

Abstract
Autologous cardiosphere-derived cells (CDCs) were the first therapeutic modality to demonstrate myocardial regeneration with a decrease in scar size and an increase in viable, functional tissue. Widespread applicability of autologous CDC therapy is limited by the need for patient-specific myocardial biopsy, cell processing, and quality control, resulting in delays to therapy and inherent logistical and economic constraints. Preclinical data had demonstrated equivalent efficiency of allogeneic to autologous CDCs. The ALLogeneic Heart STem Cells to Achieve Myocardial Regeneration (ALLSTAR) trial is a multicenter randomized, double-blind, placebo-controlled phase 1/2 safety and efficacy trial of intracoronary delivery of allogeneic CDCs (CAP-1002) in patients with myocardial infarction (MI) and ischemic left ventricular dysfunction. The phase 1 safety cohort enrolled 14 patients in an open-label, nonrandomized, dose-escalation safety trial. The phase 2 trial is a doubleblind, randomized, placebo-controlled trial that will compare intracoronary CDCs to placebo in a 2:1 allocation and will enroll up to 120 patients. The primary endpoint for both phases is safety at 1 month. For phase 2, the primary efficacy endpoint is relative change from baseline in infarct size at 12 months, as assessed by magnetic resonance imaging. The ALLSTAR trial employs a “seamless” WOVE 1 design that enables continuous enrollment from phase 1 to phase 2 and will evaluate the safety of intracoronary administration of allogeneic CDCs and its efficacy in decreasing infarct size in post-MI patients.
Keywords: Stem cells, ST elevation MI, Trial design, Myocardial infarction (MI), Regenerative medicine
Introduction
Cardiosphere-derived cells (CDCs) were the first therapeutic modality to demonstrate a reduction in scar tissue associated with an increase in the presence of viable, functional tissue in the randomized, placebo-controlled CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction (CADUCEUS) trial1,2. The intracoronary (IC) administration of autologous CDCs also improved regional function of infarcted myocardium with an excellent safety profile1,2. However, widespread applicability of autologous CDC therapy is challenged by the need for patient-specific tissue harvesting with myocardial biopsy, cell processing, and quality control resulting in delays to therapy and imposing significant logistic and economic constraints2. In addition, cell potency has also been reported to be affected by age and presence of comorbidities3–6.
The use of allogeneic stem cells, if proven safe and effective, has the potential to overcome the limitations of autologous cardiac stem cell therapy with the ability to provide a ready-to-use, “off-the-shelf” product for widespread clinical usage. Allogeneic stem cell therapy with mesenchymal stem cells (MSCs), or a more selected subset of mesenchymal precursor cells (MPCs), has been utilized in clinical studies for cardiac repair7–10. Although the safety profile of allogeneic MSCs or MPCs has been excellent, efficacy results have been mixed. Allogeneic MSCs delivered intravenously in acute myocardial infarction (AMI) patients resulted in modest functional improvements in an early phase, randomized, placebo-controlled dose-finding study7. In 60 patients with heart failure, intramyocardial (IM) delivery of increasing doses of allogeneic MPCs was well tolerated and resulted in a significant reduction in major adverse cardiac events (MACE) compared to medical control patients, but had limited impact on the parameters of cardiac function examined8. In the phase 1/2 POSEIDON trial, IM delivery of increasing doses of allogeneic MSCs was well tolerated with results similar to autologous MSCs9. Allogeneic MSCs delivered IM during left ventricular assist device (LVAD) implantation in patients with severe heart failure were well tolerated with a trend toward improvement in LVAD wearing parameters10.
In a “proof-of-concept” study of allogeneic CDCs in a rat AMI model, allogeneic CDC transplantation without immunosuppression was noted to be safe and resulted in cardiac regeneration and improvement in cardiac function11. Similarly, administration of allogeneic cardio spheres in a rat model resulted in increased viable myocardium, decreased scar, and improved cardiac function with attenuation of adverse remodeling12. The cardioprotective effects of allogeneic CDCs and cardiospheres were durable for at least 6 months, although allogeneic cells disappeared within 4 weeks11,12. IC allogeneic CDC infusion in an AMI study in Yucatan minipigs was associated with attenuation of remodeling, improvement in global and regional function, decrease in scar size, and an increase in viable myocardium compared with placebo 2 months posttreatment13. Therefore, we designed the ALLogeneic Heart STem Cells to Achieve Myocardial Regeneration (ALLSTAR) trial as a randomized, double-blind, placebo-controlled phase 1/2 safety and efficacy study of IC delivery of allogeneic CDCs (CAP-1002) in patients with post-MI ischemic left ventricular dysfunction.
Materials and Methods
CAP-1002
CAP-1002 CDCs were manufactured from donors unrelated to the recipients. All organs were procured through regional organ procurement organizations that are certified by the Centers for Medicare and Medicaid Services (CMS) and abide by CMS regulations for organ donation for research purposes. CAP-1002 consists of 25 million human allogeneic CDCs in 10 ml of a cryogenic cell preservation solution (CryoStor® CS10; Biolife Solutions, Bothell, WA, USA), which contains 10% dimethyl sulfoxide (DMSO), with heparin added. CDCs were manufactured as previously described with several modifications for purposes of regulatory compliance and commercial potential13,14. Briefly, donor hearts were utilized as a starting tissue source, tissue was cultured as explants, explant-derived cells comprised a master cell bank (MCB), cardiospheres (CSps) were generated from the MCB, and CDCs were expanded from CSps. CAP-1002 was formulated and filled into cell storage bags (PermaLifeTM; Origen Biomedical, Austin, TX, USA), subjected to controlled-rate freezing, and stored in liquid nitrogen prior to use. Vehicle consisted simply of CS10 and heparin and was similarly frozen.
CAP-1002 CDCs were identity and purity tested by flow cytometry to confirm the presence of CD105 as a surface marker and the absence of CD45 that would identify contaminating hematopoietic cells. CDCs have been thoroughly characterized with respect to their surface proteins, secreted molecules, gene profile, and in vitro and in vivo bioactivity in the published literature.
Study Objectives
The primary objective of the ALLSTAR trial was to determine the safety of IC infusion of CAP-1002 in patients with ischemic left ventricular dysfunction following myocardial infarction (MI) (phases 1 and 2). The secondary objective was to explore the efficacy of IC CAP-1002 in improving cardiac structure and clinical status in post-MI patients with ischemic left ventricular dysfunction following MI (phase 2).
Study Design and Population
The target population is patients ≥ 18 years of age with a history of MI within the past 12 months and resultant ischemic left ventricular dysfunction. The study design is summarized in Figure 1, and the inclusion and exclusion criteria for the study are summarized in Tables 1 and 2, respectively. The study protocol was approved by the institutional review board (IRB) or an independent ethics committee for each participating study site, and written informed consent and release of medical information were obtained from all patients before screening. All costs related to participation in the trial are covered by the sponsors of the trial detailed below. Data were collected at each study site using a Code of Federal Regulations 21 (CFR-21)-compliant electronic data capture system.
Figure 1.
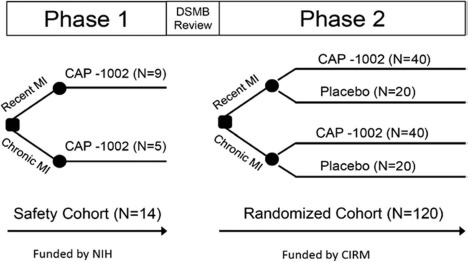
ALLSTAR study design. Abbreviations: MI, myocardial infarction; DSMB, data and safety monitoring board; NIH, National Institutes of Health; CIRM, California Institute of Regenerative Medicine.
Table 1.
Inclusion Criteria
|
TIMI, thrombolysis in myocardial infarction.
Myocardial infarction is defined by typical ischemic symptoms, serial ST-T changes (new ST elevation or new left bundle block), and elevated troponin or CK-MB >5 times the upper limit of normal with at least one of the following, based on standardly accepted definition of acute MI: development of pathological Q wave ECG changes, imaging evidence of new loss of viable myocardium, or new regional wall motion abnormalities.
Left ventricular ejection fraction can be determined by any one of the standard modalities (echocardiography, ventriculography, nuclear imaging, CT, and/or MRI).
Recent MI: assessment must be postreperfusion after index MI. Chronic MI: assessment must be at least 21 days postreperfusion after index MI.
In patients with infarcts in >1 myocardial wall, >50% of the total LV scar should be in the infarcted regions.
Table 2.
Exclusion Criteria
|
MRI, magnetic resonance imaging; VEGF, vascular endothelial growth factor; ICD, implantable cardioverter defibrillator; SGPT, serum glutamic pyruvic transaminase; MI, myocardial infarction; CT, computed tomography.
Including, but not limited to, giant cell myocarditis, cardiac or systemic sarcoidosis, Dressler's syndrome, chronic recurrent, or persistent pericarditis.
Including chronic systemic corticosteroids, biologic agents targeting the immune system, antitumor and antineoplastic drugs, anti-VEGF, or chemotherapeutic agents within 3 months prior to enrollment.
The phase 1 trial (the “safety cohort”), funded by the National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health and Capricor, enrolled 14 patients at three sites in an open-label, nonrandomized manner. The phase 2 trial, funded by the California Institute of Regenerative Medicine (CIRM) and Capricor, will randomize up to 120 patients from up to 35 clinical sites in North America in a double-blind fashion to receive IC infusion of either CAP-1002 or placebo in a 2:1 ratio favoring CAP-1002. Investigators, patients, central reviewers interpreting magnetic resonance imaging (MRI) and laboratory data, and all sponsor staff were blinded to treatment group assignment. Enrollment in phase 2 will be closed when either 120 patients are randomized or the interim analysis time point is met (6 months following infusion of the 80th evaluable patient), whichever occurs first. Two cohorts of patients will be enrolled in phase 2 and randomized separately: the primary randomized cohort consisting of patients who can be matched [have no antibodies against a donor's human leucocyte antigens, or donor-specific antibodies (DSAs)] to receive CAP-1002 from one or more donor MCBs, and the exploratory randomized cohort consisting of patients with DSAs to all available MCBs but who met all trial eligibility criteria. The primary population for the phase 2 trial is the primary randomized cohort. The purposes of the exploratory randomized cohort were to assess the prevalence of mismatched patients and to evaluate the potential effect of DSA, if any, on safety and efficacy. For both cohorts, randomization is performed within two strata: recent MI defined as index MI greater than 4 weeks but within 90 days prior to randomization, and chronic MI defined as index MI at least 90 days and no more than 1 year prior to randomization. Recent and chronic MI strata will be evaluated both separately and combined. In summary, the ALLSTAR phase 1/2 trial will include a total of up to 134 patients: approximately 83 will be treated with CAP-1002 in either the safety cohort (n = 14) or the primary randomized cohort (n = 69), approximately 34 will receive placebo in the primary randomized cohort, and up to approximately 17 additional patients will be treated in the exploratory randomized cohort. The size of the exploratory randomized cohort was estimated based on a 14% rate of mismatch in the safety cohort.
IC infusion of the investigational product is the infarct-related artery performed on day 0, within 4 weeks (28 days) of initial screening procedures and at least 4 weeks after the index MI, and was followed by 20–24 h of hospitalization with continuous cardiac (telemetry) monitoring. Patients underwent safety procedures including DSA and efficacy assessments at 2 weeks and at 1, 2, 3, 6, and 12 months postinfusion. In addition, patients will be contacted annually by telephone over the 5 years following infusion for ascertainment of data on MACE.
Investigational Product Administration
Ten of the 14 phase 1 patients and those randomized to receive CAP-1002 in phase 2 received an IC administration of 25 million cells suspended in 11.5 ml of cryopreservation solution (CryoStor® CS10; BioLife Solutions Inc.) containing 10% DMSO, heparin (1,800 U total), and nitroglycerin (450 μg total). The initial four phase 1 patients received a half dose (12.5 million cells) of CAP-1002. The placebo consisted of 11.5 ml of cryopreservation solution containing 10% DMSO, heparin (1,800 U total), and nitroglycerin (450 μg total). Normal saline containing heparin (1,200 U total) and nitroglycerin (600 μg total) were used as the intermediate wash solution between boluses of CAP-1002 and placebo.
Patients were required to have a patent infarct-related coronary artery with thrombolysis in myocardial infarction (TIMI) 3 flow prior to infusion. The investigational product was administered by IC infusion using the Abbott Trek® “over-the-wire” balloon angioplasty catheter (Abbott Vascular, Santa Clara, CA, USA) using a stop-flow technique. A balloon catheter with a diameter up to 0.5 mm larger than the stent and shorter in length (preferably 8–12 mm) was positioned in the stented segment and inflated to achieve occlusion of blood flow in the infarct-related artery; after the guidewire was removed, investigational product was injected through the wire lumen of the balloon catheter. The balloon was inflated for 2 min 15 s and deflated for 3 min in three cycles for a total procedure time of 12 min 45 s. One third of the 11.5 ml total volume of investigational product was infused during each balloon inflation cycle over a total of 1 min 45 s. Two milliliters of the wash solution was infused before and after each investigational product bolus, each over 15 s, to wash any remaining investigational product solution from the catheter into the artery (Fig. 2). Epicardial coronary flow and myocardial perfusion quantitative assessment were required at the beginning and at the completion of the infusion procedure, using the validated TIMI flow score. If ST elevation or angina symptoms occur and persist for 3 min with the balloon deflated, a diagnostic contrast injection is performed, and TIMI flow is assessed. If electrocardiography (ECG) abnormalities or symptoms occur due to no reflow (impairment of blood flow at the myocardial level or microvascular obstruction), then cell infusion is terminated.
Figure 2.
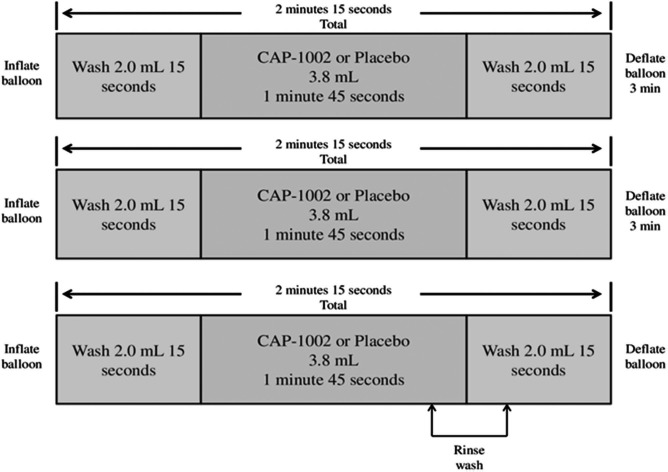
Schematics of intracoronary infusion of CAP-1002 or placebo.
Cardiac MRI Protocol
Patients underwent contrast-enhanced MRI at screening and at 6 and 12 months postinfusion. A cine imaging protocol was utilized for global ejection fraction and left ventricular volumes and mass determination. Delayed contrast-enhancement protocol was used for the assessment of infarct scar size. High-resolution delayed enhancement images were obtained using an inversion recovery-prepared gated fast gradient echo pulse sequence. The images were deidentified and sent for central analysis to an independent blinded imaging core laboratory at Johns Hopkins University. Following a protocol amendment, patients with MRI-compatible implantable cardioverter–defibrillator or pacemaker placement were eligible for enrollment.
Study Assessments and Endpoints
The primary safety endpoint is the occurrence of any of the following during the 1 month post IC infusion: acute myocarditis attributable to CAP-1002, ventricular tachycardia-related or ventricular fibrillation-related death, sudden unexpected death, or a MACE. The diagnosis of acute myocarditis will be made based on clinical presentation, with or without a clinically indicated endomyocardial biopsy. In order for acute myocarditis to be considered related to CAP-1002, humoral or cellular immune reaction specific to CAP-1002 must also be documented. Secondary safety endpoints will include later onset events and will be evaluated over the 12 months post-IC infusion, as detailed in Table 3.
Table 3.
ALLSTAR Study Endpoints
| Primary Safety Endpoint |
| Occurrence of acute myocarditis attributable to CAP-1002; ventricular tachycardia-related or ventricular fibrillation-related death∗; sudden unexpected death†; or major adverse cardiac events‡ during the 1 month post intracoronary infusion. |
| Secondary Safety Endpoints |
During the 12 months post intracoronary infusion:
These events are mutually exclusive and hierarchical; i.e., a MACE involving hospitalization would not also be counted as a cardiovascular-related hospitalization. Secondary safety endpoints do not include events counted as primary safety endpoints. |
| Primary Efficacy Endpoint |
|
Relative change from baseline in infarct size at 12 months post intracoronary infusion as assessed by MRI. |
| Secondary Efficacy Endpoints |
|
At 6 and 12 months post intracoronary infusion:
|
MRI, magnetic resonance imaging; TIMI, thrombolysis in myocardial infarction; ECG, electrocardiogram; HLA, human leucocyte antigen; BNP, brain natriuretic peptide.
Death occurring with ECG documentation of these arrhythmias during ambulatory ECG monitoring in an outpatient setting or during routine ECG monitoring while hospitalized.
Death occurring within 1 h of symptom onset or unwitnessed death in a person previously observed to be well within the preceding 24 h without an identified cause.
Composite of death, nonfatal recurrent myocardial infarction, hospitalization for heart failure, emergency room treatment for heart failure (NT-proBNP >450 pg/ml or BNP >100 pg/ml, with treatment including intravenous diuretic administration), left ventricular assist device placement, or heart transplant. Myocardial infarction is defined as the presence of troponin or CK-MB levels >5 times the upper reference limit during the 24 h following infusion. These elevations must be accompanied by symptoms of ischemia >20 min in duration and EKG changes indicative of new ischemia (new ST-T changes or new left bundle branch block), development of pathological Q waves on the EKG, imaging evidence of new loss of viable myocardium, or new regional wall motion abnormality.
Hospitalization with heart failure listed as the primary discharge diagnosis.
For phase 2, the primary efficacy endpoint is the relative percentage change in infarct size at 12 months postinfusion as assessed by contrast-enhanced MRI. The secondary efficacy endpoints will be assessed 6 and 12 months postinfusion and will include change from baseline in other MRI parameters, the 6-min walk test, New York Heart Association (NYHA) class, N-terminal pro b-type natriuretic peptide (NT-proBNP), and quality of life (QoL) parameters (Table 3). In addition, centralized analysis of immunologic studies at baseline and follow-up will be performed.
Statistical Methods
Adaptive Interim Analysis
For phase 2 of the trial, an adaptive interim analysis will be conducted when month 6 MRI data are available for approximately 80 patients in the primary randomized cohort. The objective of the analysis is to select the study population to be used for the primary analysis of the primary efficacy endpoint: either all patients combined or only patients in the recent MI stratum. The selection will be based on conditional power and will preserve the overall type 1 error rate for the primary efficacy analysis through use of an intersection hypothesis and the inverse normal method for combining p values15.
Analysis Populations
The primary analysis population for safety for both phase 1 and phase 2 will be all treated patients. For phase 2, a modified intent-to-treat analysis population will be used for the primary efficacy analysis and will include either all treated patients or only treated patients in the recent MI stratum, depending on the outcome of the adaptive interim analysis.
Analytical Methods
For the open-label phase 1 trial, the exact 90% confidence interval (CI) for the proportion of patients experiencing the primary safety endpoint will be calculated, and incidence of secondary safety endpoints will be reported. For the double-blind, placebo-controlled phase 2 trial, 90% CIs for the proportion experiencing the primary safety endpoint will be calculated for both CAP-1002 and placebo treatment groups, as well as a 90% CI for the difference in proportions between treatment groups. A test of equivalence with a 0.2 margin will be done using Wald's test. For secondary safety endpoints, event rates (number of events divided by total patient-years of follow-up) will be compared between treatment groups using negative binomial or Poisson regression (if data are too scarce for analysis, descriptive statistics—event rates and/or incidence—will be reported).
For the phase 2 efficacy analysis, percent change from baseline in infarct size will be analyzed using mixed-effects linear regression, with subject as a random effect and month (6 and 12) and treatment group as fixed effects. The baseline value will be included as a covariate. Percent change from baseline will be evaluated for substantial departure from normality, and an appropriate transformation (e.g., log, rank) will be applied if necessary. An unstructured covariance will be the default covariance structure, but other structures (e.g., compound symmetry) may be used to evaluate sensitivity to the choice of structure. Secondary efficacy endpoints will be analyzed similarly, with the exception of change from baseline in NYHA class, which will be treated as a categorical outcome and will be analyzed using a chi-square test of association or a two-sided Fisher's exact test.
Study Hypotheses, Power, and Sample Size
For the phase 2 primary efficacy analysis in which an adaptive interim analysis will be performed, an intersection hypothesis with the inverse normal method for combining p values will be used at the final analysis so that strong control of type 1 error will be maintained. Two one-sided null hypotheses will have to be rejected at the 0.025 significance level in order for the trial to be declared a success: (1) the treatment group difference in mean percent change from baseline in infarct size = 0 in the study population selected at the interim analysis (either all patients combined or only patients in the recent MI stratum), and (2) the global null hypothesis that treatment group difference in mean percent change from baseline in infarct size = 0 in both all patients combined and in only the recent MI stratum. A simulation of the adaptive interim analysis of the month 6 percent change from baseline in infarct size and the subsequent final analysis of month 12 data indicated that phase 2 will have at least 80% power to be declared successful with a total sample size of 80 and at least 90% power to be declared successful with a total sample size of 120. Simulation parameters were based on the results from the CADUCEUS trial1,2 and ALLSTAR phase 1 (Table 4). It is expected that the actual phase 2 sample size for the primary efficacy analysis will be between 80 and 120.
Table 4.
Mean and SD Relative Change From Baseline in Infarct Size Used for Simulation-Based Sample Size Estimation
| Month 6 |
Month 12 |
|||
|---|---|---|---|---|
| Mean | SD | Mean | SD | |
| CAP-1002 recent | 6.20 | 10.00 | 14.70 | 10.00 |
| CAP-1002 chronic | 3.10 | 10.00 | 7.35 | 10.00 |
| Placebo | 0.00 | 10.00 | 0.00 | 10.00 |
Handling of Missing Data
Since the primary safety endpoint is based on events that occur immediately after infusion and safety analyses are done only in patients who were infused, handling of missing data is not applicable for the primary safety endpoint. Since the secondary safety analysis involves calculation of event rates, which accounts for differential length of follow-up, handling of missing data is also not applicable for the secondary safety endpoints.
Under the assumption of ignorable missingness (i.e., missingness can at least be controlled by another variable on which data have been collected), the mixed-effects regression approach (maximum likelihood) that will be used for the phase 2 efficacy analyses is able to handle patients with either month 6 or month 12 changes from baseline missing, but not both. Therefore, for analyses based on the mITT population, missing data will be addressed using maximum likelihood. For analyses based on the ITT population, in which some subjects may have both month 6 and month 12 changes from baseline missing, multiple imputation will be used.
Study Oversight
Two separate DSMBs will oversee ALLSTAR: a DSMB appointed by NHLBI for phase 1, and an independent DSMB appointed by the sponsor, separate from the NHLBI DSMB, for phase 2. For phase 1, interim safety data reviews were conducted at times coincident with regularly scheduled meetings of the NHLBI DSMB in accordance with reaching accrual and follow-up milestones. The NHLBI DSMB review after all 14 phase 1 patients had completed the month 1 study visit triggered the initiation of phase 2. The phase 2 DSMB safety data reviews occur at prescheduled intervals not to exceed 6 months apart.
For both phases 1 and 2, interim DSMB safety reviews are unblinded and include review of primary and secondary safety endpoints, all adverse events, anti-human leukocyte antigen (HLA) antibody immunoassay, ELISpot immuno assay, cardiac biomarkers, and other safety laboratory evaluations. Data are presented to the DSMB in the form of summary tables, individual patient listings, and narratives. In addition, the DSMB chair is notified each time a serious adverse event (SAE) occurs. Preset thresholds for event rates or event rate differences between treatment groups are used by the DSMB as study stopping guidelines. Per written charter, these guidelines serve as a trigger for consultation with the DSMB for additional review and are not formal “stopping rules” that would mandate automatic closure of study enrollment. Stopping enrollment is at the discretion of the sponsor after consideration of DSMB recommendations.
An independent clinical event adjudication committee, independent DSMB, and a steering committee chaired by an independent cardiologist with deep clinical and domain expertise in biologics and clinical trial design and conduct oversee trial progress. The IRB at each participating center approved the protocol. All the patients provided written informed consent.
Discussion
The ALLSTAR trial is the first randomized, doubleblind, placebo-controlled trial to determine the safety and efficacy of IC delivery of allogeneic CDCs (CAP-1002) in patients with ischemic left ventricular dysfunction following MI. The CADUCEUS study provided the foundation for the ALLSTAR trial with major difference between CADUCEUS and ALLSTAR being the source of the CDCs (autologous vs. allogeneic CAP-1002)1,2. The principal goal of ALLSTAR is to establish the safety of CAP-1002. In parallel, signals of potential efficacy will be evaluated by a primary efficacy endpoint (infarct size by MI, similar to the CADUCEUS study) and other structural and functional measures.
The scientific rigor of the study is ensured with the inclusion of a placebo group and blinding of both participants and treatment providers during the course of the study. The absence of a placebo arm or, conversely, mere inclusion of an active control arm meeting the same inclusion criteria but receiving just standard medical care, can result in overestimation of treatment effect of the intervention. This phenomenon was recently demonstrated by Jeong et al. in a meta-analysis of 17 studies comparing IC bone marrow stem cell treatment with a control group (9 studies with a placebo control arm and 8 studies with a standard therapy control arm)16. Trials that performed IC placebo administration in the control group did not show significant changes in left ventricle ejection fraction (LVEF) at 6 months (0.92%; 95% CI, −0.61 to 2.44), whereas trials with controls receiving standard therapy without placebo administration showed significant LVEF changes (4.45%; 95% CI, 2.48 to 6.43).
Previous research suggests that markers of the biological process of left ventricular hypertrophy and enlargement (remodeling) and the factors that contribute to this process may be viewed as surrogates for progression of post-MI myocardial injury as functional measures and neurohormones17. The primary efficacy endpoint in ALLSTAR phase 2 is change in infarct size (relative to LV size) under a common imaging protocol to ensure high measurement reliability. Infarct size represents the extent of myocardial injury sustained after an MI and is likely the single most important defining factor of the remodeling process, leading to change in function, volume, and geometry of the LV. It has been shown that infarct size as measured by MRI under a common imaging protocol can be a valid surrogate endpoint for clinical outcomes18. In addition, the ability of infarct size to predict clinical endpoints has been demonstrated in epidemiological studies19,20. Anand et al. also suggest that a composite of surrogate endpoints may be more appropriate than any single one, due to the complexity of the heart failure syndrome17. This trial will explore the utility of a composite endpoint that will include the domains of clinical function, cardiac function, symptoms/QoL, and safety.
Conclusions
In summary, results from the ALLSTAR phase 1 trial demonstrated that IC infusion of allogeneic CDCs (CAP-1002) was safe and feasible. These results led to enrollment of the randomized, double-blind, placebo-controlled ALLSTAR phase 2 trial to further assess safety and also evaluate efficacy of allogeneic CDCs in reducing scar size in ischemic cardiomyopathy.
References
- 1.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marbán L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marbán E. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012; 379 (9819): 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malliaras K, Makkar RR, Smith RR, Cheng K, Wu E, Bonow RO, Marbán L, Mendizabal A, Cingolani E, Johnston PV, Gerstenblith G, Schuleri KH, Lardo AC, Marbán E. Intracoronary cardiosphere-derived cells after myocardial infarction: Evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dyS-function). J Am Coll Cardiol. 2014; 63(2): 110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhuo Y, Li SH, Chen MS, Wu J, Kinkaid HY, Fazel S, Weisel RD, Li RK. Aging impairs the angiogenic response to ischemic injury and the activity of implanted cells: Combined consequences for cell therapy in older recipients. J Thorac Cardiovasc Surg. 2010; 139(5): 1286–94, 1294.e1–2. [DOI] [PubMed] [Google Scholar]
- 4.Kinkaid HY, Huang XP, Li RK, Weisel RD. What's new in cardiac cell therapy? Allogeneic bone marrow stromal cells as “universal donor cells”. J Card Surg. 2010; 25(3): 359–66. [DOI] [PubMed] [Google Scholar]
- 5.Perin EC, Silva GV, Henry TD, Cabreira-Hansen MG, Moore WH, Coulter SA, Herlihy JP, Fernandes MR, Cheong BY, Flamm SD, Traverse JH, Zheng Y, Smith D, Shaw S, Westbrook L, Olson R, Patel D, Gahremanpour A, Canales J, Vaughn WK, Willerson JT,. A randomized study of transendocardial injection of autologous bone marrow mononuclear cells and cell function analysis in ischemic heart failure (FOCUS-HF). Am Heart J. 2011; 161(6): 1078–87.e3. [DOI] [PubMed] [Google Scholar]
- 6.Perin EC, Willerson JT, Pepine CJ, Henry TD, Ellis SG, Zhao DX, Silva GV, Lai D, Thomas JD, Kronenberg MW, Martin AD, Anderson RD, Traverse JH, Penn MS, Anwaruddin S, Hatzopoulos AK, Gee AP, Taylor DA, Cogle CR, Smith D, Westbrook L, Chen J, Handberg E, Olson RE, Geither C, Bowman S, Francescon J, Baraniuk S, Piller LB, Simpson LM, Loghin C, Aguilar D, Richman S, Zierold C, Bettencourt J, Sayre SL, Vojvodic RW, Skarlatos SI, Gordon DJ, Ebert RF, Kwak M, Moyé LA, Simari RD,; Cardiovascular Cell Therapy Research Network (CCTRN). Effect of transendocardial delivery of autologous bone marrow mononuclear cells on functional capacity, left ventricular function, and perfusion in chronic heart failure: The FOCUS-CCTRN trial. JAMA 2012; 307(16): 1717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP, Gerstenblith G, DeMaria AN, Denktas AE, Gammon RS, Hermiller JB Jr, Reisman MA, Schaer GL, Sherman W,. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009; 54(24): 2277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perin EC, Borow KM, Silva GV, DeMaria AN, Marroquin OC, Huang P, Traverse JH, Krum H, Skerrett D, Zheng Y, Willerson JT, Itescu S, Henry TD. A phase II dose-escalation study of allogeneic mesenchymal precursor cells in patients with ischemic or non-ischemic heart failure. Circ Res. 2015; 117(6): 576–84. [DOI] [PubMed] [Google Scholar]
- 9.Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY, Tracy M, Ghersin E, Johnston PV, Brinker JA, Breton E, Davis-Sproul J, Schulman IH, Byrnes J, Mendizabal AM, Lowery MH, Rouy D, Altman P, Wong Po Foo C, Ruiz P, Amador A, Da Silva J, McNiece IK, Heldman AW, George R, Lardo A,. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: The POSEIDON randomized trial. JAMA 2012; 308(22): 2369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ascheim DD, Gelijns AC, Goldstein D, Moye LA, Smedira N, Lee S, Klodell CT, Szady A, Parides MK, Jeffries NO, Skerrett D, Taylor DA, Rame JE, Milano C, Rogers JG, Lynch J, Dewey T, Eichhorn E, Sun B, Feldman D, Simari R, O'Gara PT, Taddei-Peters WC, Miller MA, Naka Y, Bagiella E, Rose EA, Woo YJ,. Mesenchymal precursor cells as adjunctive therapy in recipients of contemporary left ventricular assist devices. Circulation 2014; 129(22): 2287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malliaras K, Li TS, Luthringer D, Terrovitis J, Cheng K, Chakravarty T, Galang G, Zhang Y, Schoenhoff F, Van Eyk J, Marbán L, Marbán E. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 2012; 125(1): 100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tseliou E, Pollan S, Malliaras K, Terrovitis J, Sun B, Galang G, Marbán L, Luthringer D, Marbán E. Allogeneic cardiospheres safely boost cardiac function and attenuate adverse remodeling after myocardial infarction in immunologically mismatched rat strains. J Am Coll Cardiol. 2013; 61(10): 1108–19. [DOI] [PubMed] [Google Scholar]
- 13.Malliaras K, Smith RR, Kanazawa H, Yee K, Seinfeld J, Tseliou E, Dawkins JF, Kreke M, Cheng K, Luthringer D, Ho CS, Blusztajn A, Valle I, Chowdhury S, Makkar RR, Dharmakumar R, Li D, Marbán L, Marbán E. Validation of contrast-enhanced magnetic resonance imaging to monitor regenerative efficacy after cell therapy in a porcine model of convalescent myocardial infarction. Circulation 2013; 128(25): 2764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 2007; 115(7): 896–908. [DOI] [PubMed] [Google Scholar]
- 15.Wassmer G. On sample size determination in multi-armed confirmatory adaptive designs. J Biopharm Stat. 2011; 21(4): 802–17. [DOI] [PubMed] [Google Scholar]
- 16.Jeong H, Yim HW, Cho Y, Park HJ, Jeong S, Kim HB, Hong W, Kim H. The effect of rigorous study design in the research of autologous bone marrow-derived mononuclear cell transfer in patients with acute myocardial infarction. Stem Cell Res Ther. 2013; 4(4): 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anand IS, Florea VG, Fisher L. Surrogate end points in heart failure. J Am Coll Cardiol. 2002; 39(9): 1414–21. [DOI] [PubMed] [Google Scholar]
- 18.Desch S, Eitel I, de Waha S, Fuernau G, Lurz P, Guttberlet M, Schuler G, Thiele H,. Cardiac magnetic resonance imaging parameters as surrogate endpoints in clinical trials of acute myocardial infarction. Trials 2011; 12: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larose E, Rodés-Cabau J, Pibarot P, Rinfret S, Proulx G, Nguyen CM, Déry JP, Gleeton O, Roy L, Noël B, Barbeau G, Rouleau J, Boudreault JR, Amyot M, De Larochellière R, Bertrand OF,. Predicting late myocardial recovery and outcomes in the early hours of ST-segment elevation myocardial infarction traditional measures compared with microvascular obstruction, salvaged myocardium, and necrosis characteristics by cardiovascular magnetic resonance. J Am Coll Cardiol. 2010; 55(22): 2459–69. [DOI] [PubMed] [Google Scholar]
- 20.Wu E, Ortiz JT, Tejedor P, Lee DC, Bucciarelli-Ducci C, Kansal P, Carr JC, Holly TA, Lloyd-Jones D, Klocke FJ, Bonow RO,. Infarct size by contrast enhanced cardiac magnetic resonance is a stronger predictor of outcomes than left ventricular ejection fraction or end-systolic volume index: Prospective cohort study. Heart 2008; 94(6): 730–6. [DOI] [PubMed] [Google Scholar]