

Abstract
Transforming growth factor-β (TGF-β) belongs to a group of pleiotropic cytokines that are involved in a variety of biological processes, such as inflammation and immune reactions, cellular phenotype transition, extracellular matrix (ECM) deposition, and epithelial–mesenchymal transition. TGF-β is widely distributed throughout the body, including the nervous system. Following injury to the nervous system, TGF-β regulates the behavior of neurons and glial cells and thus mediates the regenerative process. In the current article, we reviewed the production, activation, as well as the signaling pathway of TGF-β. We also described altered expression patterns of TGF-β in the nervous system after nerve injury and the regulatory effects of TGF-β on nerve repair and regeneration in many aspects, including inflammation and immune response, phenotypic modulation of neural cells, neurite outgrowth, scar formation, and modulation of neurotrophic factors. The diverse biological actions of TGF-β suggest that it may become a potential therapeutic target for the treatment of nerve injury and regeneration.
Keywords: Transforming growth factor-β (TGF-β), TGF-β signaling, Nerve regeneration, Cellular behavior, Scar formation
Introduction
The transforming growth factor-β (TGF-β) family is a large and still growing group of structurally related cytokines. The members of the TGF-β superfamily are highly conserved through evolution and are present in nearly all multicellular organisms1. More than 30 pleiotropic ligands that belong to the TGF-β superfamily have been identified in mammalian genomes, including TGF-βs, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), activins, inhibins, nodal, dorsalin, and the Müllerian inhibiting substance/anti-Müllerian hormone (MIS/AMH)2.
A member of the TGF-β subfamily was first discovered as a polypeptide growth factor in the medium of murine sarcoma virus-transformed mouse fibroblasts by de Larco and Todaro in 19783. The sarcoma growth factor secreted and released by mouse fibroblasts was able to convert cells to anchorage-independent growth in soft agar and thus was named TGF-β because of its transforming activity. Other members of the TGF-β subfamily were discovered later on. Until now, five isoforms of the TGF-β subfamily, TGF-β1 to TGF-β5, have been identified. Among these five isoforms, TGF-β1, TGF-β2, and TGF-β3 are present in mammals, TGF-β4 is present in birds, and TGF-β5 is present in amphibians. TGF-β1, TGF-β2, and TGF-β3 are the primary three isoforms of the TGF-β subfamily, and TGF-β1 is the most abundant isoform.
Members of the TGF-β subfamily regulate a variety of cellular functions, such as cell fate, growth, proliferation, apoptosis, differentiation, polarity, movement, invasion, and adhesion. Normally expressed TGF-β plays critical roles in numerous biological behaviors, such as inflammation and immune response, embryonic development, wound healing, extracellular matrix (ECM) formation and remodeling, and epithelial–mesenchymal transition (EMT)4,5. Excessive production and/or activation of TGF-β may modulate cellular survival, growth, migration, invasion, and other phenotypes. Therefore, the expression levels of TGF-β are often upregulated in a wide spectrum of pathological conditions, including autoimmune diseases, cancer, myelodysplastic syndrome, Marfan syndrome, scleroderma, postoperative scarring in ocular conditions, fibrotic disorders, and restenosis following coronary artery bypass and angioplasty. Emerging studies have suggested that TGF-β is also critical for tissue regeneration. In the current review, we describe the production, activation, expression, and signaling pathways of TGF-β and then specifically discuss the involvement of TGF-β in nerve repair and regeneration.
The Production of TGF-β
TGF-β1, TGF-β2, and TGF-β3 are highly similar in their structures and their biological activities in vitro. The peptide sequences of TGF-β1, TGF-β2, and TGF-β3 share 70%–80% homology. They are all encoded as large precursor proteins called pre-pro-TGF-βs. The precursor protein of TGF-β1 contains 390 amino acids, while the precursor proteins of TGF-β2 and TGF-β3 each contain 412 amino acids. The precursor proteins of TGF-β1, TGF-β2, and TGF-β3 are all monomers with three conserved components: an N-terminal signal peptide, an intermediate latency-associated peptide (LAP), and a C-terminal mature TGF-β peptide.
The pre-pro-TGF-β undergoes several processing steps and then transforms into its mature form. First, the signal peptide guides the translocation of pre-pro-TGF-β and is cleaved off in the endoplasmic reticulum. After the removal of the signal peptide, two monomers of the TGF-β precursor interact with each other by disulfide bonds in the LAP region to form a homodimer called pro-TGF-β. Subsequently, downstream of a basic amino acid target sequence [Arg-X-(Arg/Lys)-Arg], the pro-TGF-β is cleaved by protease furin, a pro-protein convertase, in the Golgi apparatus. Cleaved pro-TGF-β is called the small latent complex (SLC). It has a molecular weight of ~100 kDa and contains a noncovalent association of the dimer of LAP and the dimer of mature TGF-β. Last, the LAP dimer of SLC covalently binds to the latent TGF-β-binding protein (LTBP) through disulfide linkage and then forms a large, latent complex. The formed complex has a molecular weight of ~220 kDa and is called the large latent complex (LLC)6,7. LTBP belongs to a group of extracellular multidomain proteins. It associates with TGF-β and promotes the folding, assembly, and secretion of the latent TGF-β8. Additionally, LTBP directly interacts with other ECM components, such as fibronectin and fibrillin microfibrils, and thus localizes LLC to the specific site in the ECM for the storage of TGF-β9,10.
The Activation of TGF-β
Under certain circumstances, latent TGF-β is cleaved and activated into the biologically active mature TGF-β dimer with a molecular weight of ~25 kDa. Mature TGF-β binds to its receptors and elicits a series of biological activities. Two steps are needed for the activation of latent TGF-β. First, LTBP is cleaved from the LLC, which releases the LLC from the ECM, leaving the SLC. Second, in the SLC, the dimer of LAP undergoes proteolysis or a conformational change. The noncovalent linkage between the dimer of LAP and the dimer of mature TGF-β is disrupted, and the dimer of mature TGF-β is then released11.
The activation of latent TGF-β into mature TGF-β can be mediated by several factors, such as the cellular microenvironment, reactive oxygen species (ROS), protease, metalloprotease, thrombospondin-1 (TSP-1), and αv-containing integrins.
Numerous microenviroment factors, including pH, tem perature, and chaotropic agents, can break the linkage between the dimer of LAP and the dimer of mature TGF-β. Therefore, extreme acidification or alkalinization, heating, and chaotropic agent treatment (e.g., urea) can be used to activate TGF-β. Extreme acidic or basic conditions (a pH of 1.5 or 12) could significantly activate latent TGF-β, while mild acidic conditions (a pH of 4.5) could activate latent TGF-β to a lesser extent12. Detailed studies of the activation of three different isoforms of TGF-β suggest that acidification treatment with a pH between 4.1 and 3.1 as well as alkalinization treatment with a pH between 11.0 and 11.9 could transit human recombinant latent TGF-β1 and TGF-β2 and that slightly shifted pH conditions, acidification treatment with a pH between 3.1 and 2.5, and alkalinization treatment with a pH between 10.0 and 12.3 could activate chicken recombinant latent TGF-β313. Thermal energy could also induce the activation of latent TGF-β. A temperature between 65°C and 100°C could activate recombinant latent TGF-β1, TGF-β2, and TGF-β3, while a temperature between 75°C and 100°C could activate native latent TGF-β113. Besides acidification, alkalinization, and heating to 100°C for 3 min, exposure to urea could also promote the transition of spontaneously released latent TGF-β in serum-free medium by chicken embryo fibroblasts into its active, mature form14.
Besides microenviroment factors, ROS, through hydroxyl radicals, could also modify and disturb the interaction between the dimer of LAP and the dimer of mature TGF-β and thus activate TGF-β. For example, the exposure to 100-Gray radiation or metal-catalyzed ascorbate oxidation-generated ROS could induce the activation of recombinant human TGF-β15.
The activation of latent TGF-β can also be induced by enzymes, such as plasmin and matrix metalloproteinase (MMP). Plasmin is a serine protease that dissolves fibrin blood clots and promotes the degradation of the ECM. In the activation of latent TGF-β, plasmin advances the proteolytic cleavage of LTBP from ECM, accelerates the degradation of LAP, and thus promotes the release of active mature TGF-β. In both rat kidney fibroblastic cell-conditioned medium and Chinese hamster ovary (CHO) cells transfected with the pre-pro-TGF-β1 cDNA-conditioned medium, plasma treatment led to TGF-β activation to an extent, and the result was similar to acid treatment with a pH of 4.512,16. Meanwhile, treatment of plasmin inhibitors, such as aprotinin, ε-amino-n-caproic acid, and α2 plasmin inhibitor, suppressed the inhibitory effect of TGF-β on cell movement in cocultures of bovine aortic endothelial cells and pericytes, suggesting that plasmin mediates TGF-β activation17. MMPs are a large family of zinc-containing endopeptidases that regulate tissue remodeling and dynamic changes of the ECM. Some members of the MMP family, such as MMP-9 and MMP-2, could also regulate TGF-β activation18.
TSP-1, a multidomain adhesive matrix glycoprotein that mediates cell-to-cell and cell-to-matrix interactions, can also mediate TGF-β activation. TSP-1 interacts with both the dimer of LAP and the dimer of mature TGF-β, forms a trimolecular complex, induces a structural change of SLC, and is thus responsible for a large proportion of TGF-β activation. Compared with wild-type mice, TSP-1 null mice showed histological abnormalities in most organs (e.g., lung, bronchial tube, stomach, liver, and pancreas), and the abnormalities were extremely similar to those observed in young TGF-β1 null mice19. Moreover, continuous intravenous infusion of blocking peptides, which could specifically interfere with TSP-1-induced activation of TGF-β, did not alter the amount of total TGF-β but significantly reduced the expression of active TGF-β and decreased the accumulation of glomerular ECM accumulation20.
Besides the above-mentioned factors, αv-containing integrins, such as αvβ6 integrin, are also able to activate latent TGF-β. Integrins are transmembrane factors that mediate the cell–cell and cell–ECM attachment, interaction, and signal transduction. αv-containing integrins may exhibit their activating ability through two possible mechanisms. The first mechanism is the initiation of conformation change. Through the hinge region, integrins bind to the arginyl-glycyl-aspartic acid (RGD) motif of the dimer of LAP, pull LAP from the dimer of mature TGF-β, and thus promote the functional release of active TGF-β21. The second mechanism is the protease-dependent activation. Integrins create a close connection between the latent TGF-β and proteinases such as MMPs and then stimulate the activation of the latent TGF-β through proteolytic degradation. Although the specific mechanism has not been determined, the effect of integrin-induced TGF-β activation has been identified. The spatial activation of TGF-β could be induced by αvβ6 integrin-expressing cells and could also be blocked by antibodies against active αvβ6 integrin22. Additionally, mice with a nonfunctional mutation of the RGD-binding sequence of αvβ6 and αvβ8 integrins in the LAP domain of latent TGF-β developed vasculogenesis defects, multiorgan inflammation, and lack of Langerhans cells, exhibiting major features of TGF-β null mice, despite their normal production of the latent TGF-β. This suggests that RGD-binding integrins are essential for the activation of latent TGF-β23.
TGF-β SIGNALING PATHWAYS
Activated TGF-β regulates multiple cellular processes through a general mechanism. First, members of the TGF-β family bind to TGF-β type I and type II receptors. Ligand binding induces the formation of heteromeric complexes of type I and type II receptors and activates serine/threonine kinase type I and type II receptors. Activated TGF-β receptors regulate intracellular signaling pathways, including the canonical Smad signaling pathway and numerous noncanonical signaling pathways, and thus contribute to diverse biological effects (Fig. 1).
Figure 1.
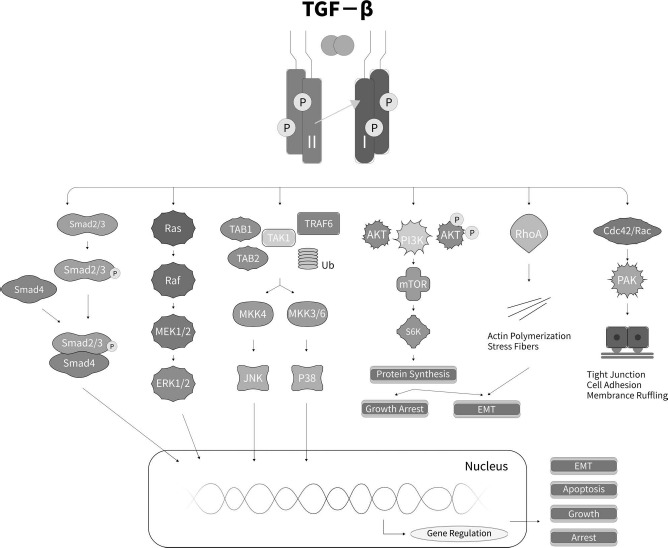
Schematic representation of TGF signaling pathways. Transforming growth factor-β (TGF-β) binds to the TGF-β type II receptor, phosphorylates the type I and II complex, and then activates the canonical and noncanonical Smad signaling pathways, including extracellular signal-related kinase (Erk), Jun N-terminal kinase (JNK)/p38, small GTPase, and phosphoinositide 3-kinase (PI3K)/Akt signaling pathways. RAF, rapidly accelerated fibrosarcoma; TAB1/2, TGF-β-activated kinase 1/2; TAK1, TGF-β-activated kinase 1; TRAF6, TNF receptor-associated factor 6; MKK, mitogen-activated protein kinase; mTOR, mechanistic target of rapamycin; S6K, ribosomal protein S6 kinase; RhoA, Ras homolog gene family, member A; PAK, p21-activated kinase; EMT, epithelial–mesenchymal transition.
The Ligand Interaction and Activation of TGF-β Receptors
The signal transduction of TGF-β is via a heteromeric complex of two related transmembrane kinase receptors: TGF-β type I and type II receptors. TGF-β type I and type II receptors are glycoproteins with molecular weights of ~55 and ~70 kDa, respectively. Both of them contain a cysteine-rich extracellular region that is relatively short in length (~150 amino acids), a transmembrane region, and a cytoplasmic juxtamembrane region that contains a serine/threonine protein kinase domain. The serine/threonine protein kinase domain is critical for receptor and substrate phosphorylation. The TGF-β type I receptor, via the protein kinase domain, is able to phosphorylate intracellular signal cascades (e.g., Smad proteins) on serine residues. The TGF-β type II receptor, via the protein kinase domain, could phosphorylate itself as well as TGF-β type I receptor on serine and threonine residues, but not tyrosine residues2. The TGF-β type I receptor also contains a unique glycine- and serine-rich (GS) domain in its intracellular region, preceding the protein kinase domain. The GS domain is a highly conserved region with ~30 amino acids and is rich in glycine and serine. The GS domain is important for TGF-β signaling pathways because it is the presence of the GS domain that enables TGF-β type II receptor to phosphorylate TGF-β type I receptor, thus mediating subsequent signal activation2.
Besides the TGF-β type I and type II receptors, there exist accessory receptors of TGF-β, namely, TGF-β type III receptors. The accessory receptors mainly consist of two membrane glycoproteins: betaglycan and endoglin24. Betaglycan can bind to all three isoforms of TGF-β–-TGF-β1, TGF-β2, and TGF-β3–with high affinity. Endoglin can only bind to TGF-β1 and TGF-β3, but not TGF-β2. Betaglycan and endoglin do not have an intrinsic signaling function by themselves but are able to interact with TGF-β type I and type II receptors and then facilitate the binding of TGF-β to its receptors.
Prior to ligand–receptor binding, mature TGF-β, TGF-β type I receptor, and TGF-β type II receptor all exist as homodimers. Activated mature TGF-β dimers can directly bind to the TGF-β type II receptor but are not able to directly bind to isolated type I receptor6. In cell mutants lacking the TGF-β type I receptor, TGF-β can still associate with the type II receptor. On the contrary, in cell mutants lacking the TGF-β type II receptor, TGF-β cannot successfully associate with the type I receptor. Additionally, the transfection of cDNA coding for the TGF-β type II receptor in the TGF-β type II receptor mutant cells can recover the binding of TGF-β to the type I receptor, suggesting that the TGF-β type II receptor is required for the binding to the type I receptor25. The association of TGF-β to the type I and type II receptors is sequential. Upon the binding of TGF-β to the extracellular domains of the TGF-β type II receptor dimer, the TGF-β type I receptor dimer is recruited to form a stable heterotetrameric receptor complex. The TGF-β type II receptor is a constitutively active kinase and is phosphorylated on serine residues in its protein kinase domain even in the basal state. The formed receptor complex induces a close structural proximity. Therefore, the phosphorylated TGF-β type II receptor is able to transactivate the recruited type I receptor by phosphorylation at serine and threonine residues in the GS domain7. The activated TGF-β type I receptor then mediates downstream signal propagation by the canonical Smad signaling pathway and/or the noncanonical signaling pathways.
The Canonical Smad Signaling Pathway
Smad proteins are the first identified downstream signaling transducers of TGF-β. Therefore, the TGF-β-induced Smad signaling pathway is called the canonical Smad signaling pathway. The proteins of the Smad family are the vertebrate homologs of the Drosophila protein mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein small body size (SMA). Until now, eight members of the Smad family have been identified in mammals. They have been subdivided into three distinct groups based on their different functions: (1) the receptor-regulated Smads (R-Smads) including Smads 1, 2, 3, 5, and 8; (2) the common-partner Smad (Co-Smad) including Smad4; and (3) the inhibitory Smads (I-Smads or anti-Smads) including Smads 6 and 7. These Smads have highly similar structures and are all composed of three domains: an N-terminal Mad homology (MH1) domain with ~130 amino acids, an intervening linker region with variable size and sequence, and a C-terminal MH2 domain with ~200 amino acids. The MH1 and MH2 domains have an affinity for each other. Therefore, in the basal state, the MH1 and MH2 domains of SMADs interact with each other to form inactivated homooligomers. The eight members of the Smad family are mediators of different signaling pathways induced by different members of the TGF-β superfamily. For instance, Smads 1, 5, and 8 are involved in BMP signaling, while Smads 2 and 3 are involved in TGF-β signaling.
In the canonical TGF-β/Smad signaling pathway, following TGF-β binding, receptor complex formation, and the consequent phosphorylation of the TGF-β type I receptor, Smad2 and Smad3 were recruited to the receptor complex. The association of Smad2/3 with TGF-β receptor complex is facilitated by the Smad anchor for receptor activation (SARA), an anchoring protein that binds to both the MH2 domains of R-Smads and the TGF-β type I receptor. After the association with the TGF-β receptor complex, Smad2/3 are directly activated by type I receptor-mediated phosphorylation at the C-terminal SS(V/M)S motif in the MH2 domain. Upon phosphorylation, Smad2/3 is released from the receptor complex and SARA and then initiate the canonical Smad signaling pathway. Activated Smad2/3 binds to partner protein Smad4 through the MH2 domain to form an R-Smad/Co-Smad complex with the assistance of scaffolding proteins, such as heat shock protein TGF-β type I receptor-associated protein-1 (TRAP-1). The R-Smad/Co-Smad complex then translocates to the nucleus; binds to the Smad-binding elements (SBEs) in promoters of target genes in a sequence-specific manner; interacts with some additional transcriptional coregulators, such as activator protein-1 (AP-1), forkhead box protein H1 (FOXH1), and CBP/p300; and then regulates transcriptional responses26,27.
The canonical Smad signaling pathway is antagonistically regulated by I-Smads. I-Smads do not contain a C-terminal SSXS phosphorylation motif. Therefore, they can bind to the TGF-β receptor complex and competitively inhibit the phosphorylation of R-Smads. The major I-Smad that blocks TGF-β-mediated canonical Smad signaling pathway is Smad7. Specifically, Smad7 competes with Smad2/3 for association with the TGF-β receptor complex, interferes with the phosphorylation and activation of Smad2/3, and thus antagonizes Smad signaling28. Another I-Smad, Smad6, mainly inhibits BMP signaling. When it is overexpressed, however, Smad6 is also able to partially inhibit TGF-β signaling29.
Besides the Co-Smad-facilitated nucleus translocation, recent studies have suggested that R-Smads can also mediate the biosynthesis of several microRNAs (miRNAs), such as miR-21 and miR-84. R-Smads facilitate the processing of primary miRNAs into precursor miRNAs in the nucleus, regulate the miRNA expression, and affect the expression of a variety of target genes at the transcriptional and posttranscriptional levels30,31.
It is worth mentioning that besides directly modulating the expressions of target genes, Smad proteins can also activate other signaling pathways, for example, the protein kinase A (PKA) signaling pathway, to propagate the physiological and pathological activities of TGF-β. The typical biological effect of PKA on cellular growth, differentiation, and apoptosis is normally initiated by the elevated level of cellular cyclic adenosine monophosphate (cAMP). Smad proteins regulate PKA signaling through a mechanism independent of cAMP. The activated complex of Smad3 and Smad4 interacts with the regulatory subunit of PKA, promotes the release of the catalytic subunit of PKA, and increases the activity of PKA and the PKA-dependent transcription factor cAMP-response element-binding protein (CREB)32–34.
The Noncanonical Signaling Pathways
In addition to the canonical Smad signaling pathway, many noncanonical signaling pathways, including the extracellular signal-regulated kinase (Erk) signaling pathway, the phosphoinositide 3-kinase (PI3K) and the Akt signaling pathway, the small GTPase signaling pathway, and the c-Jun N-terminal kinase (JNK) and the p38 mitogen-activated protein kinase (p38 MAPK) signaling pathway, are also affected by active TGF-β35.
The TGF-β type II receptor, as previously mentioned, undergoes autophosphorylation on serine residues in its protein kinase domain. The tyrosine residues of the TGF-β type II receptor can also be phosphorylated by receptor tyrosine kinase (RTK) and/or non-RTK protein proto-oncogene tyrosine–protein kinase Src. These phosphorylated tyrosine residues serve as docking sites for growth factor receptor-binding protein 2 (Grb2). Grb2 then interacts with son of sevenless (Sos), which further catalyzes the Ras exchange of GDP binding for GTP binding. GTP-bound Ras sequentially activates c-Raf, followed by the activation of Erk and a mitogen-activated protein (MAP) kinase kinase (MEK, MKK, MAPKK, or MAP2K). Activated Erk affects downstream transcription factors, such as zinc finger protein Snail, a repressor of E-cadherin transcription and an inducer of the EMT; regulates the transcription of target genes; and eventually mediates TGF-β-induced biological functions, such as EMT35–37. In addition, activated Erk signaling pathway can also activate AP-1, a transcriptional coregulator of the Smad signaling pathway, and participates in the regulation of the canonical Smad signaling pathway38.
Besides the Erk signaling pathway, the PI3K/Akt signaling pathway is also involved in TGF-β-induced EMT. Upon TGF-β activation and the subsequent formation of the TGF-β receptor complex, the TGF-β type II receptor constitutively associates with p85, the regulatory subunit of PI3K, activates PI3K and phosphorylates its downstream effector, Akt39. Activated Akt, independent of Smad proteins, activates mechanistic target of rapamycin (mTOR) and phosphorylates S6 kinase (S6K), regulates protein synthesis, and contributes to TGF-β-induced cell migration, actin filament reorganization, and EMT35. Activated Akt can also phosphorylate and inhibit the activity of glycogen synthase kinase 3β (GSK3β), an inactivating agent of glycogen synthase, stimulate the transcription of Snail, and then promote EMT40,41. On the other hand, Akt may have a negative effect on the canonical Smad signaling pathway. Akt binds to Smad3 directly, prevents the interaction of TGF-β receptor com plex with Smad3, inhibits the subsequent activation and nuclear localization of Smad3, and antagonizes Smad-mediated cellular functions, such as apoptosis and growth inhibition42.
Members of the Ras homolog (Rho)-like GTPase family, such as RhoA and Cdc42/Rac, also contribute to several TGF-β-induced biological processes during EMT, such as actin stress fiber formation and tight junction dissolution. The TGF-β type I receptor associates with partitioning defective 6 (Par6), a scaffold protein that mediates cell polarization at tight junctions. Following TGF-β activation and receptor phosphorylation, Par6 is phosphorylated by the TGF-β type II receptor at serine residue 345. Phosphorylated Par6 recruits E3 ubiquitin–protein ligase Smurf1 to activate the TGF-β receptor complex and to form a complex with Smurf1. The Par6-Smurf1 complex induces the ubiquitination and degradation of RhoA and leads to the dissociation of tight junction, the modulation of cell adhesion and membrane ruffling, and the rearrangement of the actin cytoskeleton43. In addition to RhoA, TGF-β also affects Cdc42/Rac. The activated TGF-β receptor complex induces a physical interaction with Cdc42 GTPase, activates Cdc42/Rac and its downstream cascade p21-activated kinase (PAK), forms a Cdc42 and PAK network containing tight junction protein occludin, and thus participates in the tight junction dissolution during EMT44,45.
Moreover, the p38 MAPK- and JNK-activated cascades are also activated by TGF-β and tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), a ubiquitin ligase that is able to ubiquitinate itself and other molecules upon binding. TGF-β type I receptor has a physical association with TRAF6 both in the basal and activated states. The phosphorylated TGF-β type I receptor induces the lysine 63 residue-linked autopolyubiquitination of TRAF6, which then recruits TGF-β-activated kinase 1 (TAK1) and its binding proteins TAB1 and TAB2, and promotes the linkage among TRAF6, TAK1, TAB1, and TAB246–48. TRAF6 then triggers the polyubiquitination and activation of TAK1. Activated TAK1 further phosphorylates MAP kinase kinase 4 (MKK4) and MAP kinase kinase 3/6 (MKK3/6), as well as their downstream cascades JNK and p38 MAPK. Activated JNK and p38 MAPK, either by themselves or in conjunction with the Smad proteins, modulate their target genes and thus contribute to various biological functions, such as cellular apoptosis and EMT49.
Besides the above-mentioned noncanonical signaling pathways, TGF-β signaling can also be modulated by other signaling pathways, such as Wnt, hedgehog, notch, interferon (IFN), and TNF signaling pathways6. Taken together, the full spectrum of cellular functions and biological activities of TGF-β depends on the dynamic combination of the canonical Smad signaling pathway, numerous noncanonical signaling pathways, and other signaling pathways.
Expressions of TGF-β Following Nerve Injury
TGF-β is ubiquitously expressed in most tissue microenvironments. In adult mice, there exists an abundance of TGF-β1 in the adrenal gland, bone marrow, kidneys, ovaries, and placenta, and there exists widespread expression of TGF-β1 in the cartilage, heart, pancreas, skin, and uterus50. Besides its ubiquitous expression in tissues and organs, TGF-β1 is also present at measurable levels in body fluids and secretions, such as plasma, urine, synovial effusion, cerebrospinal fluid (CSF), bronchoalveolar lavage fluid, and seminal plasma, suggesting an endocrine or lumicrine function of TGF-β151.
Emerging evidence suggests that TGF-β exhibits differential expression patterns in the central nervous system (CNS) after neural injury. The prompt and sustained change in TGF-β expression after brain injury leads to an assumption that TGF-β may serve as a biomarker for brain injury52. For example, following a localized cerebral injury, the mRNA and protein expressions of TGF-β1 in the rat brain, mainly at regions corresponding to astrocytes and macrophages, were increased53. After a penetrating brain injury, the mRNA expression levels of TGF-β1 were increased in the rat cerebral cortex54. After hypoxia–ischemia in the rat brains, the expressions of TGF-β1 in cortical layer 3 and the hippocampus of the ligated hemisphere were markedly increased from 72 to 120 h after exposure to hypoxia55. In the injured rat spinal cord, the mRNA expressions of TGF-β as well as TGF-β receptors were also altered. TGF-β and its receptors were significantly upregulated at the epicenter, rostral, and caudal to the injury, especially in regions known to contain activated microglia and degenerating axon profiles, suggesting that microglia and macrophages might be a major source of TGF-β1 production after spinal cord injury (SCI)56. The temporal and spatial expressions of TGF-β1 and TGF-β2 were reported not only in the rat spinal cord but also in the human spinal cord57. Transection of rat facial nerves would also lead to a biphasic increase of TGF-β1 mRNA expression. However, it failed to affect the expression of TGF-β3 mRNA in activated microglia around regenerating motoneurons and astrocytes58. It was also reported that the protein expressions of TGF-β type II receptor and Smad3 were rapidly upregulated in neurons of the ipsilateral cortex and CA1 region of the hippocampus after stab wound injury59.
Similar to the findings following central nerve injury, an elevated expression of TGF-β was also observed following peripheral nerve injury. After sciatic nerve crush, the mRNA expression of TGF-β1 was upregulated in both the crushed segment and the distal segment. Upregulated TGF-β1 was localized in the regions containing infiltrating macrophages60. Besides mRNA expressions, the protein expressions of TGF-β1 were also elevated in the distal nerve stump after sciatic nerve injury61,62. A biphasic expression of TGF-β protein was also observed at the lumbar segment of the spinal cord following peripheral nerve injury63.
Following nerve injury, functional TGF-β was elevated in addition to total TGF-β. Recruited and proliferating macrophages and glial cells could produce TSP, which then activated the latent TGF-β64. Furthermore, TGF-β itself could increase the amount of TSP-1 and thus enhance its own activation, suggesting the existence of a possible positive feedback loop after nerve injury65,66.
Effects of TGF-β on Nerve Regeneration
TGF-β plays critical roles in many physiological processes, including the immune response, cellular differentiation, cellular proliferation and apoptosis, cellular adhesion and migration, ECM production, EMT, and wound healing. The increased expression of TGF-β also modulates nerve regeneration by suppressing the immune response, affecting cellular behavior, regulating neurite outgrowth, and promoting glial scar formation. Moreover, TGF-β also modulates the expressions and/or activities of several biochemical cues that are important for nerve regeneration, thus indirectly affecting the regenerative process. The specific effects of TGF-β on nerve regeneration are illustrated in Figure 2 and are reviewed as follows.
Figure 2.
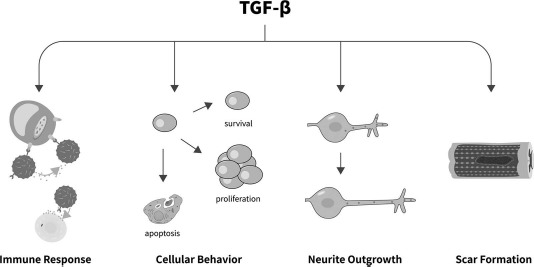
The specific effect of TGF-β on nerve regeneration. TGF-β regulates the immune response, cellular behavior, neurite outgrowth, and scar formation, playing regulatory roles in nerve regeneration.
Inflammation and Immune Response
TGF-β is an “anti-inflammatory” cytokine that regulates the proliferation, differentiation, and survival of lymphocytes; reduces the synthesis of proinflammatory cytokines; and inhibits the functions of other immune cells, such as natural killer cells (NKCs), dendritic cells, and macrophages67. Following injury, TGF-β may be secreted by immune cells surrounding the injury site and then function as an immunosuppressant68.
The protective effect of TGF-β against many autoimmune diseases has been well documented. In the nervous system, TGF-β is also associated with immune cells to mediate nerve injury-induced inflammation and the immune response. Compared with wild-type mice, mice with homozygous deletion of TGF-β1 in uninjured brain parenchyma exhibited an extensive inflammatory response. Those TGF-β1 null mice exhibited increased neuronal cell death, decreased central axonal sprouting, and delayed functional recovery following facial axotomy69. It was also reported that the TGF-β signaling pathway was activated in astrocytes following toxoplasma infection. Inhibiting TGF-β signaling could largely augment toxoplasma infection-induced immune cell infiltration and neuronal damage70. Outcomes from an in vivo study further suggested that TGF-β1 coordinated with adipose-derived mesenchymal stem cells (ADSCs) to enhance nerve regeneration by reducing inflammation and impairing the host's immune response71.
Cellular Behavior
The effects of TGF-β1 on the nervous system, just like those on many other tissues and organs, are embodied in phenotypic modulation of neurons and glial cells, including cellular survival, differentiation, proliferation, and activation.
Some studies showed that TGF-β1 inhibited the growth and proliferation of primary cultured cortical astrocytes, C6 astrocytoma cells, as well as primary cultured cortical neurons72,73. In primary neurons, TGF-β1 stimulated the expression of cell adhesion proteins to promote the movement and outgrowth of postmitotic neurons73. However, other studies suggested that TGF-β benefits neuron survival and proliferation. For example, TGF-β2 perfusion attenuated the injury-induced death of mature motoneurons, inducing a more potent effect than glial cell-derived neurotrophic factor (GDNF) or brain-derived neurotrophic factor (BDNF)74. In contrast, facial nerve avulsion treatment with an adenovirus encoding TGF-β2 prevented the loss of lesioned facial motoneurons75. In RGC-5, a retinal ganglion cell line, TGF-β1 treatment increased the expression of neural cell markers neurofilament-M (NF-160) and PGP9.5 (UCH-L1) and decreased cellular apoptosis in serum-free medium, suggesting that TGF-β1 was critical for the differentiation and survival of retinal ganglion cells76. Smad3 null mice also exhibited a more pronounced loss of neuronal viability to cortical stab wound injury compared with wild-type mice, suggesting that TGF-β, via the Smad3 signaling pathway, protects neurons in the cortex and hippocampus at early time points against injury59. The neuronal differentiation mechanism of deferoxamine, a G1/S phase cell cycle blocker, was also found to be mediated by TGF-β177.
TGF-β not only affects the survival of neuronal cells but also regulates the phenotype of glial cells. For example, TGF-β1 could suppress the mitotic effects of fibroblast growth factor (FGF) and epithelial growth factor (EGF) on astrocytes and inhibit astrocyte proliferation54. TGF-β1 could also inhibit the proliferative response of astrocyte cultures to serum and growth factors78,79. It was also demonstrated that TGF-β1 was a chemotactic factor for astrocytes in a dose-dependent fashion79. In the peripheral nerve system, TGF-β regulates Schwann cells. The treatment of purified Schwann cells with exogenous TGF-β1 increased Schwann cell proliferation and differentiation and promoted a pre- or nonmyelinating Schwann cell phenotype. Addition of TGF-β1 to cocultures of Schwann cells and neurons inhibited the effects of axons on Schwann cells while blocking Schwann cell myelination80. TGF-β1 could also reduce the expressions of the myelin-related molecules, including galactocerebroside, P0, myelin-associated glycoprotein, and myelin basic protein, and block Schwann cell myelination81. Taken together, these results suggested that TGF-β1 promoted the transition of Schwann cells to a nonmyelinating phenotype with high proliferation and differentiation capabilities, and therefore significantly enhanced the regenerative abilities after nerve injury.
In addition, TGF-β plays beneficial roles in preventing cytotoxicity and protecting neurons and glial cells. In vitro treatment with TGF-β1 reduced the excitotoxic neuronal damage, and in vivo pretreatment with TGF-β1 prior to vessel occlusion reduced the area of ischemia, suggesting that TGF-β1 played neuroprotective roles82. In another study, the same research group induced different types of excitotoxic injury on cultured hippocampal neurons and noted that TGF-β1 could protect neurons against rapidly triggered, Ca2+-mediated excitotoxic injury, but induced an opposite effect on slowly triggered excitotoxic injury83. TGF-β1 was also capable of protecting motor neurons against excitotoxic degeneration and inhibiting natural, IFN-γ-, or phorbol myristate acetate (PMA)-induced cytotoxicity of oligoendrocytes by microglia84,85.
Neurite Outgrowth
The effect of TGF-β on neurite outgrowth is controversial. It was shown that TGF-β1 could increase neurite outgrowth in dopaminergic cells after scratch lesion86. The promoting effect of secreted protein acidic and rich in cysteine (SPARC), a matricellular protein derived from olfactory ensheathing cells, on dorsal root ganglion outgrowth was through a TGF-β-dependent signaling pathway87. Moreover, TGF-β1 could also significantly increase the length of neurites extended from differentiated RGC-5 cells76. On the other hand, some studies suggested that only TGF-β2 could increase neurite length and branching pattern in cultured myenteric neurons, while TGF-β1 and TGF-β3 had no significant effect77,84,88. There have also been several studies suggesting that TGF-β inhibited neurite outgrowth. For example, the application of TGF-β1 to primary culture of cerebellar granule neurons could suppress neurite outgrowth, while further treatment with LY364947, a blocker of TGF-β1 type I receptor, abrogated the inhibitory effects89.
Of note, the promoting or inhibiting effect of TGF-β on neurite outgrowth might be influenced by the microenvironment. For example, if astrocyte culture was pre-treated with basic FGF plus interleukin-1 (IL-1), the promoting effects of TGF-β on neurite outgrowth were dramatically attenuated90.
Glial Scar Formation
TGF-β stimulates the production of the ECM and accelerates wound healing and repair. In the nervous system, TGF-β is involved in glial scar formation. In cultured astrocytes, TGF-β increased the expression of neurocan, a chondroitin sulfate proteoglycan that mediates glial scar formation and inhibits axon growth91. Similarly, latent TGF-β was brought to the injury sites through the binding effect of fibrinogen and was then activated by astrocytes. Activated TGF-β serves as a molecular linkage between vascular permeability and scar formation92. The injection of TGF-β1 caused a severe scarring response, while administration of an antibody against TGF-β1 successfully attenuated the deposition of fibrous scar tissue and the formation of glial-limiting membranes that border the lesion93. The treatment of cerebral wounds with an antibody against TGF-β2 could also attenuate matrix deposition, diminish the formation of glial-limiting membranes, lessen inflammation and angiogenesis, and, finally, robustly reduce the scarring of the CNS94. Antibodies against TGF-β1 and TGF-β2 attenuated the response of glial fibrillary acidic protein (GFAP)-immunoreactive astrocytes and oligodendrocyte progenitor (NG2-glia) cells and reduced the scar formation. However, antibodies against TGF-β1 and TGF-β2 failed to attenuate the response of CR3-immunoreactive microglia and macrophages95. Intrathecal administration of an antibody against TGF-β1 after thoracic spinal cord contusion suppressed glial scar formation, upregulated microglia/macrophage activation, and enhanced the loco-motor recovery96.
Numerous studies have also been conducted to identify the involvement of the TGF-β-activated Smad signaling pathway in glial scar formation. It was shown that Smad3 null mice had fewer neutrophils, macrophages/microglia, NG2+ cells, and GFAP+ cells, and the animals exhibited altered glial scar formation and immune response and more rapid healing of cerebral cortex wounds97. To investigate the scarring mechanisms of TGF-β1, the addition of TGF-β1 to cocultured cerebral astrocytes and meningeal fibroblasts was noted to enhance the fibroblast proliferation and promote the formation of cell clusters, thus remarkably inhibiting the neurite outgrowth of cerebellar neurons98. Given the central critical role of TGF-β/Smad signaling in scar formation, blocking TGF-β has been suggested as an effective antifibrotic therapy in treating CNS injuries. For example, dampening the TGF-β/Smad signaling pathway is enough to decrease scar formation and facilitate intrinsic axonal growth and regeneration99.
Modulation of Neurotrophic Factors
TGF-β affects the amounts and/or activities of other nerve regeneration-related cytokines. For instance, TGF-β1 markedly increased its own expression as well as the mRNA expressions of nerve growth factor (NGF) in cultured rat astrocytes, and intraventricular injection TGF-β1 increased the mRNA expressions of NGF in the rat hippocampus100. TGF-β1 regulated the mRNA and protein expressions of NGF in rat and mouse glia101. It was also found that although treatment with TGF-β1 did not change the amount of NGF in the cultures or in the medium, treatment with TGF-β1 increased neuronal survival and levels of peptide neurotransmitter substance P (SP) through mediation by exogenous NGF. Additionally, treatment with an antibody against NGF eliminated the effect of TGF-β1, which further suggested that the neurotrophic action of TGF-β1 was synergistic with NGF102.
Another neurotrophic factor, GDNF, was shown to have a rescue effect on target-deprived sympathetic spinal cord neurons, and this effect was dependent on the concomitant expression of TGF-β103. A similar regulatory effect of TGF-β on GDNF was observed in Krieglstein et al.'s study, which demonstrated that GDNF alone was unable to promote neuron survival, but GDNF could act as a neurotrophic factor when it was supplemented together with TGF-β104.
Applications of TGF-β for Repairing Nerve Injury
As the above-mentioned reviewed studies have suggested, TGF-β is involved in a variety of biological activities, such as suppressing the immune response, regulating phenotypic modulation of neuronal and glial cells, and affecting glial scar formation following nerve injury. However, whether TGF-β promotes nerve regeneration has not been determined. Some studies showed that TGF-β has a negative role on neuronal growth and proliferation, while other studies indicated that TGF-β plays a positive role72,73,76. Similarly, TGF-β either promotes or restrains neurite outgrowth, as revealed by different studies86,89. The diverse effects of TGF-β on the immune response, cellular behavior, and modulation of growth factors and cytokines suggest that TGF-β may be beneficial for nerve repair and regeneration. On the other hand, TGF-β increases the formation of fibrotic scars at the lesion site, which is normally considered as an inhibitory factor for nerve regeneration and prevents subsequent growth, plasticity, and recovery of damaged neurons. A recent breakthrough study, however, indicated that a glial scar may not be totally harmful, and, on the contrary, astrocytes at lesions of the spinal cord may express multiple axon growth-supporting molecules and may aid rather than prevent axon regeneration in the CNS105. On the basis of the obscure effect of astrocytic scars, the effect of TGF-β on nerve regeneration may be even more complex.
A number of attempts have been made to directly deliver TGF-β to injured sites. The results may provide some cues for the particular roles of TGF-β on nerve regeneration. For instance, treatment with TGF-β plus forskolin for 6 weeks resulted in a robust increased number of regenerating axons in rats that underwent tibial nerve injury, probably via upregulating growth-associated proteins and reactivation of Schwann cells106. The direct effect of TGF-β3 on facial nerve regeneration was also observed. In rabbits with facial nerve injuries, treatment with TGF-β3 significantly increased the total number and diameter of the regenerated nerve fibers and promoted morphological repair and functional recovery to a certain degree107. Despite these initial attempts that suggest the therapeutic roles of TGF-β in nerve regeneration, more numerous and intensive studies are required to further investigate the details of the clinical application of TGF-β, including treatment time, safe dose levels, and possible side effects, etc.
Concluding Remarks
In the current review article, we provide a concise summary of the biological aspects of TGF-β, mainly focusing on its production, activation, and related signaling pathways. We particularly focused on recent progress in research, which has elucidated the involvement of TGF-β in nerve repair and regeneration. It has been widely observed that following nerve injury, differentially expressed TGF-β modulates cellular survival, growth, proliferation, differentiation, and migration of neurons and glial cells. These observations suggest that TGF-β is critical for other biological events during nerve injury and regeneration, including the immune response, neurite outgrowth, scar formation, and regulation of neurotrophic factors. Importantly, several promising pre-clinical studies suggest that direct application of TGF-β to animals with nerve injury promotes nerve repair and regeneration. Obviously, treatments with exogenous TGF-β are designed on the basis of the understanding of TGF-β as a potential therapeutic target. Current preclinical studies of TGF-β for nerve regeneration, however, are still limited in number and limited to small animals. Considering the complex and even controversial biological effects of TGF-β on the nervous system, it is assumed that just like its dual effects on carcinogenesis, TGF-β may also play dual roles in nerve repair and regeneration. It has been reported that TGF-β exhibits a biphasic expression pattern in the regenerating nerve of infant rats over a period of 1 month after sciatic nerve injury108. This interesting finding may suggest that the in vivo effects of TGF-β on nerve regeneration are likely to be dependent on many potentially unknown factors both at the conceptual and technical levels. Accordingly, prior to clinical translation, insightful studies on the mechanistic aspects of TGF-β are warranted to optimize its therapeutic outcomes.
Acknowledgment
The authors thank Professor Jie Liu for his help in the revision of the manuscript. This work was supported by the Natural Science Foundation of Jiangsu Province, P.R. China (No. BK20150409) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The authors declare no conflicts of interest.
References
- 1.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007; 8(12): 970–82. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. TGF-βeta signal transduction. Annu Rev Biochem. 1998; 67: 753–91. [DOI] [PubMed] [Google Scholar]
- 3.de Larco JE, Todaro GJ. Growth factors from murine sarcoma virus-transformed cells. Proc Natl Acad Sci USA 1978; 75(8): 4001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012; 13(10): 616–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-βeta signalling. Nature 1997; 389(6651): 631–5. [DOI] [PubMed] [Google Scholar]
- 6.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012; 11(10): 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-βeta superfamily signaling. FASEB J. 1999; 13(15): 2105–24. [PubMed] [Google Scholar]
- 8.Miyazono K, Olofsson A, Colosetti P, Heldin CH. A role of the latent TGF-βeta 1-binding protein in the assembly and secretion of TGF-βeta 1. EMBO J. 1991; 10(5): 1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-βeta-binding proteins. Matrix Biol. 2015; 47: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY, Rifkin DB. Specificity of latent TGF-βeta binding protein (LTBP) incorporation into matrix: Role of fibrillins and fibronectin. J Cell Physiol. 2012; 227(12): 3828–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-βeta structure and activation. Nature 2011; 474(7351): 343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol. 1988; 106(5): 1659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown PD, Wakefield LM, Levinson AD, Sporn MB. Physicochemical activation of recombinant latent transforming growth factor-beta's 1, 2, and 3. Growth Factors 1990; 3(1): 35–43. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence DA, Pircher R, Jullien P. Conversion of a high molecular weight latent beta-TGF from chicken embryo fibroblasts into a low molecular weight active beta-TGF under acidic conditions. Biochem Biophys Res Commun. 1985; 133(3): 1026–34. [DOI] [PubMed] [Google Scholar]
- 15.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996; 10(9): 1077–83. [DOI] [PubMed] [Google Scholar]
- 16.Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol. 1990; 110(4): 1361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato Y, Rifkin DB. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: Activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J Cell Biol. 1989; 109(1): 309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-βeta and promotes tumor invasion and angiogenesis. Genes Dev. 2000; 14(2): 163–76. [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N,. Thrombospondin-1 is a major activator of TGF-βeta1 in vivo. Cell 1998; 93(7): 1159–70. [DOI] [PubMed] [Google Scholar]
- 20.Daniel C, Wiede J, Krutzsch HC, Ribeiro SM, Roberts DD, Murphy-Ullrich JE, Hugo C,. Thrombospondin-1 is a major activator of TGF-βeta in fibrotic renal disease in the rat in vivo. Kidney Int. 2004; 65(2): 459–68. [DOI] [PubMed] [Google Scholar]
- 21.Keski-Oja J, Koli K, von Melchner H. TGF-βeta activation by traction? Trends Cell Biol. 2004; 14(12): 657–9. [DOI] [PubMed] [Google Scholar]
- 22.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Mtthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96(3): 319–28. [DOI] [PubMed] [Google Scholar]
- 23.Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol. 2007; 176(6): 787–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-βeta receptor system. Cell 1991; 67(4): 785–95. [DOI] [PubMed] [Google Scholar]
- 25.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992; 71(6): 1003–14. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y, Massague J. Mechanisms of TGF-βeta signaling from cell membrane to the nucleus. Cell 2003; 113(6): 685–700. [DOI] [PubMed] [Google Scholar]
- 27.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005; 19(23): 2783–810. [DOI] [PubMed] [Google Scholar]
- 28.Chaudhury A, Howe PH. The tale of transforming growth factor-beta (TGFbeta) signaling: A soigne enigma. IUBMB Life 2009; 61(10): 929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-βeta superfamily. Nature 1997; 389(6651): 622–6. [DOI] [PubMed] [Google Scholar]
- 30.Blahna MT, Hata A. Smad-mediated regulation of micro-RNA biosynthesis. FEBS Lett. 2012; 586(14): 1906–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis-Dusenbery BN, Hata A. Smad-mediated miRNA processing: A critical role for a conserved RNA sequence. RNA Biol. 2011; 8(1): 71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Lee CJ, Zhang L, Sans MD, Simeone DM. Regulation of transforming growth factor beta-induced responses by protein kinase A in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008; 295(1): G170–G8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, Li G, Wu JJ, Wang L, Uhler M, Simeone DM. Protein kinase A modulates transforming growth factor-beta signaling through a direct interaction with Smad4 protein. J Biol Chem. 2013; 288(12): 8737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Duan CJ, Binkley C, Li G, Uhler MD, Logsdon CD, Simeone DM. A transforming growth factor beta-induced Smad3/Smad4 complex directly activates protein kinase A. Mol Cell Biol. 2004; 24(5): 2169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang YE. Non-Smad pathways in TGF-βeta signaling. Cell Res. 2009; 19(1): 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-βeta type II receptor and regulates TGF-βeta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007; 67(8): 3752–8. [DOI] [PubMed] [Google Scholar]
- 37.Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene 2001; 20(44): 6322–30. [DOI] [PubMed] [Google Scholar]
- 38.Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-βeta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem. 2005; 95(5): 918–31. [DOI] [PubMed] [Google Scholar]
- 39.Yi JY, Shin I, Arteaga CL. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J Biol Chem. 2005; 280(11): 10870–6. [DOI] [PubMed] [Google Scholar]
- 40.Kattla JJ, Carew RM, Heljic M, Godson C, Brazil DP. Protein kinase B/Akt activity is involved in renal TGF-βeta1-driven epithelial-mesenchymal transition in vitro and in vivo. Am J Physiol Renal Physiol. 2008; 295(1): F215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM,. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial-mesenchymal transition. J Cell Biol. 2005; 168(1): 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conery AR, Cao Y, Thompson EA, Townsend CM Jr, Ko TC, Luo K,. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-βeta induced apoptosis. Nat Cell Biol. 2004; 6(4): 366–72. [DOI] [PubMed] [Google Scholar]
- 43.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL,. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005; 307(5715): 1603–9. [DOI] [PubMed] [Google Scholar]
- 44.Wilkes MC, Murphy SJ, Garamszegi N, Leof EB. Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol Cell Biol. 2003; 23(23): 8878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, Luga V, Przulj N, Robinson M, Suzuki H. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005; 307(5715): 1621–5. [DOI] [PubMed] [Google Scholar]
- 46.Landstrom M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol. 2010; 42(5): 585–9. [DOI] [PubMed] [Google Scholar]
- 47.Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 2001; 20(44): 6482–91. [DOI] [PubMed] [Google Scholar]
- 48.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M,. The type I TGF-βeta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008; 10(10): 1199–207. [DOI] [PubMed] [Google Scholar]
- 49.Choi ME, Ding Y, Kim SI. TGF-βeta signaling via TAK1 pathway: Role in kidney fibrosis. Semin Nephrol. 2012; 32(3): 244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. 1989; 108(2): 661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oppenheim JJ, Feldmann M, Durum SK. Cytokine reference: A compendium of cytokines and other mediators of host defense. San Diego (CA): Academic Press; 2001. [Google Scholar]
- 52.Liao CW, Fan CK, Kao TC, Ji DD, Su KE, Lin YH, Cho WL. Brain injury-associated biomarkers of TGF-βeta1, S100B, GFAP, NF-L, tTG, AbetaPP, and tau were concomitantly enhanced and the UPS was impaired during acute brain injury caused by Toxocara canis in mice. BMC Infect Dis. 2008; 8: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Logan A, Frautschy SA, Gonzalez AM, Sporn MB, Baird A. Enhanced expression of transforming growth factor beta 1 in the rat brain after a localized cerebral injury. Brain Res. 1992; 587(2): 216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992; 117(2): 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh K, Williams C, Gluckman PD. Hypoxia-ischemia induces transforming growth factor beta 1 mRNA in the infant rat brain. Brain Res Mol Brain Res. 1992; 13(1–2): 93–101. [DOI] [PubMed] [Google Scholar]
- 56.McTigue DM, Popovich PG, Morgan TE, Stokes BT. Localization of transforming growth factor-beta1 and receptor mRNA after experimental spinal cord injury. Exp Neurol. 2000; 163(1): 220–30. [DOI] [PubMed] [Google Scholar]
- 57.Buss A, Pech K, Kakulas BA, Martin D, Schoenen J, Noth J, Brook GA. TGF-βeta1 and TGF-βeta2 expression after traumatic human spinal cord injury. Spinal Cord 2008; 46(5): 364–71. [DOI] [PubMed] [Google Scholar]
- 58.Kiefer R, Lindholm D, Kreutzberg GW. Interleukin-6 and transforming growth factor-beta 1 mRNAs are induced in rat facial nucleus following motoneuron axotomy. Eur J Neurosci. 1993; 5(7): 775–81. [DOI] [PubMed] [Google Scholar]
- 59.Yadav R, Samuni Y, Abramson A, Zeltser R, Casap N, Kabiraj TK, MLB, Samuni U. Pro-oxidative synergic bactericidal effect of NO: Kinetics and inhibition by nitroxides. Free Radic Biol Med. 2014; 67: 248–54. [DOI] [PubMed] [Google Scholar]
- 60.La Fleur M, Underwood JL, Rappolee DA, Werb Z. Basement membrane and repair of injury to peripheral nerve: Defining a potential role for macrophages, matrix metalloproteinases, and tissue inhibitor of metalloproteinases-1. J Exp Med. 1996; 184(6): 2311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li M, Zhang P, Guo W, Li H, Gu X, Yao D. Protein expression profiling during Wallerian degeneration after rat sciatic nerve injury. Muscle Nerve 2014; 50(1): 73–8. [DOI] [PubMed] [Google Scholar]
- 62.Li M, Zhang P, Li H, Zhu Y, Cui S, Yao D. TGF-βeta1 is critical for Wallerian degeneration after rat sciatic nerve injury. Neuroscience 2015; 284: 759–67. [DOI] [PubMed] [Google Scholar]
- 63.Kritis A, Kapoukranidou D, Michailidou B, Hatzisotiriou A, Albani M. Sciatic nerve crush evokes a biphasic TGF-βeta and decorin modulation in the rat spinal cord. Hippokratia 2010; 14(1): 37–41. [PMC free article] [PubMed] [Google Scholar]
- 64.Chamak B, Morandi V, Mallat M. Brain macrophages stimulate neurite growth and regeneration by secreting thrombospondin. J Neurosci Res. 1994; 38(2): 221–33. [DOI] [PubMed] [Google Scholar]
- 65.Ikeda H, Miyatake M, Koshikawa N, Ochiai K, Yamada K, Kiss A, Donlin MJ, Panneton WM, Churchill JD, Green M, Siddiqui AM, Leinweber AL, Crews NR, Ezerskiy LA, Rendell VR, Belcheva MM, Coscia CJ. Morphine modulation of thrombospondin levels in astrocytes and its implications for neurite outgrowth and synapse formation. J Biol Chem. 2010; 285(49): 38415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scott-Drew S, ffrench-Constant C. Expression and function of thrombospondin-1 in myelinating glial cells of the central nervous system. J Neurosci Res. 1997; 50(2): 202–14. [DOI] [PubMed] [Google Scholar]
- 67.Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest 2000; 117(4): 1162–72. [DOI] [PubMed] [Google Scholar]
- 68.Vidal PM, Lemmens E, Dooley D, Hendrix S. The role of “anti-inflammatory” cytokines in axon regeneration. Cytokine Growth Factor Rev. 2012; 24(1): 1–12. [DOI] [PubMed] [Google Scholar]
- 69.Makwana M, Jones LL, Cuthill D, Heuer H, Bohatschek M, Hristova M, Friedrichsen S, Ormsby I, Bueringer D, Koppius A, Bauer K, Doetschman T, Raivich G. Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. J Neurosci. 2007; 27(42): 11201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, Koshy AA, Buckwalter MS. Astrocytic TGF-βeta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J Immunol. 2014; 193(1): 139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo H, Zhang Y, Zhang Z, Jin Y. The protection of MSCs from apoptosis in nerve regeneration by TGFbeta1 through reducing inflammation and promoting VEGF-dependent angiogenesis. Biomaterials 2012; 33(17): 4277–87. [DOI] [PubMed] [Google Scholar]
- 72.Miller MW, Luo J. Effects of ethanol and basic fibroblast growth factor on the transforming growth factor beta1 regulated proliferation of cortical astrocytes and C6 astrocytoma cells. Alcohol Clin Exp Res. 2002; 26(5): 671–6. [PubMed] [Google Scholar]
- 73.Miller MW, Luo J. Effects of ethanol and transforming growth factor beta (TGF beta) on neuronal proliferation and nCAM expression. Alcohol Clin Exp Res. 2002; 26(8): 1281–5. [DOI] [PubMed] [Google Scholar]
- 74.Jiang Y, Zhang M, Koishi K, McLennan IS. TGF-βeta 2 attenuates the injury-induced death of mature motoneurons. J Neurosci Res. 2000; 62(6): 809–13. [DOI] [PubMed] [Google Scholar]
- 75.Sakamoto T, Kawazoe Y, Shen JS, Takeda Y, Arakawa Y, Ogawa J, Oyanagi K, Ohashi T, Watanabe K, Inoue K, Eto Y, Watabe K. Adenoviral gene transfer of GDNF, BDNF and TGF beta 2, but not CNTF, cardiotrophin-1 or IGF1, protects injured adult motoneurons after facial nerve avulsion. J Neurosci Res. 2003; 72(1): 54–64. [DOI] [PubMed] [Google Scholar]
- 76.Walshe TE, Leach LL, D'Amore PA. TGF-βeta signaling is required for maintenance of retinal ganglion cell differentiation and survival. Neuroscience 2011; 189: 123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Misumi S, Kim TS, Jung CG, Masuda T, Urakawa S, Isobe Y, Furuyama F, Nishino H, Hida H. Enhanced neurogenesis from neural progenitor cells with G1/S-phase cell cycle arrest is mediated by transforming growth factor beta1. Eur J Neurosci. 2008; 28(6): 1049–59. [DOI] [PubMed] [Google Scholar]
- 78.Vergeli M, Mazzanti B, Ballerini C, Gran B, Amaducci L, Massacesi L. Transforming growth factor-beta 1 inhibits the proliferation of rat astrocytes induced by serum and growth factors. J Neurosci Res. 1995; 40(1): 127–33. [DOI] [PubMed] [Google Scholar]
- 79.Morganti-Kossmann MC, Kossmann T, Brandes ME, Mergenhagen SE, Wahl SM. Autocrine and paracrine regulation of astrocyte function by transforming growth factor-beta. J Neuroimmunol. 1992; 39(1–2): 163–73. [DOI] [PubMed] [Google Scholar]
- 80.Einheber S, Hannocks MJ, Metz CN, Rifkin DB, Salzer JL. Transforming growth factor-beta 1 regulates axon/Schwann cell interactions. J Cell Biol. 1995; 129(2): 443–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guenard V, Gwynn LA, Wood PM. Transforming growth factor-beta blocks myelination but not ensheathment of axons by Schwann cells in vitro. J Neurosci. 1995; 15(1 Pt 1): 419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prehn JH, Backhauss C, Krieglstein J. Transforming growth factor-beta 1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemic injury in vivo. J Cereb Blood Flow Metab. 1993; 13(3): 521–5. [DOI] [PubMed] [Google Scholar]
- 83.Prehn JH, Miller RJ. Opposite effects of TGF-βeta 1 on rapidly- and slowly-triggered excitotoxic injury. Neuropharmacology 1996; 35(3): 249–56. [DOI] [PubMed] [Google Scholar]
- 84.Ho TW, Bristol LA, Coccia C, Li Y, Milbrandt J, Johnson E, Jin L, Bar-Peled O, Griffin JW, Rothstein JD,. TGFbeta trophic factors differentially modulate motor axon outgrowth and protection from excitotoxicity. Exp Neurol. 2000; 161(2): 664–75. [DOI] [PubMed] [Google Scholar]
- 85.Merrill JE, Zimmerman RP. Natural and induced cytotoxicity of oligodendrocytes by microglia is inhibitable by TGF beta. Glia 1991; 4(3): 327–31. [DOI] [PubMed] [Google Scholar]
- 86.Knoferle J, Ramljak S, Koch JC, Tonges L, Asif AR, Michel U, Wouters FS, Heermann S, Krieglstein K, Zerr I, Bahr M, Lingor P. TGF-βeta 1 enhances neurite outgrowth via regulation of proteasome function and EFABP. Neurobiol Dis. 2010; 38(3): 395–404. [DOI] [PubMed] [Google Scholar]
- 87.Au E, Richter MW, Vincent AJ, Tetzlaff W, Aebersold R, Sage EH, Roskams AJ. SPARC from olfactory ensheathing cells stimulates Schwann cells to promote neurite outgrowth and enhances spinal cord repair. J Neurosci. 2007; 27(27): 7208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hagl C, Schafer KH, Hellwig I, Barrenschee M, Harde J, Holtmann M, Porschek S, Egberts JH, Becker T, Wedel T, Bottner M. Expression and function of the transforming growth factor-b system in the human and rat enteric nervous system. Neurogastroenterol Motil. 2013; 25(7): 601–e464. [DOI] [PubMed] [Google Scholar]
- 89.Jaskova K, Pavlovicova M, Cagalinec M, Lacinova L, Jurkovicova D. TGFbeta1 downregulates neurite outgrowth, expression of Ca2+ transporters, and mitochondrial dynamics of in vitro cerebellar granule cells. Neuroreport 2014; 25(5): 340–6. [DOI] [PubMed] [Google Scholar]
- 90.Fok-Seang J, DiProspero NA, Meiners S, Muir E, Fawcett JW. Cytokine-induced changes in the ability of astrocytes to support migration of oligodendrocyte precursors and axon growth. Eur J Neurosci. 1998; 10(7): 2400–15. [DOI] [PubMed] [Google Scholar]
- 91.Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci. 2000; 20(7): 2427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-βeta after vascular damage. J Neurosci. 2010; 30(17): 5843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur J Neurosci. 1994; 6(3): 355–63. [DOI] [PubMed] [Google Scholar]
- 94.Logan A, Green J, Hunter A, Jackson R, Berry M. Inhibition of glial scarring in the injured rat brain by a recombinant human monoclonal antibody to transforming growth factor-beta2. Eur J Neurosci. 1999; 11(7): 2367–74. [DOI] [PubMed] [Google Scholar]
- 95.Moon LD, Fawcett JW. Reduction in CNS scar formation without concomitant increase in axon regeneration following treatment of adult rat brain with a combination of antibodies to TGFbeta1 and beta2. Eur J Neurosci. 2001; 14(10): 1667–77. [DOI] [PubMed] [Google Scholar]
- 96.Kohta M, Kohmura E, Yamashita T. Inhibition of TGF-βeta1 promotes functional recovery after spinal cord injury. Neurosci Res. 2009; 65(4): 393–401. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, Moges H, Bharucha Y, Symes A. Smad3 null mice display more rapid wound closure and reduced scar formation after a stab wound to the cerebral cortex. Exp Neurol. 2007; 203(1): 168–84. [DOI] [PubMed] [Google Scholar]
- 98.Kimura-Kuroda J, Teng X, Komuta Y, Yoshioka N, Sango K, Kawamura K, Raisman G, Kawano H. An in vitro model of the inhibition of axon growth in the lesion scar formed after central nervous system injury. Mol Cell Neurosci. 2010; 43(2): 177–87. [DOI] [PubMed] [Google Scholar]
- 99.Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011; 331(6019): 928–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lindholm D, Hengerer B, Zafra F, Thoenen H. Transforming growth factor-beta 1 stimulates expression of nerve growth factor in the rat CNS. Neuroreport 1990; 1(1): 9–12. [DOI] [PubMed] [Google Scholar]
- 101.Yu G, Fahnestock M. Differential expression of nerve growth factor transcripts in glia and neurons and their regulation by transforming growth factor-beta1. Brain Res Mol Brain Res. 2002; 105(1–2): 115–25. [DOI] [PubMed] [Google Scholar]
- 102.Chalazonitis A, Kalberg J, Twardzik DR, Morrison RS, Kessler JA. Transforming growth factor beta has neurotrophic actions on sensory neurons in vitro and is synergistic with nerve growth factor. Dev Biol. 1992; 152(1): 121–32. [DOI] [PubMed] [Google Scholar]
- 103.Schober A, Hertel R, Arumae U, Farkas L, Jaszai J, Krieglstein K, Saarma M, Unsicker K. Glial cell line-derived neurotrophic factor rescues target-deprived sympathetic spinal cord neurons but requires transforming growth factor-beta as cofactor in vivo. J Neurosci. 1999; 19(6): 2008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K. TGF-βeta and the regulation of neuron survival and death. J Physiol Paris 2002; 96(1–2): 25–30. [DOI] [PubMed] [Google Scholar]
- 105.Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016; 532(7598): 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sulaiman W, Dreesen TD. Effect of local application of transforming growth factor-beta at the nerve repair site following chronic axotomy and denervation on the expression of regeneration-associated genes. Laboratory investigation. J Neurosurg. 2014; 121(4): 859–74. [DOI] [PubMed] [Google Scholar]
- 107.Wang Y, Zhao X, Huojia M, Xu H, Zhuang Y. Transforming growth factor-beta3 promotes facial nerve injury repair in rabbits. Exp Ther Med. 2016; 11(3): 703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cozzolino M, Malindretos P. The role of vitamin D receptor activation in chronic kidney disease. Hippokratia 2010; 14(1): 7–9. [PMC free article] [PubMed] [Google Scholar]