

Abstract
The concept of neurovascular unit (NVU), formalized at the 2001 Stroke Progress Review Group meeting of the National Institute of Neurological Disorders and Stroke, emphasizes the intimate relationship between the brain and its vessels. Since then, the NVU has attracted the interest of the neuroscience community resulting in considerable advances in the field. Here the current state-of-knowledge of the NVU will be assessed, focusing on one of its most vital roles: the coupling between neural activity and blood flow. The evidence supports a conceptual shift in the mechanisms of neurovascular coupling, from a unidimensional process involving neuronal-astrocytic signaling to local blood vessels, to a multidimensional one in which mediators released from multiple cells engage distinct signaling pathways and effector systems across the entire cerebrovascular network in a highly orchestrated manner. The recently appreciated NVU dysfunction in neurodegenerative diseases, although still poorly understood, supports emerging concepts that maintaining neurovascular health promotes brain health.
Keywords: Functional hyperemia, cerebral blood flow, endothelium, astrocytes, pericytes, neurodegeneration
Introduction
The brain, an organ of unparalleled sophistication, seems to have a fundamental design glitch: it consumes a large amount of energy, but lacks a reservoir to store fuel for use when needed. Therefore, the brain receives energy substrates, primarily oxygen and glucose, “on the fly” through its blood supply. Considering the dynamic and regionally diverse energy requirements imposed by brain activity, blood flow needs to reach the brain at the right time and place, and in the right amount. Failure to do so has disastrous consequences. Complete interruption of the cerebral blood supply for more than a few minutes, for example when a cerebral artery is occluded in stroke or after pump failure in cardiac arrest, causes irretrievable brain damage and death. On the other hand, if flow is not completely stopped, but is reduced or not well matched to the energy demands of the tissue, more subtle brain alterations ensue, leading to chronic brain injury in vulnerable areas often associated with cognitive impairment (Iadecola, 2013).
There has been a long-standing interest in how the brain regulates its own blood supply, driven not only by the desire to gain a better understanding of the harmful effects of cerebrovascular insufficiency, but also by the early realization that regional changes in cerebral blood flow (CBF) may provide a window on brain function. This large body of work spanning over almost two centuries revealed a remarkable complexity in the interaction between the brain and its vessels, unmatched by the vasculature in other organs.
In this context, the concept of neurovascular unit (NVU) emerged from the first Stroke Progress Review Group meeting of the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (July 2001) to emphasize the unique relationship between brain cells and the cerebral vasculature (https://www.ninds.nih.gov/About-NINDS/Strategic-Plans-Evaluations/Strategic-Plans/Stroke-Progress-Review-Group). Although the vital importance of cerebral blood vessels in brain health had long been appreciated, hard-core neuroscientists considered brain cells and cerebral blood vessels as distinct entities. Such dichotomy led to the tacit assumption that, unless the delivery of blood flow to the brain was critically compromised, neurons had little to do with the vasculature, and vice-versa. Similarly, a rigid distinction was placed between “neurodegenerative diseases”, e.g., Alzheimer’s disease (AD), and cerebrovascular diseases, e.g., stroke, such that these conditions were considered mutually exclusive and mechanistically unrelated. The NVU concept challenged these assumptions and emphasized the symbiotic relationship between brain cells and cerebral blood vessels, calling attention to their developmental, structural and functional interdependence in health and disease.
The neuroscience community embraced the concept enthusiastically, and the NVU has drawn increasing interest, as attested by the dramatic rise in the number of yearly citations over the past decade (Figure 1). New technologies have enabled investigators to delve deeper into the functions of the NVU in health and disease. Consequently, the NVU has taken center stage in all facets of normal brain function, and in the pathobiology of a wide variety of brain diseases.
Figure 1. Citations of the neurovascular unit in the literature from 1997 to 2016.
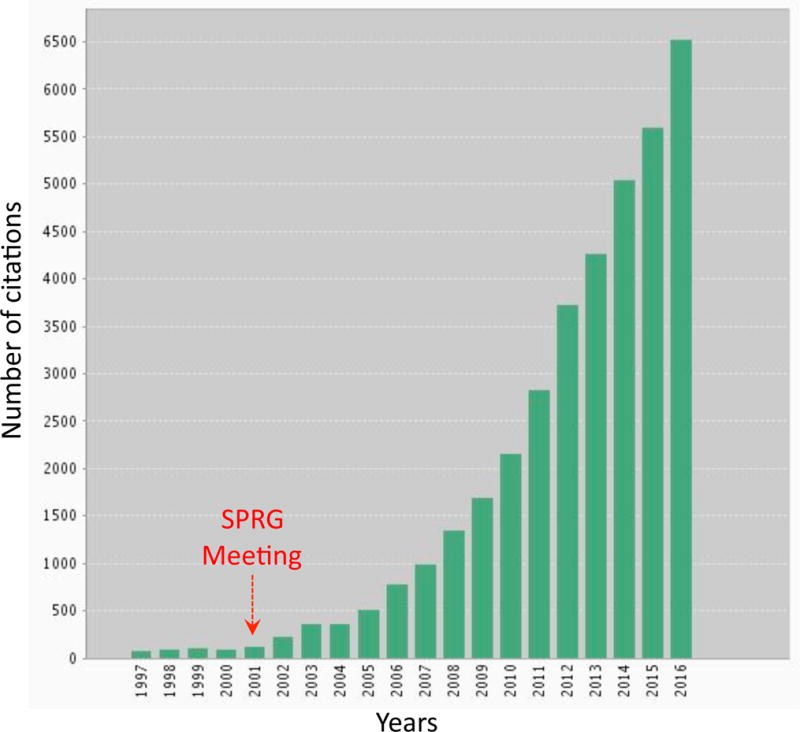
The search term “neurovascular unit” was used in the Web of Science database (Thompson Reuters). Since the SPRG meeting in 2001, a dramatic rise in the number of citations was observed. Prior to 2001, the term was often used for critical care units combining neurology and neurosurgery.
Here we will assess the current state-of-knowledge of the NVU, focusing on one of its prototypical functions: the coupling between neural activity and CBF (neurovascular coupling). To this end, we will review and integrate observations related to the contribution of different NVU cells to neurovascular coupling, in an attempt to provide a comprehensive picture of how activation of restricted groups of neurons is able to generate hemodynamic responses engaging the entire cerebrovascular tree, from capillaries deep in the substance of the brain to pial arteries on the brain surface. The contribution of NVU dysfunction to brain pathologies will also be examined, focusing on neurodegenerative diseases, highly prevalent conditions previously considered unrelated to vascular factors. Finally, a critical assessment of the evidence will be provided in an effort to flesh out unresolved issues and outstanding questions.
Neurovascular interactions through the ages
Throughout the 18th and 19th century the prevailing view was that the brain was not involved in regulating its own blood supply, thought to be controlled exclusively by the systemic circulation (Friedland and Iadecola, 1991). Hints of change can be appreciated at the end of the 19th century in the work of Roy and Sherrington in animals and Angelo Mosso in humans (Figure 2). Roy and Sherrington, based on the effect of injecting brain extracts in the carotid arteries of animals equipped with a device to monitor intracranial pressure, proposed that metabolites produced by neuronal activity diffuse to local blood vessels to increase blood flow to the activated brain (Roy and Sherrington, 1890). Even earlier, Mosso, studying individuals with skull defects, had discovered that somatic, mental or emotional stimuli induce changes in brain volume attributable to hemodynamic factors (Mosso, 1880). This body of work, albeit based on indirect indices of CBF, not measurable at the time, raised the possibility that brain activity was able to elicit changes in cerebral perfusion. These prescient observations were dismissed and quickly forgotten, and the idea that the brain could regulate its own blood supply did not reemerge until decades later (Friedland and Iadecola, 1991). In the late 1930s, Schmidt and Hendrix (Schmidt and Hendrix, 1938) demonstrated in the cat that illuminating the eye increases visual cortex temperature, a proxy for cerebral perfusion (Figure 2). Remarkably, the work of Roy and Sherrington was not mentioned in this and related publications of this period, resurfacing in the literature only in the 1960s (Friedland and Iadecola, 1991). A separate line of investigations, in the early 20th century, revealed that vascular density differs across the brain, and suggested that the density of vessels correlated with regional levels of energy consumption and functional activity (Craigie, 1945). Consistent with this hypothesis, it was found that sustained increases in neural activity lead to increases in local vascular density (Craigie, 1945). These early observations led to the idea that the vascular changes induced by neural activity were needed to fulfill the increased metabolic needs of active brain regions, providing initial evidence of the critical relationship linking brain function to the cerebral blood supply.
Figure 2. Historical overview of the concept of neurovascular coupling.

Pioneers in establishing the concept of neurovascular coupling are shown on top and examples of their work presented below the timeline. Studies in the late 1800s, by Angelo Mosso and Charles Roy and Charles Sherrington, hinted at the possibility that brain activity increases cerebral blood flow. The lower panels illustrate the apparatus used by Mosso to record changes in brain volume in patients with skull defects (Mosso, 1880), and Roy and Sherrington’s recordings of brain expansion in response to intracarotid infusion of a brain extract (Roy and Sherrington, 1890). In the 1930s Carl Schmidt recorded increases in temperature in the cat visual cortex by shining light into the eye. The original recording is shown in the lower panel (Schmidt and Hendrix, 1938). In the 1950–60s Seymour Kety and Lou Sokoloff developed autoradiographic methods to image regional CBF during neural activation. The lower panel shown the increase in CBF produced in the calcarine cortex and superior colliculus by visual stimulation (Freygang and Sokoloff, 1958). In the 1960-70s David Ingvar and Niels Lassen developed methods to measure regional CBF in the human brain using radioactive tracers and external detection. The lower panel illustrates the increase in CBF produced by hand movement in the contralateral sensory-motor cortex and supplementary motor area (Lassen et al., 1978), marking the birth of functional brain imaging.
Kety and Schmidt pioneered a method to measure CBF quantitatively using nitrous oxide as a tracer, and demonstrated that CBF increases with global brain hyperactivity, for example, in anxiety or hyperthyroidism, and is reduced when brain activity is suppressed, e.g., in coma (Kety, 1950). However, this method provided an average of CBF for the whole brain and could not assess regional cerebral perfusion and its relationship to local neural activity. The development of autoradiographic techniques using diffusible tracers to measure CBF quantitatively and in multiple brain regions, provided evidence that neural activity evokes CBF increases highly restricted to the activated network (Figure 2). Building on this work, Ingvar and Lassen developed methods to measure regional CBF in the human brain using intracarotid injection of radioactive tracers and external detection by a γ-camera (Figure 2). The subsequent development of positron emission tomography and MRI-based methods allowed investigators to monitor CBF in humans with great spatial resolution (Raichle and Mintun, 2006). In particular, the discovery of the blood oxygenation-level dependent (BOLD) effect, reflecting excess CBF delivery relative to local oxygen consumption, enabled the non-invasive detection of activity-dependent hemodynamic signals across the behaving human brain (Raichle and Mintun, 2006). While advancing our understanding of the neurobiology of human behavior, MRI-based functional brain imaging has also firmly established the concept that cerebrovascular function is intimately related to brain activity.
Structural diversity of the NVU along the cerebrovascular tree
The association between neurons and vessels varies significantly across the cerebrovascular network. Branching off the circle of Willis at the base of the brain, cerebral vessels run on the brain surface within the subarachnoid space (pial arteries and arterioles) forming a highly collateralized network (Blinder et al., 2013; Cipolla, 2010) (Figures 3, 4). Pial arterioles have multiple layers of smooth muscle cells (SMC) separated from the endothelium by a prominent elastic lamina (Roggendorf and Cervos-Navarro, 1977) (Figure 4). Although not in direct contact with brain cells, pial vessels are richly innervated by nerve fibers originating from peripheral autonomic and sensory ganglia, including the superior cervical, the sphenopalatine and the trigeminal ganglia (Hamel, 2006) (Figure 4). These nerve fibers, which express a wide variety of neurotransmitters and neuropeptides, run at the adventitia-media border forming an intricate mesh enveloping the vessels (Iadecola et al., 1993). Pial arteries dive into the substance of the brain surrounded by an extension of the subarachnoid space, the perivascular space, a virtual space delimited by the vascular basement membrane and the glia limitans (Virchow-Robin space)(Jones, 1970; E. T. Zhang et al., 1990) (Figure 4). Penetrating arterioles are endowed with a thinner smooth muscle cell layer, which eventually becomes a single layer (Dahl, 1973; Roggendorf and Cervos-Navarro, 1977). At this level, perivascular nerves are more sparse and the elastic lamina becomes less prominent (Roggendorf and Cervos-Navarro, 1977). The perivascular compartment contains several cell types, including perivascular macrophages (PVM), Mato cells, pial cells, and mast cells, among others, as well as nerve and collagen fibers (E. T. Zhang et al., 1990) (Figure 4). As arterioles penetrate deeper into the brain (intraparenchymal arterioles), the glial membrane and the vascular basement membrane fuse together obliterating the perivascular space (Jones, 1970; E. T. Zhang et al., 1990) (Figure 5). At this level, arterioles have a single or discontinuous layer of SMC, lack perivascular nerves and are encased in astrocytic end-feet (Roggendorf and Cervos-Navarro, 1977) (Figure 5). Occasionally axonal terminals or dendrites are seen in close apposition to the vascular basement membrane, often with an intervening glial leaflet (Iadecola et al., 1993; H. Wang et al., 2005). These neural processes originate from interneurons or from subcortical and brainstem nuclei projecting to the cortex, such as the basal forebrain cholinergic nuclei, locus coeruleus, and raphe magnus (Cohen et al., 1996; Hamel, 2006; Iadecola, 1993). Endothelial cells extend protrusions through the basal lamina into SMC (myoendothelial projections) enriched with gap junctions (Dahl, 1973; Longden et al., 2016). As arterioles transition into cerebral capillaries (Figure 5), SMC are replaced by pericytes, mural cells embedded into the endothelial basement membrane, covering approximately 30% of the vascular surface, and occasionally extending “peg and socket” protrusions into endothelial cells (Armulik et al., 2011; Dahl, 1973; Damisah et al., 2017). Like in arterioles, the circumference of the capillaries is covered by astrocytic end-feet, although neural processes can be at times observed next to the capillary basal lamina.
Figure 3. Anatomy of the cerebrovascular tree and segmental vascular resistance.

The internal carotid artery (ICA) enters the skull and merges with branches of the vertebral arteries to form the circle of Willis at the base of the brain. The middle cerebral artery (MCA) takes off from the circle of Willis and supplies a large territory of the cerebral cortex. The MCA gives rise to pial arteries and arterioles that run on the surface of the brain forming a heavily interconnected network from which arterioles penetrating into the substance of the brain originate (penetrating arterioles). Penetrating arterioles give rise to the capillary network, which feeds into the venous system returning the blood to the heart. The component (%) of the total vascular resistance that each cerebrovascular segment offers to blood flow, which reflects their potential for flow control, is indicated. Therefore, vessels outside the brain are responsible for 60% of the resistance, and vessels within the substance of the brain for 40% (based on data from (Stromberg and Fox, 1972) and (De Silva and Faraci, 2016)).
Figure 4. Neurovascular associations along the cerebrovascular tree: Pial arteries and penetrating arterioles.

A pial arteriole on the cortical surface, giving rise to a penetrating arteriole is shown on the right. Pial arterioles have thick coat of SMC, are surrounded by the subarachnoid space, and are densely innervated by nerve fibers originating form cranial autonomic and sensory ganglia, such as the sympathetic, parasympathetic and trigeminal ganglia. Penetrating arterioles enter the substance of the brain and are surrounded by a perivascular space containing several cell types including perivascular macrophages (PVM). In Figures 3 and 4, the vasculature (green) was visualized with the lipophilic dye DiO injected into the circulation. PVM and meningeal macrophages (blue), located in the perivascular and subarachnoid space, respectively, are visualized by CD206 immunocytochemistry.
Figure 5. Neurovascular associations along the cerebrovascular tree: Intraparenchymal arterioles and capillaries.
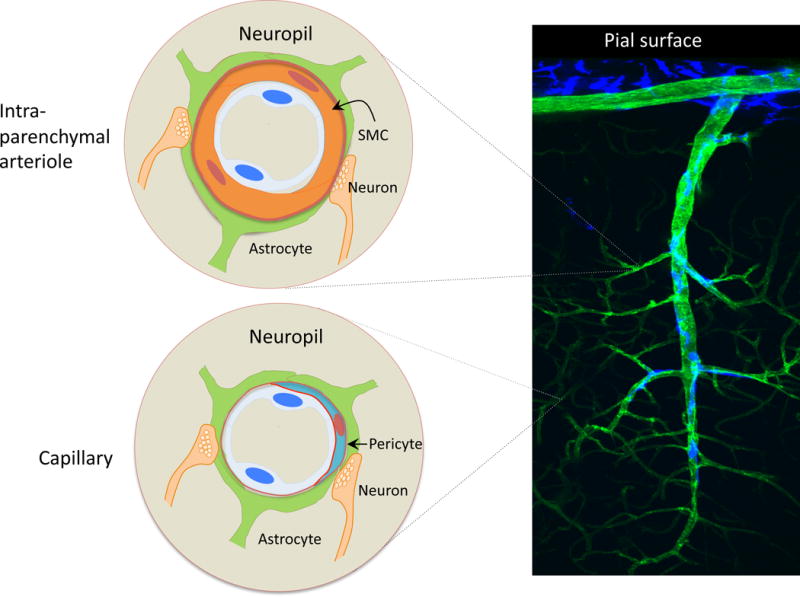
As the arterioles penetrate deeper into the brain the perivascular space disappears and the vessels become encased in astrocytic end feet (intraparenchymal arterioles). Endowed with a single layer of SMC, intraparenchymal arterioles lack perivascular nerves. Occasionally, neural processes originating from local neurons or subcortical pathways projecting to the cerebral cortex terminate near the vessel. Capillaries are devoid of SMC but are endowed with pericytes, which are fully enclosed by the basement membrane of the endothelium. Occasional neurovascular contacts similar to those seen in intraparenchymal arterioles are also observed.
Neurovascular coupling
One of the better-appreciated and most studied roles of the NVU pertains to the link between neural activity and CBF. In the “resting” brain, CBF varies in proportion to the energy consumption of each brain region. Thus, flow is higher in regions with higher energy utilization, e.g., inferior colliculus, and lower in regions with lower energy use, like the white matter (Sokoloff, 2013). Furthermore, increases in neural activity lead to increases in CBF highly restricted to the activated areas (functional hyperemia) (Chaigneau et al., 2003; Cox et al., 1993; Freygang and Sokoloff, 1958; Iadecola, 1993; LeDoux et al., 1983). The spatial and temporal correspondence between neural activity and the associated hemodynamic response is sufficiently precise, at least at the regional level, that the flow response can be used to map brain function (functional brain imaging) (Raichle and Mintun, 2006). However, at the microvascular level, the CBF increase in some regions exceeds the area of activation. For example, in the auditory, visual and cerebellar cortex the vascular response does not faithfully match the activated area (Harrison et al., 2002; Iadecola et al., 1997; O’Herron et al., 2016), while in the olfactory bulb there is close overlap between the two (Chaigneau et al., 2003). Non-overlapping vascular and neuronal topology in neocortex (Blinder et al., 2013) and retrograde vasodilatation (discussed later in this review) are likely responsible for such lack of fidelity (Chen et al., 2014; Iadecola et al., 1997; Longden et al., 2017). Since hemodynamic signals are the most powerful tool at our disposal for functional brain mapping, their spatial alignment with neural activity is becoming increasingly relevant as the resolution of fMRI increases.
The close relationship between neuronal and vascular function is also illustrated by the profound changes in neurovascular coupling that take place in the developing brain. In rodents before post-natal day 11, brain activation is not associated with sustained CBF increases, leading to an absent or negative BOLD signal (Colonnese et al., 2008; Kozberg et al., 2013). The lack of a flow response may be important for vascular development since the resulting hypoxia is a critical stimulus for cerebral angiogenesis (Lacoste and Gu, 2015). However, in the second and third week of life neural activity leads to increasingly larger hemodynamic responses resulting in progressively larger BOLD signals (Colonnese et al., 2008). The transition between the second and third post-natal week corresponds to dramatic neurovascular and systemic changes, including increases in vascular density, synaptogenesis, myelination and connectivity, energy metabolism, resting CBF and sensitivity of the cerebral microcirculation to vasoactive stimuli (Colonnese et al., 2008; Engl et al., 2017; Goyal et al., 2014; Nehlig et al., 1989), as well as systemic blood pressure (Kozberg et al., 2013). Although the factors responsible for the developmental shift in neurovascular coupling remain to be elucidated, the findings attest to the profound influence of key structural, metabolic and functional changes in the developing brain on shaping cerebral perfusion.
Why does CBF increase during brain activity?
The tight coupling between neural activity and CBF is thought to reflect the lack of energy reserves in the brain requiring a well-timed delivery of oxygen and glucose restricted to the activated areas. The flow increase may also be needed to clear the brain of potentially toxic by-products of brain activity, for example, lactate, CO2, the amyloid-β peptide (Aβ), and tau, as well as for brain temperature regulation (Tarasoff-Conway et al., 2015; Zhu et al., 2006). These considerations have led to a “feed-back” model in which the metabolic and clearance needs of the tissue drive the delivery of blood flow. Indeed, metabolic by-products of brain activity include potent vasodilators, such as adenosine, CO2, H+, and lactate, which could potentially initiate the flow response (Freeman and Li, 2016; Ko et al., 1990). However, other evidence supports a “feed-forward” model in which the increase in CBF is not driven by the tissue metabolic state. As revealed by the transformative work of Fox and Raichle, the increase in CBF is greater than the need of the tissue oxygen (Raichle and Mintun, 2006), resulting in an excess delivery of O2. In addition, increases in CBF occur also in conditions of excess oxygen and glucose, suggesting that activation-induced depletion of these energy substrates does not drive the flow increase (reviewed in (Attwell and Iadecola, 2002)). Based on this evidence, it was suggested that the CBF delivery is regulated by a feed-forward mechanism driven by neurovascular signaling pathways resulting in the release of vasoactive by-products of synaptic activity, such as K+, nitric oxide (NO) and prostanoids (Attwell et al., 2010; Attwell and Iadecola, 2002; Drake and Iadecola, 2007).
In the end, these two models may not be mutually exclusive (Figure 6). Microvascular studies showing that at the onset of neural activity there is a reduction in O2 and/or glucose preceding the CBF increase have rekindled the view that metabolic factors may also play a role in neurovascular coupling (Freeman and Li, 2016), but there might be regional diversity in their involvement. Basal oxygen levels vary widely in different brain regions (Lyons et al., 2016), and, depending on local vascular topology and intensity of the activating stimulus, regional hypoxia may develop, promoting vasodilatation of local vessels. Another hint that intravascular hypoxia may contribute to the flow response is provided by experiments demonstrating that at the beginning of activation a dip in intravascular O2 leads to increased deformability of red blood cells which improves microvascular rheology and increases capillary flow (Wei et al., 2016). These lines of evidence, collectively, suggest that a feed-forward mechanism may trigger an exaggerated flow response driven by neurovascular signaling, but feedback mechanisms may also be in place to adjust the CBF delivery more closely to the metabolic needs of the tissue. In support of this hypothesis, the vascular response during sustained neural activation tends to peak at the onset, subsequently readjusting toward a lower level (Drew et al., 2011; Freeman and Li, 2016; Ngai et al., 1988). Therefore, both metabolism-dependent (feed-back) and independent (feed-forward) mechanisms may be involved in functional hyperemia, depending on the timing, intensity and duration of the activation, as well as the brain region and the brain’s developmental stage. Although the mechanistic details remain incomplete, the evidence suggests that the main function of neurovascular coupling is to maintain the homeostasis of the cerebral microenvironment by delivering the energy substrates needed to initiate and sustain neural activity, while clearing potentially toxic by-products of brain metabolism including heat.
Figure 6. Potential feed-forward and feed-back mechanisms driving the local vascular response evoked by synaptic activity.
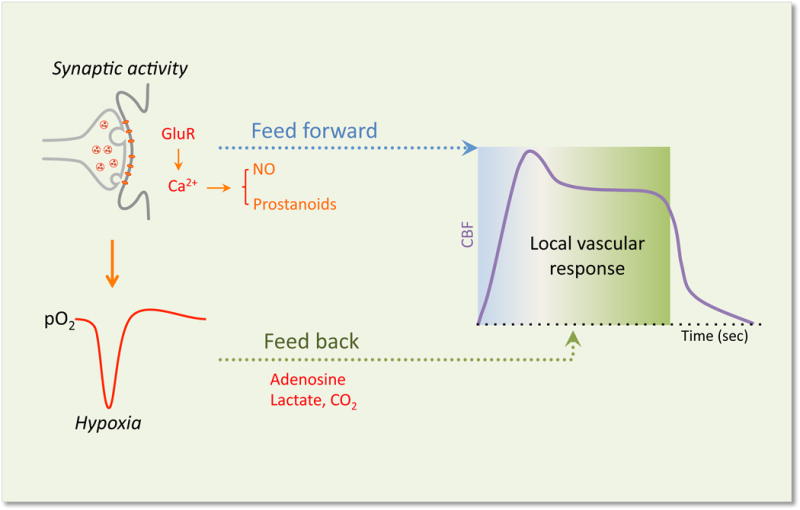
Glutamate released by synaptic activity activates postsynaptic glutamate receptors (GluR) leading to activation of Ca2+-dependent signaling pathways resulting in the release of vasoactive factors that may drive the initial “feed-forward” component (metabolism independent) of the local vascular response in arterioles and capillaries. At the same time a reduction in tissue O2 caused by the increase energy consumption induced by activation leads to the accumulation of vasoactive metabolic byproducts that may drive a secondary feed-back component (metabolism dependent) to better match the flow response to the metabolic needs of the tissue.
It has also been suggested that, in a reversal of roles, the hemodynamic response could influence the neural response via mechanical, thermal or chemical effects on astrocytes (hemo-neural hypothesis or vasculo-neuronal coupling) (Kim et al., 2016; Moore and Cao, 2008). This concept received support from a study in brain slices in which increases or decreases in transmural pressure/flow in penetrating arterioles was found to suppress or enhance, respectively, pyramidal cell activity through mechanosensitive transient receptor potential vanilloid receptor type 4 (TRPV4) channels in astrocytes (Kim et al., 2016). These data suggest an autoregulatory mechanism whereby intravascular pressure, and possibly tissue pressure, would directly modulate resting neural activity via astrocytic mechanosensors. The mechanisms and implications of these intriguing observations need further exploration.
Cellular bases of neurovascular coupling
During the previous decades, most studies focused on the mediators responsible for neurovascular coupling. This work suggested that multiple agents released by neural activity are involved. However, it was also recognized that local release of vasoactive mediators could not account for hemodynamic changes in upstream vascular segments remote from the activated site. The availability of tools to investigate neurovascular interactions at the cellular level has provided greater insight into the cellular processes initiating, transmitting, propagating and implementing the vascular response (Table 1). These will be examined next.
Table 1.
Key cellular steps underlying neurovascular coupling in neocortex
| Step | Cell type | Mediator/Mechanism | Cellular targets | Vascular segment |
|---|---|---|---|---|
| Initiation |
|
|
|
|
|
| ||||
| Modulation/ Spatial shaping |
|
|
|
|
|
| ||||
| Neurovascular transmission |
|
|
|
|
|
| ||||
| Retrograde propagation |
|
|
|
|
|
| ||||
| Implementation |
|
|
– |
|
Abbreviations: BF: basal forebrain; FMV: flow-mediated vasodilatation; LC: locus coeruleus; MLC: myosin light chain; RM: raphe magnus; SMC: smooth muscle cells
Neurons: initiation of the local vascular response
Neurons regulate CBF by generating signals, which, either directly or through interposed cells, act on local blood vessels to initiate the vascular response. Glutamatergic synaptic activity activates post-synaptic NMDA and AMPA receptors, leading to increases in intracellular Ca2+ and activation of Ca2+ dependent enzymes such as neuronal NO synthase (nNOS) and cyclooxygenase-2 (COX-2), which produce potent vasodilators, respectively, NO and prostanoid (Attwell et al., 2010; Lecrux and Hamel, 2016) (Figure 6). At the same time, glutamate acts on metabotropic glutamate receptors in astrocytes, triggering Ca2+ increases in these cells and production of vasoactive agents (see below). Adenosine, a potent vasodilator produced from ADP by ecto-nucleotidases, also contributes to the increase in CBF (Iadecola, 1993; Ko et al., 1990). In addition, a role for ATP acting on purinergic receptors has been postulated both in retina and neocortex (Biesecker et al., 2016; Mishra et al., 2016). Finally, ionic changes evoked by neural activity, in particular the increase in extracellular K+, a vasodilator at concentrations <12mM (Filosa et al., 2006), could also participate (see section on astrocytes and endothelium).
Recent studies have begun to shed light on the role of specific neuronal types and their mediators in neurovascular coupling. In whisker barrel cortex hyperemia induced by stimulation of the contralateral facial whiskers, pyramidal cells expressing the prostanoid-synthesizing enzyme COX-2 are activated from thalamic afferents (Lecrux et al., 2011) and contribute to the vascular response through prostaglandin E2 (PGE2) acting on EP2 and EP4 receptors (Lecrux and Hamel, 2016). These pharmacological observations are consistent with earlier findings in transgenic mice that COX-2, but not COX1, is required for the full expression of the vascular response (Niwa et al., 2000a; 2001a). Somatosensory stimuli also activate GABA interneurons (Lecrux et al., 2011). Increasing evidence suggests that interneurons may be critical for neurovascular coupling. Interneurons are enriched with vasoactive neurotransmitters and neuromodulators and have close associations with cerebral microvessels (Cauli and Hamel, 2010). In cerebral cortex and cerebellum, inhibitory interneurons are able to modify vascular diameter in brain slices through the vasoconstrictor NPY or the vasodilator NO (Cauli et al., 2004; Rancillac et al., 2006). In vivo, optogenetic stimulation of GABAergic interneurons induces biphasic hemodynamic responses, dilatation followed by constriction, closely resembling those induced by somatosensory activation in this preparation (Uhlirova et al., 2016b). While the constrictor response was attributed to NPY, the mediator(s) responsible for the dilatation could not be identified, but a role of NO or VIP was suggested (Uhlirova et al., 2016b). Interestingly, optogenetic activation of GABAergic interneurons increases CBF even if glutamatergic or GABAergic activity is pharmacologically blocked (Anenberg et al., 2015), suggesting that these cells can influence flow independently of the activity of their neuronal network, but the mechanisms of the vasodilatation remain to be established. While interneurons have the potential to modulate CBF, evidence for their participation in neurovascular coupling to physiological stimuli is limited. In mice deficient in the cell-cycle enzyme D2 cyclin, which lack stellate interneurons in the cerebellar molecular layer, the increase in blood flow produced by somatosensory activation of the cerebellar crus II is suppressed (Yang et al., 2000). Considering that stellate interneurons are enriched with nNOS and that nNOS-derived NO is the major determinant of functional hyperemia in cerebellum (Rancillac et al., 2006; Yang et al., 1999; Yang and Iadecola, 1998; Yang et al., 2003), these findings are consistent with the hypothesis that stellate interneurons mediate the response by releasing NO. However, more direct evidence in cerebellum or elsewhere in brain is lacking.
Another aspect of the involvement of neurons in neurovascular coupling pertains to neurovascular projections arising from subcortical nuclei. Locus coeruleus, raphe, and basal forebrain send diffuse adrenergic, serotoninergic and cholinergic projection, respectively, to the neocortex (Cohen et al., 1996; Hamel, 2006). These projections often terminate on astrocytic end-feet close to cerebral microvessels, and are able to powerfully modulate cerebral perfusion (Cohen et al., 1996; Toussay et al., 2013; F. Zhang et al., 1995). In particular, activation of the basal forebrain cholinergic system increases neocortical CBF diffusely, an effect attributable to acetylcholine release and endothelial NO production, and to activation of COX-2 expressing pyramidal cells and GABAergic interneurons (Lecrux and Hamel, 2016; F. Zhang et al., 1995). Although its lesion does not alter the changes in CBF induced by neural activity (Ibayashi et al., 1991), this pathway may contribute to maintain resting CBF and modulate the increases in CBF produced by somatosensory activation (Iadecola et al., 1983; Lecrux and Hamel, 2016; Lecrux et al., 2017). On the other hand, cortical norepinephrine levels, which depend in large part on the noradrenergic innervation from the locus coeruleus, have been shown to increase the vasoconstrictor tone in cerebral cortex and help focus oxygen delivery to the activated areas (Bekar et al., 2012). Therefore, these neurovascular pathways do not directly mediate the vasodilatation in response to activation, but, consistent with their broader role in neocortical neuromodulation (Lee and Dan, 2012), may contribute to shape the spatial and temporal features of the hemodynamic response (Table 1).
Little is known about the participation of perivascular nerves arising from the cranial ganglia in neurovascular coupling, but surgical ablation or pharmacological inactivation of the sympathetic and parasympathetic innervation does not affect the magnitude of the pial arterial dilatation evoked by somatosensory activation (Ibayashi et al., 1991). However, these nerves may protect cerebral blood vessels from forced vasodilatation during seizures or acute hypertension (Bill and Linder, 1976; Mueller et al., 1979). Furthermore, they may also contribute to the development and integrity of the vascular wall (Eichmann and Brunet, 2014), a process requiring molecular signals distinct from those in peripheral arteries (Brunet et al., 2014). Interestingly, the perivascular innervation has been implicated in generating resting state fMRI connectivity signals in surgically disconnected brain regions in humans (Warren et al., 2017). This finding, if confirmed, would suggest a role of perivascular nerves in the synchronization of hemodynamic signals across the brain and require a re-evaluation of the significance of fMRI-based assessments of functional neural connectivity.
Astrocytes: signal transmission to the local vasculature
Owing to their intimate association with both synapses and local microvessels, astrocytes are well positioned to link neural activity to microvascular function. Paulson and Newman proposed that astrocytes could influence blood flow by “siphoning” extracellular K+ released by neural activity into end-feet abutting cerebral microvessels (Paulson and Newman, 1987). However, subsequent studies in the retina of KIR4.1 channel null mice, in which glia K+ siphoning is suppressed, showed that neurovascular coupling was still present suggesting that this mechanism may not be involved in retinal arteriolar dilatation (Metea et al., 2007).
Zonta et al. provided evidence that Ca2+ increases in astrocytes, evoked by electrical stimulation, could influence the diameter of adjacent microvessels in brain slices through metabotropic glutamate receptors (mGluR) and COX reaction products (Zonta et al., 2003). Subsequent studies using pharmacological approaches also in brain slices extended these initial observations demonstrating that increases in astrocytic Ca2+, triggered by neural activity, photolysis of caged Ca2+ or activation of mGluR5, have the potential to modulate microvascular diameter, producing constriction or dilatation depending on the oxygen levels (Gordon et al., 2008), resting vascular tone (Blanco et al., 2008) or extracellular K+ (Girouard et al., 2010). The postulated sequence of events leading to vasodilation is that glutamate released by synaptic activity activates mGluR on astrocytes leading to inositol 3-phosphate (IP3) mobilization and Ca2+ release from intracellular stores, phospholipase-A2 (PLA2) activation, release of arachidonic acid and production of PGE2 and epoxyeicosatrienoic acids (EETs) via the COX and cytochrome p450 epoxygenase pathways, respectively (Attwell et al., 2010; Mishra, 2016). An alternative mechanism is that Ca2+ transients in astrocytes activate large conductance Ca2+ activated K+ (BK) channels in astrocytic end-feet leading to K+ release and vascular SMC relaxation through KIR channels (Filosa et al., 2006). However, the relevance of this mechanism remains unclear because mice lacking a subunit of the BK channel have normal neurovascular coupling (Girouard et al., 2010), probably reflecting intervening compensatory mechanisms. Similarly, experiments in brain slices suggest that activity-induced release of D-serine by astrocytes may induce endothelial NO production (Stobart et al., 2013), a mechanism that also needs further study since neurovascular coupling is preserved in mice lacking endothelial NO synthase (Girouard et al., 2007).
Subsequent in vivo studies challenged the role of astrocytic Ca2+ transients and attendant upstream and downstream signaling in neurovascular coupling (reviewed in (Mishra, 2016; Petzold and Murthy, 2011; Uhlirova et al., 2016a)). The increases in Ca2+ may be too slow (Takano et al., 2006), often occurring after the onset of the hemodynamic response (Bonder and McCarthy, 2014; Nizar et al., 2013). Faster responses have been described (Lind et al., 2013), but questions have been raised about spillover of signals from neurons due to bulk labeling with organic Ca2+ dyes (Otsu et al., 2015; Petzold and Murthy, 2011; Uhlirova et al., 2016a). In experiments using genetically encoded astrocytic Ca2+ sensors in the visual cortex, no arteriolar blood flow changes were observed when astrocytic Ca2+ was modulated by chemogenetic approaches (Bonder and McCarthy, 2014). In contrast, in the activated olfactory bulb rapid increases in astrocytic Ca2+ preceding the vascular response were observed even with genetically encoded sensors (Otsu et al., 2015), suggesting regional differences in the role of astrocytic Ca2+ surge in neurovascular coupling. Questions have also been raised about the role of mGluR5 and IP3 receptors. mGluR5 expression is developmentally regulated and suppressed after the third week of life in mice (Sun et al., 2013), challenging the hypothesized mechanism that the Ca2+ increase in astrocytes in adult mice depends on these receptors. Indeed, in the olfactory bulb, mGluR5 were responsible for the Ca2+ increase in astrocytes of juvenile, but not adult mice (Otsu et al., 2015). In mice lacking IP3 type 2 receptors, the main regulators of Ca2+ release from intracellular stores in astrocytes, neurovascular coupling occurs in the absence of astrocytic Ca2+ elevations (Bonder and McCarthy, 2014; Jego et al., 2014; Nizar et al., 2013), although persistence of Ca2+ transients in astrocytic processes has been described in these mice (Srinivasan et al., 2015). The role of COX-1, a rate-limiting enzyme for PGE2 synthesis, in astrocytic neurovascular coupling also remains unclear. The expression of COX-1 in astrocytes has been questioned (Anrather et al., 2011; Lau et al., 2014; Lecrux et al., 2011), and neurovascular coupling is not altered by COX-1 inhibitors or in COX-1 null mice (Liu et al., 2012; Niwa et al., 2001a; Rosenegger et al., 2015). Therefore, whatever the source and timing of the Ca2+ signal in astrocytes, the downstream signaling pathways leading to the vasodilatation remain unclear.
Recent data may help reconcile, at least in part, these discordant observations. The role of astrocytes in neurovascular coupling may be restricted to capillaries and may not involve arterioles, and the signaling pathways regulating the response of these vascular segments may be distinct (Biesecker et al., 2016; Mishra et al., 2016). In brain slices, electrical stimulation increases the diameter of cerebral capillaries and astrocytic Ca2+, an effect suppressed by AMPA or P2X1 receptor inhibition, suggesting that ATP released from neurons acts on astrocytic P2X1 receptors leading to the Ca2+ rise (Mishra et al., 2016). Contradicting previous evidence implicating PLA2 and cytochrome p450 epoxygenase pathways (Attwell et al., 2010; Mishra, 2016), pharmacological studies indicated that the Ca2+ rise leads to activation of phospholipase-D2, arachidonic acid production, COX-1 activation and PGE2 release, which, in turn mediates the vasodilatation by acting on EP4 receptors, presumably on pericytes (Mishra et al., 2016). In contrast, neurovascular coupling in arterioles does not involve astrocytes, but is mediated by NMDA receptors and neuronal NO production (Mishra et al., 2016). Accordingly, the arteriolar response to activation is blocked by NOS inhibition or NMDA receptor antagonism, which does not affect the response of capillaries (Mishra et al., 2016). Findings supporting a dual neurovascular regulation of capillaries and arterioles were also obtained in the retina. Ca2+ transients in Muller cells, assessed with genetically encoded sensors, were linked to dilatation of capillaries, although the timing between Ca2+ rise and vascular response could not be consistently resolved (Biesecker et al., 2016). The Ca2+ transients were not linked to dilatation of arterioles, and conditional deletion of IP3 type 2 receptors in Muller cells abolished the dilatation of capillaries but not arterioles. Although there is still considerable controversy about the role of capillaries and pericytes (discussed in subsequent sections) and COX-1 in neurovascular coupling (discussed above), the data may help explain some of the negative studies on the role of Ca2+ transients in neurovascular coupling, which focused on arteriolar responses. Taken together, these findings support the concept that astrocytic signals may be a critical link between neural activity and the microvasculature, at least in certain segments of the cerebrovascular network.
Endothelial cells: retrograde propagation of vasomotor responses
Endowed with powerful vasoactive agents (NO, prostanoids, endothelin, etc.), endothelial cells have long been known to regulate CBF in response to chemical and mechanical signals (Andresen et al., 2006; Seals et al., 2011), but their role in neurovascular coupling has not received attention until recently. Emerging evidence suggests a crucial role of the endothelium in the retrograde propagation of activity-induced neurovascular signals.
Hemodynamic considerations dictate that in a vascular network there has to be a coordinated dilatation of downstream and upstream vessels in order to increase flow while avoiding a “flow steal” from interconnected vascular territories (Segal, 2015). Pial arterioles are an important site of flow control (Figure 3), and signals generated by neural activity deep in the brain tissue have to be conveyed to upstream arterioles remote from the area of activation to increase flow efficiently. Indeed, visualization of pial arterioles in the somatosensory cortex revealed branch-selective vasodilatation supplying the activated site during somatosensory stimuli activating layer IV neurons, e.g., (Ngai et al., 1988). Since diffusion of vasoactive agents released by active neurons could not explain these remote vascular adjustments, it was proposed that, in analogy with the retrograde vasodilation observed in hamster cheek pouch by Duling (Segal, 2015), the vascular response generated in or near the activated area is conducted retrogradely along blood vessels engaging larger pial arteries upstream (Iadecola, 1993; Woolsey and Rovainen, 1991). Evidence was subsequently provided that isolated cerebral arterioles exhibit conducted vasodilation (Dietrich et al., 1996), and neural activity-induced retrograde propagation of vasodilatation was described in cerebellar cortex pial arterioles in vivo (Iadecola et al., 1997). A role for brain-derived vasoactive factors released in the subarachnoid space and acting on pial arterioles was also excluded (Ngai and Winn, 2002). At the same time, vascular mapping and fMRI studies revealed that during somatosensory activation vascular responses are first observed in deep cortical laminae, where the thalamic afferents terminate, and then more superficially, suggesting retrograde propagation of the vascular response (Silva and Koretsky, 2002; Uhlirova et al., 2016b). These observations supported the idea that activation restricted to a specific site generates a vascular response that propagates retrogradely to the upstream vascular segment supplying the activated region, but the cellular bases of the effect remained unclear.
In systemic vessels the endothelium is well known to participate in the retrograde propagation of vascular signals (Segal, 2015). In brain, however, damage of the pial vessel endothelium using the light-and-dye technique did not attenuate the peak pial dilatation produced by somatosensory activation (Xu et al., 2008), but the temporal profile of the vasodilatation was not resolved. More recently, Chen et al. used a similar approach to produce a highly localized lesion of the endothelium of a single somatosensory cortex pial arteriole, and found that the conducted vasodilation induced by somatosensory activation failed to propagate beyond the lesion site (Chen et al., 2014). Furthermore, wide-field endothelial lesions markedly altered the amplitude and temporal dynamics of the hemodynamic response, with a slower rise and no initial peak (Chen et al., 2014), implicating endothelial cells in the full expression and temporal coordination of the vasomotor response induced by somatosensory activation.
What drives the endothelial propagation of vasomotor signals? In peripheral vessels, conducted vasomotor responses have two components: a fast component mediated by Ca2+-activated K+ channels (KCa) and spreading through electrical coupling between endothelial cells, and a slow component mediated by Ca2+ waves triggering the endothelial release of NO and prostanoids (Segal, 2015; Tallini et al., 2007). Although much less in know about the mechanisms of retrograde vasodilatation in the cerebral vasculature, a recent study implicates endothelial KIR channels, rather than KCa, in the mechanisms of the fast component. Enriched with KIR channels, but not KCa channels, capillary cerebral endothelial cells are highly sensitive to K+ generated during neural activity, either from the abutting astrocytic end-feet or diffusion form nearby synapses (Longden et al., 2017). Owing to a unique capillary-parenchymal arteriole preparation, this study showed that micropipette application of 6–10mM K+ to capillaries generated a robust hyperpolarization in endothelial cells, transmitted retrogradely to penetrating arterioles at an estimated speed of 2mm/sec, and leading to hyperpolarization and relaxation of SMC (Longden et al., 2017). K+ application did not induce capillary dilatation, suggesting that the capillary was the K+ sensor and the upstream penetrating arteriole the effector of the vasodilation. This conclusion was supported by in vivo experiments demonstrating that the increase in capillary flow produced by K+ application is not due to local effects on capillary diameter but is linked to conducted dilatation of upstream arterioles (Longden et al., 2017). The propagation of the vasodilatation was blocked by KIR inhibition with barium or endothelial deletion of KIR1.2 channels, pointing to KIR channels as key mediators of the conducted hyperpolarization (Longden et al., 2017). This rapid propagation mechanism could be mediated by ionic currents traveling through the endothelium via gap junctions and from endothelium to SMC via myoendothelial junctions (Segal, 2015; Tallini et al., 2007). Consistent with their role in neurovascular coupling, endothelial deletion of KIR1.2 suppressed the increase in CBF produced by somatosensory activation (Longden et al., 2017). However, in contrast with the endothelial injury model in which the time course of the hemodynamic response was altered (Chen et al., 2014), in KIR1.2 null mice the CBF increase evoked by whisker stimulation was uniformly reduced by ≈50% without major effects on its temporal profile (Longden et al., 2017). Although methodological differences cannot be excluded, the suppression of the flow response may suggest functions of endothelial KIR1.2 channels beyond conducted vasodilatation. Conducted vasomotor responses, both vasodilation and vasoconstriction, can be generated also by other agents, for example acetylcholine and catecholamines, respectively (Bagher and Segal, 2011). In brain arterioles, ATP and prostaglandin F2α generate constriction followed by conducted vasodilatation (Dietrich et al., 1996), but their role in neurovascular coupling has not been explored.
Smooth muscle cells and pericytes: the vasomotor apparatus
Signals generated by neurons, astrocytes and endothelial cells ultimately engage the vasomotor apparatus to induce vasodilatation, reduce vascular resistance and increase blood flow. In brain as in other organs, SMC wrapping around resistance vessels are considered the major effectors of vasomotor responses and flow regulation (Cipolla, 2010), and neural activity leads to SMC relaxation in pial and penetrating arterioles (Drew et al., 2011; Ngai et al., 1988; Uhlirova et al., 2016b). SMC constrict and relax in response to a wide variety of vasoactive agents and to conducted vasodilatatory stimuli originating from downstream vessels (see previous section). Another key physiological characteristic of SMC is their ability to constrict or relax in response to increases or decreases in intravascular pressure, a phenomenon termed myogenic response (Cipolla, 2010; Longden et al., 2016). Cerebral arteries, especially penetrating arterioles, have a strong propensity to generate myogenic tone, a property that is essential for cerebral blood vessels to keep CBF stable during changes in arterial pressure within a certain range (cerebrovascular autoregulation) (Koller and Toth, 2012). Thus, blood pressure increases lead to vasoconstriction and decreases to vasodilatation. The resulting changes in vascular resistance keep CBF relatively stable. Cerebrovascular autoregulation and myogenic tone set the level of resting CBF and may contribute to neurovascular coupling (see next section). The changes in the contractile state of SMC are mediated by the interplay between, membrane potential and intracellular Ca2+, and Ca2+ sensitivity of the contractile apparatus, which ultimately controls the assembly of contractile proteins (Cipolla, 2010; Longden et al., 2016).
In capillaries, SMC are replaced by pericytes (Figure 5). Pericyte have an important role in establishing and maintaining vascular structure and the blood-brain barrier (BBB) (Armulik et al., 2011; 2010; Daneman et al., 2010), but have long been implicated also in flow regulation (Krueger and Bechmann, 2010). Some studies have shown that a proportion of pericytes are contractile, respond to brain-generated vasoactive signals, and are able to influence capillary diameter both in vitro and in vivo (Hall et al., 2014; Mishra et al., 2016; Peppiatt et al., 2006). In addition, a 30% pericyte loss in mice with haploinsufficiency of platelet-derived growth factor receptor-β (Pdgfr-β) is associated with reduced neurovascular coupling and brain oxygenation levels (Kisler et al., 2017b). However, other studies have failed to demonstrate a role of pericytes in flow regulation (Cudmore et al., 2016; Fernández-Klett et al., 2010; Hill et al., 2015; Wei et al., 2016). A major problem is that, due to the lack of specific markers, it has been difficult to distinguish pericytes from SMC at the arteriolar-capillary transition. Vessel size is not a reliable indicator of capillaries and the commonly used Pdgfr-β and proteoglycan NG2 are not specific pericyte markers (Armulik et al., 2011). Consequently, due to different criteria for pericyte identification, there has been considerable confusion about their functional roles (Attwell et al., 2016; Krueger and Bechmann, 2010). Pericytes are morphologically heterogeneous, but the functional significance of such diversity remains unclear (Armulik et al., 2011). One interpretation is that mural cells next to feeding arterioles, which encircle the vessel like SMC and contain SMC actin, are “contractile pericytes”, whereas cells running along the major axis of the capillary with short stubby processes projecting sideways are “non-contractile pericytes” (Attwell et al., 2016; Hartmann et al., 2015). Another point of view is that “contractile pericytes” are SMC located at the arteriole-capillary transition, and that “true” pericytes have no contractile function (Hill et al., 2015). A limitation of these studies is that the ultrastructural features of these cells, for example their relationship to the endothelial basement membrane which envelops the pericyte, a key feature for their identification (Armulik et al., 2011; Park et al., 2013), was not determined. A tracer recently introduced may provide a new tool to study pericytes (Damisah et al., 2017), but, as discussed in the final section of this review, other approaches would also be needed to reliably phenotype mural cells. Irrespective of whether these contractile cells are pericytes or SMC, the larger issue concerns the role of cerebral capillaries in the regulation of CBF during neural activity, which will be addressed next.
Which segment of the cerebrovascular network mediates neurovascular coupling?
Neural activity evokes coordinated vasomotor responses distributed over the vascular network supplying the activated area. The structural diversity of the NVU along the cerebrovascular tree (Figures 4,5) suggests differences in the role of each vascular segment in CBF regulation. In the cat neocortex, pooled measurements of pressure gradients between vascular segments indicated that 39% of the total resistance to flow is attributable to vessels upstream of pial arteries 300μm in diameter, 21% across the pial microcirculation (300 to 50μm pial arterioles), and 40% downstream of these vessels (De Silva and Faraci, 2016; Stromberg and Fox, 1972) (Figure 3). This distribution of vascular resistance seems to be conserved across species (De Silva and Faraci, 2016). Therefore, while the pial microcirculation is responsible for a sizable fraction of the total resistance, hence potential for flow control, an even larger component is attributable to downstream intracerebral vessels. However, the breakdown of the vascular resistance among penetrating arterioles, precapillary arterioles, capillaries and venules has been difficult to assess because intravascular pressure measurements in smaller vessels embedded deep in the brain is not feasible. Capillaries are closer to neurons than arteries and, due to their large surface area, minimal changes in their diameter could produce large changes in flow (Attwell et al., 2010). Consequently, capillaries are well suited to regulate CBF in response to the metabolic needs of the brain. Computational studies attempting to assess segmental cerebrovascular resistance have given varying results due to difficulties in setting boundary parameters in idealized networks (Schmid et al., 2017). In two recent studies, in which realistic vascular networks of the mouse somatosensory cortex were used, one attributed the bulk of resistance to arterioles in deep cortical layers and capillaries in superficial layers (Schmid et al., 2017), and the other to capillaries “adjacent to feeding arterioles” (Gould et al., 2017), highlighting the uncertainty of estimating segmental vascular resistance.
Experiments investigating capillary vasoactivity have also led to inconsistent results. In brain slices neural activity was found to induce capillary dilatation due to relaxation of pericytes (Hall et al., 2014). Related in vivo experiments demonstrated that the capillary hemodynamic response precedes arteriolar dilatation, suggesting that the increase in capillary flow is not a passive consequence of the increased flow in upstream arterioles (Hall et al., 2014; Kisler et al., 2017b). Based on these findings and on the mouse cerebrovascular tree topology (Blinder et al., 2013) it was estimated that capillaries are responsible for 84% of the flow increase produced by neural activity (Hall et al., 2014). However, other studies demonstrated that somatosensory activation does not change capillary diameter, and that relaxation of upstream arterioles accounts for the bulk of the flow response (Drew et al., 2011; Hill et al., 2015; Wei et al., 2016). A lack of capillary vasoactivity was also shown with local application of K+ (Longden et al., 2017), seizures (Fernández-Klett et al., 2010), ischemia due to carotid artery occlusion or carotid stenosis (Damisah et al., 2017; Hill et al., 2015), and isoflurane anesthesia (Cudmore et al., 2016). One possible explanation for these discordant results is that studies reporting capillary vasoactivity may have focused on transitional microvessels closer to arterioles and endowed with α-actin-containing mural cells (SMC?), whereas negative studies on capillary segments with “non-contractile” mural cells (pericytes?). Furthermore, considering the small activity-induced increases in capillary diameter, e.g., 1% in (Kisler et al., 2017b), the resolution of the imaging approach used could also be a factor (Drew et al., 2011). However, as discussed in the next section, whether or not capillaries are major contributors to cerebrovascular resistance, does not rule out their participation in neurovascular coupling.
Local and conducted vasomotor responses in neurovascular coupling
Capillaries are uniquely positioned to detect neuronal and astroglial signals, which led to the hypothesis that neurovascular coupling could be initiated at the microvascular level and conducted upstream (Cox et al., 1993; Iadecola, 2004; 1993; Woolsey and Rovainen, 1991). As reviewed in the previous section, the evidence indicates that, irrespective of their ability to directly regulate vascular resistance, capillaries are equipped with the signaling mechanisms to detect neural activity and transmit the signal to α-actin-containing mural cells and SMC in vessels upstream. A likely scenario for the transmission and coordination of the vascular response across the cerebrovascular tree is the following. Activation-induced increases in extracellular K+ triggers hyperpolarization of capillary endothelial cells and, possibly, pericytes via KIR channels (Longden et al., 2017) (Figure 7). The hyperpolarization propagates upstream through inter-endothelial gap junctions and reaches SMC in penetrating arterioles, most likely through myoendothelial junctions, producing their relaxation (Longden et al., 2017) (Figure 7). At the same time, capillary hypoxia increases the deformability of red blood cells, reducing blood viscosity, and increasing capillary flow independently of their diameter (Wei et al., 2016) (Figure 7). The resulting shear stress on the endothelium of feeding arterioles produces SMC relaxation by releasing endothelial vasorelaxing factors (flow-mediated vasodilation) (Koller and Toth, 2012) (Figure 8). Owing to their strong myogenic response (Longden et al., 2016), penetrating arterioles may also relax further in response to the pressure drop produced by the retrograde vasodilatation. The SMC relaxation produced by conducted hyperpolarization and myogenic tone could be complemented by the direct action on SMC of vasoactive agents released by neurons and glia during neural activity (NO, adenosine, ATP, prostanoids, etc.) (Table 1) (Figure 8). Finally, in upstream pial arterioles remote from the site of activation, the vasodilatation is likely to result from two mechanisms: the conducted vasodilatation propagating from arterioles downstream and flow-mediated vasodilatation/myogenic response locally (Figure 9). Accordingly, the pial arteriolar relaxation observed on the brain surface may not be driven directly by neural signals, but by intrinsic vascular signals traveling retrogradely from capillaries and arterioles at the activated site deep in the substance of the brain. It must be noted that lesion of the glia limitans near pial arterioles with L-2-aminoadipidic acid attenuates the vasodilatation produced by neural activity, implicating astrocytes in the transmission of the vascular response (Xu et al., 2008). However, this observation has been difficult to interpret since damaging effects of this harsh treatment on other cell types cannot be ruled out. Concerning the role of conducted vasodilatation, a caveat is that there are likely to be regional variations in this mechanism related to vascular topology, modality of activation, and location of the site of activation relative to resistance vessels. Furthermore, as mentioned above, there are regional differences in the cellular bases neurovascular response, e.g., olfactory bulb, neocortex, cerebellum, etc., which may also play a role. Regional variations not withstanding, the available evidence suggests that neurovascular coupling reflects a coordinated multicellular response acting at different levels of the cerebrovascular network through segment-specific mechanisms.
Figure 7. Neurovascular coupling at the capillary level.

Synaptic activity leads to release of extracellular K+ and increase O2 utilization. In turn, K+ activates KIR channels on endothelial cells, and pericytes leading to endothelial cell hyperpolarization, which is conducted retrogradely via gap junctions linking adjacent endothelial cells. At the same time the reduction in O2 leads to increase deformability of red blood cells (RBC), reducing their viscosity and increasing capillary flow. The resulting shear stress on capillary endothelium may also contribute to their hyperpolarization. The role of astrocytes is less clear, but activation of K+ channels in astrocytes may also contribute to increase K+ in the vicinity of endothelial cells, and due to their link to adjacent astrocytes could also play a role in the retrograde propagation of the hyperpolarization. ATP released during neural activity could increase astrocytic Ca2+ via P2X1 receptors and relax contractile mural cells via reaction products of the COX pathways (Mishra et al., 2016), but as discussed in the text there is no consensus on the ability of capillaries to dilate. Neurovascular contacts from interneurons or subcortical pathways are not depicted (see figure 5).
Figure 8. Neurovascular coupling at the level of intraparenchymal arterioles.

The propagated endothelial hyperpolarization arising from capillaries is transferred to smooth muscle cells (SMC) via myoendothelial junctions, resulting in their relaxation and vasodilatation. The signal continues to be transferred retrogradely to vessels upstream through gap junctions linking SMC as well as endothelial cells. The vasodilatation is complemented by vasoactive factors released by nearby activated neurons and astrocytes, which contributes to sustain the vasodilatation in its propagation to upstream vessels. In addition, the drop in intravascular pressure and increased flow velocity caused by the vasodilatation downstream may contribute to smooth muscle hyperpolarization and relaxation by activating the myogenic response/flow-mediated vasodilatation (FMV). Therefore, at the arteriolar level the vasodilatation has two components: propagated response from capillaries and local response from activated neurons and astrocytes. Neurovascular contacts from interneurons or subcortical pathways are not depicted (see figure 5). Ado: adenosine; Glu: glutamate; Prost: prostanoids.
Figure 9. Neurovascular coupling at the level of pial arteriole.
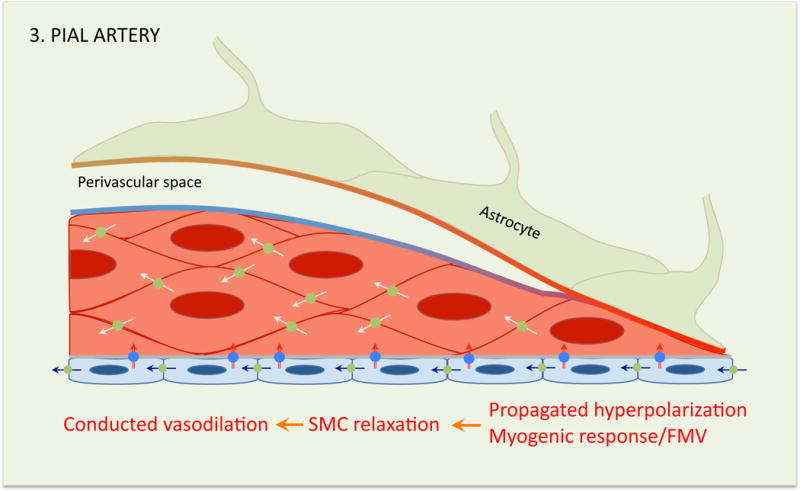
At this level the vasodilation depends on the propagated endothelial and SMC hyperpolarization, and, possibly, activation of the myogenic response/flow mediated vasodilation (FMV). Therefore, at the level of pial arterioles the hemodynamic response is not directly related to synaptic activity, but depends on the retrograde propagation of vasodilatation arising from smaller arterioles and capillaries surrounding the activated area. Perivascular nerves are not depicted (see figure 4).
The neurovascular unit: beyond blood flow
Neurovascular coupling provides a striking example of the close interaction between the brain and its vessels. However, the NVU has also other structural components and functional attributes not directly related to cerebral perfusion that are critically important for brain health. These are briefly reviewed in the following sections.
Brain development
Despite their different embryonic origin, the proliferation, fate determination, migration and terminal differentiation of neural progenitors is intertwined closely with that of vascular cells (Wälchli et al., 2015). A remarkably common array of signaling molecules is involved in both brain and vascular development (Raab and Plate, 2007). Early in brain development, chemoattractant signals, e.g., VEGF-A, secreted by neural progenitors in the subventricular zone guide the in-growth of blood vessels from the perineural vascular plexus, the primitive vascular network surrounding the brain (Raab et al., 2004). In turn, endothelial cells in the subventricular zone and hippocampal subgranular zone provide instructive cues critical for adult neurogenesis, which continues throughout life (Raab and Plate, 2007). The vasculature acts as scaffold for the migration of neuronal progenitors through BDNF produced by endothelial cells (Snapyan et al., 2009), a process that also depends on astrocytic VEGF signaling (Bozoyan et al., 2012). Blood vessels also guide the migration of oligodendrocyte precursors (Tsai et al., 2016), and, in turn, oligodendrocyte precursors are essential for postnatal white matter vascularization through HIF1-dependent transcription (Yuen et al., 2014). The vital trophic and metabolic interaction between brain cells and vessels continues into adulthood, when endothelial metabolism and growth factors contribute to the survival of neurons, astrocytes and oligodendrocytes (Brix et al., 2012; Carmeliet and Ruiz de Almodovar, 2013), and in repair processes following acute brain injury (Hayakawa et al., 2012).
Blood-brain barrier
Neurovascular interactions are also critical for the formation and maintenance of the BBB (Andreone et al., 2015; Zhao et al., 2015a). Cerebral endothelial cells are joined by impermeable tight junctions, express unique molecular transporters and are endowed with endocytotic systems for transcytosis, the transfer of molecules through cells in membrane-bound vesicles (Zhao et al., 2015a). While Wnt/β-catenin signaling from neuronal precursors is critical for vascular patterning and BBB development prenatally (Andreone et al., 2015), after birth astrocytes contribute to the maintenance of the BBB through sonic hedgehog as well as β-catenin, which promote tight junction expression and integrity (Alvarez et al., 2011; Tran et al., 2016). Pericytes are also required for normal BBB development, and pericyte deficiency increases BBB permeability by promoting endothelial transcytosis (Armulik et al., 2010; Daneman et al., 2010). Suppression of transcytosis may depend on the endothelial expression of the omega-3 fatty acid transporter mfsd2a, a member of the major facilitator superfamily of secondarily-active transporters (Andreone et al., 2015). Mfsd2a establishes a unique lipid environment in cerebral endothelial cells that suppresses caveolae-mediated transcytosis (Andreone et al., 2017). These data suggest that the two key features of the BBB, transcytosis and tight junctions, are regulated independently, which may have disease relevance. For example in ischemic stroke, a condition association with BBB disruption, the early opening of the BBB, which is pathogenically relevant, is mediated by an increases in transcytosis and followed by tight junction disruption days later after tissue damage sets in (Knowland et al., 2014).
Cerebrovascular matrix: from structural support to signal transduction
An integral part of the NVU, the matrix embeds the neurovascular cellular assembly and is comprised of: (a) collagen subunits, constituting the structural framework, (b) proteoglycans, which bind water and secreted regulatory factors, such as growth factors, and (c) glycoproteins, which subserve a wide variety of functions (Haffner et al., 2016; Joutel et al., 2015). Matrix proteins are involved in signal transduction by binding to cell-surface receptors, such as integrins, and regulating the distribution secreted factors with growth promoting and regulatory function. Initial studies suggest that the matrix of cerebral vessels varies in composition depending on the specific segment of the cerebrovascular tree. For example, collagen and glycoproteins are more abundant in pial arteries, possibly reflecting their ability to withstand higher intravascular pressures, whereas proteoglycans, rich in regulatory factors, predominate in microvessels (Badhwar et al., 2014; Chun et al., 2011). A significant proportion of matrix proteins is located in the basal lamina that separates the different cellular compartment of the NVU (endothelial, smooth muscle, astrocytes), collagen IV being most abundant (Joutel et al., 2015). Highlighting the critical role of the matrix in cerebrovascular structure and function, mutations of matrix and matrix related proteins are associated with hereditary cerebral arteriopathies affecting small vessels, including collagen IV-related small vessel diseases, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), and cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) (Haffner et al., 2016; Joutel et al., 2015). These conditions, although uncommon, have provided valuable insight into sporadic small vessels disease, a major cause of cognitive impairment on vascular basis and a contributor to AD (Iadecola, 2013).
Perivascular compartment: clearance pathways and immune surveillance
The perivascular compartment has attracted much interest as a major avenue for the removal of potentially toxic byproducts of brain activity released in the extracellular space, including Aβ and tau (Tarasoff-Conway et al., 2015) (Figure 10). Several vascular clearance pathways have been proposed, with particular reference to factors involved in neurodegenerative diseases. A transvascular pathway has been described through which BBB transporters transfer Aβ from the perivascular space to the circulating blood and vice-versa, involving the low density lipoprotein receptor-related protein-1 (LRP1) and the receptor for advanced glycation end-products (RAGE), respectively (Deane et al., 2012; 2004; Zhao et al., 2015b) (Figure 10). Pathological levels of Aβ lead to LRP1 degradation via the proteasome, promoting vascular Aβ accumulation (Deane et al., 2004; Park et al., 2013). Pericytes may also be involved since brain Aβ clearance is markedly reduced Pdgfr-β-deficient mice crossed with mice overexpressing APP, resulting in Aβ accumulation in brain and blood vessels (Sagare et al., 2013). Interestingly, tight junction proteins at the BBB may hinder Aβ clearance, because their disruption enhances the disposal of brain Aβ into the blood (Keaney et al., 2015).
Figure 10. The perivascular space and its clearance pathways.
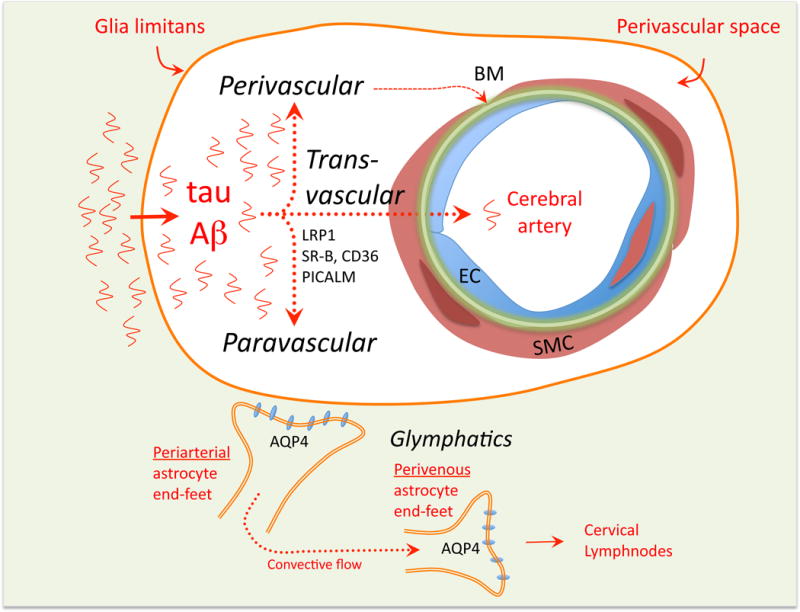
The perivascular space surrounding penetrating arterioles is involved in the clearance of unwanted molecules form the brain, including, but not limited to, Aβ and tau. Products of cerebral activity and metabolism reach the perivascular space by diffusion and can be disposed of through different pathways. A transvascular pathway is thought to rely on scavenger receptors and molecular transporters that carry the unwanted molecules across the vascular wall. A perivascular pathway has long been proposed in which the molecules are transported retrogradely along vascular basement membranes reaching the subarachnoid space and eventually discharging into the cervical nodes. A paravascular (glymphatic) pathway relies on aquaporin-4 channels (AQP4) on astrocytic end-feet to allow the CSF to enter the interstitial space, creating a convective flow that clears unwanted molecules from the brain and feeds into the perivascular venous side eventually draining into dural lymphatics or the cribriform plate. Abbreviations: CD36: cluster of differentiation 36; LRP1: low-density lipoprotein receptor-related protein-1; PICALM: Phosphatidylinositol Binding Clathrin Assembly Protein; SR-B: scavenger receptor B.
A perivascular pathway has long been proposed in which solutes leave the brain parenchyma by traveling retrogradely along the vascular basal lamina, eventually reaching cerebral arteries on the brain surface and draining into cervical lymphnodes (Hladky and Barrand, 2014; Morris et al., 2016). The routes linking the arterial wall to cervical lymphnodes have not been clearly defined, but they may involve the dural lymphatic system and the CSF outflow pathway through the nasal cribriform plate (Hladky and Barrand, 2014; Louveau et al., 2016).
A paravascular pathway (glymphatic system) runs in opposite direction to the perivascular pathway and involves the aquaporin-4 water channel enriched in astrocytic end-feet. By imaging the movement of fluorescent dyes injected into the CSF of the cisterna magna, an expansion of the subarachnoid space between the cerebellum and the dorsal medulla, the dye is seen first around arteries on the brain surface, then in the parenchyma, and, later, in draining veins (Iliff et al., 2012). Lack of aquaporin-4 channels slows the movement of the dye dramatically (Iliff et al., 2012). On these bases, it was proposed that the CSF penetrates into the brain substance by tracking around arteries, enters the interstitial space through aquaporin-4 channels in perivascular astrocytic end-feet, and, by exchanging with the interstitial fluid, carries the waste away from the brain parenchyma, exiting the interstitial space through aquaporin-4 channels surrounding the venous perivascular space (Nedergaard, 2013). Eventually, the CSF loaded with waste collected from the interstitial space drains into the dural lymphatics or other CSF exit pathways (Nedergaard, 2013). This system may be involved in the clearance of brain Aβ and tau (Iliff et al., 2012), and is modulated by the brain’s physiological state, particularly sleep (Iliff et al., 2014). Aging, brain trauma, and brain microinfarcts impede this clearance pathway (Kress et al., 2014; Plog et al., 2015; M. Wang et al., 2017), raising the possibility that failure of the glymphatic system is involved in the brain dysfunction associated with these conditions.
These studies raise several questions. In the paravascular pathway, it is unclear how protein waste enters the perivenous compartment and how it reaches the lymphatics. The perivascular and paravascular clearance models are based on different methodological approaches, i.e., injection of markers either in the brain parenchyma (perivascular) or CSF (paravascular), which may introduce artifacts (Hladky and Barrand, 2014). Furthermore, the anatomical relationships between the perivascular and paravascular pathways, which run in opposite directions, and how they share the periarterial compartment need further exploration (Bedussi et al., 2017; Morris et al., 2016). Finally, the relative contribution of these pathways vs. endogenous clearance mechanisms, e.g., enzymatic degradation, in the human brain remain to be established (J. S. Miners et al., 2014).
The perivascular space also houses innate immune cells, which are involved in immune homeostasis and have a profound influence on the response of the brain to infectious or immune challenges. PVM are a subgroup of resident brain macrophages located in the perivascular space, juxtaposed to the outer wall of penetrating arteries and veins (Prinz et al., 2017) (Figure 4). PVM originate from erithromyeloid precursors in the yolk sac and, like microglia, enter the brain early in development (Prinz et al., 2017). PVM promote hypothalamic-adrenal axis activation and autonomic responses in models of cardiac ischemia, stress, and inflammation, may harbor the HIV virus infecting the brain, and participate in tumor angiogenesis and cerebrovascular repair (Prinz et al., 2017). Capable of producing large amounts of free radicals in close proximity to the vessel wall, PVM are a major source of vascular oxidative stress and key contributors to neurovascular and/or cognitive dysfunction in models of chronic hypertension (Faraco et al., 2016b) and Aβ overproduction (Park et al., 2017). On the other hand, owing to their scavenging function, PVM may prevent cerebrovascular accumulation of amyloid in models of cerebral amyloid angiopathy (CAA) (Hawkes and McLaurin, 2009; Thanopoulou et al., 2010). Overall, PVM are emerging as a novel constituent of the NVU that awaits further exploration.
The NVU in neurodegenerative disease
Due to their age dependence and lack of effective treatments, neurodegenerative diseases are predicted to grow to epidemic proportions over the next several decades (Gammon, 2014). Since alterations of the NVU are anticipated to lead to brain dysfunction and damage (Figure 11), there has been a growing interest in exploring the potential contribution of neurovascular dysfunction to neurodegeneration (De-la-Torre, 2017). The resulting body of work has provided evidence for a wide variety of neurovascular alterations in neurodegenerative diseases (Table 2), reviewed in the next sections.
Figure 11. Potential pathogenic mechanisms by which neurovascular dysfunction can cause brain dysfunction.

Alterations of the NVU may lead to reductions in CBF below the threshold required for normal brain oxygenation leading to hypoxia. BBB dysfunction may alter the homeostasis of the brain internal milieu by limiting the delivery of glucose and other nutrients and impairing the clearance of unwanted metabolites through efflux transporters. Reduction in trophic factor production by NVU cells may increase neuronal and glial vulnerability and susceptibility to disease. Alterations of clearance pathways may promote the accumulation of molecules, such as Aβ and tau, leading to proteinopathies.
Table 2.
Evidence of neurovascular dysfunction in major neurodegenerative diseases
| Disease | Vascular morphology | Blood-brain barrier | Cerebral perfusion | Selected references |
|---|---|---|---|---|
| Alzheimer’s disease |
|
|
|
Janelidze et al, 2017 Toledo et al., 2013 Roher et al., 2004 Montagne et al, 2015 Kisler et al., 2017a |
|
| ||||
| Frontotemporal dementia |
|
|
|
Sudre et al., 2017 Janelidze et al., 2017 Dopper et al., 2016 Martin et al., 2001 Thal et al., 2015 |
|
| ||||
| Amyotrophic lateral sclerosis |
|
|
|
Murphy et al., 2012 Rule et al., 2010 Abrahams et al.,1996 Henkel et al., 2009 Winkler et al., 2013 |
|
| ||||
| Idiopathic Parkinson disease |
|
|
|
Janelidze et al., 2015 Kortekaas et al., 2005 Pisani et al., 2012 Rosengarten et al., 2010 Al-Bachari et al., 2014 Guan et al., 2013 Faucheux et al., 1999 |
|
| ||||
| Dementia with Lewy bodies |
|
|
|
Janelidze et al., 2017 Roquet et al., 2016 Ghebremedhin et al., 2010 Miners et al., 2014 |
Abbreviations: ATS: atherosclerosis; Qalb: CSF/Plasma albumin ratio (index of BBB permeability); Gd: gadolinium; Pgp: P-glycoprotein efflux transporter; SN: substantia nigra; TJ: tight junctions; WM: white matter.
Alzheimer’s disease
The most common cause of dementia in the elderly, AD is characterized pathologically by the accumulation of the Aβ in extracellular amyloid plaques and hyperphosphorylated tau in intracellular neurofibrillary tangles, which are thought to induce neuronal dysfunction and damage (De Strooper and Karran, 2016). It has long been known that cerebrovascular alterations are associated with AD (De-la-Torre, 2017), but only recently the contribution of vascular factors has received direct attention. AD brains have microvascular alterations (increased capillary tortuosity, capillary rarefaction, thickened basement membrane, “string vessels” etc.) (Love and J. S. Miners, 2016) and more atherosclerosis in intracranial vessels than age matched controls (Roher et al., 2004). Community-based pathological studies of clinically diagnosed AD have revealed that AD pathology (plaques and tangles) and ischemic lesions frequently coexist in the same brain (Schneider et al., 2007; Toledo et al., 2013), and that ischemic lesions lower the threshold for clinical dementia (Kapasi et al., 2017). Resting CBF is reduced and hemodynamic responses to neural activation are attenuated in the presymptomatic phase of AD (reviewed in (Kisler et al., 2017a)), suggesting early involvement in the disease process. Similarly, neurovascular dysfunction is observed in patients with CAA (Charidimou et al., 2017). BBB permeability is increased in patients with prodromal AD, an effect associated with increased soluble Pdgfr-β in the CSF, suggesting pericyte degeneration (Montagne et al., 2015). A pathogenic role for vascular factors in AD has also been suggested by epidemiological studies indicating that cerebrovascular diseases and AD have similar risk factors (hypertension, diabetes, obesity, etc.), and that improved cardiovascular health may have contributed to the recently reported reduction in AD incidence (Pase et al., 2017). Indeed, vascular risk factors, such as hypertension, promote amyloid deposition (Gottesman et al., 2017; Petrovitch et al., 2000; Rodrigue et al., 2013), attributable to reduced Aβ clearance and/or enhanced amyloid precursor protein (APP) processing (Carnevale et al., 2012; Faraco et al., 2016a).
Most studies on the mechanisms of neurovascular dysfunction in AD have focused on the role of Aβ (Kisler et al., 2017a). Thomas et al. first reported that Aβ, in addition to causing neuronal dysfunction, also impairs the ability of endothelial cells to relax systemic vessels in vitro (Thomas et al., 1996). Using mice overexpressing mutated APP (Tg2576 mice), it was subsequently established that Aβ impairs all the major factors regulating the cerebral circulation: neurovascular coupling, endothelial function and cerebrovascular autoregulation (Iadecola et al., 1999; Niwa et al., 2002a; 2000b). Aβ was also found to reduce resting CBF and to promote vasoconstriction (Niwa et al., 2002b; 2001b), resulting in increased susceptibility to ischemic brain injury (F. Zhang et al., 1997). These neurovascular alterations were observed before amyloid plaques and behavioral deficits, suggesting an early pathogenic role, and were subsequently confirmed in other APP overexpression models (Beckmann et al., 2003; Tong et al., 2005). Mechanistically, Aβ may act on the innate immunity receptor CD36 in PVM, inducing vascular oxidative stress through the superoxide synthesizing enzyme NADPH oxidase (Han et al., 2008; Park et al., 2017; 2013; 2008; Tong et al., 2005). Radicals, in turn, cause endothelial dysfunction via DNA damage, poly-ADP ribose activation, ADP-ribose-mediated TRPM2 channel opening and Ca2+ overload (Park et al., 2014b). In addition to failure in LRP1-mediated transvascular Aβ clearance (Deane et al., 2004), innate immunity receptors, such as CD36, also promote CAA (Park et al., 2013), in which Aβ accumulation leads to vascular oxidative stress, neurovascular dysfunction, and, with aging, irreversible damage to the vasomotor apparatus (Park et al., 2014a).
Frontotemporal dementia
Frontotemporal dementia (FTD), the leading cause of dementia in middle age, comprises a heterogeneous group of neurodegenerative conditions characterized by diverse cognitive and behavioral manifestations (Olney et al., 2017). Familial forms of the disease are often caused by mutations of the progranulin, tau, or C9orf72 gene, but sporadic forms are more common (Olney et al., 2017). Resting CBF is reduced in the frontal, parietal and temporal cortex of presymptomatic patients, an effect independent of atrophy (Dopper et al., 2016). Pathological studies have described microvascular damage, microhemorrhages and white matter ischemic lesions, more pronounced in older individuals, suggesting that the vascular disease may be independent of FTD pathology and age related (Sudre et al., 2017; Thal et al., 2015). BBB alterations have also been reported (Janelidze et al., 2017). On the other hand, astrogliosis and astrocytic damage, prominent pathological features of FTD, correlate regionally with reductions in CBF (Martin et al., 2001), implicating astrocyte loss in the flow reduction and, possibly, in the mechanisms of the disease. Studies in mouse models reflecting selected aspects of FTD have confirmed some of these neurovascular alterations. In agreement with pathological studies showing microhemorrhages, progranulin-deficient mice exhibit increases BBB permeability and hemorrhages after stroke, but resting CBF or cerebrovascular reactivity were not reduced (Jackman et al., 2013). The BBB dysfunction was attributed to alterations in the structure of endothelial tight junctions, which appeared shorter and simplified (Jackman et al., 2013). BBB alterations have also been reported in mice overexpressing tau mutations found in FTD, e.g., P301L (Blair et al., 2015). However, in these mice the cerebrovascular reactivity to pCO2 was reported to be increased (Wells et al., 2015), ruling out global suppression of vasomotor function.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is characterized by dysfunction and degeneration of upper and lower motor neurons resulting in progressive weakness and paralysis (van Es MD et al., 2017). Up to 50% of cases have cognitive dysfunction of the type observed in FTD, and up to 15% of ALS cases also develop FTD (van Es MD et al., 2017). Alterations in the BBB and blood-spinal cord barrier were initially suggested to play a role in the disease based on increased plasma protein in the CSF and on microvascular alterations in spinal cord microvessels in autopsy specimens, e.g., reduced tight junction proteins, endothelial and pericyte damage, capillary rarefaction, plasma protein extravasation (Evans et al., 2013; Winkler et al., 2013). Studies in a mouse model of familial ALS caused by mutation of the free radical scavenging enzyme SOD1 have demonstrated uncoupling between spinal cord blood flow and glucose metabolism, as well as increased permeability of spinal cord capillaries and microhemorrhages, which precede motor neuron degeneration (Miyazaki et al., 2012; Zhong et al., 2008). Prevention of the barrier alterations with an activated protein C analog delayed the development of disease (Winkler et al., 2014). Although frank microhemorrhages were not observed by high resolution MRI (Verstraete et al., 2010), BBB dysfunction and red blood cell extravasation may be present in a subset of patients (Winkler et al., 2013). A separate line of investigations also suggests the involvement of neurovascular factors in motor neuron degeneration. In mice, deficiency of VEGF leads to motor neuron dysfunction and degeneration (Poesen et al., 2008), and administration of VEGF rescues motor neuron dysfunction in SOD1 mutant mice (Storkebaum et al., 2005). Initial genetic studies suggested a link of ALS with several VEGF haplotypes resulting in low VEGF production, which were not later confirmed except for a minor gender-specific effect (Lambrechts et al., 2009). The protection by VEGF is likely to result from its neurotrophic effects, although a microvascular contribution cannot be ruled out (Quaegebeur et al., 2011). However, vasoprotective effects are difficult to reconcile with the observation that increased vascular permeability may be damaging in ALS, since VEGF increases vascular permeability (Quaegebeur et al., 2011). Decreases in cortical perfusion, including non-motor areas, are well described early in the disease and correlate with disease progression (Murphy et al., 2012; Rule et al., 2010). In ALS patients with frontal cognitive dysfunction, the increase in CBF evoked by activation tasks in frontal association areas is reduced, an effect that is more widespread in patients with more severe cognitive deficits (Abrahams et al., 1996). These findings point to a more global alteration in neurovascular regulation, which could play a role in the cognitive manifestation of the disease.
Parkinson disease
Idiopatic Parkinson disease (PD) is a neurodegenerative disorder characterized by movement and cognitive deficits in which the pathology starts in the brainstem, particularly the nigrostriatal pathway, and spreads to the cortex (Przedborski, 2017). Postmortem studies of advanced cases have shown capillary damage and fragmentation in frontal cortex and brainstem nuclei (Guan et al., 2013), as well as increased endothelial cell nuclei in the substantia nigra (Faucheux et al., 1999). Increases in VEGF in the CSF have been reported (Janelidze et al., 2015), together with evidence of increased BBB permeability in the late stages of the disease (Kortekaas et al., 2005; Pisani et al., 2012). On the other hand, CBF reductions have been observed early in the disease course in neocortical regions without overt pathology (Borghammer et al., 2010; Fernández-Seara et al., 2012). In PD with cognitive impairment CBF reductions in parietal regions correlated with the cognitive dysfunction (Al-Bachari et al., 2014), suggesting a link between cognitive deficits and CBF reductions. However, cerebrovascular reactivity seems to be preserved since the hemodynamic response to visual stimulation or to increases in arterial pCO2 (hypercapnia) are not attenuated (Al-Bachari et al., 2014; Rosengarten et al., 2010). An interesting hypothesis is that the CBF reductions is due alterations of the cholinergic, serotoninergic and noradrenergic neurovascular innervation of the neocortex (Al-Bachari et al., 2014; Fernández-Seara et al., 2012). The experimental observation that these subcortical pathways may influence resting CBF and not neurovascular coupling (see section on neurovascular coupling), would be consistent with this hypothesis.
Dementia with Lewy Bodies
CBF is reduced in frontal, insular, temporal and occipital cortex in dementia with Levy bodies (DLB), a condition characterized by fluctuations in cognition, hallucinations and parkinsonism (Galasko, 2017). The changes in CBF occur in the prodromal stage of the disease (Roquet et al., 2016), and may be due to a reduction in energy metabolism (Ishii et al., 2015). While vascular alterations, such as reduced VEGF, small vessel disease, and atherosclerosis, have been described in the occipital cortex (Ghebremedhin et al., 2010; S. Miners et al., 2014), there might be an inverse relationship between Lewy body pathology and vascular pathology (Ghebremedhin et al., 2010). Therefore, Levy body pathology may stave off vascular pathology and vice-versa.
Where do we go from here?
The concept of NVU has come a long way since 2001. Central to brain health, neurovascular interactions have emerged as essential contributors to brain development and vital for maintaining the homeostasis of the brain internal milieu. Neurovascular coupling has evolved from a unidimensional process driven by the direct effect of neuronal mediators on local blood vessels, to a multi-dimensional one in which a multitude of mediators released from multiple cell types engages unique signaling pathways and effector systems on different cerebrovascular segments in a coordinated fashion (Table 1). The resulting carefully orchestrated vascular response involves the entire cerebrovascular tree and leads to spatially and temporally focused increases in CBF. The evidence implicates neurons and astrocytes in the generation, modulation and transmission of the signal to local microvessels, capillary endothelial cells in the retrograde propagation of vasomotor signals, and contractile mural cells and SMC of progressively larger vascular segments as the main effector of the CBF increase (Table 1). Therefore, neural activity initiates the process, but the implementation of the hemodynamic response requires the coordinated interaction of other NVU cells across the whole cerebrovascular network. Significant progress has also been made in the pathobiology of the NVU. Increasing evidence implicates NVU dysfunction in major neurodegenerative diseases, in which the role of vascular factors was traditionally not considered. This new knowledge has raised a number of questions pertaining to the inner workings of the NVU and to its role in brain diseases.
Knowledge gaps in neurovascular coupling
The contribution of feed-forward and feedback mechanisms to the flow increase remains unclear. In particular, the region-specific factors determining the recruitment of one mechanism versus the other and the cells and mediators driving the attendant hemodynamic response have not been identified. The cellular bases of neurovascular interactions at the different levels of the cerebrovascular tree have not been established. Initial studies have highlighted the role of endothelial KIR channel induced hyperpolarization at the capillary level and of neuronal NO in arterioles, but how these responses are generated, transmitted and appropriately timed requires further study. Furthermore, whether or not astrocytes contribute to the K+ surge at the capillary level and their potential contribution to the release of vasoactive factors, metabolic adaptation and retrograde vasodilatation at the arteriolar level has not been fully elucidated.
The role of capillaries in neurovascular coupling remains controversial. Are capillaries neural activity sensors or also effectors of the initial hemodynamic response? Are the small changes in capillary diameter observed in some studies physiologically relevant? Answering these questions would require a reexamination of what a capillary is, and a better characterization of the cells associated with them and their hemodynamic impact. Using vessel size and isolated cell markers as the sole identification tools for mural cells has generated considerable confusion. A different approach is needed based on: (a) establishing the ontogeny of mural cells, (b) defining the transcription and growth factors driving their development and localization, and (c) identifying their molecular signature. This effort would require a comprehensive reevaluation of the brain’s vascular development focusing specifically on mural cells, e.g., (Jung et al., 2017).
Relatively little is known about the upstream propagation of vasoactive signals from capillaries to pial arterioles. Are gap junctions and myoendothelial junctions the sole mode of inter-endothelial and endothelial-SMC transmission, or are diffusible factors also involved? Do mural cells, e.g., pericytes, play a role? The mechanisms focusing the hemodynamic response by selectively engaging arteriolar branches supplying the activated region, e.g., (Ngai et al., 1988), and the possible role of glial limitans astrocytes require further study. In systemic arteries perivascular sympathetic (noradrenergic) nerves control propagated responses (Segal, 2015), but it is unclear whether they play a similar role in cerebral arteries. In this regard, it is of interest that the noradrenergic innervation of the neocortex may play a role in focusing the vascular response (Bekar et al., 2012), a concept that requires further exploration.
Answering these questions will be facilitated by the use of new models and approaches to study the cerebral vasculature. For example, optical coherence tomography (Baran and R. K. Wang, 2016), 3-photon microscopy (Ouzounov et al., 2017), and ultrafast ultrasound localization microscopy (Errico et al., 2015), may offer an unprecedented look at the cerebral microcirculatory network at a depth, spatial extent and/or resolution previously unattainable. Whenever feasible, coupling these imaging tools with genetically-encoded activity sensors and cell specific reporters, as well as chemo- and opto-genetic approaches to activate specific neural pathways may provide novel insights into the spatiotemporal relationships linking neural activity and segmental vascular responses. Using conscious animals will eliminate anesthesia as a major confounder and will allow investigators to probe neurovascular coupling beyond the realm of olfactory, somatosensory or visual stimuli and towards complex behaviors in multiple brain regions (Gao et al., 2017). The introduction of more realistic multicellular in vitro models of the NVU, such as the 3-D microfluidics organ-on-chip technology for the BBB (Adriani et al., 2017) and cerebrovascular organoids derived from human induced pluripotent stem cells (IPS) (Appelt-Menzel et al., 2017), provides the opportunity to explore the molecular interactions among NVU cells beyond what can be achieved with conventional co-culture systems. Large-scale single-cell RNA sequencing applied to the cerebral vasculature (He et al., 2016; Tasic et al., 2016) will provide a glimpse into the molecular signature of specific cerebrovascular cells, and provide insight into the molecular bases of the functional differences of vascular and perivascular cells in the different segments of the cerebrovascular tree.
NVU dysfunction and neurodegeneration
Our understanding of the role of neurovascular dysfunction in neurodegenerative diseases is still rudimentary. Although there is overwhelming evidence that neurodegeneration is associated with structural and functional alterations of the NVU, often early in the disease course (Table 2), several questions remain to be addressed. Is the NVU dysfunction an epiphenomenon of the degenerative process or a pathogenic contributor? In most cases, the reductions in CBF are not severe enough to induce ischemic injury. However, it cannot be excluded that reduced cerebral perfusion, in concert with neurovascular uncoupling, may have deleterious effects on the brain in the long run. BBB dysfunction, neurotrophic failure and impaired clearance associated with NVU dysfunction may also contribute (Figure 11), but their pathogenic impact remains to the established. Which brings us to a related question. Does the neurovascular dysfunction promote the neurodegenerative process? In AD, experimental and clinical studies indicate that vascular dysfunction is an early manifestation of the disease and may promote AD pathology. Is this the case also in other neurodegenerative diseases? Considering that oxidative stress and inflammation, major causes of neurovascular dysfunction, have been implicated in virtually all neurodegenerative conditions (Ransohoff, 2016), these factors could be driving both the vascular and neurodegenerative pathologies independently, amplifying their respective pathogenic impact. In diseases like AD, in which the coexistence of neurodegeneration with cerebrovascular lesions is well documented, what is the role of the cerebrovascular pathology in the clinical expression of the disease? Vascular lesions could lower the threshold for cognitive deficits by increasing the overall lesion burden on the brain. Alternatively, the two processes could be synergistic, amplifying each other, as suggested by evidence that vascular dysfunction promotes AD pathology (see above). Although ischemic injury is not inconsequential to the clinical expression of AD (Kapasi et al., 2017), there is no definitive evidence of a synergistic interaction, e.g., (Vemuri et al., 2015). However, the reduction in dementia incidence attributed to better cardiovascular health (Pase et al., 2017) suggests a contribution of vascular pathology to neurodegeneration, an attractive hypothesis that requires further testing in randomized clinical trials.
The factors by which neurodegenerative pathology leads to neurovascular dysfunction remain to be defined. Are selected NVU cells and specific cerebrovascular segments particularly vulnerable, and, if so, why? Are there common signaling pathways and mediators responsible for the dysfunction in different diseases? What is the contribution of neuronal, glial and vascular cell metabolism to the vascular dysfunction? Difficulties in performing mechanistic studies in human patients, the long interval between onset of pathology and clinical diagnosis, lack of reliable biomarkers, and lack of animal models recapitulating in full disease phenotypes have been major impediments to progress. However, developments in both the clinical and experimental arenas may help overcome some of these barriers. Large-scale “omic” studies, advanced imaging with markers of disease, new molecular biomarkers, and IPS cells from patients, may provide mechanistic clues to be explored further in experimental models. In turn, new animal models incorporating neurodegenerative pathology with vascular co-morbidities and new cell-specific approaches to probe the NVU promise a dramatic expansion of knowledge that can then be verified in clinical studies. Therefore, there is a strong rationale for bolstering efforts to clarify the role of the NVU dysfunction in neurodegenerative diseases (Corriveau et al., 2016). New knowledge in this field may open new avenues in the prevention, early diagnosis and treatment of some of the most devastating illnesses affecting humankind.
Acknowledgments
Dr. Iadecola is supported by NIH grants R37-NS89323, R01-NS34179, R01-NS95441, R01-NS37853 and R01-NS100447. Additional support was provided by the Feil Family Foundation and the Fondation Leducq. Dr. Marcus Raichle provided the picture of Kety, Sokoloff, Ingvar and Lassen in Figure 2, and Dr. Labaik Park the picture of the penetrating arteriole in Figures 4 and 5. The help of Dr. Giuseppe Faraco in the preparation of the figures is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams S, Goldstein LH, Kew JJ, Brooks DJ, Lloyd CM, Frith CD, Leigh PN. Frontal lobe dysfunction in amyotrophic lateral sclerosis. A PET study. Brain. 1996;119(Pt 6):2105–2120. doi: 10.1093/brain/119.6.2105. [DOI] [PubMed] [Google Scholar]
- Adriani G, Ma D, Pavesi A, Kamm RD, Goh ELK. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab on a Chip. 2017;17:448–459. doi: 10.1039/C6LC00638H. [DOI] [PubMed] [Google Scholar]
- Al-Bachari S, Parkes LM, Vidyasagar R, Hanby MF, Tharaken V, Leroi I, Emsley HCA. Arterial spin labelling reveals prolonged arterial arrival time in idiopathic Parkinson’s disease. Neuroimage Clin. 2014;6:1–8. doi: 10.1016/j.nicl.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonnière L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, Deik AA, Ginty DD, Clish CB, Gu C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron. 2017;94:581–594.e5. doi: 10.1016/j.neuron.2017.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Lacoste B, Gu C. Neuronal and Vascular Interactions. Annu Rev Neurosci. 2015;38:25–46. doi: 10.1146/annurev-neuro-071714-033835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen J, Shafi N, Bryan R. Endothelial influences on cerebrovascular tone. J Appl Physiol. 2006;100:318–327. doi: 10.1152/japplphysiol.00937.2005. [DOI] [PubMed] [Google Scholar]
- Anenberg E, Chan AW, Xie Y, LeDue JM, Murphy TH. Optogenetic stimulation of GABA neurons can decrease local neuronal activity while increasing cortical blood flow. J Cereb Blood Flow Metab. 2015;35:1579–1586. doi: 10.1038/jcbfm.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, Gallo EF, Kawano T, Orio M, Abe T, Gooden C, Zhou P, Iadecola C. Purinergic signaling induces cyclooxygenase-1-dependent prostanoid synthesis in microglia: roles in the outcome of excitotoxic brain injury. PLoS ONE. 2011;6:e25916. doi: 10.1371/journal.pone.0025916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelt-Menzel A, Cubukova A, Günther K, Edenhofer F, Piontek J, Krause G, Stüber T, Walles H, Neuhaus W, Metzger M. Establishment of a Human Blood-Brain Barrier Co-culture Model Mimicking the Neurovascular Unit Using Induced Pluri- and Multipotent Stem Cells. Stem Cell Reports. 2017;8:894–906. doi: 10.1016/j.stemcr.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–625. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab. 2016;36:451–455. doi: 10.1177/0271678X15610340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badhwar A, Stanimirovic DB, Hamel E, Haqqani AS. The proteome of mouse cerebral arteries. J Cereb Blood Flow Metab. 2014;34:1033–1046. doi: 10.1038/jcbfm.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P, Segal SS. Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf) 2011;202:271–284. doi: 10.1111/j.1748-1716.2010.02244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran U, Wang RK. Review of optical coherence tomography based angiography in neuroscience. Neurophoton. 2016;3:010902–13. doi: 10.1117/1.NPh.3.1.010902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann N, Schuler A, Mueggler T, Meyer E, Wiederhold K, Staufenbiel M, Krucker T. Age-dependent cerebrovascular abnormalities and blood flow disturbances in APP23 mice modeling Alzheimer’s disease. J Neurosci. 2003;23:8453–8459. doi: 10.1523/JNEUROSCI.23-24-08453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedussi B, van der Wel NN, de Vos J, van Veen H, Siebes M, VanBavel E, Bakker EN. Paravascular channels, cisterns, and the subarachnoid space in the rat brain: A single compartment with preferential pathways. J Cereb Blood Flow Metab. 2017;37:1374–1385. doi: 10.1177/0271678X16655550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekar LK, Wei HS, Nedergaard M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab. 2012;32:2135–2145. doi: 10.1038/jcbfm.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker KR, Srienc AI, Shimoda AM, Agarwal A, Bergles DE, Kofuji P, Newman EA. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J Neurosci. 2016;36:9435–9445. doi: 10.1523/JNEUROSCI.1782-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A, Linder J. Sympathetic control of cerebral blood flow in acute arterial hypertension. Acta Physiol Scand. 1976;96:114–121. doi: 10.1111/j.1748-1716.1976.tb10176.x. [DOI] [PubMed] [Google Scholar]
- Blair LJ, Frauen HD, Zhang B, Nordhues BA, Bijan S, Lin YC, Zamudio F, Hernandez LD, Sabbagh JJ, Selenica MLB, Dickey CA. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun. 2015;3:8. doi: 10.1186/s40478-015-0186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco VM, Stern JE, Filosa JA. Tone-dependent vascular responses to astrocyte-derived signals. 2008;294:H2855–63. doi: 10.1152/ajpheart.91451.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H, Kleinfeld D. The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci. 2013;16:889–897. doi: 10.1038/nn.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder DE, McCarthy KD. Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J Neurosci. 2014;34:13139–13150. doi: 10.1523/JNEUROSCI.2591-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghammer P, Chakravarty M, Jonsdottir KY, Sato N, Matsuda H, Ito K, Arahata Y, Kato T, Gjedde A. Cortical hypometabolism and hypoperfusion in Parkinson’s disease is extensive: probably even at early disease stages. Brain Struct Funct. 2010;214:303–317. doi: 10.1007/s00429-010-0246-0. [DOI] [PubMed] [Google Scholar]
- Bozoyan L, Khlghatyan J, Saghatelyan A. Astrocytes control the development of the migration-promoting vasculature scaffold in the postnatal brain via VEGF signaling. J Neurosci. 2012;32:1687–1704. doi: 10.1523/JNEUROSCI.5531-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brix B, Mesters JR, Pellerin L, Jöhren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1α-mediated target gene activation. J Neurosci. 2012;32:9727–9735. doi: 10.1523/JNEUROSCI.0879-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet I, Gordon E, Han J, Cristofaro B, Broquères-You D, Liu C, Bouvrée K, Zhang J, del Toro R, Mathivet T, Larrivée B, Jagu J, Pibouin-Fragner L, Pardanaud L, Machado MJC, Kennedy TE, Zhuang Z, Simons M, Lévy BI, Tessier-Lavigne M, Grenz A, Eltzschig H, Eichmann A. Netrin-1 controls sympathetic arterial innervation. J CLin Invest. 2014;124:3230–3240. doi: 10.1172/JCI75181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ruiz de Almodovar C. VEGF ligands and receptors: implications in neurodevelopment and neurodegeneration. Cell Mol Life Sci. 2013;70:1763–1778. doi: 10.1007/s00018-013-1283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, Pallante F, Zlokovic B, Yan SS, Lembo G. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension. 2012;60:188–197. doi: 10.1161/HYPERTENSIONAHA.112.195511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Hamel E. Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics. 2010;2:1–9. doi: 10.3389/fnene.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, Hamel E. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24:8940–8949. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigneau E, Oheim M, Audinat E, Charpak S. Two-photon imaging of capillary blood flow in olfactory bulb glomeruli. Proc Natl Acad Sci U S A. 2003;100:13081–13086. doi: 10.1073/pnas.2133652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140:1829–1850. doi: 10.1093/brain/awx047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EMC. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc. 2014;3 doi: 10.1161/JAHA.114.000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HB, Scott M, Niessen S, Hoover H, Baird A, Yates J, Torbett BE, Eliceiri BP. The proteome of mouse brain microvessel membranes and basal lamina. J Cereb Blood Flow Metab. 2011;31:2267–2281. doi: 10.1038/jcbfm.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ. The Cerebral Circulation. 2010 doi: 10.4199/C00005ED1V01Y200912ISP002. [DOI] [Google Scholar]
- Cohen Z, Bonvento G, Lacombe P, Hamel E. Serotonin in the regulation of brain microcirculation. Prog Neurobiol. 1996;50:335–362. doi: 10.1016/s0301-0082(96)00033-0. [DOI] [PubMed] [Google Scholar]
- Colonnese MT, Phillips MA, Constantine-Paton M, Kaila K, Jasanoff A. Development of hemodynamic responses and functional connectivity in rat somatosensory cortex. Nat Neurosci. 2008;11:72–79. doi: 10.1038/nn2017. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Bosetti F, Emr M, Gladman JT, Koenig JI, Moy CS, Pahigiannis K, Waddy SP, Koroshetz W. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A Framework for Advancing Research Priorities in the Cerebrovascular Biology of Cognitive Decline. Cell Mol Neurobiol. 2016;36:281–288. doi: 10.1007/s10571-016-0334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox SB, Woolsey TA, Rovainen CM. Localized dynamic changes in cortical blood flow with whisker stimulation corresponds to matched vascular and neuronal architecture of rat barrels. Journal of Cerebral Blood Flow & Metabolism. 1993;13:899–913. doi: 10.1038/jcbfm.1993.113. [DOI] [PubMed] [Google Scholar]
- Craigie EH. The architecture of the cerebral capillary bed. Biol Rev Camb Philos Soc. 1945;20:133–146. doi: 10.1111/j.1469-185x.1945.tb00446.x. [DOI] [PubMed] [Google Scholar]
- Cudmore RH, Dougherty SE, Linden DJ. Cerebral vascular structure in the motor cortex of adult mice is stable and is not altered by voluntary exercise. J Cereb Blood Flow Metab. 2016;2:271678. doi: 10.1177/0271678X16682508. X16682508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E. The fine structure of intracerebral vessels. Z Zellforsch Mikrosk Anat. 1973;145:577–586. doi: 10.1007/BF00306725. [DOI] [PubMed] [Google Scholar]
- Damisah EC, Hill RA, Tong L, Murray KN, Grutzendler J. A fluoro-Nissl dye identifies pericytes as distinct vascular mural cells during in vivo brain imaging. Nat Neurosci. 2017;79:559–12. doi: 10.1038/nn.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva TM, Faraci FM. Microvascular Dysfunction and Cognitive Impairment. Cell Mol Neurobiol. 2016;36:241–258. doi: 10.1007/s10571-015-0308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E. The Cellular Phase of Alzheimer’s Disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- De-la-Torre JC. Are Major Dementias Triggered by Poor Blood Flow to the Brain? Theoretical Considerations. J Alzheimers Dis. 2017;57:353–371. doi: 10.3233/JAD-161266. [DOI] [PubMed] [Google Scholar]
- Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J CLin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Dietrich HH, Kajita Y, Dacey RG. Local and conducted vasomotor responses in isolated rat cerebral arterioles. Am J Physiol. 1996;271:H1109–16. doi: 10.1152/ajpheart.1996.271.3.H1109. [DOI] [PubMed] [Google Scholar]
- Dopper EGP, Chalos V, Ghariq E, den Heijer T, Hafkemeijer A, Jiskoot LC, de Koning I, Seelaar H, van Minkelen R, van Osch MJP, Rombouts SARB, van Swieten JC. Cerebral blood flow in presymptomatic MAPT and GRN mutation carriers: A longitudinal arterial spin labeling study. Neuroimage Clin. 2016;12:460–465. doi: 10.1016/j.nicl.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Iadecola C. The role of neuronal signaling in controlling cerebral blood flow. Brain Lang. 2007;102:141–152. doi: 10.1016/j.bandl.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Drew PJ, Shih AY, Kleinfeld D. Fluctuating and sensory-induced vasodynamics in rodent cortex extend arteriole capacity. Proc Natl Acad Sci USA. 2011;108:8473–8478. doi: 10.1073/pnas.1100428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichmann A, Brunet I. Arterial innervation in development and disease. Sci Transl Med. 2014;6:252ps9–252ps9. doi: 10.1126/scitranslmed.3008910. [DOI] [PubMed] [Google Scholar]
- Engl E, Jolivet R, Hall CN, Attwell D. Non-signalling energy use in the developing rat brain. J Cereb Blood Flow Metab. 2017;37:951–966. doi: 10.1177/0271678X16648710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico C, Pierre J, Pezet S, Desailly Y, Lenkei Z, Couture O, Tanter M. Ultrafast ultrasound localization microscopy for deep super-resolution vascular imaging. Nature. 2015;527:499–502. doi: 10.1038/nature16066. [DOI] [PubMed] [Google Scholar]
- Evans MC, Couch Y, Sibson N, Turner MR. Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2013;53:34–41. doi: 10.1016/j.mcn.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, Iadecola C. Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab. 2016a;36:241–252. doi: 10.1038/jcbfm.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, Racchumi G, Murphy M, van Rooijen N, Anrather J, Iadecola C. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J CLin Invest. 2016b;126:4674–4689. doi: 10.1172/JCI86950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucheux BA, Bonnet AM, Agid Y, Hirsch EC. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. The Lancet. 1999;353:981–982. doi: 10.1016/S0140-6736(99)00641-8. [DOI] [PubMed] [Google Scholar]
- Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci USA. 2010;107:22290–22295. doi: 10.1073/pnas.1011321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Seara MA, Mengual E, Vidorreta M, Aznárez-Sanado M, Loayza FR, Villagra F, Irigoyen J, Pastor MA. Cortical hypoperfusion in Parkinson’s disease assessed using arterial spin labeled perfusion MRI. Neuroimage. 2012;59:2743–2750. doi: 10.1016/j.neuroimage.2011.10.033. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- Freeman RD, Li B. Neural-metabolic coupling in the central visual pathway. Philosophical Transactions of the Royal Society B: Biological Sciences. 2016;371:20150357. doi: 10.1098/rstb.2015.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freygang W, Sokoloff L. Quantitative measurement of regional circulation in the central nervous system by the use of radioactive inert gas. Adv Biol Med Phys. 1958;6:263–279. doi: 10.1016/b978-1-4832-3112-9.50011-6. [DOI] [PubMed] [Google Scholar]
- Friedland RP, Iadecola C. Roy and Sherrington (1890): a centennial reexamination of “On the regulation of the blood-supply of the brain”. Neurology. 1991 doi: 10.1212/wnl.41.1.10. [DOI] [PubMed] [Google Scholar]
- Galasko D. Lewy Body Disorders. Neurol Clin. 2017;35:325–338. doi: 10.1016/j.ncl.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammon K. Neurodegenerative disease: brain windfall. Nature. 2014;515:299–300. doi: 10.1038/nj7526-299a. [DOI] [PubMed] [Google Scholar]
- Gao YR, Ma Y, Zhang Q, Winder AT, Liang Z, Antinori L, Drew PJ, Zhang N. Time to wake up_ Studying neurovascular coupling and brain-wide circuit function in the un-anesthetized animal. Neuroimage. 2017:1–0. doi: 10.1016/j.neuroimage.2016.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghebremedhin E, Rosenberger A, Rüb U, Vuksic M, Berhe T, Bickeböller H, de Vos RAI, Thal DR, Deller T. Inverse relationship between cerebrovascular lesions and severity of lewy body pathology in patients with lewy body diseases. J Neuropathol Exp Neurol. 2010;69:442–448. doi: 10.1097/NEN.0b013e3181d88e63. doi:10.1097/NEN.0b013e3181 d88e63. [DOI] [PubMed] [Google Scholar]
- Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci USA. 2010;107:3811–3816. doi: 10.1073/pnas.0914722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:303–309. doi: 10.1161/01.ATV.0000253885.41509.25. [DOI] [PubMed] [Google Scholar]
- Gordon GRJ, Choi HB, Rungta RL, Ellis-Davies GCR, Macvicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman RF, Schneider ALC, Zhou Y, Coresh J, Green E, Gupta N, Knopman DS, Mintz A, Rahmim A, Sharrett AR, Wagenknecht LE, Wong DF, Mosley TH. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. JAMA. 2017;317:1443–1450. doi: 10.1001/jama.2017.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould IG, Tsai P, Kleinfeld D, Linninger A. The capillary bed offers the largest hemodynamic resistance to the cortical blood supply. J Cereb Blood Flow Metab. 2017;37:52–68. doi: 10.1177/0271678X16671146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19:49–57. doi: 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Pavlovic D, Dalkie N, Waldvogel HJ, O’Carroll SJ, Green CR, Nicholson LFB. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013;23:154–164. doi: 10.1111/j.1750-3639.2012.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner C, Malik R, Dichgans M. Genetic factors in cerebral small vessel disease and their impact on stroke and dementia. J Cereb Blood Flow Metab. 2016;36:158–171. doi: 10.1038/jcbfm.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Han BH, Zhou ML, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J, Cirrito JR, Milner E, Holtzman DM, Zipfel GJ. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J Neurosci. 2008;28:13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RV, Harel N, Panesar J, Mount RJ. Blood capillary distribution correlates with hemodynamic-based functional imaging in cerebral cortex. Cereb Cortex. 2002;12:225–233. doi: 10.1093/cercor/12.3.225. [DOI] [PubMed] [Google Scholar]
- Hartmann DA, Underly RG, Grant RI, Watson AN, Lindner V, Shih AY. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophoton. 2015;2:041402. doi: 10.1117/1.NPh.2.4.041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci USA. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Pham LDD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci USA. 2012;109:7505–7510. doi: 10.1073/pnas.1121146109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Vanlandewijck M, Raschperger E, Andaloussi Mäe M, Jung B, Lebouvier T, Ando K, Hofmann J, Keller A, Betsholtz C. Analysis of the brain mural cell transcriptome. Sci Rep. 2016;6:35108. doi: 10.1038/srep35108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, Grutzendler J. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron. 2015;87:95–110. doi: 10.1016/j.neuron.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11:26. doi: 10.1186/2045-8118-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Regulation of the cerebral microcirculation during neural activity: Is nitric oxide the missing link? Trends Neurosci. 1993;16:206–214. doi: 10.1016/0166-2236(93)90156-g. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Beitz AJ, Renno W, Xu X, Mayer B, Zhang F. Nitric oxide synthase-containing neural processes on large cerebral arteries and cerebral microvessels. Brain Res. 1993;606:148–155. doi: 10.1016/0006-8993(93)91583-e. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Mraovitch S, Meeley M, Reis D. Lesions of the basal forebrain in rat selectively impair the cortical vasodilation elicited from cerebellar fastigial nucleus. Brain Res. 1983;279:41–52. doi: 10.1016/0006-8993(83)90161-0. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Yang G, Ebner TJ, Chen G. Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J Neurophysiol. 1997;78:651–659. doi: 10.1152/jn.1997.78.2.651. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Ibayashi S, Ngai AC, Howard MA, Meno JR, Mayberg MR, Winn HR. Lack of sympathetic and cholinergic influences on cerebral vasodilation caused by sciatic nerve stimulation in the rat. Journal of Cerebral Blood Flow & Metabolism. 1991;11:678–683. doi: 10.1038/jcbfm.1991.120. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111–147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K, Hosokawa C, Hyodo T, Sakaguchi K, Usami K, Shimamoto K, Hosono M, Yamazoe Y, Murakami T. Regional glucose metabolic reduction in dementia with Lewy bodies is independent of amyloid deposition. Ann Nucl Med. 2015;29:78–83. doi: 10.1007/s12149-014-0911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman K, Kahles T, Lane D, Garcia-Bonilla L, Abe T, Capone C, Hochrainer K, Voss H, Zhou P, Ding A, Anrather J, Iadecola C. Progranulin deficiency promotes post-ischemic blood-brain barrier disruption. J Neurosci. 2013;33:19579–19589. doi: 10.1523/JNEUROSCI.4318-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze S, Hertze J, Nägga K, Nilsson K, Nilsson C, Wennström M, van Westen D, Blennow K, Zetterberg H, Hansson O, group, T.S.B.S Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol Aging. 2017;51:104–112. doi: 10.1016/j.neurobiolaging.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze S, Lindqvist D, Francardo V, Hall S, Zetterberg H, Blennow K, Adler CH, Beach TG, Serrano GE, van Westen D, Londos E, Cenci MA, Hansson O. Increased CSF biomarkers of angiogenesis in Parkinson disease. Neurology. 2015;85:1834–1842. doi: 10.1212/WNL.0000000000002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego P, Pacheco-Torres J, Araque A, Canals S. Functional MRI in mice lacking IP3-dependent calcium signaling in astrocytes. J Cereb Blood Flow Metab. 2014;34:1599–1603. doi: 10.1038/jcbfm.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E. On the mode of entry of blood vessels into the cerebral cortex. J Anat. 1970;106:507–520. [PMC free article] [PubMed] [Google Scholar]
- Joutel A, Haddad I, Ratelade J, Nelson MT. Perturbations of the cerebrovascular matrisome: a convergent mechanism in small vessel disease of the brain? J Cereb Blood Flow Metab. 2015 doi: 10.1038/jcbfm.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B, Arnold TD, Raschperger E, Gaengel K, Betsholtz C. Visualization of vascular mural cells in developing brain using genetically labeled transgenic reporter mice. J Cereb Blood Flow Metab. 2017;126:271678. doi: 10.1177/0271678X17697720. X17697720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134:171–186. doi: 10.1007/s00401-017-1717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaney J, Walsh DM, O’Malley T, Hudson N, Crosbie DE, Loftus T, Sheehan F, McDaid J, Humphries MM, Callanan JJ, Brett FM, Farrell MA, Humphries P, Campbell M. Autoregulated paracellular clearance of amyloid- across the blood-brain barrier. Science Advances. 2015;1:e1500472–e1500472. doi: 10.1126/sciadv.1500472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kety S. Circulation and metabolism of the human brain in health and disease. Am J Med. 1950;8:205–217. doi: 10.1016/0002-9343(50)90363-9. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Ramiro Diaz J, Iddings JA, Filosa JA. Vasculo-Neuronal Coupling: Retrograde Vascular Communication to Brain Neurons. J Neurosci. 2016;36:12624–12639. doi: 10.1523/JNEUROSCI.1300-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017a;12:1–16. doi: 10.1038/nrn.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, Boas DA, Sakadžić S, Zlokovic BV. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017b:1–15. doi: 10.1038/nn.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, Agalliu D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82:603–617. doi: 10.1016/j.neuron.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko KR, Ngai AC, Winn HR. Role of adenosine in regulation of regional cerebral blood flow in sensory cortex. Am J Physiol. 1990;259:H1703–8. doi: 10.1152/ajpheart.1990.259.6.H1703. [DOI] [PubMed] [Google Scholar]
- Koller A, Toth P. Contribution of flow-dependent vasomotor mechanisms to the autoregulation of cerebral blood flow. J Vasc Res. 2012;49:375–389. doi: 10.1159/000338747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortekaas R, Leenders KL, van Oostrom JCH, Vaalburg W, Bart J, Willemsen ATM, Hendrikse NH. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- Kozberg MG, Chen BR, DeLeo SE, Bouchard MB, Hillman EMC. Resolving the transition from negative to positive blood oxygen level-dependent responses in the developing brain. Proc Natl Acad Sci USA. 2013;110:4380–4385. doi: 10.1073/pnas.1212785110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger M, Bechmann I. CNS pericytes: concepts, misconceptions, and a way out. Glia. 2010;58:1–10. doi: 10.1002/glia.20898. [DOI] [PubMed] [Google Scholar]
- Lacoste B, Gu C. Control of cerebrovascular patterning by neural activity during postnatal development. Mech Dev. 2015;138(Pt 1):43–49. doi: 10.1016/j.mod.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Poesen K, Fernández-Santiago R, Al-Chalabi A, Del Bo R, Van Vught PWJ, Khan S, Marklund SL, Brockington A, van Marion I, Anneser J, Shaw C, Ludolph AC, Leigh NP, Comi GP, Gasser T, Shaw PJ, Morrison KE, Andersen PM, Van den Berg LH, Thijs V, Siddique T, Robberecht W, Carmeliet P. Meta-analysis of vascular endothelial growth factor variations in amyotrophic lateral sclerosis: increased susceptibility in male carriers of the -2578AA genotype. J Med Genet. 2009;46:840–846. doi: 10.1136/jmg.2008.058222. [DOI] [PubMed] [Google Scholar]
- Lassen NA, Ingvar DH, Skinhoj E. Brain function and blood flow. Sci Am. 1978;239:62–71. doi: 10.1038/scientificamerican1078-62. [DOI] [PubMed] [Google Scholar]
- Lau YM, Wong SC, Tsang SW, Lau WK, Lu AP, Zhang H. Cellular sources of cyclooxygenase-1 and -2 up-regulation in the spinal dorsal horn after spinal nerve ligation. Neuropathol Appl Neurobiol. 2014;40:452–463. doi: 10.1111/nan.12078. [DOI] [PubMed] [Google Scholar]
- Lecrux C, Hamel E. Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philosophical Transactions of the Royal Society B: Biological Sciences. 2016;371:20150350. doi: 10.1098/rstb.2015.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrux C, Sandoe CH, Neupane S, Kropf P, Toussay X, Tong XK, Lacalle-Aurioles M, Shmuel A, Hamel E. Impact of Altered Cholinergic Tones on the Neurovascular Coupling Response to Whisker Stimulation. J Neurosci. 2017;37:1518–1531. doi: 10.1523/JNEUROSCI.1784-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, Lévesque M, Plaisier F, Shmuel A, Cauli B, Hamel E. Pyramidal neurons are “neurogenic hubs” in the neurovascular coupling response to whisker stimulation. J Neurosci. 2011;31:9836–9847. doi: 10.1523/JNEUROSCI.4943-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux J, Thompson M, Iadecola C, Tucker L, Reis D. Local Cerebral Blood Flow Increases during Auditory and Emotional Processing in the Conscious Rat. ScienceNew Series. 1983;221:576–578. doi: 10.1126/science.6867731. [DOI] [PubMed] [Google Scholar]
- Lee SH, Dan Y. Neuromodulation of brain states. Neuron. 2012;76:209–222. doi: 10.1016/j.neuron.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind BL, Brazhe AR, Jessen SB, Tan FCC, Lauritzen MJ. Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc Natl Acad Sci USA. 2013;110:E4678–87. doi: 10.1073/pnas.1310065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Harder DR, Koehler RC. Relative contribution of cyclooxygenases, epoxyeicosatrienoic acids, and pH to the cerebral blood flow response to vibrissal stimulation. 2012;302:H1075–85. doi: 10.1152/ajpheart.00794.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci. 2017;275:H1489–12. doi: 10.1038/nn.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden TA, Hill-Eubanks DC, Nelson MT. Ion channel networks in the control of cerebral blood flow. J Cereb Blood Flow Metab. 2016;36:492–512. doi: 10.1177/0271678X15616138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Da Mesquita S, Kipnis J. Lymphatics in Neurological Disorders: A Neuro-Lympho-Vascular Component of Multiple Sclerosis and Alzheimer’s Disease? Neuron. 2016;91:957–973. doi: 10.1016/j.neuron.2016.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Miners JS. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016;131:645–658. doi: 10.1007/s00401-015-1522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DG, Parpaleix A, Roche M, Charpak S. Mapping oxygen concentration in the awake mouse brain. Elife. 2016;5:e12024. doi: 10.7554/eLife.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Craft DK, Su JH, Kim RC, Cotman CW. Astrocytes degenerate in frontotemporal dementia: possible relation to hypoperfusion. Neurobiol Aging. 2001;22:195–207. doi: 10.1016/s0197-4580(00)00231-1. [DOI] [PubMed] [Google Scholar]
- Metea MR, Kofuji P, Newman EA. Neurovascular coupling is not mediated by potassium siphoning from glial cells. J Neurosci. 2007;27:2468–2471. doi: 10.1523/JNEUROSCI.3204-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Palmer JC, Tayler H, Palmer LE, Ashby E, Kehoe PG, Love S. Aβ degradation or cerebral perfusion? Divergent effects of multifunctional enzymes. Front Aging Neurosci. 2014;6:238. doi: 10.3389/fnagi.2014.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners S, Moulding H, de Silva R, Love S. Reduced vascular endothelial growth factor and capillary density in the occipital cortex in dementia with Lewy bodies. Brain Pathol. 2014;24:334–343. doi: 10.1111/bpa.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A. Binaural blood flow control by astrocytes: listening to synapses and the vasculature. J Physiol. 2016:1–55. doi: 10.1113/JP270979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci. 2016:1–12. doi: 10.1038/nn.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Masamoto K, Morimoto N, Kurata T, Mimoto T, Obata T, Kanno I, Abe K. Early and progressive impairment of spinal blood flow-glucose metabolism coupling in motor neuron degeneration of ALS model mice. J Cereb Blood Flow Metab. 2012;32:456–467. doi: 10.1038/jcbfm.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jabcobs RE, Liu C, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic B. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron. 2015 doi: 10.1016/j.neuron.2014.12.032. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CI, Cao R. The hemo-neural hypothesis: on the role of blood flow in information processing. J Neurophysiol. 2008;99:2035–2047. doi: 10.1152/jn.01366.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AWJ, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, Weller RO, Carare RO. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016;131:725–736. doi: 10.1007/s00401-016-1555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosso A. Sulla circolazione del cervello dell’uomo. Atti della R. Accademia dei Lincei. 1880;5:237–358. [Google Scholar]
- Mueller SM, Heistad DD, Marcus ML. Effect of sympathetic nerves on cerebral vessels during seizures. Am J Physiol. 1979;237:H178–84. doi: 10.1152/ajpheart.1979.237.2.H178. [DOI] [PubMed] [Google Scholar]
- Murphy MJ, Grace GM, Tartaglia MC, Orange JB, Chen X, Rowe A, Findlater K, Kozak RI, Freedman M, Lee TY, Strong MJ. Widespread cerebral haemodynamics disturbances occur early in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2012;13:202–209. doi: 10.3109/17482968.2011.625569. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340:1529–1530. doi: 10.1126/science.1240514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehlig A, Pereira-de Vasconcelos A, Boyet S. Postnatal changes in local cerebral blood flow measured by the quantitative autoradiographic [14C]iodoantipyrine technique in freely moving rats. Journal of Cerebral Blood Flow & Metabolism. 1989;9:579–588. doi: 10.1038/jcbfm.1989.83. [DOI] [PubMed] [Google Scholar]
- Ngai AC, Ko KR, Morii S, Winn HR. Effect of sciatic nerve stimulation on pial arterioles in rats. Am J Physiol. 1988;254:H133–9. doi: 10.1152/ajpheart.1988.254.1.H133. [DOI] [PubMed] [Google Scholar]
- Ngai AC, Winn HR. Pial arteriole dilation during somatosensory stimulation is not mediated by an increase in CSF metabolites. 2002;282:H902–7. doi: 10.1152/ajpheart.00128.2001. [DOI] [PubMed] [Google Scholar]
- Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000a;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001a;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. 2002a;283:H315–23. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis. 2002b;9:61–68. doi: 10.1006/nbdi.2001.0460. [DOI] [PubMed] [Google Scholar]
- Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. 2001b;281:H2417–24. doi: 10.1152/ajpheart.2001.281.6.H2417. [DOI] [PubMed] [Google Scholar]
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, Ashe KH, Carlson GA, Iadecola C. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A. 2000b;97:9735–9740. doi: 10.1073/pnas.97.17.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizar K, Uhlirova H, Tian P, Saisan PA, Cheng Q, Reznichenko L, Weldy KL, Steed TC, Sridhar VB, MacDonald CL, Cui J, Gratiy SL, Sakadžić S, Boas DA, Beka TI, Einevoll GT, Chen J, Masliah E, Dale AM, Silva GA, Devor A. In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J Neurosci. 2013;33:8411–8422. doi: 10.1523/JNEUROSCI.3285-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Herron P, Chhatbar PY, Levy M, Shen Z, Schramm AE, Lu Z, Kara P. Neural correlates of single-vessel haemodynamic responses in vivo. Nature. 2016;534:378–382. doi: 10.1038/nature17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney NT, Spina S, Miller BL. Frontotemporal Dementia. Neurol Clin. 2017;35:339–374. doi: 10.1016/j.ncl.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 2015;18:210–218. doi: 10.1038/nn.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounov DG, Wang T, Wang M, Feng DD, Horton NG, Cruz-Hernández JC, Cheng YT, Reimer J, Tolias AS, Nishimura N, Xu C. In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nature methods. 2017 doi: 10.1038/nmeth.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Koizumi K, El Jamal S, Zhou P, Previti ML, Van Nostrand WE, Carlson G, Iadecola C. Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke. 2014a;45:1815–1821. doi: 10.1161/STROKEAHA.114.005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pitstick R, Younkin LH, Younkin SG, Zhou P, Carlson GA, Anrather J, Iadecola C. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ Res CIRCRESAHA. 2017;117:311054–37. doi: 10.1161/CIRCRESAHA.117.311054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Wang G, Moore J, Girouard H, Zhou P, Anrather J, Iadecola C. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nat Commun. 2014b;5:5318. doi: 10.1038/ncomms6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, Pierce J, Arreguin A, Anrather J, Younkin SG, Carlson GA, McEwen BS, Iadecola C. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci USA. 2013;110:3089–3094. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pase MP, Satizabal CL, Seshadri S. Role of Improved Vascular Health in the Declining Incidence of Dementia. Stroke. 2017;48 doi: 10.1161/STROKEAHA.117.013369. STROKEAHA. 117.013369-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson O, Newman E. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237:896–898. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovitch H, White LR, Izmirilian G, Ross GW, Havlik RJ, Markesbery W, Nelson J, Davis DG, Hardman J, Foley DJ, Launer LJ. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: the HAAS. Honolulu-Asia aging Study. Neurobiol Aging. 2000;21:57–62. doi: 10.1016/s0197-4580(00)00106-8. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–797. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Pisani V, Stefani A, Pierantozzi M, Natoli S, Stanzione P, Franciotta D, Pisani A. Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson’s disease. J Neuroinflammation. 2012;9:188. doi: 10.1186/1742-2094-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, Deane R, Nedergaard M. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci. 2015;35:518–526. doi: 10.1523/JNEUROSCI.3742-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poesen K, Lambrechts D, Van Damme P, Dhondt J, Bender F, Frank N, Bogaert E, Claes B, Heylen L, Verheyen A, Raes K, Tjwa M, Eriksson U, Shibuya M, Nuydens R, Van Den Bosch L, Meert T, D’Hooge R, Sendtner M, Robberecht W, Carmeliet P. Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. J Neurosci. 2008;28:10451–10459. doi: 10.1523/JNEUROSCI.1092-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. 2017;18:385–392. doi: 10.1038/ni.3703. [DOI] [PubMed] [Google Scholar]
- Przedborski S. The two-century journey of Parkinson disease research. Nat Rev Neurosci. 2017;18:251–259. doi: 10.1038/nrn.2017.25. [DOI] [PubMed] [Google Scholar]
- Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron. 2011;71:406–424. doi: 10.1016/j.neuron.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Raab S, Beck H, Gaumann A, Yüce A, Gerber HP, Plate K, Hammes HP, Ferrara N, Breier G. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91:595–605. doi: 10.1160/TH03-09-0582. [DOI] [PubMed] [Google Scholar]
- Raab S, Plate KH. Different networks, common growth factors: shared growth factors and receptors of the vascular and the nervous system. Acta Neuropathol. 2007;113:607–626. doi: 10.1007/s00401-007-0228-3. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–476. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- Rancillac A, Rossier J, Guille M, Tong XK, Geoffroy H, Amatore C, Arbault S, Hamel E, Cauli B. Glutamatergic Control of Microvascular Tone by Distinct GABA Neurons in the Cerebellum. J Neurosci. 2006;26:6997–7006. doi: 10.1523/JNEUROSCI.5515-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Diaz-Arrastia R, Park DC. Risk factors for β-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol. 2013;70:600–606. doi: 10.1001/jamaneurol.2013.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggendorf W, Cervos-Navarro J. Ultrastructure of arterioles in the cat brain. Cell Tissue Res. 1977;178:495–515. doi: 10.1007/BF00219571. [DOI] [PubMed] [Google Scholar]
- Roher A, Esh C, Rahman A, Kokjohn T, Beach T. Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke. 2004;35:2623–2627. doi: 10.1161/01.STR.0000143317.70478.b3. [DOI] [PubMed] [Google Scholar]
- Roquet D, Sourty M, Botzung A, Armspach JP, Blanc F. Brain perfusion in dementia with Lewy bodies and Alzheimer’s disease: an arterial spin labeling MRI study on prodromal and mild dementia stages. Alzheimers Res Ther. 2016;8:29. doi: 10.1186/s13195-016-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CHT, Wamsteeker Cusulin JI, Gordon GR. Tonic Local Brain Blood Flow Control by Astrocytes Independent of Phasic Neurovascular Coupling. J Neurosci. 2015;35:13463–13474. doi: 10.1523/JNEUROSCI.1780-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengarten B, Dannhardt V, Burr O, Pöhler M, Rosengarten S, Oechsner M, Reuter I. Neurovascular Coupling in Parkinson’s Disease Patients: Effects of Dementia and Acetylcholinesterase Inhibitor Treatment. JAD. 2010;22:415–421. doi: 10.3233/JAD-2010-101140. [DOI] [PubMed] [Google Scholar]
- Roy C, Sherrington C. On the regulation of the blood-supply of the brain. J Physiol (Lond) 1890;11:85–108. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rule RR, Schuff N, Miller RG, Weiner MW. Gray matter perfusion correlates with disease severity in ALS. Neurology. 2010;74:821–827. doi: 10.1212/WNL.0b013e3181d3e2dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Schmid F, Tsai PS, Kleinfeld D, Jenny P, Weber B. Depth-dependent flow and pressure characteristics in cortical microvascular networks. PLoS Comput Biol. 2017;13:e1005392. doi: 10.1371/journal.pcbi.1005392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CF, Hendrix JP. The action of chemical substances on cerebral blood vessels. Res Publ Assoc Res Nerv Ment Dis 1938 [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci. 2011;120:357–375. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS. Integration and Modulation of Intercellular Signaling Underlying Blood Flow Control. J Vasc Res. 2015;52:136–157. doi: 10.1159/000439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AC, Koretsky AP. Laminar specificity of functional MRI onset times during somatosensory stimulation in rat. Proc Natl Acad Sci U S A. 2002;99:15182–15187. doi: 10.1073/pnas.222561899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapyan M, Lemasson M, Brill MS, Blais M, Massouh M, Ninkovic J, Gravel C, Berthod F, Götz M, Barker PA, Parent A, Saghatelyan A. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J Neurosci. 2009;29:4172–4188. doi: 10.1523/JNEUROSCI.4956-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L. In: Circulation in the central nervous system. Windhorst U, editor. Springer Science & Business Media; Berlin, Heidelberg: 2013. pp. 561–578. [DOI] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS. Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci. 2015;18:708–717. doi: 10.1038/nn.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobart JLL, Lu L, Anderson HDI, Mori H, Anderson CM. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2013;110:3149–3154. doi: 10.1073/pnas.1215929110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Stromberg DD, Fox JR. Pressures in the pial arterial microcirculation of the cat during changes in systemic arterial blood pressure. Circ Res. 1972;31:229–239. doi: 10.1161/01.res.31.2.229. [DOI] [PubMed] [Google Scholar]
- Sudre CH, Bocchetta M, Cash D, Thomas DL, Woollacott I, Dick KM, van Swieten J, Borroni B, Galimberti D, Masellis M, Tartaglia MC, Rowe JB, Graff C, Tagliavini F, Frisoni G, Laforce R, Finger E, de Mendonça A, Sorbi S, Ourselin S, Cardoso MJ, Rohrer JD. White matter hyperintensities are seen only in GRN mutation carriers in the GENFI cohort. NeuroImage: Clinical. 2017 doi: 10.1016/j.nicl.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS, Kotlikoff MI. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res. 2007;101:1300–1309. doi: 10.1161/CIRCRESAHA.107.149484. [DOI] [PubMed] [Google Scholar]
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Ménard J, Zetterberg H, Wisniewski T, de Leon MJ. Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, Bertagnolli D, Goldy J, Shapovalova N, Parry S, Lee C, Smith K, Bernard A, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19:335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, von Arnim CAF, Griffin WST, Mrak RE, Walker L, Attems J, Arzberger T. Frontotemporal lobar degeneration FTLD-tau: preclinical lesions, vascular, and Alzheimer-related co-pathologies. J Neural Transm. 2015;122:1007–1018. doi: 10.1007/s00702-014-1360-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci USA. 2010;107:20816–20821. doi: 10.1073/pnas.1005888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Monsell SE, Kukull WA, Trojanowski JQ. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136:2697–2706. doi: 10.1093/brain/awt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussay X, Basu K, Lacoste B, Hamel E. Locus coeruleus stimulation recruits a broad cortical neuronal network and increases cortical perfusion. J Neurosci. 2013;33:3390–3401. doi: 10.1523/JNEUROSCI.3346-12.2013. doi:10.1523/JNEUROSCI .3346-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Göthert JR, Malik AB, Valyi-Nagy T, Zhao YY. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood-Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation. 2016;133:177–186. doi: 10.1161/CIRCULATIONAHA.115.015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H, Tien AC, Kuo CJ, Chan JR, Daneman R, Fancy SPJ. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 2016;351:379–384. doi: 10.1126/science.aad3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlirova H, Kılıç K, Tian P, Sakadžić S, Gagnon L, Thunemann M, Desjardins M, Saisan PA, Nizar K, Yaseen MA, Hagler DJ, Vandenberghe M, Djurovic S, Andreassen OA, Silva GA, Masliah E, Kleinfeld D, Vinogradov S, Buxton RB, Einevoll GT, Boas DA, Dale AM, Devor A. The roadmap for estimation of cell-type-specific neuronal activity from non-invasive measurements. Philosophical Transactions of the Royal Society B: Biological Sciences. 2016a;371:20150356. doi: 10.1098/rstb.2015.0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlirova H, Kılıç K, Tian P, Thunemann M, Desjardins M, Saisan PA, Sakadžić S, Ness TV, Mateo C, Cheng Q, Weldy KL, Razoux F, Vandenberghe M, Cremonesi JA, Ferri CG, Nizar K, Sridhar VB, Steed TC, Abashin M, Fainman Y, Masliah E, Djurovic S, Andreassen OA, Silva GA, Boas DA, Kleinfeld D, Buxton RB, Einevoll GT, Dale AM, Devor A. Cell type specificity of neurovascular coupling in cerebral cortex. Elife. 2016b;5:155. doi: 10.7554/eLife.14315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es MA, MD, H PO, MD, C PA, MD, A-C PA, MD, P PRJ, PhD, V PJH, MD, van den Berg PLH., MD Seminar Amyotrophic lateral sclerosis. The Lancet. 2017:1–15. doi: 10.1016/S0140-6736(17)31287-4. [DOI] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, Raman MR, Machulda MM, Mielke MM, Lowe VJ, Senjem ML, Gunter JL, Rocca WA, Roberts RO, Petersen RC, Jack CR. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain. 2015;138:761–771. doi: 10.1093/brain/awu393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraete E, Biessels GJ, van Den Heuvel MP, Visser F, Luijten PR, van Den Berg LH. No evidence of microbleeds in ALS patients at 7 Tesla MRI. Amyotrophic Lateral Sclerosis. 2010;11:555–557. doi: 10.3109/17482968.2010.513053. [DOI] [PubMed] [Google Scholar]
- Wang H, Hitron IM, Iadecola C, Pickel VM. Synaptic and vascular associations of neurons containing cyclooxygenase-2 and nitric oxide synthase in rat somatosensory cortex. Cereb Cortex. 2005;15:1250–1260. doi: 10.1093/cercor/bhi008. [DOI] [PubMed] [Google Scholar]
- Wang M, Ding F, Deng S, Guo X, Wang W, Iliff JJ, Nedergaard M. Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. J Neurosci. 2017;37:2870–2877. doi: 10.1523/JNEUROSCI.2112-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren DE, Sutterer MJ, Bruss J, Abel TJ, Jones A, Kawasaki H, Voss M, Cassell M, Howard MA, Tranel D. Surgically disconnected temporal pole exhibits resting functional connectivity with remote brain regions. bioRxiv. 2017:1–17. doi: 10.1101/127571. [DOI] [Google Scholar]
- Wälchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, Gerhardt H, Engelhardt B. Wiring the Vascular Network with Neural Cues: A CNS Perspective. Neuron. 2015;87:271–296. doi: 10.1016/j.neuron.2015.06.038. [DOI] [PubMed] [Google Scholar]
- Wei HS, Kang H, Rasheed IYD, Zhou S, Lou N, Gershteyn A, McConnell ED, Wang Y, Richardson KE, Palmer AF, Xu C, Wan J, Nedergaard M. Erythrocytes Are Oxygen-Sensing Regulators of the Cerebral Microcirculation. Neuron. 2016;91:851–862. doi: 10.1016/j.neuron.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, Holmes HE, O’Callaghan JM, Colgan N, Ismail O, Fisher EM, Siow B, Murray TK, Schwarz AJ, O’Neill MJ, Collins EC, Lythgoe MF. Increased cerebral vascular reactivity in the tau expressing rTg4510 mouse: evidence against the role of tau pathology to impair vascular health in Alzheimer’s disease. J Cereb Blood Flow Metab. 2015;35:359–362. doi: 10.1038/jcbfm.2014.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, Cleveland DW, Zlokovic BV. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci USA. 2014;111:E1035–42. doi: 10.1073/pnas.1401595111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolsey TA, Rovainen CM. Whisker barrels: a model for direct observation of changes in the cerebral microcirculation with neuronal activity. In: Lassen NA, editor. Brain Work and Mental Activity: Quantitative Studies with Radioactive Tracers : Proceedings of the Alfred Benzon Symposium 31 Held at the Premises of the Royal Danish Academy of Sciences and Letters; Copenhagen. August 12–16, 1990; John Wiley\& Sons, Limited; 1991. [Google Scholar]
- Xu HL, Mao L, Ye S, Paisansathan C, Vetri F, Pelligrino DA. Astrocytes are a key conduit for upstream signaling of vasodilation during cerebral cortical neuronal activation in vivo. 2008;294:H622–32. doi: 10.1152/ajpheart.00530.2007. [DOI] [PubMed] [Google Scholar]
- Yang G, Chen G, Ebner T, Iadecola C. Nitric oxide is the predominant mediator of cerebellar hyperemia during somatosensory activation in rats. Am J Physiol. 1999;277:R1760–R1770. doi: 10.1152/ajpregu.1999.277.6.R1760. Regulatory Integrative Comp. Physiol. 46. [DOI] [PubMed] [Google Scholar]
- Yang G, Huard JM, Beitz AJ, Ross ME, Iadecola C. Stellate neurons mediate functional hyperemia in the cerebellar molecular layer. The Journal of Neuroscience. 2000;20:6968–6973. doi: 10.1523/JNEUROSCI.20-18-06968.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Iadecola C. Activation of cerebellar climbing fibers increases cerebellar blood flow: role of glutamate receptors, nitric oxide, and cGMP. Stroke. 1998;29:499–507. doi: 10.1161/01.str.29.2.499. - discussion 507–8. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhang Y, Ross ME, Iadecola C. Attenuation of activity-induced increases in cerebellar blood flow in mice lacking neuronal nitric oxide synthase. 2003;285:H298–304. doi: 10.1152/ajpheart.00043.2003. [DOI] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SPJ, Zahed H, Maltepe E, Rowitch DH. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. 2014;158:383–396. doi: 10.1016/j.cell.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ET, Inman CB, Weller RO. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat. 1990;170:111–123. [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Eckman C, Younkin S, Hsiao K, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Xu S, Iadecola C. Role of nitric oxide and acetylcholine in neocortical hyperemia elicited by basal forebrain stimulation: evidence for an involvement of endothelial nitric oxide. Neuroscience. 1995;69:1195–1204. doi: 10.1016/0306-4522(95)00302-y. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015a;163:1064–1078. doi: 10.1016/j.cell.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, Owens NC, Rege SV, Si G, Ahuja A, Zhu D, Miller CA, Schneider JA, Maeda M, Maeda T, Sugawara T, Ichida JK, Zlokovic BV. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015b;18:978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O’Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, Zlokovic BV. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Ackerman JJH, Sukstanskii AL, Yablonskiy DA. How the body controls brain temperature: the temperature shielding effect of cerebral blood flow. J Appl Physiol. 2006;101:1481–1488. doi: 10.1152/japplphysiol.00319.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]