

Summary
Dendritic cell (DC) activation by toll-like receptor (TLR) agonists causes rapid glycolytic reprogramming that is required to meet the metabolic demands of their immune activation. Recent efforts in the field have identified an important role for extracellular glucose sourcing to support DC activation. However, the contributions of intracellular glucose stores to these processes have not been well characterized. We demonstrate that DCs possess intracellular glycogen stores and that cell-intrinsic glycogen metabolism supports the early effector functions of TLR-activated DCs. Inhibition of glycogenolysis significantly attenuates TLR-mediated DC maturation and impairs their ability to initiate lymphocyte activation. We further report that DCs exhibit functional compartmentalization of glucose and glycogen – derived carbons, where these substrates preferentially contribute to distinct metabolic pathways. This work provides novel insights into nutrient homeostasis in DCs, demonstrating that differential utilization of glycogen and glucose metabolism regulates their optimal immune function.
Keywords: Dendritic cells, glycolysis, glycogen, glycogenolysis, PYG, glycogen shunt
ETOC BLURB
Thwe et al. show that dendritic cells (DCs) possess intracellular glycogen stores that fuel their activation-associated glycolysis induction and immune effector function. They uncover a novel mechanism of metabolic regulation in DCs by which glucose and glycogen –derived carbons preferentially contribute to distinct metabolic pathways.
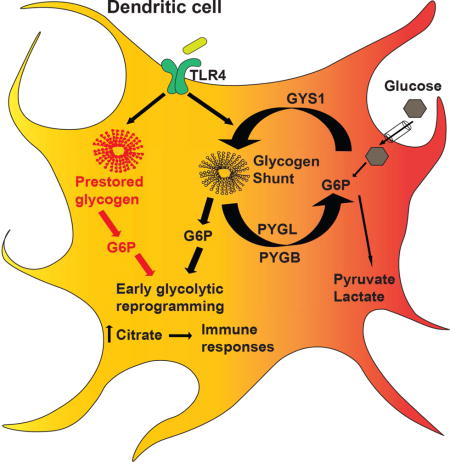
Introduction
Dendritic cells (DCs) are canonical “professional antigen presenting cells” of the immune system and play a central role in coordinating both innate and adaptive immune responses (Banchereau and Steinman, 1998; Lee and Iwasaki, 2007; Lipscomb and Masten, 2002). DCs recognize microbial pathogens and other inflammatory stimuli through the expression of innate immune receptors including the Toll-like receptor (TLR) family (Akira and Takeda, 2004; Amati et al., 2006; Barton and Medzhitov, 2002). DC activation by TLR signaling initiates a complex set of transcriptional and translational events that are characterized by the up-regulation of surface co-stimulatory molecule expression, inflammatory cytokine secretion, and the ability to stimulate T lymphocytes via antigen presentation by major histocompatibility (MHC) molecules.
TLR stimulation initiates a shift in DC metabolism characterized by upregulation of aerobic glycolysis, which plays a vital role in supporting the immune effector function and survival of both human and mouse DCs (Amiel et al., 2012; Amiel et al., 2014; Everts et al., 2012; Krawczyk et al., 2010). Rapid glycolysis induction supports the metabolic requirements associated with the high levels of protein synthesis that contribute to DC immune activity. The TLR-mediated “glycolytic burst” drives de novo fatty acid synthesis via glucose-dependent citrate metabolism, which supports the synthesis and secretion of inflammatory cytokines (Amiel et al., 2014; Rehman et al., 2013). Interrupting the glucose-to-citrate pathway significantly impairs DC maturation, cytokine secretion, and T cell stimulatory capacity (Amiel et al., 2014; Everts et al., 2012; Krawczyk et al., 2010).
Immune cells are thought to primarily support activation-associated glycolysis via increased expression of glucose transporters (Everts and Pearce, 2014; Fox et al., 2005; Pearce and Everts, 2015; Pearce and Pearce, 2013). Consistent with this, the role of the inducible glucose transporter, GLUT1, in regulating activation-associated glucose flux in both myeloid and lymphoid immune cells has been a major focus in the field (Freemerman et al., 2014; Macintyre et al., 2014). In DCs however, GLUT1 upregulation occurs several hours after TLR stimulation, while TLR-mediated glycolytic reprogramming happens within minutes of activation. Thus, the source of glucose supporting the earliest events in DC activation, namely whether glucose is sourced from the extracellular environment or from intracellular pools, has not been fully defined. We propose that the DCs utilize intracellular glycogen reserves to fuel their metabolic needs during early immune activation and that glycogen metabolism is required by these cells to initiate proper immune effector responses.
Glycogen, a large branch-chained glucose polymer, has been extensively characterized in hepatocytes, muscle cells, and neuronal tissue where it serves as an intracellular carbon reservoir (Adeva-Andany et al., 2016; Roach et al., 2012; Voet et al., 2013). By expressing tissue-specific enzymes for glycogen synthase (GYS) and glycogen phosphorylase (PYG), rate-limiting enzymes of glycogen synthesis and break down respectively, cells in the liver, muscle, and brain store glucose in the form of glycogen to be utilized according to their specific metabolic demands (Adeva-Andany et al., 2016; Roach et al., 2012; Voet et al., 2013). During glycogenolysis, PYG isozymes break down glycogen into glucose-1-phosphate (G1P), which is subsequently converted into glucose-6-phosphate (G6P) and can serve as a direct substrate for further catabolism via glycolysis. In this manner, glycogen-storing cells, such as those in muscle and brain tissue, can maintain intracellular glycogen reserves for cell-intrinsic metabolic requirements (Adeva-Andany et al., 2016; Voet et al., 2013). The significance of cell-intrinsic glycogen metabolism in immune cells has not been well-characterized.
We demonstrate that DCs express specific isoforms of enzymes essential for glycogen synthesis and breakdown and that these cells require glycogen metabolism to support their immune function. Although the presence of glycogen in DCs has been previously implicated (Maroof et al., 2005), the direct role for glycogen in DC metabolism and immune function has not been described. We propose that DCs use intracellular glycogen reserves to support early glycolytic metabolism that accompanies their activation. We show that disruption of glycogen metabolism significantly impairs DC maturation and immune effector function, particularly at early stages of activation and in glucose-restricted conditions. We further show that glycogen-derived carbons preferentially contribute to the TCA-dependent citrate pool compared to glucose catabolized directly by the cell. These findings elucidate a novel metabolic regulatory pathway in DCs, and provide new insights into energy and nutrient homeostasis in these cells in support of their immune activation.
Results and Discussion
DCs express glycogen metabolic machinery and utilize cell-intrinsic glycogen metabolism upon activation
TLR stimulation drives DCs to undergo glycolytic reprogramming in order to meet cellular anabolic demands associated with activation (Amiel et al., 2014; Krawczyk et al., 2010). We performed a nutrient screening assay using single-carbon-source defined media and found that DCs can catabolize both short- and long-chain glucose polymers (Fig 1A). The ability of DCs to generate NADH from glycogen (Fig 1A) is of particular interest given its role as the predominant form of glucose macromolecule storage in normal physiology. While cells are unlikely to encounter extracellular glycogen in vivo, these assays demonstrate that DCs exhibit the capability to catabolize glycogen and are likely to express the key enzymes of glycogen metabolism.
Figure 1. DCs utilize intracellular glycogen metabolism upon LPS stimulation.
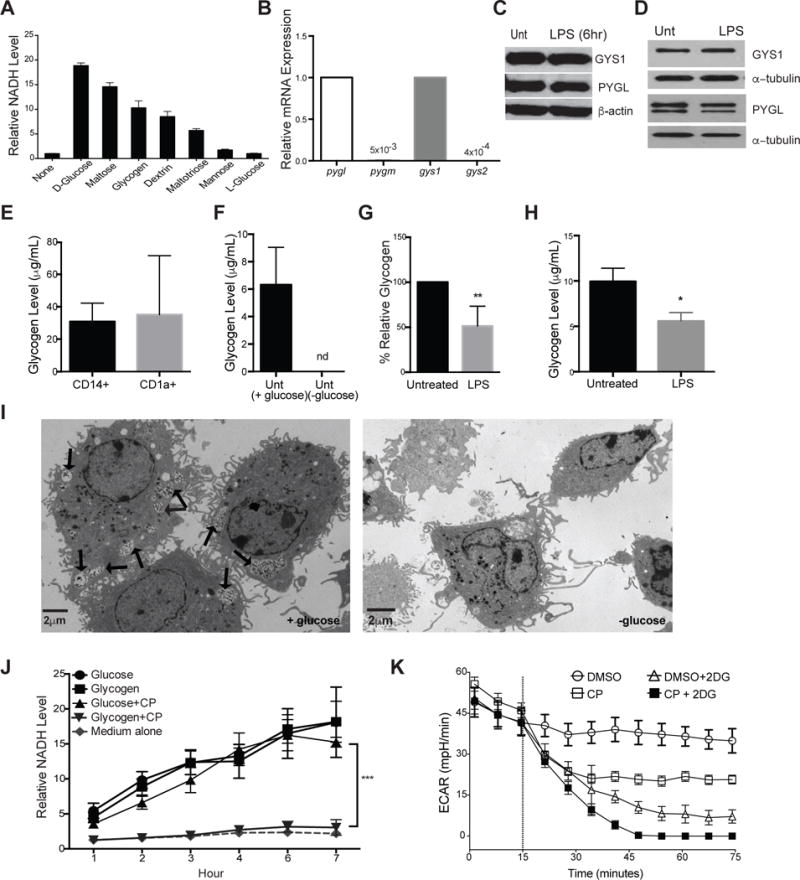
(A) BMDCs were cultured in the indicated substrates as the sole nutrient sources and measured for ability to produce NADH as described in the methods. Data indicate relative NADH production at 6 hrs normalized to no carbon source controls, n=3. (B) Relative mRNA expression of pyg and gys isoforms in naive BMDCs. (C–D) PYGL, GYS1, and β-actin protein expression in unactivated and 6hr LPS-stimulated BMDCs (C) and 24hr LPS-stimulated moDCs (D). (E–H) Intracellular glycogen levels of: human peripheral blood CD14+ monocytes and CD1a+ DCs (E), untreated BMDCs cultured overnight in ±glucose (F), BMDCs (G) and moDCs (H) stimulated ±LPS in 5mM glucose (n=3–6, mean ±SD, student’s t-test, *P<0.05, ** P=0.0021, nd= not detected). Glycogen levels were normalized to 10ˆ5 cells. (I) TEM images of unactivated BMDCs in 5mM glucose (left) and 0mM glucose (right), with arrows indicating intracellular glycogen deposits identified by tannic acid staining. (J) NADH levels over time in BMDCs cultured in glucose or glycogen containing media (as in A) ±CP (n=4, mean±SD, ***P <0.0001). (K) Basal ECAR of resting BMDCs treated with CP, 2DG, or both (treatment introduced at dotted line), representative of at least 3 replicates.
We analyzed mRNA levels of glycogen phosphorylase (PYG) and glycogen synthase (GYS), the rate-limiting enzymes of glycogen breakdown and synthesis pathways, respectively in DCs. Glycogenolysis is executed by three different tissue-specific PYG isozymes in mice and humans: PYGL in the liver, PYGM in muscle, and PYGB in brain tissue. Glycogen synthesis is controlled by two different tissue-specific GYS isozymes: GYS1 in muscles and other peripheral tissue, and GYS2 in the liver. Both mRNA and protein analysis in BMDCs (Fig 1B, 1C) and moDCs (Fig 1D) showed that DCs express PYGL and GYS1 isozymes. These enzymes were not appreciably regulated following 6hr stimulation with LPS (Fig 1C, 1D). Detection of intracellular glycogen in freshly isolated human CD14+ monocytes and CD1a+ dendritic cells (Fig 1E) indicates a physiological role for glycogen in these cells. Unactivated DCs contain intracellular glycogen pools that are fully depleted when cells are cultured in glucose-free media (Fig 1F), and partially depleted by LPS stimulation in BMDCs (Fig 1G) and moDCs (Fig 1H). TEM images of BMDCs show distinct glycogen deposits by tannic acid stain that are absent in cells grown without glucose (Fig 1I) (Afzelius, 1992).
To validate the efficacy and specificity of the PYG inhibitor, CP91149 (CP), we incubated BMDCs with CP in the nutrient screening assay (as in Fig 1A) and assessed inhibition of glucose or glycogen catabolism. Glycogen-dependent NADH levels were fully attenuated in the presence of CP, while glucose-dependent NADH levels were unaffected (Fig 1J), demonstrating the specificity of this inhibitor. PYG inhibition caused a reduction in basal glycolysis rates in unactivated BMDCs (Fig 1K), indicating that DCs utilize intracellular glycogen to support basal glycolytic demands. Importantly, the effect of glycolysis inhibitor 2-deoxyglucose (2DG) was non-redundant with CP, showing that free glucose and intracellular glycogen stores make distinct contributions to DC metabolism (Fig 1K).
PYGL inhibition impacts DC survival in hypoglycemic conditions
Glycogen metabolism supports cancer cell growth, proliferation, and cellular lifespan (Favaro et al., 2012). We tested the effect of PYG inhibition on the survival of BMDCs at early (6hrs) and late (24hrs) time points after LPS activation. PYG inhibition resulted in modest increases in cell death at early time points under low-glucose conditions (Fig 2A). This phenotype was increased after 24hrs of inhibition (Fig 2B). In contrast, the viability of human moDCs was not impacted at all glucose concentrations tested (Fig 2C).
Figure 2. Glycogen metabolism supports survival and early maturation of TLR-activated DCs.
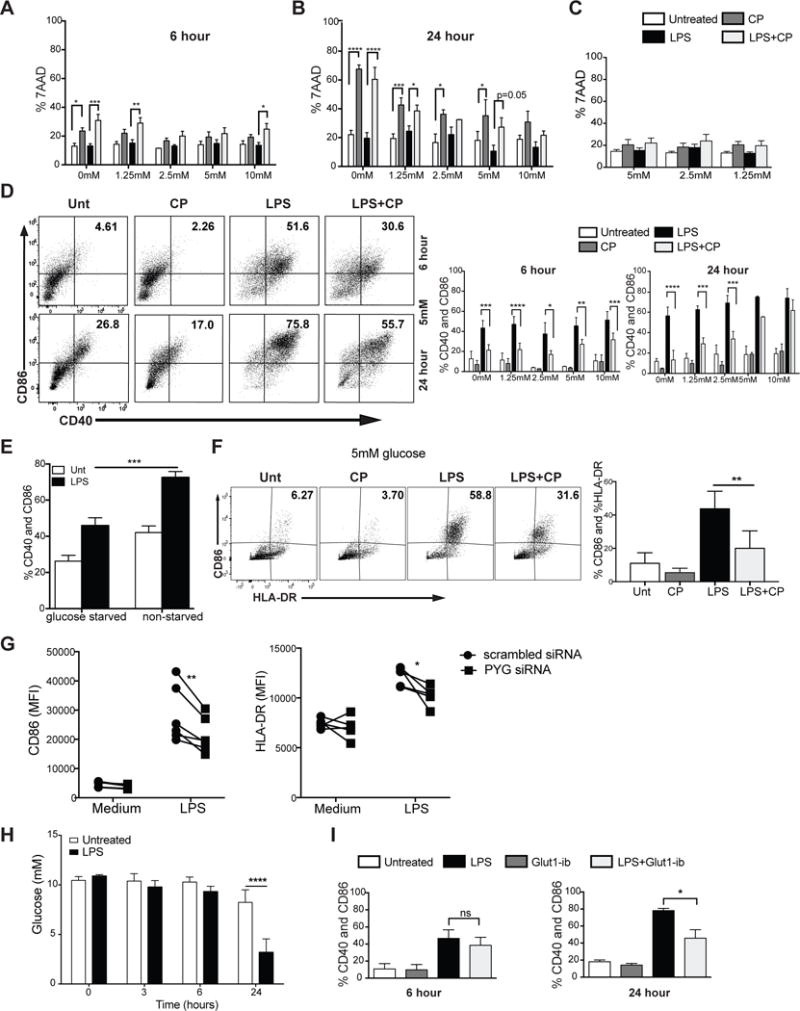
(A–B) 7AAD viability staining of BMDCs stimulated with LPS ± CP for 6hrs (A) and 24hrs (B) at 5mM glucose. (C) 7AAD viability staining of moDCs stimulated with LPS ± CP for 24hrs. (D) BMDCs were stimulated for 6 and 24 hours and analyzed for CD40 and CD86 surface expression. (E) CD40 and CD86 expression of BMDCs stimulated for 6 hrs in free glucose medium with and without glucose starvation. (F) CD86 and HLA-DR expression of moDCs stimulated with LPS±CP for 24hrs in 5mM glucose. (G) CD86 and HLA-DR surface expression of 24hr LPS-stimulated moDCs silenced with control (scrambled) or PYG-targeted siRNA. (H) Glucose measurements from supernatant of BMDCs stimulated with LPS for 3, 6, and 24 hrs. (I) CD40 and CD86 surface expression of BMDCs stimulated ±GLUT1-inhibitor in normal glucose for 6 and 24hr. (A–F, H–I) n=3–6, mean±SD, Two-way ANOVA with Tukey Post-test, *P≤0.05 ***P=0.0006 ****P<0.0001. (G) n=5, Paired t-test, *P=0.04, **P=0.0093.
Glycogen metabolism preferentially supports early DC maturation
TLR-driven early glycolytic burst is a metabolic hallmark of activated DCs (O’Neill, 2014), and both lymphoid and myeloid cells depend heavily on extracellular glucose for glycolysis-dependent effector responses (Everts et al., 2012; Krawczyk et al., 2010; Pearce and Pearce, 2013; Pearce et al., 2009). This may pose a limitation on the abundance of glucose in highly inflamed tissues and secondary lymphoid organs where DCs likely experience nutrient competition with proliferating lymphocytes (Lawless et al., 2017). We hypothesized that glycogen metabolism supports early TLR-mediated glycolysis and activation in DCs by providing an intracellular source of glucose carbons. We examined the surface expression of CD40 and CD86 in BMDCs stimulated with LPS for 6 and 24hrs in the presence or absence of PYG inhibitor over a range of glucose concentrations representing both hyper- and hypoglycemic states. CD40 and CD86 expression was attenuated by CP treatment (Fig 2D and S1a), with a more pronounced effect at 6hrs and in hypoglycemic conditions (Fig 2D and S1a). PYG inhibition with an alternative inhibitor, DAB, at 6hrs after stimulation gave similar outcomes (Fig S1b). Reduced CD40 and CD86 expression was observed both in BMDCs starved of intracellular sugar (Fig 2E) and in moDCs inhibited by CP (Fig 2F) further suggesting a role for glycogen pools in sustaining DC maturation. In addition, PYG-targeted siRNA was used to silence PYG expression in moDCs. As mRNA expression data indicated that both PYGB and PYGL isoenzymes are expressed in human moDCs (Fig S1c), both isoforms were silenced simultaneously in moDCs using targeted siRNA (Fig S1d). Genetic silencing of PYG in moDCs resulted in attenuation of LPS-induced expression of maturation markers (Fig 2G).
While the importance of GLUT1 has been conclusively delineated in both myeloid and lymphoid immune cells (Amiel et al., 2014; Freemerman et al., 2014; Macintyre et al., 2014; Wieman et al., 2007), the kinetics of GLUT1 regulation do not account for the acute glycolytic reprogramming that occurs in activated DCs. GLUT1 upregulation in activated DCs is not detected before 6 hours of LPS stimulation (Fig S1e), which correlates with the finding that extracellular glucose is depleted only after 6hrs of stimulation (Fig 2H). To confirm that the cells are less dependent on imported glucose for early activation, we assessed DC maturation at 6 and 24hrs after LPS stimulation while blocking GLUT1 activity with inhibitor STF31 (Fig 2I and S1f). In contrast to PYG inhibition, GLUT1 inhibition had a significant impact on the maturation at 24hrs but not 6hrs after activation. These data provide strong evidence that cell-intrinsic glycogen metabolism plays a central role in driving DC maturation, particularly during early time points and in glucose-restricting conditions.
PYG inhibition impacts DC immune effector function
Blocking glycolysis in TLR-activated DCs impairs their ability to produce inflammatory cytokines and stimulate T cells (Amiel et al., 2014; Krawczyk et al., 2010). We tested whether these responses are also affected by PYG inhibition. Intracellular cytokine staining for TNF-α and IL-12 after 4hrs of LPS stimulation showed that PYG inhibition attenuates inflammatory cytokine production, with a larger effect in low-glucose conditions (Fig 3A–B, S2a). Multiplex cytokine analysis of LPS-stimulated DCs showed reduced proinflammatory cytokines and chemokine production in PYG-inhibited cells compared to controls in both BMDCs (Fig 3C) and moDCs (Fig 3D). CP did not globally impact all LPS-mediated protein production as other cytokines were unaffected (Fig S2b–c). siRNA-mediated knockdown of PYG expression in moDCs recapitulated the inhibitor data, as LPS-driven IL-12 production was attenuated in PYG-silenced moDCs (Fig 3E).
Figure 3. PYGL inhibition attenuates immune effector functions of DC.
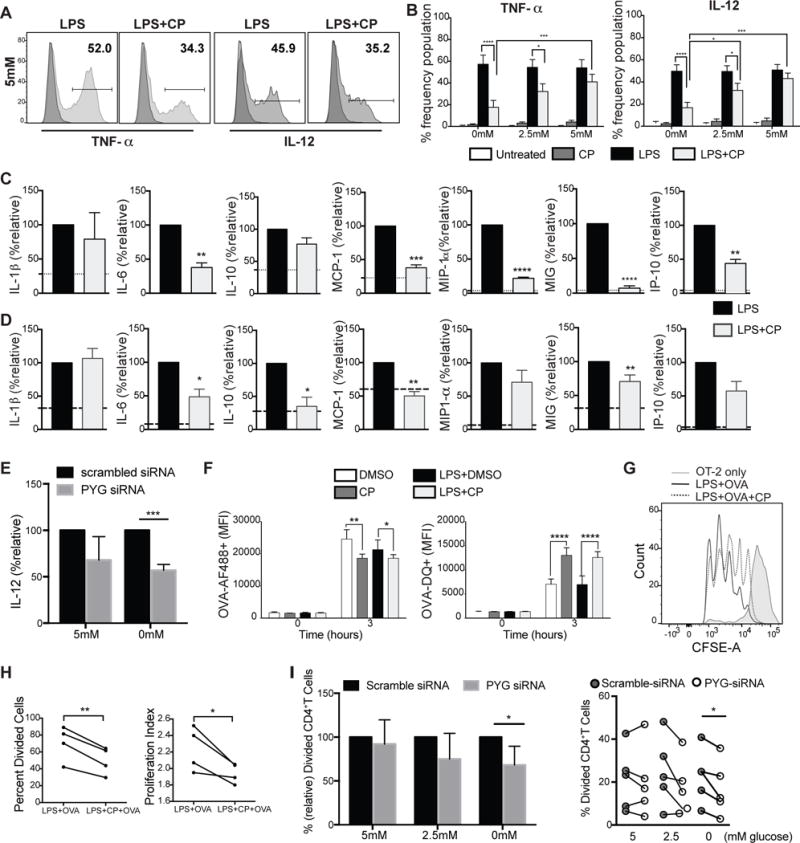
(A–B) Intracellular staining of TNF-α and IL-12 of BMDCs stimulated with LPS for 4 hrs in 5mM glucose. (C–D) Multiplex panels of cytokine and chemokine measurements from the supernatant of BMDCs (C)and moDCs (D)activated with LPS for 6 hrs. Dotted lines represent unstimulated levels. (E) Relative IL-12 production by moDCs LPS-stimulated for 24hrs transfected with control or PYG-targeted siRNA. (F) BMDCs treated with LPS ±CP plus OVA-AF488 or OVA-DQ for 3 hours and analyzed by flow cytometry for antigen uptake and processing. (G) BMDCs were pulsed for 6 hours with indicated treatments and subsequently co-cultured with CFSE-labeled OT-II Tcells. CFSE dilution was measured on day 3 post co-culture. (H) Measurements of proliferation of OT-II T cells (from (G)) stimulated by BMDCs pre-treated with indicated conditions. (I) si-RNA transfected moDCs were co-cultured with CellTrace Violet –labeled human naïve CD4+T cells for 4 days. Data were normalized to scrambled siRNA. Proliferation was measured after 4 days. (A–I) n=3–5, mean±SD, (B, F) 2-way ANOVA Tukey Posttest. (C–E, H–I) student’s t-test, *P≤0.05, **P<0.001, ***P=0.0004, ****P<0.0001.
To examine the ability of DCs to take up and process antigens, we stimulated BMDCs with LPS plus OVA-AF488 or OVA-DQ for 3 hours (Fig 3F). OVA-AF488 allows tracking of antigen uptake, while OVA-DQ only fluoresces upon antigen uptake and processing. PYG-inhibited DCs showed reduced antigen uptake regardless of LPS stimulation (Fig 3F while antigen processing was unexpectedly enhanced by PYG inhibition. We next tested the effect of PYG inhibition or silencing on DC ability to stimulate CD4+ T cells. PYG-inhibited BMDCs exhibited significantly reduced capacity to stimulate T cells (Fig 3G and 3H). PYG-silenced moDCs exhibited similar impairments in CD4+ T cell stimulation (Fig 3I). These data demonstrate that cell-intrinsic glycogen metabolism contributes to the regulation of the multifaceted dimensions of DC immune effector function.
Glycogen-derived carbons fuel both glycolytic reprogramming and mitochondrial respiration in activated DCs
We proposed that glycogen-derived glucose drives early glycolytic flux in TLR-activated DCs prior to GLUT1 upregulation. To test this, we performed a real-time extracellular flux analysis on BMDCs and moDCs. LPS-driven glycolytic burst was significantly attenuated by PYG inhibition in both BMDCs (Fig 4A, S3a) and moDCs (Fig 4B). We further tested whether cell-intrinsic glycogen metabolism also contributes to mitochondrial respiration during early activation. Concomitant with glycolysis reduction, PYG inhibition attenuated the oxygen consumption rate (OCR) in BMDCs regardless of activation (Fig 4C). These data suggest that pre-existing glycogen pools contribute metabolic substrates for mitochondrial respiration. Consistent with this, PYG inhibition accelerates LPS-mediated ATP depletion during early activation in a time-dependent manner (Fig 4D). The synergistic effect of combined CP and ATP-synthase inhibitor oligomycin in reducing ATP production (Fig 4E) indicates that glycogen catabolism contributes to both cytosolic and mitochondrial ATP generation. These findings indicate the intriguing possibility that there may be distinct roles for glucose and glycogen-derived carbon molecules in DC metabolism.
Figure 4. Glycogen-derived carbons fuel early glycolytic reprogramming and mitochondrial respiration in activated DCs.
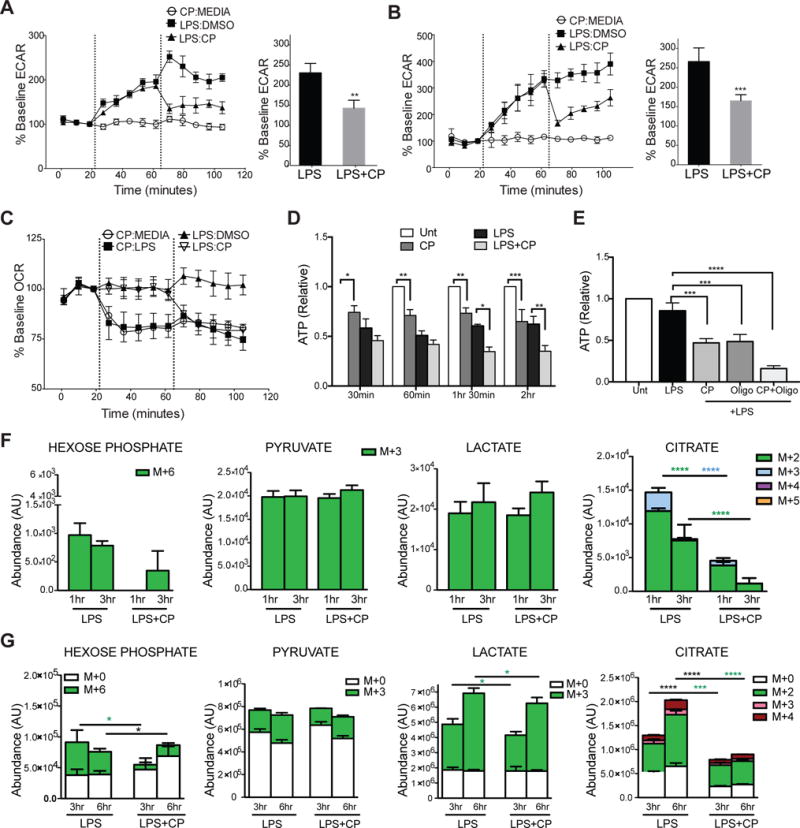
(A–B) Real-time changes in ECAR of BMDCs (A) and moDCs (B). (C) Real-time changes in OCR of BMDCs. (A–C) treatments introduced at dotted lines (1st injection: 2nd injection). (D–E) ATP levels of BMDCs in 30 minute intervals (D) and at 2 hrs (E) after stimulation with indicated treatments. (F) BMDCs cultured and differentiated in 13C6-glucose were switched to normal glucose at the time of stimulation with LPS±CP for 1 and 3 hours and 13C-labeled metabolites were detected by LC-MS spectrometry. (G) Inverse metabolomics of F, where BMDC were differentiated in normal 12C glucose and switched to 13C6-glucose at the time of stimulation with LPS±CP for 3 and 6hrs. Data represent n=4, mean ±SD, (A–B) paired students t-test. (D–G) n=5, Two-way ANOVA, Tukey Posttest. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001. (F–G) Statistical significance of each color * represents color-coded 13C6- or black * for 12C groups. White bars indicate l2C-glucose and all color bars denote 13C6-glucose.
Since PYG inhibition resulted in reduced intracellular ATP levels (Fig 4D & E), we assessed the effect of PYG inhibition on the activation of AMPK, a key metabolic sensor of intracellular nutrient and ATP levels (Hardie et al., 2012). PYG inhibition resulted in increased phosphorylation of AMPK (Fig S3b), which is reported to antagonize BMDC activation (Krawczyk et al., 2010). This is consistent with reports showing that inhibition of glycolysis induces compensatory activation of AMPK (Wang et al., 2011; Wu et al., 2015). However, PYG inhibition had no impact on LPS-mediated GLUT1 upregulation (Fig S3c), suggesting that AMPK regulation of glucose transport is not a significant mechanism at play in our model. Nevertheless, LKB1 deficient BMDCs, which are incapable of activating AMPK, show decreased sensitivity to PYG inhibition during maturation at normal glucose concentrations, suggesting that AMPK compensatory activation during PYG inhibition may be involved in regulating maturation in these conditions (Fig S3d).
Previous work has demonstrated that glucose consumed by activated DCs enters the TCA cycle to generate citrate, which is preferentially translocated from the mitochondria into the cytosol via the citrate shuttle to support de novo fatty acid synthesis. This process is linked to ER and Golgi membrane expansion, which is hypothesized to enhance the production of effector molecules central to DC activation (Amiel et al., 2014; Rehman et al., 2013). To examine the role of glycogenolysis in citrate metabolism explicitly, we performed metabolic tracing experiments in which BMDCs were differentiated in 13C-labeled glucose to label all intracellular metabolites. Cell were subsequently switched to normal glucose at the time of LPS stimulation in the presence or absence of CP for 1 and 3hrs. As previously published, LPS stimulation induces substantial metabolic flux through glycolysis and TCA citrate production ((Everts et al., 2014) and data not shown). PYG inhibition significantly reduced 13C-labeled citrate while no statistically significant impact on hexose phosphate, pyruvate, lactate, and post-citrate metabolites fumarate and malate was observed (Fig 4F and S3e). Hexose phosphate refers to any 6-carbon sugar since our metabolite tracing approach could not distinguish individual sugars among this group. These data indicate that intracellular glycogen reserves preferentially support the generation of citrate following LPS stimulation.
Glutamine can also serve as an important carbon source for the TCA cycle. However, the findings that nearly the entire glutamine pool is derived from 12C-labeled sources (Fig S3f) and that CP has very little effect on glutamine levels (Fig S3f), suggests that glutamine metabolism is not directly impacted by PYG inhibition. This is further supported by observations that: 1) CP attenuates the maturation of BMDCs stimulated in the presence or absence of glutamine (Fig S3g); and 2) glutaminolysis inhibitor DON has no significant impact on glycolytic burst or OCR (Fig S3h).
To identify the role of glycogen metabolism in regulating extracellular glucose flux, the reverse metabolomics experiment was performed in which BMDCs differentiated in normal glucose were switched to 13C-glucose at the time of LPS activation and analyzed at 3 and 6 hours post stimulation (Fig 4G). PYG inhibition minimally affected the 13C-glucose contribution to cytoplasmic hexose phosphate, lactate, and pyruvate, while it severely attenuated both 12C and 13C-glucose contributions to citrate production (Fig 4G).
The metabolite tracing data are consistent with previously published work (Everts et al., 2014), in which extracellular glucose contributes heavily to cytoplasmic glycolytic metabolites and citrate production from the TCA cycle (Fig 4F–G). However, these data also uncover two previously unappreciated aspects of glucose metabolism in DCs: 1) glycogen-derived carbons from basal glycogen stores (CP-sensitive 13C metabolites in Fig 4F) preferentially support initial glycolytic intermediates and citrate synthesis; 2) a significant amount of glucose imported from the extracellular environment gets rapidly converted into glycogen (CP-sensitive 13C metabolites in Fig 4G). The finding that extracellular 13C-glucose incorporation into citrate (Fig 4G), succinate, fumarate, and malate (Fig S3i) is sensitive to PYG inhibition suggests that a significant portion of extracellular glucose destined for mitochondrial oxidation is metabolically routed via a glycogen-dependent pathway during DC activation. The routing of glucose carbons via a rapid sequence of glycogen synthesis and glycogenolysis is characteristic of a metabolic pathway described in astrocytes and muscle cells as the “glycogen shunt” (Shulman et al., 2001; Shulman and Rothman, 2001). Our metabolic profiling studies support a model where glucose processing in TLR-stimulated DCs undergoes three functionally distinct pathways: 1) the catabolism of pre-activation intracellular glycogen stores; 2) the catabolism of imported glucose directly; 3) the incorporation of imported glucose into synthesis and breakdown of glycogen via the glycogen shunt (modeled in Fig S4).
While the glycogen shunt is clearly inefficient from an energetic perspective, others have argued that glycogen breakdown and synthesis may occur in separate spatial pools within brain and muscle cells to fuel rapid bursts of metabolic activity required in these cells that override the total energetic cost of this process (Calder and Geddes, 1992; Elsner et al., 2002; Obel et al., 2012). DCs may employ a similar strategy of compartmentalized glycogen metabolism in order to fuel early immune activation. However, how this occurs and how it may be regulated in DCs remains an important question. Precedent for distinct and parallel sugar metabolism has been previously reported, whereby granulocyte phagocytic capability is driven by glycogen-derived carbons, while their chemotaxis is fueled by catabolism of free glucose carbons (Weisdorf et al., 1982). We propose that the source of carbons in activated DCs, namely whether it is glucose or glycogen –derived, may dictate differential functional responses. We speculate that spatial compartmentalization of these processes in the cytoplasm may be an important component of how glycogen metabolism is regulated.
While glycogen metabolism has been previously implicated in myeloid cells of the immune system (Maroof et al., 2005; Scott, 1968; Weisdorf et al., 1982; Yunis and Arimura, 1964, 1966), the role of glycogen metabolism in specific immune effector functions of DCs has not been previously defined. We show here a definitive role for glycogen metabolism in regulating immune effector functions of both human and mouse DCs. We further demonstrate that glucose- and glycogen-derived carbons exhibit distinct metabolic fates, a phenomenon that we suspect is not DC-specific and likely occurs in other cells that utilize cell-intrinsic glycogen metabolism. Ongoing studies are focused on elaborating the mechanistic details of how glycogen-dependent compartmentalization of metabolic pathways occurs in response to different immune stimuli. With a growing interest in understanding how metabolic regulation controls the functional effector responses of immune cells, this work delineates an intricate and novel layer of complexity to how metabolic pathways operate at a subcellular level, which may be exploited in cell-based therapeutic applications in the future.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-alpha tubulin (clone DM-1A) | Abcam | Cat#ab7291 |
| PYGL rabbit polyclonal | Proteintech | Cat#15851-1-AP |
| Rabbit monoclonal anti-GYS1 (clone: EP817Y) | Abcam | Cat#ab40810 |
| Rabbit polyclonal Anti-Glut1 (AF488) | Novusbio | NB110-39113AF488 |
| Hexokinae I (C35C4) Rabbit mAb | Cell Signaling | Cat#2024 |
| Hexokinase II (C64G5) Rabbit mAb | Cell Signaling | Cat#2867 |
| PKFP (D4B2) Rabbit mAb | Cell Signaling | Cat#8164 |
| Pyruvate Dehydrogenase (C54G1) Rabbit mAb | Cell Signaling | Cat#3205 |
| AMPKα (D63G4) Rabbit mAb | Cell Signaling | Cat#5382 |
| Phospho-AMPKα (Thr172) (40H9) Rabbit mAb | Cell Signaling | Cat#2535 |
| Goat anti-mouse IgG-HRP | Santa Cruz | Cat#sc-2005 |
| PECy7 Anti-mouse CD11c (Clone: N418) | BioLegend | Cat#117318 |
| FITC Anti-mouse CD40 (Clone: 3/23) | BioLegend | Cat#124068 |
| Pacific Blue Anti-mouse CD86 (Clone: GL-1) | BioLegend | Cat#105022 |
| PE Anti-mouse IA-b (Clone: AF6-120.1) | BD Biosciences | Cat#553552 |
| PE-Cyanine Anti-mouse TNF-α (Clone: MP6-XT22) | eBiosciences | Cat#25-7321-80 |
| APC Anti-mouse IL-12 (Clone: C15.6) | BioLegend | Cat#505205 |
| Miltenyi human CD14 beads | Miltenyi | Cat#130-050-201 |
| Miltenyi mouse CD4 beads (L3T4) | Miltenyi | Cat#130-049-201 |
| Pacific Blue anti-mouse CD4 (Clone: GK1.5) | BioLegend | Cat#100427 |
| FITC anti-human CD1a (Clone: HI 149) | BioLegend | Cat#300103 |
| APC anti-human CD40 (Clone: 5C3) | BioLegend | Cat#334309 |
| Pacific Blue anti-human CD86 (Clone: IT2.2) | BioLegend | Cat#305417 |
| PE HLA-DR (Clone: L24.3) | BioLegend | Cat#307605 |
| FITC Mouse Anti-human CD86 (Clone 2331) | BD Biosciences | Cat#555657 |
| Human FC receptor Binding Inhibitor | eBiosciences | Cat#14-9161 |
| APC Mouse Anti-human CD40 (Clone 5C3) | BD Biosciences | Cat#555591 |
| APC Mouse Anti-human HLA-DR-eF780 | eBiosciences | Cat#47-9956 |
| V450 Mouse Anti-human CD80 (Clone: L307.4) | Bd Biosciences | Cat#560442 |
| FITC Mouse Anti-human CD4 (Clone: RTA-T4) | BD Biosciences | Cat#555346 |
| APC Mouse Anti-human CD3 (Clone: SK7) | BD Biosciences | Cat#641397 |
| ELISA ab: Purified Rat Anti-human IL-12p70 | BD Biosciences | Cat#555065 |
| ELISA ab: Biotin Mouse Anti-human IL-12p40/p70 | BD Biosciences | Cat#554660 |
| Human Naïve Pan T cell isolation kit | Miltenyi | Cat#130-097-095 |
| Human CD8 Microbeads | Miltenyi | Cat#130-045-201 |
| Biological Samples | ||
| Human Peripheral blood LeucoPak | CVPH Medical Center Blood Bank, Plattsburg, NY | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Endotoxin free LPS (Escherichia coli Serotype O) | InvivoGen | Cat#tlrl-eblps |
| Glycogen from Bovine Liver | Sigma Aldrich | Cat#G0885-5G; CAS: 9005-79-2 |
| CP-91149 | Selleckhem | Cat#S2717 |
| 1,4-Dideoxy-1,4-imino-D-arabinitol hydrochloride (DAB) | Santa Cruz | Cat#220553; CAS 100991-92-2 |
| STF31-glut1 inhibitor | Tocris Biosciences | Cat#4484; CAS 724741-75-7 |
| D-Glucose-13C6 | Sigma Aldrich | Cat#389374 |
| Golgi plug: Protein Transport Inhibitor | BD Biosciences | Cat#51-2301KZ |
| Recombinant murine GM-CSF | Peprotech | Cat#315-03 |
| 7-AAD | BD Pharmingen | Cat#51-68981E |
| Recombinant Human GM-CSF | Peprotech | Cat#300-03 |
| Recombinant Human IL-4 | Peprotech | Cat#200-04 |
| Taqman Fast Advanced Master Mix | Applied Biosystems | Cat#4444557 |
| OVA-AF 488 | Molecular Probes | Cat# O-34781 |
| OVA-DQ | Molecular Probes | Cat# D-12053 |
| Biolog Inc redox dye mix MA | Biolog Inc. | Cat#NC0781517 |
| CellTrace Violet | Thermo Fisher | Cat#C34557 |
| StrepAvidin Poly HRP | Sanquin | M1942 |
| DON (6-diazo-5-oxo-L-norleucine) | Sigma Aldrich | D2141 |
| Critical Commercial Assays | ||
| Glucose Assay | Eton Biosciences | Cat#SKU120003400 |
| Glycogen Assay Hydrolysis Enzyme | Biovision | K646-100-5 |
| Seahorse Bioassay | Agilent Technologies | Cat#102416 |
| ATP Determination Kit | Invitrogen | Cat#A22066 |
| iScript cDNA synthesis Kit | Biorad | Cat#170-8891 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Mouse Cytokine Magnetic 20-Plex Panel | Invitrogen | Cat#LMC0006M |
| Human Cytokine Magnetic 30-Plex Panel | Invitrogen | Cat#LHC6003M |
| PM-M1 Panel: Phenotype Microarrays | Biolog Inc. | Cat#13101 |
| Experimental Models: Organisms/Strains | ||
| C57/Bl6J | Jackson Laboratory | N/A |
| B6.Cg-Tg (TcraTcrb)425Cbn/J | Jackson Laboratory | N/A |
| Software and Algorithms | ||
| FlowJo Software | TreeStar | N/A |
| Prism V 7.0 | Graphpad Prism | N/A |
| Other | ||
| Neon Transfection System | Invivogen | MPK5000 |
| PYGL human si-RNAs | Dharmacon | M-009569-02-0005 5 |
| PYGB human si-RNAs | Dharmacon | M-009587-01-0005 5 |
| pygl Taqman Gene Expression Assay primer | Thermo Fisher | Mm01289790-m1 |
| pygm Taqman Gene Expression Assay primer | Thermo Fisher | Mm00478582-m1 |
| gys1 Taqman Gene Expression Assay primer | Thermo Fisher | Mm01962575-s1 |
| gys2 Taqman Gene Expression Assay primer | Thermo Fisher | Mm01267381-g1 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Eyal Amiel (eamiel@uvm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
The University of Vermont’s Animal Facility is a barrier facility housing only mice. All animals are housed in autoclaved Lab Products™ microisolator cages on ventilated racks and handled using aseptic technique in laminar flow work stations and are provided with sterile water and irradiated rodent chow (Lab Diets™ Isopro RMH 3000). Mice are maintained in a pathogen free environment at a constant temperature and humidity, with 12-hour light and 12-hour dark cycle. Personnel wear shoe covers, isolation gowns, masks, bouffant and exam gloves. In addition, animal husbandry personnel wear dedicated scrubs and footwear. Health monitoring of colony and sentinel animals is performed quarterly. The University’s program of animal care has been fully-accredited by AAALAC, International for over 25 years. OT-II (B6.Cg-Tg (TcraTcrb) 425Cbn/J and C57/Bl6J mice mice were purchased from Jackson Laboratory and were maintained at the University of Vermont animals care facility under protocols approved by Institutional Animal Care and Use Committee. For most experiments, adult mice (2–6 months of age) were used. Mouse experiments include data from both male and female mice, however the specific sex distribution for each individual experiment was not explicitly tracked. Itgaxcre LKB1fl/fl mice PubMed: 21124450 were housed and bred at the LUMC, Leiden, Netherlands, under SPF conditions. All animal experiments were performed in accordance with local government regulations, and the EU Directive 2010/63EU and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes and approved by the CCD, animal license number AVD116002015253.
METHOD DETAILS
Mouse DC culture and Activation
Bone marrow-derived DCs (BMDCs) were generated as follows: BM cells were flushed from femurs of 9–18-week-old mice and the cells were differentiated in GM-CSF (20ng/mL; Peprotech) in complete DC medium (CDCM), comprised of RPMI1640, 10% FCS, 2mM L-glutamine, 1IU/mL Pen-Strep, 1mM beta-mercaptoethanol, for 7 days, with a medium change every 2 days. On day 7, DCs were washed in CDCM and cultured at 2×10ˆ5 cells per 200μL of media alone, STF31 (12.5 μM), CP91149 (75–100μM), DAB (1mM), LPS (100ng/mL), LPS plus STF31 or CP91149, or DAB, or OVA (from whole egg white) at indicated time points. Where appropriate, DCs were stimulated in CDCM containing 0mM, 1.25mM, 2.5mM, or 5mM glucose.
Glucose starvation experiment
BMDCs were starved for glucose overnight, with a non-starved group as a control. On the next day, DCs from both groups were washed with sugar free RPMI and stimulated with LPS in glucose free medium ±CP for 6hours. CD40 and CD86 expression was analyzed by Flow cytometry.
Human DC culture and activation
Human monocyte-derived DCs (moDCs) were differentiated from peripheral blood monocytes as follows: Blood filters from de-identified blood donors were provided by CVPH Medical Center Blood Bank in Plattsburgh, NY. Filters were reverse-flushed in sterile PBS, and PBMCs were prepared by Ficoll-Paque (density gradient of 1.0772) centrifugal separation using LSM media (MP biochemical; Fisher). Resulting monocytes were enriched using CD14 positive selection beads per manufacturer instructions (Miltenyi Bioscience) and cultured in complete DC medium (CDCM) supplemented with human recombinant GM-CSF (20ng/mL) plus human recombinant IL-4 (20ng/mL) (Peprotech) for 7 days. On day 7, moDC were harvested, stimulated as indicated, and analyzed by FACS for maturation and by multiplex panels (Life Technologies) for cytokine production.
Quantitative Real-time PCR of pygl, pygm, gys1, and gys2 expression
RNA was isolated with an RNAeasy Kit (Qiagen) and cDNA was synthesized with an iScript cDNA Synthesis Kit (Biorad). pygl, pygm, gys1, gys2, and slc2a1 Taqman primer probes (Applied Bioscience system) and AB7500 sequence detection system or QuantStudio 3.0 were used for relative mRNA expression. mRNA relative quantitative values were calculated based on 2(−ΔΔCT) and normalized to untreated samples.
Glycogen phosphorylase knockdown by siRNA transfection of moDC
For knockdown of glycogen phosphorylase isoforms, moDCs were generated as mentioned above. At day 4 of the culture, the cells were harvested, washed with PBS, brought to a concentration of 1×10ˆ6 cells/100 μL resuspension buffer, and finally, transfected by electroporation with either 10 nM anti-PGYL siRNA in combination with 10 nM anti-PYGB siRNA or 20 nM scrambled siRNA (Dharmacon). Electroporation was performed using a Neon Transfection System (Invivogen) with the following settings: 1600 V, 20 ms and one pulse. Immediately after electroporation, 1×10ˆ6 cells were taken up in 5 mL 10% HI-FCS basal media, containing no antibiotics, and plated at 200 cells/μL. The next morning, the media was re-supplemented with penicillin, streptomycin, rGM-CSF and rIL-4. At day 6, the cells were harvested, stimulated as indicated, and analyzed by FACS for maturation and by ELISA for cytokine production. Silencing efficiency was determined by qPCR on 6 day-old cells. The transfection efficiency was routinely greater than 80%.
Antigen Uptake, Processing, and in-vitro T cell responses
BMDCs were stimulated ±LPS with OVA-AF488 (5μg/mL) and OVA-DQ (5μg/mL) for antigen uptake and processing, respectively. For in vitro T cell responses, T cells were generated using mouse CD4 positive selection beads from spleens of 6–10-week-old transgenic OT-II mice and age-matched Wild-type B6 mice. BMDCs were pulsed with whole Ovalbumin protein (OVA), extracted from egg white, and LPS in the presence or absence of CP for 6 hours, washed 3 times, and co-cultured with CFSE-labeled OT-II T cells at a 1:5 ratio for 72 hours. T cell proliferation (CFSE dilution) was analyzed by flow cytometry. For alloreaction studies of siRNA transfected moDC, the cells were washed 2 times, and co-cultured with CellTrace Violet-labelled human naïve CD4+ T cells, which were isolated using a naïve pan T cell isolation kit (Miltenyi) followed by negative selection using CD8 MicroBeads (Miltenyi), at a 1:4 ratio for 4 days. T cell proliferation was analyzed by flow cytometry.
Metabolism assays
Extracellular glucose and intracellular glycogen levels were measured with a Glucose assay kit (Eton Biosciences) and a Glycogen assay kit (Biovision), respectively. For Biolog assays, (Metabolic phenotypic screening assays), IFM-1 reagent, Biolog MA redox dye, and Biolog plates were purchased from Biolog Inc. Fully differentiated BMDC were plated overnight at 50,000 cells per well in specified nutrient sources in basal MC-0 medium (IFM1 media with 5%FCS, 0.3mM L-glutamine, 100I/U Pen Strep). 20μL of Biolog MA dye was added to each well the next morning. The assays were measured at 592 nm absorbance as indicated. Data were normalized to the readings at time 0. Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured with Metabolic Flux Analyzer (Seahorse Bioscience, North Billerica, MA 24XP and/or 96XP). ATP concentrations were measured with an ATP Determination Kit (Invitrogen) according to the manufacturer’s instructions.
BMDC cultures and activation for metabolomics
For metabolomics tracing in Fig 4F, BMDCs were differentiated in 13C6-glucose containing CDCM. On day 7, the cells were switched to 12C6-glucose medium, with LPS and LPS+CP added at the time of media switch and stimulated for 1 and 3 hours. At each time point, cells were harvested, counted, pelleted, and frozen for the UHPLC-MLS metabolomics processing below. For inverse metabolomics in Fig 4G, BMDCs normally differentiated in regular CDCM were switched to 13C6-glucose medium at the time of stimulation, with and without CP for 3 and 6 hours. Cells were harvested and processed as above. Supernatant from the 6 hour stimulation groups was collected for Multiplex Cytokine analysis.
UHPLC-MS Metabolomics
Frozen cell pellets were extracted at 2e6 cells/mL in ice cold lysis/extraction buffer (methanol:acetonitrile:water 5:3:2). Samples were agitated at 4 °C for 30 min followed by centrifugation at 10,000 g for 10 min at 4 °C. Protein and lipid pellets were discarded, and supernatants were stored at −80 °C prior to metabolomic analysis. Ten μL of extracts were injected into an UHPLC system (Vanquish, Thermo, San Jose, CA, USA) and run on a Kinetex C18 column (150 × 2.1 mm, 1.7 μm – Phenomenex, Torrance, CA, USA) at 250 μl/min (phase A: Optima H2O, 0.1% formic acid; phase B: acetonitrile, 0.1% formic acid). The autosampler was held at 7 °C and the column compartment at 25 °C. The UHPLC system was coupled online with a Q Exactive mass spectrometer (Thermo, Bremen, Germany), scanning in Full MS mode (2 μscans) at a 70,000 resolution in the 60–900 m/z range in negative and then positive ion mode (separate runs). Eluate was subjected to electrospray ionization (ESI) with 4 kV spray voltage, 15 sheath gas and 5 auxiliary gas. Metabolite assignments and isotopologue distributions were determined using the software Maven (Princeton, NJ, USA)1, upon conversion of .raw files into .mzXML format through MassMatrix (Cleveland, OH, USA). Chromatographic and MS technical stability were assessed by determining CVs for heavy and light isotopologues in a technical mixture of extract run every 10 injections. Relative quantitation was performed by exporting the values for integrated peak areas of light metabolites and their isotopologues into Excel (Microsoft, Redmond, CA, USA) for statistical analysis including t-test and ANOVA (significance threshold for p-values < 0.05).
Electron Microscopy
Samples were fixed in Karnovsky’s Fixative for 1hr at 4°C, washed in 0.1M Cacodylate Buffer, and post-fixed in 1% OsO4 for 1hr at 4°C followed by an extensive rinse with Cacodylate buffer. Samples were then dehydrated in a graded series of ethanol, and embedded in Spurr. Sections were cut with a Reichert Ultracut Microtome and stained with toluidine blue. For contrast, 1% tannic acid was added to the cut sections of the grids for 10 minutes, followed by 6 minutes of uranyl acetate and 4 minutes of lead citrate. Cells were examined with a JEM1400 transmission electron microscope (JEOL USA).
Immunoblot analysis
Cell lysates were prepared using 2X NP-40 lysis buffer. 20μg protein was loaded into each well of a 12.5% polyacrylamide gel, transferred onto activated nitrocellulose membrane (BioRad). Electrophoretic transfer was performed using Trans-Blot Turbo RTA mini Nitrocellulose transfer kit. Membranes were blocked in 2%milk in 1×TBST at RT for 1hr, and incubated in indicated antibody at 4°C overnight. Blots were washed 3× in 1×TBST at RT, probed with secondary antibodies at RT for 45–60 minutes, and washed 3–4× with 1×TBST. Proteins were visualized by SuperSignal West Pico Chemiluminescent substrate and exposed with GeneXpert System imager. Trans-Blot Turbo Transfer system and secondary antibodies for Western blots were generously provided by Dr. Paula Deming, Medical Laboratory and Radiation Science Department, UVM.
Flow Cytometry and Cytokine measurements
The following fluorescently labeled antibodies were used for flow cytometry: anti-CD11c (N418), anti-CD40 (3/23), anti-CD86 (GL1), IA-b (AF6-120.1), anti-CD1a (HI149), anti-CD40 (5C3), anti-CD86 (IT2.2), anti-TNFα (MP6-XT22), anti-IL-12p40 (C15.6). Stimulated cells as indicated were harvested and washed in 1% FACS buffer (PBS plus 1% FBS), stained with specific antibodies, and incubated on ice for 30 minutes. All samples were acquired using a LSRII flow cytometer (BD Biosciences). For intracellular cytokine expression, cells were activated with indicated treatment groups for a total of 4 hours with an addition of Golgi plug (1:1000) (Biolegend) after the first hour of stimulation. For intracellular staining of TNF-α and IL-12 (Biolegend), cells were fixed in 4% Paraformaldehyde, permeabilized in 0.2% saponin, and stained with antibodies in FACS buffer (1%FBS in PBS). Samples were analyzed on a BD LSRII flow cytometer. For cytokine levels of BMDCs and moDCs, supernatants were collected as indicated time points and measured with Mouse Cytokine Magentic 20-Plex and Human Cytokine Magnetic 30-Plex panels (Life Technologies) per manufacture instructions using Bio-Plex array suspension system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Throughout the manuscript, “n” refers to independent cell cultures from individual mice or human samples. All the experiments reported in Fig 1 were repeated n=3–6 mice per condition, with the following detailed n values: Figure 1A–E and 1I, n=3; Fig 1F–G, n=6; Fig 1H, n=4; and Fig 1J, n=4. The survival and maturation experiments in Fig 2A–F and S1 were repeated with n=6. Intracellular cytokine experiments in Fig 3 and S2 were repeated with n=4. Multiplex analyses in Fig 3 and S2 were repeated with n=6. All metabolomics experiments in Fig 4 and S3 were repeated with n=4, and the seahorse experiments in Fig 1, 4, and S3 were repeated with n=3–5. All the siRNA experiments in Fig 2G, 3I, and S1c–d were repeated with n=5. Data were analyzed with GraphPad Prism software (version 6.0). Samples were analyzed using Student’s t-test, One-way, and Two-way ANOVA where appropriate. ANOVA tests were post-calculated by Tukey’s multiple comparison test or Sidak test. Results are means +SD as indicated, and statistical values are represented significant when p values were equal or below 0.05.
Supplementary Material
Highlights.
Dendritic cells (DCs) express machinery for cell-intrinsic glycogen metabolism
Intracellular glycogen stores that are catabolized to support DC effector function
Inhibition of glycogen catabolism impairs dendritic cell activation
Glycogen-derived carbons preferentially support DC citrate synthesis
Acknowledgments
The authors would like to acknowledge the UVM core facilities (Flow Cytometry, Animal Resource, Microscopy Imaging, and Advance Genome Technology Cores) for services provided in support of this work. Special acknowledgement to Dr. Ralph Budd, Dr. Paula Deming, and the VCIID COBRE for extensive support. Funding Sources: AD is a recipient of the Boettcher Webb-Waring Biomedical Research – Early Career grant; Veni Fellowship NWO (BE), LUMC fellowship (BE), 2016 AAI Careers in Immunology Fellowship (PT and EA), UVM College of Nursing and Health Sciences Incentive Grant (EA), UVM start-up Funds (EA), P30GM118228 (EA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, P.T. and E.A.; Methodology, P.T., A.D., B.E., and E.A.; Formal Analysis, P.T., J.R., and A.D.; Investigation, P.T., L.P, R.C., B.E., S.B., J.R., A.D., and E.A.; Writing – Original Draft, P.T. and E.A.; Writing – Review & Editing, P.T., A.D., B.E., and E.A.; Funding Acquisition, E.A.; Resources, A.D., B.E., and E.A.; Supervision, A.D., B.E., and E.A.
References
- Adeva-Andany MM, Gonzalez-Lucan M, Donapetry-Garcia C, Fernandez-Fernandez C, Ameneiros-Rodriguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. doi: 10.1016/j.bbacli.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzelius BA. Section staining for electron microscopy using tannic acid as a mordant: a simple method for visualization of glycogen and collagen. Microsc Res Tech. 1992;21:65–72. doi: 10.1002/jemt.1070210110. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Amati L, Pepe M, Passeri ME, Mastronardi ML, Jirillo E, Covelli V. Toll-like receptor signaling mechanisms involved in dendritic cell activation: potential therapeutic control of T cell polarization. Curr Pharm Des. 2006;12:4247–4254. doi: 10.2174/138161206778743583. [DOI] [PubMed] [Google Scholar]
- Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, Pearce EJ. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. Journal of immunology. 2012;189:2151–2158. doi: 10.4049/jimmunol.1103741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel E, Everts B, Fritz D, Beauchamp S, Ge B, Pearce EL, Pearce EJ. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. Journal of immunology. 2014;193:2821–2830. doi: 10.4049/jimmunol.1302498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr Opin Immunol. 2002;14:380–383. doi: 10.1016/s0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- Calder PC, Geddes R. Heterogeneity of glycogen synthesis upon refeeding following starvation. Int J Biochem. 1992;24:71–77. doi: 10.1016/0020-711x(92)90231-o. [DOI] [PubMed] [Google Scholar]
- Elsner P, Quistorff B, Hansen GH, Grunnet N. Partly ordered synthesis and degradation of glycogen in cultured rat myotubes. The Journal of biological chemistry. 2002;277:4831–4838. doi: 10.1074/jbc.M108226200. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nature immunology. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, Pearce EL, Pearce EJ. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120:1422–1431. doi: 10.1182/blood-2012-03-419747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Pearce EJ. Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Frontiers in immunology. 2014;5:203. doi: 10.3389/fimmu.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Gunther UL, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell metabolism. 2012;16:751–764. doi: 10.1016/j.cmet.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. The Journal of biological chemistry. 2014;289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews. Molecular cell biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, Navarro MN, Murray J, Finlay DK. Glucose represses dendritic cell-induced T cell responses. Nature communications. 2017;8:15620. doi: 10.1038/ncomms15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Iwasaki A. Innate control of adaptive immunity: dendritic cells and beyond. Semin Immunol. 2007;19:48–55. doi: 10.1016/j.smim.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Lipscomb MF, Masten BJ. Dendritic Cells: Immune Regulators in Health and Disease. 2002 doi: 10.1152/physrev.00023.2001. [DOI] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell metabolism. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof A, English NR, Bedford PA, Gabrilovich DI, Knight SC. Developing dendritic cells become ‘lacy’ cells packed with fat and glycogen. Immunology. 2005;115:473–483. doi: 10.1111/j.1365-2567.2005.02181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA. Glycolytic reprogramming by TLRs in dendritic cells. Nature immunology. 2014;15:314–315. doi: 10.1038/ni.2852. [DOI] [PubMed] [Google Scholar]
- Obel LF, Muller MS, Walls AB, Sickmann HM, Bak LK, Waagepetersen HS, Schousboe A. Brain glycogen-new perspectives on its metabolic function and regulation at the subcellular level. Front Neuroenergetics. 2012;4:3. doi: 10.3389/fnene.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EJ, Everts B. Dendritic cell metabolism. Nature reviews. Immunology. 2015;15:18–29. doi: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A, Hemmert KC, Ochi A, Jamal M, Henning JR, Barilla R, Quesada JP, Zambirinis CP, Tang K, Ego-Osuala M, et al. Role of fatty-acid synthesis in dendritic cell generation and function. Journal of immunology. 2013;190:4640–4649. doi: 10.4049/jimmunol.1202312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. The Biochemical journal. 2012;441:763–787. doi: 10.1042/BJ20111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RB. Glycogen in human peripheral blood leukocytes. I. Characteristics of the synthesis and turnover of glycogen in vitro. J Clin Invest. 1968;47:344–352. doi: 10.1172/JCI105730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci U S A. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RG, Rothman DL. The “glycogen shunt” in exercising muscle: A role for glycogen in muscle energetics and fatigue. Proc Natl Acad Sci U S A. 2001;98:457–461. doi: 10.1073/pnas.98.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet D, Voet JG, Pratt CW. Fundamentals of biochemistry: life at the molecular level. Hoboken, NJ: Wiley; 2013. [Google Scholar]
- Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One. 2011;6:e17234. doi: 10.1371/journal.pone.0017234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisdorf DJ, Craddock PR, Jacob HS. Glycogenolysis versus glucose transport in human granulocytes: differential activation in phagocytosis and chemotaxis. Blood. 1982;60:888–893. [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Molecular biology of the cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sarkissyan M, McGhee E, Lee S, Vadgama JV. Combined inhibition of glycolysis and AMPK induces synergistic breast cancer cell killing. Breast Cancer Res Treat. 2015;151:529–539. doi: 10.1007/s10549-015-3386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunis AA, Arimura GK. ENZYMES OF GLYCOGEN METABOLISM IN WHITE BLOOD CELLS. I. GLYCOGEN PHOSPHORYLASE IN NORMAL AND LEUKEMIC HUMAN LEUKOCYTES. Cancer Res. 1964;24:489–492. [PubMed] [Google Scholar]
- Yunis AA, Arimura GK. Enzymes of glycogen metabolism in white blood cells. II. Activation and inactivation of glycogen phosphorylase of rat chloroma. Biochim Biophys Acta. 1966;118:325–334. doi: 10.1016/s0926-6593(66)80041-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.