

Abstract
Cell therapy is not only a novel medical practice but also a medicinal product [cell therapy product (CTP)]. More and more CTPs are being approved for marketing globally because of the rapid development of bio-medicine in cell culture, preservation, and preparation. However, regulation is the most important criterion for the development of CTPs. Regulations must be flexible to expedite the process of marketing for new CTPs. Recently, the Taiwan Food and Drug Administration (TFDA) updated the related regulations such as regulation of development, current regulatory framework and process, and the application and evaluation processes. When the quality of CTPs has been improved significantly, their safety and efficacy are further ensured. The treatment protocol, a new design for adaptive licensing to current clinical practice, is a rapid process for patients with life-threatening diseases or serious conditions for which there are no suitable drugs, medical devices, or other therapeutic methods available. The hospital can submit the treatment protocol to apply for cell therapy as a medical practice, which may result in easier and faster cell therapy development, and personalized treatment for individual patients will evolve quickly.
Keywords: Cell therapy, Clinical application, Investigational new drug (IND), New drug application (NDA), Treatment protocol
Introduction
Definition of Cell Therapy
Cell therapy (cytotherapy) is a kind of treatment in which live whole cells are introduced into a patient or maturation of a specific cell population for the transplantation of human autologous or allogeneic cells into the body to replace, repair, reconstruct, or supplement damaged cells/tissues. Cell therapy includes two categories: (1) mainstream medicine: intense research with potential therapeutic benefits; however, such research is controversial as it involves human embryonic stem cells (hESCs)1; (2) alternative medicine: the practice of injecting cell materials in an attempt to cure diseases. Cell therapy is different from conventional stem cell therapy in that the injected cells are already differentiated (e.g., muscle cells, gland cells, skin cells), whereas the cells that conventional stem cell therapy utilizes are usually undifferentiated (e.g., embryonic cells). Cell therapies have long been used by alternative medicine practitioners who have claimed great benefits and are very different from conventional stem cell therapy.
Autologous or allogeneic cells that are manipulated (e.g., in vitro culture) are not the same as the original cells. Cell therapy products (CTPs) are biomedicines containing cells/tissues that have been manipulated to change their biological characteristics, and these cells/tissues can be used to treat, diagnose, or prevent diseases instead of being used for the same essential functions in the body. They include cellular immunotherapies and other types of cells, such as adult and embryonic stem cells (ESCs), for certain therapeutic indications. Cell types are classified as undifferentiated stem cells, committed progenitor cells, differentiated cells with specific functions, and tissue cells. These cells can be genetically modified; furthermore, they can be used with biomolecules, biomaterials, biosynthetic substances, or structural materials belonging to medical devices. Human CTPs are the autologous or allogeneic cells used to treat, prevent, or diagnose diseases, but xenogeneic cells are not included. Their advantages include fewer side effects, no drug resistance, and specificity for personalized treatment; their risks include immunogenicity, autoimmunity, rejection, infection, or tumorigenicity.
CTPs around the World
Recently, advanced countries have considered CTPs as one kind of biologic or medical device. Various CTPs have been approved and used clinically, in the following regions: America (US and Canada), Asia (Japan, Korea, and Singapore), Europe [European Medicines Agency (EMA)], and Oceania (Australia and New Zealand)2–6 (Table 1). Many products are currently under active investigation worldwide, and their market size is expected to grow rapidly in the near future.
Table 1.
Approved Cell Therapy Products (CTPs) Around the World
| Approved Cell Therapy Products (Year) |
||
|---|---|---|
| Country | Autologous | Allogeneic |
| Australia | Cartogen (2002) | |
| ReCell/CellSpray (2006) | ||
| Canada | ReCell/CellSpray (2006) | Prochymal, MSC (2012) |
| EMA | ChondroCelect (2009) | |
| MACI (2013) | ||
| Provenge, DC (2013) | ||
| Holoclar (2015) | ||
| Cells of stromal vascular fraction of adipose tissue (2015) | ||
| Bone marrow-derived nonhematopoietic stem cells (2015) | ||
| T cells (2016) | ||
| Adipose-derived MSC (2016) | ||
| Japan | JACE (2007) | Temcell HS injection (2015) |
| JACC (2012) | ||
| HeartSheet (2015) | ||
| Korea | Chondron (2001) | Kaloderm (2005, 2010) Cartistem, MSC (2012) |
| Holoderm (2002) | ||
| Keraheal (2006) | ||
| CreaVax-RCC (2007) | ||
| Immuncell-LC (2007) | ||
| NKM (2007) | ||
| Hyakgraft-3D (2007) | ||
| Innolak (2007) | ||
| Adipocell (2008) | ||
| RMS Ossron (2009) | ||
| AutoStem (2010) | ||
| QeenCell (2010) | ||
| CureSkin (2010) | ||
| LSK Autograft (2010) Hearticellfram-AMI (2011) | ||
| Cupistem (2012) | ||
| New Zealand | ReCell/CellSpray (2006) | Prochymal, MSC (2012) |
| Singapore | Chondrotransplant (2002) | |
| Cartogen (2002) | ||
| ReCell/CellSpray (2006) | ||
| USA | Carticel (1997) | Gintuit (2012) |
| Provenge, DC (2010) | HPC, Cord Blood (2012, 2013, 2016) | |
| Laviv, fibrocell (2011) | Ducord (2012) Hemacord (2013) | |
| Allocord (2013) | ||
EMA, European Medicines Agency; Hemacord (HPC, Cord Blood); MSCs, mesenchymal stem cells; MACI, matrix applied characterized autologous cultured chondrocytes.
In Taiwan, the Taiwan Food and Drug Administration (TFDA) has not yet approved any CTPs for marketing but has sought to expedite the approval of CTPs from two developed countries for clinical application. Additionally, numerous cellular-related clinical studies are ongoing, and the amount of application continues to grow rapidly. Some CTPs have been regulated as routine medical practice executed by doctors in hospitals.
Current Regulatory Framework in Taiwan
We developed the guidelines in accordance with the needs of our country and international standards including the guidelines of the World Health Organization (WHO), United States Food and Drug Administration (US FDA), European Medicines Agency (EMA), and Japan's Pharmaceuticals and Medical Devices Agency (PMDA), etc. On the basis of the guidelines of the International Conference on Harmonization (ICH), adapted references are compared and reviewed by the advisory committee preliminarily. When the guideline is discussed by the public and reviewed by our legislative body, it will be announced by the appropriate authority. Like most of the guidelines around the world, the guidelines of Taiwan are also professional, specific, and sometimes flexible. Compared to other relevant guidelines worldwide, the defining characteristic of the guidelines of Taiwan is a new design for adaptive licensing (adapted from the PMDA)–treatment protocol (Fig. 1B).
Figure 1.
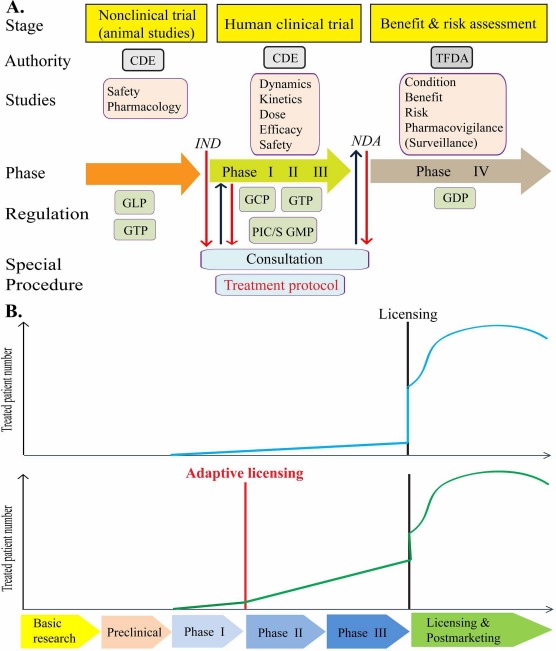
(A) Framework of cell therapy product (CTP) development and application: The discovery, testing, surveillance, and application of CTPs involve several practice performance and regulation requirements. (B) Treatment protocol: current feasibility and use of elements of “adaptive licensing” under the existing Taiwan cell therapy regulatory framework. (B) (Top) Normal procedure: before 2016, CTPs should be licensed once the clinical trial phases I, II, and III are complete. (B) (Bottom) Adaptive licensing: after 2016, if a treatment protocol is provided, phase I is complete, and CTP safety is confirmed; CTPs can be used for therapy. Treated patient number and clinical data can be applied for phases II and III. Therefore, treated patients' number in adaptive licensing is much more than normal procedures during clinical trial phases II and III. CDE, Center for Drug Evaluation; TFDA, Taiwan Food and Drug Administration; IND, investigational new drug; NDA, new drug application; GCP, good clinical practice; GDP, good distribution practice; GLP, good laboratory practice; GTP, good tissue practice; PIC/S GMP, Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme Good Manufacturing Practice.
CTPs may still be developing, and their scientific data may not be sufficient by general regulations. However, for the unmet need of specific patients, CTPs can be used as a therapeutic strategy. The challenges we encountered during the process of developing the guidelines are balancing the need of the patients with the development of medicinal products, developmental ability of the pharmaceutical industry, international tension, as well as addressing the concerns of our legislative body.
Law
All of the guidelines and regulations of CTPs are based on either the Pharmaceutical Affairs Act7 or the Medical Care Act8. In accordance with these two recent laws, CTPs are considered a “medical practice and medicinal product.”
Regulation
Nonclinical and clinical trials9 have to be in compliance with good laboratory practices (GLP) and good clinical practices (GCP)10, respectively. Additionally, application and approval for using CTPs must be in accordance with the regulation on medical product registry11.
Guidance
The harvesting and manufacturing of cell and tissue specimens from which CTPs are derived have to be in compliance with good tissue practices (GTP)12 and the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme Good Manufacturing Practice (PIC/S GMP)13,14. The transportation and application of CTPs have to conform to good distribution practices (GDP)15,16 and guidance on CTP application17, respectively. GTP, PIC/S GMP, and GDP are all inspected by the TFDA18. Each of these is defined as follows:
-
1.
GTP: A guidance to ensure safety and quality of organ-, cell-, and tissue-derived products during the process of harvesting, management, testing, and storage. Its objective is to prevent the introduction or transmission of communicable diseases, as well as to raise quality standards and establish reliable quality assurance systems.
-
2.
PIC/S GMP: A guidance to ensure that the production of CTPs can involve certain specific considerations arising from the nature of the products and manufacturing processes. The PIC/S GMP standards require taking some special precautions while CTPs are manufactured, controlled, and administered.
-
3.
GDP: A guidance to ensure that companies, institutions, and individuals involved in the distribution of CTPs can maintain them in the correct conditions. The performance of GDP is the main point, especially at the postmarketing stage. GDP stipulates that during the period from CTPs' approval for use until they are administered, all distribution processes should be controlled, including storage by the distributor and transportation to the hospital and clinic.
Regulation
Regulations are the most important criteria for the development and marketing of CTPs. The cell therapy regulatory strategy is a risk-based approach. Relevant factors include the origin of cells, level of cell manipulation, indication (valid for specific diseases), combination product (products combined with other products such as cells, medicines, and medical devices), etc. Cell therapy regulations specify and regulate manufacturing, quality control, nonclinical safety, clinical safety and efficacy, and postmarket surveillance (Fig. 1A).
Application Process
Consultation Prior to Submission
To facilitate the approval of CTPs, it is important to have early scientific advice. Applicants such as sponsors, hospitals, or contract research organizations (CROs) should apply to the Center for Drug Evaluation (CDE) of Taiwan for consultation and then submit the formal application including technical documents (e.g., protocol design, consent letter, etc.) and administrative documents, which have been verified as complete, to the TFDA. For a successful outcome, it is necessary to provide the following items in the application: data on product characterization, specification, quality control, and release; well-developed and controlled manufacturing information; preclinical studies to show safety and effects of products; and evidence to support human dosing and scientific rationale.
Registration
The applicants have to apply online to register all applicants, product, trial plan, contact information, major inclusion and exclusion conditions, trial population size, and other information.
Evaluation Procedure
The TFDA will invite experts in quality, nonclinical research, clinical research, and statistics to establish a review team upon receiving the investigational new drug (IND) or new drug application (NDA). The reviewers discuss and provide some suggestions. Finally, the TFDA evaluates the benefits and risks and decides to approve or reject the application.
If CTPs simultaneously meet the following four criteria, the TFDA will adjust the review process, called “fast track,” to facilitate investigational application approval and forego discussion by the advisory committee: (1) minimal manipulation: the process does not alter the relevant biological characteristics of cells or tissues; (2) homologous use: the repair, construction, replacement, or supplementation of cells with a biological function that performs the same basic functions in the recipient as in the donor; (3) no combination: the cells are not in combination with other products such as cells, medicines, or medical devices; and (4) no systemic effects: the cells do not have any systemic effects other than their indication, their function is not induced by the metabolism activity of living cells, or the CTPs are used only for first- and second-class relatives or reproduction in spite of having systemic effects.
Advisory Committee
An advisory committee consists of experts on the review team, consumers, and patient representatives that convene for further discussion. The TFDA takes advice provided by the advisory committee to complement the internal review process and make a regulatory decision.
Points to Consider on CTPs
Production
It is impossible for CTPs to have terminal sterilization in that their drug substances (active substance) are living cells. Large-scale purification steps, viral removal, and inactivation in the manufacturing process are also hard to perform. Therefore, the source of materials and reagents should be strictly controlled19.
Source Control.
Cell
Depending on the donor source, cells are divided into autologous cells and allogeneic cells. For autologous cells, the screening and testing of specific pathogens should be performed to avoid the spreading of viruses and adventitious agents. If there is no screening and testing, the samples and products must be particularly labeled with “no pathogen screening and testing performed; handle as a biohazard.” For allogeneic cells, the screening and testing of specific pathogens of donors should refer to the related guidelines for donor eligibility determination. The screening items include human immunodeficiency virus (HIV), high-risk hepatitis groups, degenerative spongiform encephalopathy, and tuberculosis. The tested items include HIV (types I and II), antibody of hepatitis B (surface and core antigen), hepatitis C virus (HCV), syphilis, and other specific pathogens. Additionally, the identification of genetic polymorphism for human leukocyte antigen (HLA) should be considered according to the rejection phenomenon.
Cell banks for cell therapies are different from production banks for other biologics. Cells are a kind of product but are not produced in a production factory. The cell bank is limited in size passage number; there may be only a few cell passages prior to becoming a final product. Cell bank systems can ensure that CTPs are from identical cell sources. Generally, cell banks are classified as master cell banks and working cell banks, and their tested items are as follows: (1) master cell bank: microbiological characteristics, species-specific virus, identification, composition, activity and differentiation, stability of chromosome, and other factors influencing stability such as culture conditions, storage, and viability; (2) working cell bank: sterility, mycoplasma, adventitious virus, and suitable identification test.
Reagent
Reagents are various components used in the manufacturing process but are not a part of the final product, such as fetal bovine serum (FBS), trypsin, growth factors, cell hormones, monoclonal antibodies, antibiotics, medium, and other materials. They are used in the cell growth, differentiation, selection, purification, or crucial manufacturing procedures. The safety, potency, and purity will be affected if adventitious agents in the reagent are introduced into CTPs. The reagents should be noted as follows: (1) reagents listed in detail: all reagents in the product should be listed along with their final concentration in the manufacturing process and certificate of analysis (COA); (2) removal of various reagents from the final product: the procedure for determining if there is any residual amount of reagents in the final product should be described; (3) others: antibiotics (e.g., penicillin or β-lactam) should not be used in the manufacturing process to avoid causing allergic reactions in patients.
Excipient
The excipient is an inactive ingredient in the final product, for example, human serum albumin (HSA) or dimethyl sulfoxide (DMSO). The inactive ingredient and its final concentration should be listed on the COA.
Cell Preparation
All procedures in the manufacturing and purification process should be described in detail, and a flowchart for cell preparation should be provided outlining the following seven steps: 1) Cell collection and handling; 2) Cell population isolation and purification; 3) Cell culture; 4) Manufacturing and storage; 5) Cell modification; 6) Validation of complex cell culture procedure; 7) Final harvest.
Cell Collection and Handling
The number and size of tissue/cell samples collected from the donors should be specified, and their handling procedures should be described. The standard operating procedure (SOP) for handling organ/tissue should be described in detail, and the acceptance criteria must meet the specification for final product release.
Cell Population Isolation and Purification
The instrument and/or technique for cell sorting or isolation should be provided such as density gradient or magnetic-activated cell sorting (MACS).
Cell Culture
The culture condition and batch size should be provided including temperature, incubation time, and maximum passage number. The reliability of the cell culture procedure should be confirmed.
Manufacturing Time and Intermediate Storage
The time needed for every step from the donor tissue/cell harvest to the final harvest should be estimated. The most important thing is to realize the time limit of every step to decide if testing in the manufacturing process is essential. The storage time and condition should be recorded from cell harvest to final harvest.
Cell Modification
An approach should be provided when cells are physically treated (i.e., blending, centrifugation, dialysis, separation, etc.), chemically treated, or genetically modified. If cells are genetically modified, the manufacturing and control must comply with the national and international regulations for cell therapy.
Complex Cell Product
Whether there is any influence on the growth, function, and integrity of cells when grown on a matrix/device/scaffold should be evaluated. If the matrix/device/scaffold is a biomaterial, its degradation rate should be taken into consideration. The validation of complex cell culture procedures is also required.
Final Harvest
The handling procedure, storage conditions, and time of final harvest should be provided. The conditions for cleaning and media used must be described if the final cells are harvested by centrifugation.
Radiation Treatment (Case Dependent)
If autologous cells or allogeneic CTPs need radiation treatment prior to transplantation, data should be provided to show that cells will not replicate and will maintain expected characteristics.
Final Formulation
The formulation of final products should be described in detail. If there is any excipient in the final formulation, such as growth factors or HSA, its source must be provided.
In-Process Control (IPC)
IPC should be set in the critical or intermediate step to manage the overall manufacturing process, and analytical methods and acceptance criteria should be described. The reproducibility of the manufacturing process and consistency of final products can be ensured by the control of critical steps.
Manufacturing Process Validation
The documents for the manufacturing process validation should be provided to describe the manufacturing process design, key parameters, and suitability of acceptance criteria (qualified standard or range for acceptance). The changeable factor in the manufacturing process such as cell source, reagent supplier, or parameter variation criteria (changeable range for parameters such as temperature, speed, and incubation time) should be included in the validation of the manufacturing process to verify CTP robustness.
Apparatus and Equipment
All apparatus and equipment used in manufacturing CTPs should be listed. Apparatus and equipment can be used to produce CTPs provided that they are approved by the TFDA. However, the apparatus and equipment are required to be verified if they are not approved.
Product Testing.
Microbiology
Testing items are described as follows:
-
1.
Sterility: These sterility methods are included in the pharmacopoeia such as the Chinese Pharmacopoeia of Taiwan, the European Pharmacopoeia (EP), and the US Pharmacopoeia (USP). The sterility test for final product release should be in accordance with the pharmacopoeia method or an alternative method (noncompendia method). If a noncompendia testing method is adopted, its suitability must be verified.
-
2.
Mycoplasma: The mycoplasma should be tested in the manufacturing process. The major two possible contamination sources are animal serum products and the culture environment. In long-term culture, the presence of mycoplasma should be tested in the manufacturing process.
-
3.
Foreign pathogens: An in vitro test of viruses should be performed in the master cell bank and final products. An in vivo analysis of viruses should be performed in the master cell bank.
Identity
The cell genotype and phenotype are identified according to the cell population and source. The cell phenotype can be analyzed by suitable and validated bio-markers such as cell antigens and biochemical activity. For allogeneic cells, the identification analysis includes histocompatibility markers and genetic polymorphisms. For complex CTPs, the unicellular component (e.g., frame) is identified according to material characteristic.
Purity
The specific cell population and its living cell number directly influence the efficacy and safety of CTPs. Therefore, cell population, other cellular contaminants, and the ratio of living cells/dead cells should be included in the releasing specification, and its acceptance criteria should be set: (1) pyrogen/endotoxin: limulus amebocyte lysate (LAL) assay is used to replace pyrogen test to test endotoxin; (2) process-related impurity: the peptide, protein, and reagent in the manufacturing process (i.e., the residual amount of cell hormone, growth factor, antibody, and serum); (2) cell-related impurity: the impurities from the cell source such as cell accumulation, dead cells, debris from cell degradation, and unexpected cell phenotype.
Potency
The potency of CTPs can be revealed by clinical trial results since cell activity is correlated with clinical efficacy. The potency test, stability test, and comparison test should be conducted prior to the product release. Types of tests used are the following: (1) biological test: biological tests should be conducted in the living cell system such as living animals, in vitro organs, or tissue/cell culture systems; (2) nonbiological test: nonbiological tests of cells can reveal specific bioactivity, which is closely correlated to efficacy and potency; (3) multiple tests: when the potency cannot be supported by one analytical method, multiple tests are used to complement its insufficiency.
Viability (Survival Rate)
Cell viability should be tested prior to release. Acceptance of the products is contingent upon batch production record, results of stability tests, and safety and efficacy in animal studies and human clinical trials. The minimum viability should be 70%. If it is not up to standard, data must be provided to attest that dead cells and cell debris will not influence the safety and efficacy of CTPs.
Cell Number/Dose
A condition for product testing and release is that there is a minimum number of living cells and effective cells in the products. The maximum dose for transplantation and clinical application as well as basis for the dose should be recorded.
Tumorigenicity
In vitro subculture of CTPs may lead to gene instability that induces tumorigenicity. It is impossible for stem cell amplification or differentiation to reach 100% efficiency, and undifferentiated cells cannot be removed by purification effectively. In this case, there is a risk that tumorigenicity could be induced when these undifferentiated stem cells and incompletely differentiated cells are transplanted into humans. Consequently, the chromosome integrity and tumorigenicity should be evaluated during cell culture process or final cell culture generation.
Final Product Release Testing
The destined specification (e.g., testing item, method, acceptance criterion) of final products should be listed. In some cases, every dose of product can be considered as a single lot number. Prior to administration to patients, the results of final product release testing should be disclosed; otherwise, manufacturers would have to clearly point out that certain data have not been acquired and provide a response plan. The analytical method for final product releasing should be validated.
Batch Analysis
To verify the robustness of the manufacturing process and consistency of CTPs, it is essential to have at least three validation batches to analyze results.
Reference Cell Standard
A reference cell standard is used not only to assure the consistency but also to verify manufacturing system suitability.
Well-Closed Container System
It is necessary to evaluate the suitability of container and cover for CTPs, for example, via adsorption, exudate, biological, and container integrity testing.
Product Stability
The shelf life for final products, intermediates, and opened products should be regulated and supported by stability tests as follows: (1) stability test of the manufacturing process: to establish a stability testing plan and determine its parameters to ensure product stability during the frozen storage period; (2) stability test of final products: to provide supporting data that CTPs can remain stable from dispensing to administration and to establish an expiry date.
Nonclinical Studies
The aims of nonclinical studies focus on activity, safety, and efficacy of products. It is preferable to provide information and evaluation of pharmacology and safety in the animal model. If a suitable animal model is not available, in vitro data can be used as rationale unless reasonable explanation is provided. In this phase, the following keynotes should be considered: selection of biologically relevant animal species/model, support of rationale for phase I in-human clinical trial, recommendations related to human clinical trial design, and conformability to regulatory requirements.
Pharmacology Tests
(1) Major pharmacodynamics test: A suitable animal model should be selected in accordance with the specified use (the specific purpose for which the product is being used, i.e., for what disease the product is being used as a therapeutic agent), and the minimum or suitable effective dose should be explored. (2) Minor pharmacodynamics test: Unexpected physiological function caused by product distribution, secretion of other bioactive materials, or targeting of unexpected organs should be evaluated in the animal model. (3) Safety pharmacology test: Cell characteristics or transplantation site may affect vital organs; thus, the test is case dependent. (4) Cell kinetics, migration, and persistence: The adsorption, distribution, metabolism, and excretion of molecular pharmacokinetics are not necessarily adopted by CTPs. It is necessary to explore cellular performance, distribution, viability, persistence, and transport when the products are introduced into the body. (5) Interaction: The interaction between cells and unicellular constructs/biomaterials as well as their integration into peripheral tissues should be monitored.
Safety Test
The safety test should be individualized case by case for patients. Concerns about CTP safety may arise from the change of cellular activity, residual materials, constituted components, adjuvants, cytokines or drugs, or induction of autoimmunity. The safety test includes single- and multiple-dose tests, a local tolerance test, a tumorigenicity test, a genetic test, a reproduction test, and an immunogenicity test.
Human Clinical Trials
The requirements regarding clinical trials related to CTPs are almost the same as those to common drugs. The overall consideration of these products depends on risk. For drug development, clinical trials with CTPs are sometimes different from those using conventional pharmaceuticals. Consultation with TFDA or CDE is recommended at the drug discovery stage. To achieve expected efficacy, CTPs may need a specific surgery or administration route, or combination therapy. During clinical trials, it is necessary to explore the influential factors of CTPs and establish the best SOP. In this phase, the following keynotes should be considered: study aim and design, selection of treated subject population, informed consent of recipients, administration of CTPs, establishment of a tracking system, safety monitoring and efficacy evaluation, and statistical analysis and consideration of data.
Pharmacodynamics
Though the mechanism of CTPs is poorly understood, it is required that their main function be known. For complex products, the primary mode of action (PMOA) should be described. If CTPs consist of both cellular and unicellular components, their tolerance, degradation rate, and functionality should be evaluated.
Pharmacokinetics
The conventional adsorption, distribution, metabolism, and excretion tests are usually not adopted for CTP evaluation. The possible method of testing and its feasibility should be discussed. The viability, reproduction/differentiation, distribution/migration, and function should be monitored before CTPs expire.
Dose Exploration
Dose selection is dependent on the quality, nonclinical data, and potency of CTPs. It is required to find a minimum or optimal effective dose in clinical phases I/II. The maximum safe dose should also be explored if applicable.
Efficacy
Efficacy is determined by subject population evaluation, optimum dose for best efficacy, efficacy duration, and benefit–risk evaluation. If the relationship between significant evaluation endpoint and efficacy in a clinical trial is confirmed and acknowledged by the public, this endpoint can be accepted as a surrogated endpoint.
Safety
Common adverse reactions should be detected at minimum. All safety concerns in the preclinical phase should be explored in clinical trials. In the case of CTPs with a long shelf-life, it is required that patients be surveyed to confirm their long-term efficacy and safety.
Benefit–Risk Assessment.
Condition Analysis
NDA review will decide if the application is regular or address an unmet medical need. The former (regular application) is not in serious condition and is generally accepted with standard review or abbreviated review. The latter (unmet medical need) is a serious or life-threatening disease that has no acceptable medical option or iterative treatment and is usually accepted with priority review, or priority and abbreviated review.
Benefit and Risk Evaluation
The benefit is usually determined by the efficacy of the CTPs in treating the patients. Risks are defined as possible danger in human CTPs, for example, pathogen infection, disease transmission, contamination, cross-contamination, and mix-ups. The evaluation of benefit and risk is very crucial for patients to receive safe and effective treatment.
Pharmacovigilance (Postmarket Surveillance)
Pharmacovigilance includes risk management and long-term monitoring of CTPs. Risk management contributes to risk reduction and/or avoidance, and long-term monitoring is beneficial for the efficacy and safety scrutiny. Postmarket surveillance is required for all CTPs; in some cases, CTPs may need to be monitored long term to address special safety concerns.
Treatment Protocol
Before 2016, CTPs should be licensed once the clinical trial phases I, II, and III are complete; however, a more flexible regulation treatment protocol was adopted in 2016. The treatment protocol, provided by physicians, is a new design for adaptive licensing of current clinical practices regulated by the new “Human Trials Management Regulation.” This protocol is a rapid process for patients with life-threatening diseases/serious conditions to receive cell therapy as an alternative method when no suitable drugs, medical devices, or other therapy methods are available. The hospital can submit the treatment protocol to apply for cell therapy as a medical practice if safety data for human clinical trial are sufficient (Fig. 1B).
Perspective
Freedom of Information
The improvement of doctor–patient relationships and open public platform are very crucial for the freedom of information. Informed consent should be obtained prior to CTP administration to ensure that recipients are aware of the risks, such as tumor formation or lack of proof of clinical benefit.
Consumer Protection
Consumer protection is an important topic in modern society, especially within the medical field. Guidelines for consumer protection should be enacted and observed. The consumer has the right to appeal and ask for compensation if any dispute or adverse drug reaction (ADR) occurs. It is also necessary to have a complete regulatory system in place to protect and treat patients effectively and establish a just relationship between doctors and patients.
Government Administration
The governmental operation should be more efficient, flexible, and intuitive in regard to the development of CTPs. Concise and streamline procedures are a way to foster an intuitive administrative process. It is essential to make the consultation method more practical, prompt, efficient, and succinct.
Facilitation of International Cooperation
International cooperation and exchange should be emphasized, as they can facilitate progress and development. Regular participation in international activities is conducive to acquiring up-to-date information and exchange of information.
Establishment of Online Registry
The establishment of an online system for integrating, distributing, and sharing medical resources can make medical care and consultation simple and rapid. An efficient online system for registration and consultation can save much time and associated expenses for medical professionals and patients.
Establishment of Education Systems
Development of a training program, workshop, or symposium with regulators is a feasible way to stimulate scientific principles to enhance the quality, safety, and efficacy of CTPs. For long-term development, it is necessary to establish a national forum with government, manufacturing, and academic representatives for discussions to promote the modernization of cell therapy regulation and improvement of manufacturing CTPs.
Promotion of Pharmaceutical Industry
The clinical application of cell therapy can be promoted through research facilitation, research and development (R&D), and production of CTPs. Translational medicine is a new multidisciplinary field linking basic medical research and clinical application; its process involves pathology research, characterization of diseases, specific drug discovery, and patient application. All of the processes are closely related to the introduction and cultivation of talented persons, as well as pharmaceutical industry promotion.
Regulatory Challenge
Regulatory challenges may be related to ethics, quality, safety, and efficacy. Potential hurdles related to regulation of CTPs are a major ethic issue or the fact that rapid scientific progress and a market for innovation may provide a climate for unprecedented risks (e.g., mutation or biodistribution). Quality control is highly complex with respect to CTPs because of short shelf life (cell viability) and variety of cell origin. Conventional requirements for pharmacology and toxicity may not be appropriate and sufficient to establish a safety monitoring system. CTPs are variable and cellularly heterogeneous; their efficacy is often individualized and patient specific.
Support for the Development of Innovative Medicinal Products
The division of labor and cooperation between medical technology and product development will make cell therapy a viable treatment. Medical technology should be transparent to lead to further developments in the international medicine. Expansion of the scale of the CTP enterprise is advantageous for developing medical technology, manufacturing products, and treatment.
Revision or Enactment of Related Regulations
Regulatory science is evidence based; thus, improvement is dependent on scientific data. Continuous revision or enactment of flexible and specific guidelines, regulation, and laws for CTPs will expedite new drug discovery. Conditional approval should be set up and clearly defined, requiring that the CTP source is species specific, transport is time limited, and clinical application is highly professional. Regulatory mindset should be changed from protecting public health to promoting public health.
International Conference on Harmonization (ICH)
Harmonization may be quite difficult because every country has a different environment and background for CTP development, but it is crucial for different countries to unite and cooperate with respect to cell therapies. Therefore, it is necessary to identify possible topics for harmonization and facilitate harmonization internationally.
Individual and Precise Medicine
Cell therapy can be designed to satisfy individual customers' needs and is usually patient specific and made to order. CTPs are very promising to play a key role in establishing individual and precise medicine. To specify the need of individual patients, it is necessary to evolve bioeconomics and establish human bioinformatics banks for individual diversity.
Insurance and Payment
Along with the rapid development of CTPs, cell therapy for the treatment of most diseases may come to fruition very soon. However, cell therapies usually require a very long course of treatment and have high medical costs. It is imperative that cell therapy equitably coverall insured persons and that it is affordable for all patients to avoid CTPs being privileged or prioritized for only some people.
Conclusion
This review describes current cell therapy regulation in Taiwan. Along with the rapid development of novel biotechnology, more and more CTPs have been approved for marketing and clinical application. Related regulations will be perfected and be flexible enough to provide diverse treatment options for patients. CTPs can potentially be used for personalized treatment of individual patients in the near future. The significant challenges facing cell therapies are the difficulty in evaluating the efficacy of personalized treatment and establishing a long-term follow-up strategy. In addition, it is not easy to ascertain or control the quality of CTPs because of cell complexity and difficulties in performing animal studies with human cells (e.g., rejection of xenogeneic cells). However, it is expected that cell therapies will demonstrate splendid opportunities for treatment of many diseases provided we can overcome the risks and potential challenges.
Though there are still many regulatory and technical challenges for cell therapy ahead of us, we believe that they will be solved in the near future through our endeavors. Cell therapy is promising–-a promising therapeutic approach, but it might require a significant amount of funding and time to reach the desired objectives. If the challenges are overcome and all prospects come true, the effect and impact of cell therapy will be both enormous and significant.
Acknowledgment
The authors declare no conflicts of interest.
References
- 1.Gage FH. Cell therapy. Nature 1998; 392(6679 Suppl): 18–24. [PubMed] [Google Scholar]
- 2.European Medicines Agency (EMA). Advanced therapy medicinal products. Available from http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000301.jsp&mid=WC0b01ac05800862c0
- 3.Japan Pharmaceuticals and Medical Devices Agency (PMDA). Available from https://www.pmda.go.jp/files/000214524.pdf
- 4.Korea Ministry of Food and Drug Safety (MFDS). Available from http://www.mfds.go.kr/eng/index.do?searchKeyCode=161&nMenuCode=164
- 5.Singapore Health Sciences Authority (HSA). Available from http://www.hsa.gov.sg/content/hsa/en/Health_Products_Regulation/Western_Medicines/New_Drug_Approvals.html
- 6.United States Food and Drug Administration (USFDA). Cellular & Gene Therapy Products. Available from http://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/default.htm
- 7.Laws and regulations database of the Republic of China. Pharmaceutical affair act. Taiwan: Ministry of Health and Welfare; 1970. [amended on 2015 Dec. 2]. Available from http://law.moj.gov.tw/Eng/LawClass/LawAll.aspx?PCODE=L0030001 [Google Scholar]
- 8.Laws and regulations database of the Republic of China. Medical care act. Taiwan: Ministry of Health and Welfare; 1986. [amended on 2014 Jan. 29]. Available from http://law.moj.gov.tw/Eng/LawClass/LawAll.aspx?PCODE=L0020021 [Google Scholar]
- 9.Laws and regulations database of the Republic of China. Human trial management regulation. Taiwan: Ministry of Health and Welfare; 2009. [amended 2016 Apr. 14]. Available from http://law.moj.gov.tw/Eng/LawClass/LawContent.aspx?PCODE=L0020162 [Google Scholar]
- 10.Laws and regulations database of the Republic of China. Regulations for good clinical practice. Taiwan: Ministry of Health and Welfare; 2014. Available from http://law.moj.gov.tw/Eng/LawClass/LawContent.aspx?pcode=L0030056 [Google Scholar]
- 11.Laws and regulations database of the Republic of China. Regulations for registration of medicinal products. Taiwan: Ministry of Health and Welfare; 2014. [amended on 2016 Apr. 16]. Available from http://law.moj.gov.tw/Eng/LawClass/LawContent.aspx?PCODE=L0030057 [Google Scholar]
- 12.Laws and regulations database of the Republic of China. Regulations of good tissue practice (GTP). Taiwan: Ministry of Health and Welfare; 2009[amended on 2012 Oct. 2]. Available from http://law.moj.gov.tw/Law/law_getfile.ashx?FileId=0000061450 [Google Scholar]
- 13.Laws and regulations database of the Republic of China. Pharmaceutical good manufacturing practice (GMP) regulations. Taiwan: Ministry of Health and Welfare; 2013[amended on 2013 July 30]. Available from http://law.moj.gov.tw/Eng/LawClass/LawContent.aspx?PCODE=L0030073 [Google Scholar]
- 14.Regulations database of the Taiwan Food and Drug Administration. PIC/S: Guide to good manufacturing practice for medicinal products (Part I, annexes). 2015 Oct 1. available from http://www.fda.gov.tw/TC/includes/GetFile.ashx?id=2846&chk=973b2ff6-0849-49d4-87f2–895bb4c95bec&mid=46&name=fdContent
- 15.World Health Organization (WHO). Good distribution practices (GDP) for pharmaceutical products, Annex 5, WHO Technical Report Series. 2010; No. 957. Available from http://www.who.int/medicines/areas/quality_safety/quality_assurance/GoodDistributionPracticesTRS957Annex5.pdf
- 16.Regulations database of the Taiwan Food and Drug Administration. Good distribution practice (GDP). Taiwan; 2015. Available from http://www.fda.gov.tw/TC/includes/GetFile.ashx?id=1762&chk=fd6cb69b-1011-43cc-b55f-6aeefe14b82f&mid=46&name=fdContent
- 17.Regulations database of the Taiwan Food and Drug Administration. Guidance of investigation of human cell therapy products. Taiwan; 2014. [amended on 2015 July 19]. Available from http://www.fda.gov.tw/tc/includes/GetFile.ashx?mID=19&id=38557&chk=ad2d8eb9-018f-44a0-a486-482753ffcf17
- 18.Taiwan Food and Drug Administration. Available from http://www.fda.gov.tw/TC/siteContent.aspx?sid=332
- 19.Lin Y-C, Wang P-Y, Tsai S-C, Lin C-L, Tai H-Y, Lo C-F, Wu S-I, Chiang Y-M, Liu L-L. Regulatory aspects of gene therapy and cell therapy product. 1st ed. New York (NY): Springer International Publishing; 2015. [Google Scholar]