ genes? What is the proportion of cells that must be targeted to achieve a therapeutic effect?
Box 2. Clinician’s Corner.
Treatments for the most common forms of arthritis (rheumatoid arthritis (RA), juvenile idiopathic arthritis (JIA), and osteoarthritis (OA)) are either immunosuppressive, palliative, or surgical. However, the etiology of disease likely varies from individual to individual, which may be the reason why existing treatment options are effective only in a subset of patients. Understanding the various mechanisms of disease pathogenesis should help identify distinct classes of treatments for each type of arthritis.
An individual’s genetic make-up can greatly contribute to their predisposition to arthritis. Indeed, distinct genetic loci and risk alleles are associated with various forms of arthritis.
Risk alleles may affect different factors associated with arthritis, including joint development, inflammatory signaling, or responses to environmental stimuli. A patient’s unique combinations of risk alleles may explain why she/he responds to treatment options when only targeting a single pathway.
Unraveling the mechanisms of these causal loci for arthritis may provide an avenue for the development of personalized therapeutics specific for a patient’s risk allele profile.
Trends Box.
Arthritis represents the most prevalent cause of disability in the United States, but the genetic basis of disease etiology remains poorly understood.
Recent GWAS and candidate association studies have identified several loci associated with arthritis. These studies suggest that unique loci may play distinct roles in the development of various types of arthritis.
Advances in genome editing technologies enable the precise modification of candidate causal loci and functional validation in disease pathogenesis. Recently, epigenome editing has been used to uncover the function of regulatory elements near disease susceptibility loci.
Concurrent advances in tissue engineering from pluripotent stem cells have facilitated arthritis disease modeling.
Gene editing tools have been used in other fields for both regenerative medicine and disease modeling. Given the wealth of data ascribing genetic variation to arthritis, at this time, there is significant potential for using gene editing in conjunction with tissue engineering, to discover mechanisms underlying genetic drivers of arthritis.
Acknowledgments
We would like to acknowledge the support by the Nancy Taylor Foundation for Chronic Diseases, the Arthritis Foundation, the Thorek Memorial Foundation, the Allen Distinguished Investigator Program, through The Paul G. Allen Frontiers Group, NIH grants AR061042, AR50245, AR48852, AG15768, AR48182, AR067467, AR065956, OD008586, GM119914, HG007900, DA036865,AR069085, NSF CAREER Award CBET-1151035, and the Collaborative Research Center of the AO Foundation, Davos, Switzerland
Glossary
- CCR5
co-receptor necessary for HIV-1 infection
- Chondrogenic differentiation
Differentiation of stem cells to chondrocyte-like cells as assessed by the ability of differentiated cells to produce a cartilaginous matrix rich in GAG and type II collagen
- CRISPR activator/interference screen
Use of gRNA library in conjunction with a dCAS9 effector (e.g. dCAS9-VP64, dCAS9-KRAB) to screen putative regulatory elements involved in the expression of a gene of interest
- Disease-modifying anti-rheumatic drugs (DMARDs)
class of therapeutics targeting immune pathways either through cytotoxicity, or targeted inhibition of lymphocytes. Synthetic DMARDs include methotrexate and sulfasalazine that can deplete lymphocytes and inhibit inflammatory signaling, respectively. Biologic DMARDs are antibodies or decoy receptors designed to block inflammatory signaling or lymphocyte activation
- Disease-modifying osteoarthritis drugs (DMOADs)
class of therapeutic drugs aimed at targeting causative factors of OA in a manner that alters disease progression rather than just providing pain relief
- DNMT3a
DNMT3a fusion to dCasS9 can mediate site-specific methylation which can result in transcriptional repression
- Epigenetic silencing
heritable transcriptional repression, which typically occurs through DNA or histone methylation
- Expression quantitative trait locus
region of the genome containing variants influencing gene expression. These may be in cis or trans (Fig 2A)
- FokI endonuclease domain
Catalytic domain of FokI endonuclease mediating double strand DNA cleavage
- Genome wide association study (GWAS)
analysis of a large cohort to identify genetic variants associated with a trait or disease phenotype of interest
- Glycosaminoglycan
unbranched polysaccharide consisting of amino sugars, usually bound to a proteoglycan
- Homology-directed repair (HDR)
DNA repair process using homologous DNA as a template for repair
- Hyaluronan
glycosaminoglycan found in joint synovial fluid and articular cartilage. It binds the proteoglycan core protein aggrecan and is responsible for the stiffness of articular cartilage
- Insertional mutagenesis
Random or targeted insertion of novel DNA sequences in the genome, disrupting an endogenous gene, or gene regulatory mechanisms
- Isogenic Lines
Cells lines with identical genetic backgrounds, with the exception of defined variants
- Juvenile Idiopathic Arthritis (JIA)
most common inflammatory autoimmune joint disease in children
- KRAB
Krüppel associated box domain functions as a transcriptional repressor through heterochromatin induction
- MHC loci
contain genes encoding extracellular receptors and processing proteins important in antigen presentation. Variation at these loci can mediate susceptibility to autoimmune disease
- Metabolic Syndrome
describes a combination of at least three of the following conditions: elevated fasting glucose (≥100 mg/dL), elevated blood pressure (≥130/85 mmHg), elevated triglycerides (≥150 mg/dL), low HDL (≤40 mg/dL in men; ≤50 mg/dL in women), and central obesity. These individual conditions typically result from acquired insulin resistance and excess adipose deposition
- Non-homologous end joining (NHEJ)
DNA repair process resulting in random insertions and deletions
- Osteoarthritis
painful degenerative joint disease characterized by erosion of articular cartilage, inflammation of the synovium (synovitis), and formation of bone spurs
- p300
histone acetyltransferase mediating H3K27 acetylation at enhancers, among other targets. Fusion to dCas9 generates a robust trans-activator capable of mediating activation from distal enhancers
- Polygenic inheritance
acquisition of traits influenced by two or more genes (e.g. eye color)
- Post-traumatic arthritis
form of osteoarthritis characterized by degenerative changes in the articular cartilage, subchondral bone remodeling, and synovial inflammation occurring secondary to joint injury
- Protospacer adjacent motif (PAM)
sequence flanking a target sequence necessary for Cas9-mediated cleavage. The PAM sequences vary by Cas9 variant (5′-NGG-3′ for S. pyogenes Cas9)
- Psoriatic Arthritis
inflammatory joint disease similar to RA, observed in a subset of patients with the chronic skin condition, psoriasis
- Rheumatoid Arthritis
systemic and chronic autoimmune condition characterized by synovitis, and cartilage and bone erosion. Successive flares and remissions lead to the formation of scar tissue that contributes to joint deformity
- Safe-harbor loci
regions of the genome capable of supporting sustained transgene expression without negative consequences
- Saturating mutagenesis
mutation of all DNA bases within a region of interest, typically approached using the CRISPR-Cas9 system by cloning a library of all possible gRNAs targeting a region of interest into a lentiviral vector. The library is transduced at low multiplicity of infection to create the maximum number of unique mutations within the population
- Single nucleotide polymorphism (SNP)
naturally occurring variation at one base pair in the genome
- Stickler syndrome
collagen disorder most often caused by mutations in COL2A1; results in joint pathology and hearing loss. Patients are predisposed to arthritis development
- Thanatophoric dwarfism
orthopedic disease caused by an FGFR3 mutation resulting in severe shortening of the skeleton
- Transdifferentiation
reprogramming of a somatic cell type of one lineage to another usually using overexpression of defined master transcription factors
- Variable number tandem repeats
repetitive sequences of variable number of repeats between individuals
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smolen JS, et al. Rheumatoid arthritis. The Lancet. 2016;388:2023–2038. doi: 10.1016/S0140-6736(16)30173-8. [DOI] [PubMed] [Google Scholar]
- 2.Martel-Pelletier J, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072. doi: 10.1038/nrdp.2016.72. [DOI] [PubMed] [Google Scholar]
- 3.Huemer C, et al. Patterns of joint involvement at onset differentiate oligoarticular juvenile psoriatic arthritis from pauciarticular juvenile rheumatoid arthritis. J Rheumatol. 2002;29:1531–1535. [PubMed] [Google Scholar]
- 4.Kerschbaumer A, et al. An overview of psoriatic arthritis – epidemiology, clinical features, pathophysiology and novel treatment targets. Wien Klin Wochenschr. 2016;128:791–795. doi: 10.1007/s00508-016-1111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 6.Loeser RF. Age-related changes in the musculoskeletal system and the development of osteoarthritis. Clin Geriatr Med. 2010;26:371–386. doi: 10.1016/j.cger.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mobasheri A, et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2017;13:302–311. doi: 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
- 8.Wu CL, et al. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann Rheum Dis. 2015;74:2076–2083. doi: 10.1136/annrheumdis-2014-205601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson WH, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:580–592. doi: 10.1038/nrrheum.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh JA, et al. American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care & Research. 2015;68:1–25. doi: 10.1002/acr.22783. Jan-(2016) [DOI] [PubMed] [Google Scholar]
- 11.Singh JA, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res. 2012;64:625–639. doi: 10.1002/acr.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ajeganova S, et al. Disease-modifying antirheumatic drug-free sustained remission in rheumatoid arthritis: an increasingly achievable outcome with subsidence of disease symptoms. Ann Rheum Dis. 2016;75:867–873. doi: 10.1136/annrheumdis-2014-207080. [DOI] [PubMed] [Google Scholar]
- 13.Nam JL, et al. Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2017 doi: 10.1136/annrheumdis-2016-210713. [DOI] [PubMed] [Google Scholar]
- 14.Molenaar ETH, et al. Progression of radiologic damage in patients with rheumatoid arthritis in clinical remission. Arthritis Rheumatol. 2004;50:36–42. doi: 10.1002/art.11481. [DOI] [PubMed] [Google Scholar]
- 15.Brown AK, et al. An explanation for the apparent dissociation between clinical remission and continued structural deterioration in rheumatoid arthritis. Arthritis Rheumatol. 2008;58:2958–2967. doi: 10.1002/art.23945. [DOI] [PubMed] [Google Scholar]
- 16.Choy EH, et al. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol. 2013;9:154–163. doi: 10.1038/nrrheum.2013.8. [DOI] [PubMed] [Google Scholar]
- 17.Ramos-Casals M, et al. Autoimmune diseases induced by TNF-targeted therapies. Best Pract Res Cl Rh. 2008;22:847–861. doi: 10.1016/j.berh.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Sokumbi O, et al. Vasculitis associated with tumor necrosis factor-α inhibitors. Mayo Clin Proc. 2012;87:739–745. doi: 10.1016/j.mayocp.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chevalier X, et al. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. 2009;61:344–352. doi: 10.1002/art.24096. [DOI] [PubMed] [Google Scholar]
- 20.Furman BD, et al. Targeting pro-inflammatory cytokines following joint injury: acute intra-articular inhibition of interleukin-1 following knee injury prevents post-traumatic arthritis. Arthritis Res Ther. 2014;16 doi: 10.1186/ar4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bresnihan B, et al. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–2204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 22.Tarp S, et al. Defining the optimal biological monotherapy in rheumatoid arthritis: A systematic review and meta-analysis of randomised trials. Semin Arthritis Rheum. 2016 doi: 10.1016/j.semarthrit.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Hochberg MC, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthrit Rheum-Arthr. 2012;64:465–474. doi: 10.1002/acr.21596. [DOI] [PubMed] [Google Scholar]
- 24.McAlindon TE, et al. Effect of Intra-articular Triamcinolone vs Saline on Knee Cartilage Volume and Pain in Patients With Knee Osteoarthritis: A Randomized Clinical Trial. JAMA. 2017;317:1967–1975. doi: 10.1001/jama.2017.5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yousef GM. Personalized Cancer Genomics: The Road Map to Clinical Implementation. Clin Chem. 2012;58:661–663. doi: 10.1373/clinchem.2011.181073. [DOI] [PubMed] [Google Scholar]
- 26.Matzko ME, et al. Orthogenomics: an update. J Am Acad Orthop Surg. 2012;20:536–546. doi: 10.5435/JAAOS-20-08-536. [DOI] [PubMed] [Google Scholar]
- 27.Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- 28.Urnov FD, et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 29.Stahl EA, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet. 2012;44:483–489. doi: 10.1038/ng.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panoutsopoulou K, et al. Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann Rheum Dis. 2011;70:864–867. doi: 10.1136/ard.2010.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haag S, et al. Positional identification of RT1-B (HLA-DQ) as susceptibility locus for autoimmune arthritis. J Immunol. 2015;194:2539–2550. doi: 10.4049/jimmunol.1402238. [DOI] [PubMed] [Google Scholar]
- 32.Storm EE, et al. Limb alterations in brachypodism mice due to mutations in a new member of the TGFb-superfamily. Nature. 1994;368:639–642. doi: 10.1038/368639a0. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto Y, et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat Genet. 2007;39:529–533. doi: 10.1038/2005. [DOI] [PubMed] [Google Scholar]
- 34.Southam L, et al. An SNP in the 5′-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet. 2007;16:2226–2232. doi: 10.1093/hmg/ddm174. [DOI] [PubMed] [Google Scholar]
- 35.Chapman K, et al. A meta-analysis of European and Asian cohorts reveals a global role of a functional SNP in the 5′ UTR of GDF5 with osteoarthritis susceptibility. Hum Mol Genet. 2008;17:1497–1504. doi: 10.1093/hmg/ddn038. [DOI] [PubMed] [Google Scholar]
- 36.Pan F, et al. Association between GDF5 rs143383 polymorphism and knee osteoarthritis: an updated meta-analysis based on 23,995 subjects. BMC Musculoskel Dis. 2014;15:1–11. doi: 10.1186/1471-2474-15-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen N, et al. Three single nucleotide polymorphisms of TNFAIP3 gene increase the risk of rheumatoid arthritis. Oncotarget. 2017;8:20784–20793. doi: 10.18632/oncotarget.15265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saad MN, et al. Genetic Case-Control Study for Eight Polymorphisms Associated with Rheumatoid Arthritis. PLoS ONE. 2015;10:e0131960. doi: 10.1371/journal.pone.0131960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurreeman FAS, et al. A Candidate Gene Approach Identifies the TRAF1/C5 Region as a Risk Factor for Rheumatoid Arthritis. PLoS Medicine. 2007;4:e278. doi: 10.1371/journal.pmed.0040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Begovich AB, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.arcOGEN Consortium et al. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. 2012;380:815–823. doi: 10.1016/S0140-6736(12)60681-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louka ML, et al. Expression of nucleostemin gene in primary osteoarthritis. Gene. 2016;587:27–32. doi: 10.1016/j.gene.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 43.Okada Y, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2013;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinks A, et al. Fine-mapping the MHC locus in juvenile idiopathic arthritis (JIA) reveals genetic heterogeneity corresponding to distinct adult inflammatory arthritic diseases. Ann Rheum Dis. 2016 doi: 10.1136/annrheumdis-2016-210025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson SD, et al. The susceptibility loci juvenile idiopathic arthritis shares with other autoimmune diseases extend to PTPN2, COG6, and ANGPT1. Arthritis Rheum. 2010;62:3265–3276. doi: 10.1002/art.27688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hollenbach JA, et al. Juvenile idiopathic arthritis and HLA Class I and Class II interactions and age-at-onset effects. Arthritis Rheum. 2010;62:1781–1791. doi: 10.1002/art.27424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Translational cancer research. 2013;2:130–143. doi: 10.3978/j.issn.2218-676X.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richardson CD, et al. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:1–7. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 49.Thakore PI, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Meth. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beerli RR, et al. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. PNAS. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thakore PI, et al. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Meth. 2016;13:127–137. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klann TS, et al. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol. 2017;7:46545. doi: 10.1038/nbt.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim YG, et al. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Desjarlais JR, Berg JM. Toward rules relating zinc finger protein sequences and DNA binding site preferences. PNAS. 1992;89:7345–7349. doi: 10.1073/pnas.89.16.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gersbach CA, et al. Synthetic Zinc Finger Proteins: The Advent of Targeted Gene Regulation and Genome Modification Technologies. Acc Chem Res. 2014;47:2309–2318. doi: 10.1021/ar500039w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beerli RR, et al. Positive and negative regulation of endogenous genes by designed transcription factors. PNAS. 2000;97:1495–1500. doi: 10.1073/pnas.040552697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Snowden AW, et al. Gene-Specific Targeting of H3K9 Methylation Is Sufficient for Initiating Repression In Vivo. Current Biology. 2002;12:2159–2166. doi: 10.1016/s0960-9822(02)01391-x. [DOI] [PubMed] [Google Scholar]
- 58.Boch J, Bonas U. XanthomonasAvrBs3 Family-Type III Effectors: Discovery and Function. Annu Rev Phytopathol. 2010;48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- 59.Boch J, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 60.Miller JC, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 61.Mahfouz MM, et al. De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. PNAS. 2011;108:2623–2628. doi: 10.1073/pnas.1019533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li T, et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011;39:359–372. doi: 10.1093/nar/gkq704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mercer AC, et al. Chimeric TALE recombinases with programmable DNA sequence specificity. Nucleic Acids Res. 2012;40:11163–11172. doi: 10.1093/nar/gks875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maeder ML, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137–1142. doi: 10.1038/nbt.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le Cong, et al. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perez-Pinera P, et al. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Meth. 2013;10:239–242. doi: 10.1038/nmeth.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096–1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 68.Jinek M, et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nelson CE, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2015;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tabebordbar M, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Long C, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:1–10. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu XS, et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167:233–235.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tao K, et al. Effects of combined rAAV-mediated TGF-β and sox9 gene transfer and overexpression on the metabolic and chondrogenic activities in human bone marrow aspirates. Journal of Experimental Orthopaedics. 2017;4:4. doi: 10.1186/s40634-017-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tao K, et al. rAAV- mediated combined gene transfer and overexpression of TGF-β and SOX9 remodels human osteoarthritic articular cartilage. J Orthop Res. 2016;34:2181–2190. doi: 10.1002/jor.23228. [DOI] [PubMed] [Google Scholar]
- 76.Shi S, et al. Growth factor transgenes interactively regulate articular chondrocytes. J Cell Biochem. 2013;114:908–919. doi: 10.1002/jcb.24430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Virk MS, et al. “Same Day” Ex-vivo Regional Gene Therapy: A Novel Strategy to Enhance Bone Repair. Mol Ther. 2011;19:960–968. doi: 10.1038/mt.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brunger JM, et al. Scaffold-mediated lentiviral transduction for functional tissue engineering of cartilage. Proc Natl Acad Sci USA. 2014;111:E798–E806. doi: 10.1073/pnas.1321744111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao X, et al. A comparison of bone regeneration with human mesenchymal stem cells and muscle-derived stem cells and the critical role of BMP. Biomaterials. 2014;35:6859–6870. doi: 10.1016/j.biomaterials.2014.04.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maeder ML, Gersbach CA. Genome-editing Technologies for Gene and Cell Therapy. Mol Ther. 2016;24:430–446. doi: 10.1038/mt.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glass KA, et al. Biomaterials. Biomaterials. 2014;35:5921–5931. doi: 10.1016/j.biomaterials.2014.03.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Evans CH, et al. Arthritis Gene Therapy and its Tortuous Path into the Clinic. Transl Res. 2013;161:205–216. doi: 10.1016/j.trsl.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim SH, et al. Ex Vivo Gene Delivery of IL-1Ra and Soluble TNF Receptor Confers a Distal Synergistic Therapeutic Effect in Antigen-Induced Arthritis. Mol Ther. 2002;6:591–600. [PubMed] [Google Scholar]
- 84.Khoury M, et al. Inflammation-inducible anti-TNF gene expression mediated by intra- articular injection of serotype 5 adeno-associated virus reduces arthritis. J Gene Med. 2007;9:596–604. doi: 10.1002/jgm.1053. [DOI] [PubMed] [Google Scholar]
- 85.Garaulet G, et al. IL10 Released by a New Inflammation-regulated Lentiviral System Efficiently Attenuates Zymosan-induced Arthritis. Mol Ther. 2013;21:119–130. doi: 10.1038/mt.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van de Loo FAJ, et al. An inflammation-inducible adenoviral expression system for local treatment of the arthritic joint. Gene Ther. 2004;11:581–590. doi: 10.1038/sj.gt.3302182. [DOI] [PubMed] [Google Scholar]
- 87.Rachakonda PS, et al. Application of inflammation- responsive promoter for an in vitro arthritis model. Arthritis Rheumatol. 2008;58:2088–2097. doi: 10.1002/art.23598. [DOI] [PubMed] [Google Scholar]
- 88.Müller-Kuller U, et al. A minimal ubiquitous chromatin opening element (UCOE) effectively prevents silencing of juxtaposed heterologous promoters by epigenetic remodeling in multipotent and pluripotent stem cells. Nucleic Acids Res. 2015;43:1577–1592. doi: 10.1093/nar/gkv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pfeifer A, et al. Transgenesis by lentiviral vectors: Lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. PNAS. 2002;99:2140–2145. doi: 10.1073/pnas.251682798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lombardo A, et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Meth. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- 91.Brunger JM, et al. Genome Engineering of Stem Cells for Autonomously Regulated, Closed-Loop Delivery of Biologic Drugs. Stem Cell Rep. 2017 doi: 10.1016/j.stemcr.2017.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brunger JM, et al. CRISPR/Cas9 editing of induced pluripotent stem cells for engineering inflammation-resistant tissues. Arthritis Rheumatol. 2016 doi: 10.1002/art.39982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Farhang N, et al. CRISPR-Based Epigenome Editing of Cytokine Receptors for the Promotion of Cell Survival and Tissue Deposition in Inflammatory Environments. Tissue Eng Part A. 2017 doi: 10.1089/ten.TEA.2016.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ang YS, et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell. 2016;167:1734–1749.e22. doi: 10.1016/j.cell.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Diekman BO, Christoforou N. Cartilage tissue engineering using differentiated and purified induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109:19172–19177. doi: 10.1073/pnas.1210422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kattman SJ, et al. Stage-Specific Optimization of Activin/Nodal and BMP Signaling Promotes Cardiac Differentiation of Mouse and Human Pluripotent Stem Cell Lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 97.Xin X, et al. A Site-Specific Integrated Col2.3GFP Reporter Identifies Osteoblasts Within Mineralized Tissue Formed In Vivo by Human Embryonic Stem Cells. Stem Cells Trans Med. 2014 doi: 10.5966/sctm.2013-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chakraborty S, et al. A CRISPR/Cas9-Based System for Reprogramming Cell Lineage Specification. Stem Cell Rep. 2014;3:940–947. doi: 10.1016/j.stemcr.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Black JB, et al. Targeted Epigenetic Remodeling of Endogenous Loci by CRISPR/Cas9-Based Transcriptional Activators Directly Converts Fibroblasts to Neuronal Cells. Stem Cell. 2016;19:406–414. doi: 10.1016/j.stem.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Willard VP, et al. Use of Cartilage Derived From Murine Induced Pluripotent Stem Cells for Osteoarthritis Drug Screening. Arthritis & Rheumatology. 2014;66:3062–3072. doi: 10.1002/art.38780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Craft AM, et al. Generation of articular chondrocytes from human pluripotent stem cells. Nat Biotechnol. 2015;33:638–645. doi: 10.1038/nbt.3210. [DOI] [PubMed] [Google Scholar]
- 102.Yamashita A, et al. Generation of Scaffoldless Hyaline Cartilaginous Tissue from Human iPSCs. Stem Cell Rep. 2015;4:404–418. doi: 10.1016/j.stemcr.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee J, et al. Early induction of a prechondrogenic population allows efficient generation of stable chondrocytes from human induced pluripotent stem cells. FASEB J. 2015 doi: 10.1096/fj.14-269720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reynard LN, et al. CpG methylation regulates allelic expression of GDF5 by modulating binding of SP1 and SP3 repressor proteins to the osteoarthritis susceptibility SNP rs143383. Hum Genet. 2014;133:1059–1073. doi: 10.1007/s00439-014-1447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parnas O, et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell. 2015;162:675–686. doi: 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jaitin DA, et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 2016;167:1883–1896.e15. doi: 10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 108.Wang T, et al. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shalem O, et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fulco CP, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sanjana NE, et al. High-resolution interrogation of functional elements in the noncoding genome. Science. 2016;353:1545–1549. doi: 10.1126/science.aaf7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Korkmaz G, et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol. 2016;34:192–198. doi: 10.1038/nbt.3450. [DOI] [PubMed] [Google Scholar]
- 113.Simeonov DR, et al. Discovery of an autoimmunity-associatedIL2RAenhancer by unbiased targeting of transcriptional activation. bioRxiv. 2016 doi: 10.1101/091843. [DOI] [Google Scholar]
- 114.Yamashita A, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature. 2015;513:507–511. doi: 10.1038/nature13775. [DOI] [PubMed] [Google Scholar]
- 115.Yokoyama K, et al. Enhanced Chondrogenesis of Induced Pluripotent Stem Cells From Patients With Neonatal-Onset Multisystem Inflammatory Disease Occurs via the Caspase 1-Independent cAMP/Protein Kinase A/CREB Pathway. Arthritis & Rheumatology. 2014;67:302–314. doi: 10.1002/art.38912. [DOI] [PubMed] [Google Scholar]
- 116.Kim K, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kilpinen H, et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546:370–375. doi: 10.1038/nature22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang Y, et al. CRISPR/Cas9 knockout of HAS2 in rat chondrosarcoma chondrocytes demonstrates the requirement of hyaluronan for aggrecan retention. Matrix Biol. 2016 doi: 10.1016/j.matbio.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang M, et al. CRISPR/Cas9 mediated generation of stable chondrocyte cell lines with targeted gene knockouts; analysis of an aggrecan knockout cell line. Bone. 2014;69:118–125. doi: 10.1016/j.bone.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 120.Tebas P, et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Long C, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yan H, et al. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc Natl Acad Sci USA. 2016;113:E6199–E6208. doi: 10.1073/pnas.1608245113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gibson GJ, Yang M. What rheumatologists need to know about CRISPR/Cas9. Nat Rev Rheumatol. 2017;13:205–216. doi: 10.1038/nrrheum.2017.6. [DOI] [PubMed] [Google Scholar]