

Abstract
Background
The entomopathogenic mushroom Cordyceps militaris is an important medicinal and food resource owing to its various medicinal components and pharmacological effects. However, the high frequency of strain degeneration during subculture seriously restricts the large-scale production of C. militaris, and the mechanism underlying strain degeneration remains unclear. In this study, we artificially cultured C. militaris for six generations and compared changes during fruiting body growth. The transcriptome of six generations of C. militaris strains were sequenced with the Illumine Hiseq4000.
Results
The subcultured C. militaris strains degenerated beginning at the third generation, with incomplete fruiting body growth beginning at the fourth generation. Over 9,015 unigenes and 731 new genes were identified. In addition, 35,323 alternative splicing (AS) events were detected in all samples, and more AS events occurred in the second, fourth and sixth generations. Compared with the first generation, the third generation (degenerated strain) included 2,498 differentially expressed genes (DEGs) including 1,729 up-regulated and 769 down-regulated genes. This number was higher than the number of DEGs in the second (1,892 DEGs), fourth (2,006 DEGs), fifth (2,273 DEGs) and sixth (2,188 DEGs) generations. Validation of RNA-seq by qRT-PCR showed that the expression patterns of 51 DEGs were in accordance with the transcriptome data.
Conclusion
Our results suggest that the mechanism of C. militaris strain degeneration is associated with gene involved in toxin biosynthesis, energy metabolism, and DNA methylation and chromosome remodeling.
Introduction
Cordyceps militaris, a tonic herb used in traditional Chinese medicine, is an economically valuable, edible entomopathogenic mushroom that is used clinically as a substitute of Cordyceps sinensis owing to its similar chemical components and pharmacological activities [1]. C. militaris contains several pharmacologically active ingredients, such as cordycepin, adenosine, polysaccharide, ergosterol, that exhibit significant biological activities known to protect and improve lung and kidney function [2], and to immunomodulatory [1,3], antioxidant [4], anti-tumor [5,6,7],anti-inflammatory [8], and hypotensive effects [9].
Because naturally occurring C. militaris is rare, cultivated C. militaris fruiting bodies are currently commercially available as medicinal materials and health food products in China, Korea, and Southeast Asia [10]. This artificial cultivation has improved the imbalance between the supply and the demand of wild C. militaris [11]. However, the high frequency of strain degeneration during subculture and preservationof C. militaris has limited its large-scale production and industrial process [12]. Strain degeneration results in the significant fluctuations in sporulation quantity, deficiencies in fruiting body formation, reductions in secondary metabolite yields, and other irreversible hereditary phenotypes, leadings to considerable economic losses [13].The strain degeneration of C. militaris involves complex regulatory processes and involves many factors [14]. Studies have shown that mating type change, DNA methylation, genetic mutations, harmful substance accumulation, and virus infections are all precipitating factors of C. militaris degeneration [15].
The genome sequence of C. militaris and the transcriptomes of the mycelium and fruiting body have been analyzed by next-generation sequencing (NGS) [16,17]. However, the molecular mechanism of strain degeneration remains unclear. In this study, we cultured C. militaris for six generations and compared changes in fruiting body growth during subculture. We also analyzed the transcriptomes of different generations of C. militaris using Illumine Hiseq4000 technology. Subsequently, the differentially expressed genes (DEGs) as determined by RNA-Seq were analyzed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases and were validated by quantitative real-time PCR (qRT-PCR). Our results provide a better understanding the transcriptomic changes that occur in C. militaris during subculture and the underlying molecular mechanism of strain degeneration.
Materials and methods
Strain and sample preparation
Wild-type of C. militaris, designated YCC, was collected from Maoshan hilly area (at altitude 267.5 m) without specific permissions, Jiangsu province, China and isolated by tissue separation in the asexual phase by the Laboratory of Hi-Tech Processing of Sericultural Resources, Sericultural Research Institute, Chinese Academy of Agricultural Sciences, China.
For the collection of different generations following asexual development, the strain was inoculated onto potato dextrose agar (PDA) plates (20% potato, 2% dextrose, 1.5% agar and 1% peptone, w/v) and incubated at 23°C on a shaker at 150 rpm for 7 d in the dark. Mycelia were inoculated into rice medium, cultivated in the dark at 23°C for 10 d, and then kept at 23°C under a 17:7 h dark/light cycle for fruiting body production [18]. The mycelia and fruiting body of this strain were subcultured for the acquisition of second, third, fourth, fifth, and sixth generations, designated YCCZ2, YCCZ3, YCCZ4, YCCZ5, and YCCZ6, respectively. Mycelium from all six generations were collected for DNA and RNA extraction.
RNA extraction and sequencing
Total RNA from each sample was extracted using RNeasy Plant Mini kit (Qiagen Co. LtD., Beijing, China) and the residual genomic DNA was digested using RNase-free DNase I according to the manufacturer’s protocol. Poly (A) mRNA was enriched using oligo (dT) beads and fragmented using fragmentation buffer. Finally, 100 ng purified and enriched mRNA was used to construct a cDNA library for each sample using NEBNext® UltraTM RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA). cDNA fragments of 200 bps (± 25 bps) were selected and purified by gel-electrophoresis and used as templates for amplification with PCR for end- repair and poly (A) addition. Purified library products were evaluated using the Agilent 2200 Tape Station and Qubit 2.0 software (Life Technologies, Carlsbad, CA, USA). RNA-Seq was performed at Gene Denovo Co., Ltd. (Shenzhen, China) using the HiSeq TM 4000 (Illumina, San Diego, CA, USA).
Transcriptome assembly and function annotation
Prior to sequencing, raw data were filtered to produce high-quality clean data. Clean reads were then assembled de novo into longer contigs based on overlapping regions using the Trinity platform (http://trinityrnaseq.sourceforge.net/) [19]. All subsequent analyses were performed using clean data. For annotation, clean data were mapped to the C. militaris genomic data (BioProject acession no. PRJNA225510) from the NCBI transcriptome reference database.
All splice junction sites of the same gene (≥ 5 reads) were determined and compared with the reference splice junction sites (≤ 1 base) using TopHat align to distinguish new alternative splicing (AS) events. DEGs were filtered and identified according to the edgeR criteria (http://www.Bioconductor.org/packages/release/bioc/html/edgeR.html). Functional annotation (GO terms) were downloaded from Uniprot database (http://www.uniprot.org/uniprot). For GO enrichment analysis, GO annotations for each DEG were retrieved by mapping to GO terms in the database at http://www.geneontology.org. For KEGG pathway analysis, KEGG orthology terms for DEGs were retrieved from the KEGG pathway database (http://www.genome.jp/kegg/). Cluster analysis of gene expression patterns was performed using cluster software [20] and Java Treeview software [21]. All expressed genes in the current transcriptomes were annotated based on BLAST homology searches and searched against the Swiss-Prot and TrEMBL databases by double-direction BLAST. To further explore the function of DEGs in different samples, KEGG enrichment analyses were performed using hypergeometric tests with the Blastall software.
Validation of RNA-Seq by quantitative real-time PCR
qRT-PCR was carried out to validate the quality of RNA-Seq data. Total RNA was extracted as described above. Each RNA sample was treated with RNase-free DNase I (TaKaRa, Shiga, Japan) following the manufacturer's protocol to remove any residual genomic DNA. DNase I-treated RNA (2 mg) was subjected to reverse transcriptase using oligo (dT) primer and PrimeScriptTM Reverse Transcriptase (TaKaRa, Shiga, Japan) according to the manufacturer's protocol. Total RNA (2 μg) was used to synthesize cDNA with PrimeScript™ RT reagent Kit (Perfect Real Time, TaKaRa, Japan). The C. militaris housekeeping gene, glyceraldehyde-phosphate dehydrogenase (Cmgapdh), was used as an internal control for normalization. Primers for the qRT-PCR of 51 DEGs were designed with Premier 6.0 software and are shown in S1 Table. Three biological replicates were performed per sample.
Data analysis
Sequence similarity was analyzed using Blast software. Multiple sequence alignment analysis was carried out with Clustalx W software. All experiments were repeated at least three times, and the results are expressed as mean ± S.D. Statistical evaluation was done using ANOVA (SPSS 18.0), with p < 0.05 considered significant.
Results
Changes in the morphological characteristics of the C. militaris fruiting body in different generations
The growth of C. militaris fruiting body is an important morphological characteristics in evaluating strain degeneration during subculture. C. militaris was subcultured in rice medium for six generations, and changes in morphological characteristics of fruiting body were observed after 40 days of cultivation (Fig 1). In the third generation, the fruiting body became stronger and shorter, while the size of fruiting body was significantly reduced in the fourth generation. The strain appeared to be completely degenerated in the fifth generation, and only mycelia grew in the sixth generation. As shown in Fig 1, the subculture of C. militaris induced strain degeneration beginning in the third generation.
Fig 1. Changes in the C. militaris fruiting body at different generations.

Transcriptome sequencing and assembly
As shown in Table 1, the average number of clean reads per sample was 53,118,184. After quality filtering, over 6×109 bps of clean data was obtained across the different samples (S2 Table), and more than 98% HQ clean reads were identified (S1 Fig). About 90% reads were mapped to the C. militaris genome sequence (S3 Table). In total, 9,651 genes were detected in all transcriptomes, including 9,015 known genes and 731 novel genes. The number of new gene in each generation from YCCZ1 to YCCZ6 was 668, 705, 699, 704, 702 and 699, respectively (Table 1). The six samples showed similar matching results, with ~90% of reads matching the predicted genes in the genome and ~70% being unique matches (S2 Fig). The above results revealed that our sample data were reasonable with good correlations among biological replicates and that sequencing data could be used for following analyses.
Table 1. Summary of sequencing data utilized in this study.
| Sample | Clean Reads | HQ Clean reads | Clean Base | Total mapped | Total mapped Ratio (%) | Unique Match(%) | Known Gene | New Gene | All Gene |
|---|---|---|---|---|---|---|---|---|---|
| YCCZ1 | 41009930 | 40623584 | 6062919977 | 11603568 | 70.23 | 70.12 | 8586 (88.96%) | 668 | 9254 |
| YCCZ2 | 59291792 | 58701420 | 8755948868 | 11607702 | 70.30 | 70.17 | 8735 (90.51%) | 705 | 9440 |
| YCCZ3 | 47879596 | 47274196 | 7052765933 | 14351858 | 69.17 | 69.04 | 8714 (90.29%) | 699 | 9413 |
| YCCZ4 | 56986574 | 56336928 | 8406700715 | 16735019 | 69.68 | 69.54 | 8745 (90.61%) | 704 | 9449 |
| YCCZ5 | 52911126 | 52298678 | 7800954755 | 15641003 | 69.90 | 69.77 | 8700 (90.15%) | 702 | 9402 |
| YCCZ6 | 60630086 | 60093188 | 8971315366 | 17870079 | 69.05 | 68.89 | 8746 (90.62%) | 699 | 9445 |
Note: Clean Reads = Raw Reads—Adapter—Low Quality–High N rate
HQ Clean Reads = Raw Reads—Clean Reads
Known Gene Percentage = number of detected / total genes in reference genome.
Analyses of differentially expressed genes (DEGs)
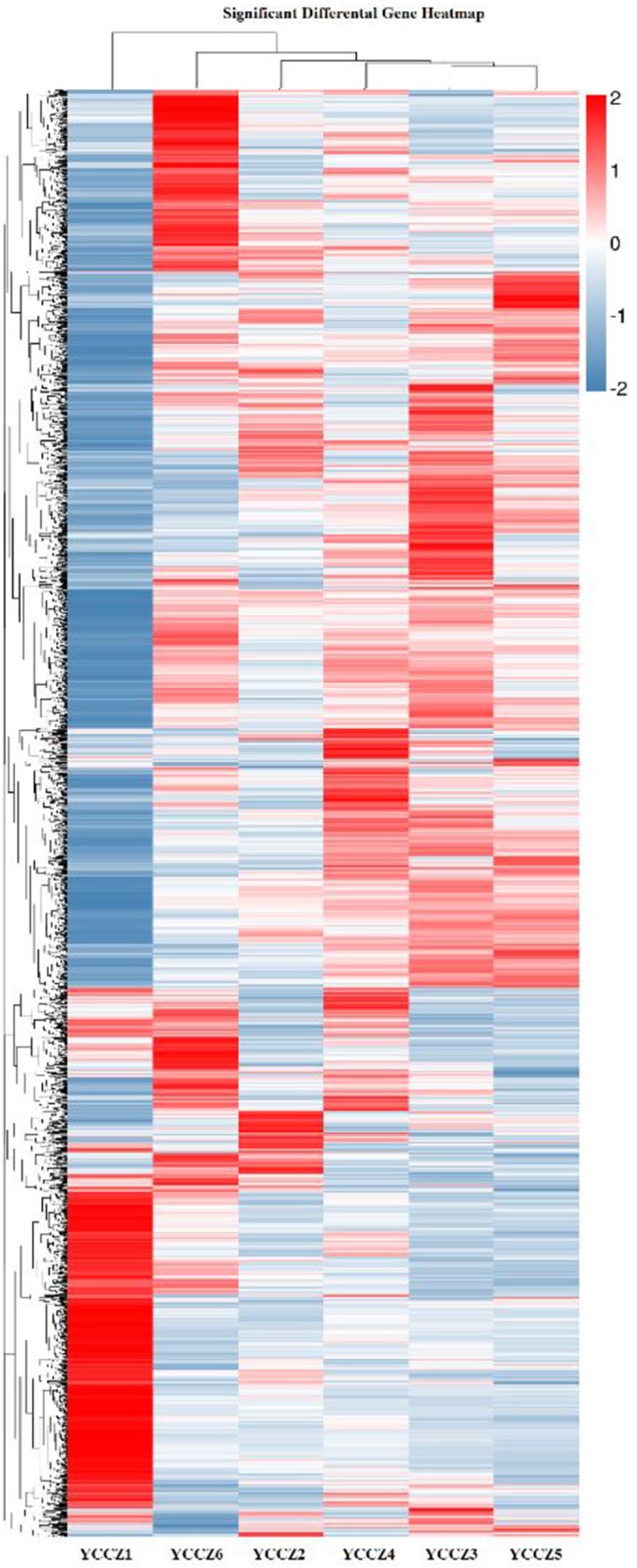
Compared with YCCZ1, 1,892 DEGs were identified in YCCZ2 (1,253 up-regulated and 639 down-regulated), 2,498 in YCCZ3 (1,729 up-regulated and 769 down-regulated), 2,006 in YCCZ4 (1,511 up-regulated and 495 down-regulated), 2,273 in YCCZ5 (1,437 up-regulated and 836 down-regulated) and 2,188 in YCCZ6 (1,602 up-regulated and 586 down-regulated) (Fig 2A). The most DEGs were thus observed in the third generation, when the C. militaris fruiting body began to experience abnormal growth (Fig 1), indicating that these DEGs may play key roles in strain degeneration. In addition, a scatter plot was protracted based on DEGs analyses. Compared with YCCZ1, difference in expression levels of the DEGs in YCCZ3 and YCCZ5 was significantly higher than those in YCCZ1 and YCCZ2 (Fig 2B–Fig 2F). Furthermore, a correlation heat map (CHM) was constructed and sample cluster analysis was performed based on DEGs in different samples. The heat map of significant DEGs shows the relationship of samples YCCZ1 to YCCZ6 (Fig 3). Taken together, all results suggest that these specific DEGs may be involved in the strain degeneration progress.
Fig 2. Differentially expressed genes (DEGs) analyses.

Note: (A) DEGs statistics of different samples. (B) Scatter plot of YCCZ1-YCCZ2. (C) Scatter plot of YCCZ1-YCCZ3. (D) Scatter plot of YCCZ1-YCCZ4. (E) Scatter plot of YCCZ1-YCCZ5. (F) Scatter plot of YCCZ1-YCCZ6.
Fig 3. DEG-based correlation heat map (CHM) and sample cluster analysis.
GO and KEGG pathway analyses of AS genes
GO analysis was performed to summarize and explore the functional categories of the genes that underwent AS in different samples. GO terms were annotated for a total of 5,014 AS genes in YCCZ1, 6,230 in YCCZ2, 5,638 in YCCZ3, 6,170 in YCCZ4, 5,951 in YCCZ5 and 6,320 in YCCZ6 (S3 Fig). The Intergenic (10,863) and IR (5,222) AS events predominated, accounting for 30.8% and 14.8%, respectively, of all AS events. The number of AS genes in YCCZ2, YCCZ4 and YCCZ6 was higher than those in other samples. The proportions of enriched GO terms among AS genes were globally similar between YCCZ1 (wild-type) and YCCZ5 (degenerated) based on biological process, molecular function, and cellular component ontolopies (Fig 4). For the biological processes category, genes with AS events were predominantly enriched in GO terms that were relevant to cellular process and metabolic process. For the molecular function category, most AS genes were annotated with the term binding, followed by the term catalytic activity.
Fig 4. GO classification map of YCCZ1 vs. YCCZ5.

Moreover, 6,944 AS genes were annotated with KEGG pathways information (Table 2, S4 Fig). Enriched pathway analysis showed that these genes were significantly enriched in 17 pathways mainly relating to antigen processing and presentation, cell adhesion molecules, phagosome, endocytosis, and natural killer cell-mediated cytotoxicity.
Table 2. Enrichment KEGG pathway analysis.
| list | Pathway | DEGs genes with pathway annotion (17) | All genes with pathway annotation (6944) | P value | Q value | Pathway ID |
|---|---|---|---|---|---|---|
| 1 | Antigen processing and presentation | 6(35.29%) | 81(1.17%) | 0.000000 | 0.000001 | ko04612 |
| 2 | Cell adhesion molecules (CAMs) | 6(35.29%) | 159(2.29%) | 0.000001 | 0.000016 | ko04514 |
| 3 | Phagosome | 6(35.29%) | 168(2.42%) | 0.000002 | 0.000016 | ko04145 |
| 4 | Endocytosis | 6(35.29%) | 231(3.33) | 0.000012 | 0.000074 | ko04144 |
| 5 | Natural killer cell mediated cytotoxicity | 5(29.41%) | 137(1.97%) | 0.000014 | 0.000074 | ko04650 |
| 6 | Complement and coagulation cascades | 3(17.65%) | 77(1.11%) | 0.000797 | 0.003454 | ko04610 |
| 7 | Regulation of actin cytoskeleton | 3(17.65%) | 216(3.11) | 0.014621 | 0.054308 | ko04810 |
| 8 | Pancreatic secretion | 2(11.76%) | 104(1.5%) | 0.026100 | 0.084825 | ko04972 |
| 9 | Oxidatie phosphorylation | 2(11.76%) | 141(2.03%) | 0.045622 | 0.131797 | ko00190 |
| 10 | Vitamin digestion and absorption | 1(5.88%) | 24(0.35%) | 0.057223 | 0.148780 | ko04977 |
| 11 | Nicotinate and nicotinnamide metabolism | 1(5.88%) | 32(0.46%) | 0.075602 | 0.178696 | ko00760 |
| 12 | Fat digestion and absorption | 1(5.88%) | 39(0.56%) | 0.091407 | 0.194538 | ko04975 |
Validation of DEGs by qRT-PCR
The expression of 51 DEGs was validated using qPCR, as shown in Fig 5. According to qRT-PCR, the DEG expression levels were consistent with those of obtained using RNA-Seq. Among these genes, 32 genes were up-regulated and 13 were down-regulated. Functional relative analysis shown that 15 of these genes are involved in toxin biosynthesis and detoxification (Fig 5C), 14 genes are related to carbohydrate and energy metabolism, nine genes are involved in DNA methylation and chromosome remodeling (Fig 5A), eight genes are involved in biosynthesis of secondary metabolites (Fig 5B), and six genes are involved in fruiting body formation, sexual development and light-induced brown film formation (Fig 5D). These results suggest that C. militaris strain degeneration involves genes related to toxin biosynthesis, energy metabolism, DNA methylation, and chromosome remodeling.
Fig 5. Validation of DEGs expression by qRT-PCR.

(A) Genes related to DNA methylation, chromosome remodeling, mitosis, and meiosis. (B) Genes related to fruiting body formation, sexual development, light-induced brown film formation, and secondary metabolites biosynthesis. (C) Genes related to toxin biosynthesis and detoxification, and transmembrane transport. (D) Genes related to cell growth and development, cell apoptosis and autophagy, and macro molecular metabolism.
Discussion
C. militaris,a model fungus belonging to the Ascomycetes,is an entomogenous fungus that forms a fruiting body on several types of media, such as silkworm pupa, solid rice medium, germinated soybean medium, and soybean broth. It also forms mycelia during submerged fermentation, producing several bioactive substances [19, 22].
Irreversible C. militaris strain degeneration is a common phenomenon that occurs with high frequency during the process of subculture and preservation. Such degeneration seriously affects large-scale industrialized production of C. militaris, leading to considerable economic losses [23]. There are many factors that may lead to fungal degeneration, including gene mutations, changes in mating type, DNA methylation, and fungal and viral infections [24]. Our previous work indicated that mutations in the 18S rRNA gene and mating-type (MAT) region, as well as down-regulated of CmMAT gene expression levels, may play important roles in C. militaris degeneration [25]. Quantitative real-time PCR analysis detected no CmMAT1-2-1 gene expression in the degenerated strain. Expression levels of the CmMAT1-1-1 and CmMAT1-1-2 genes were significantly down-regulated to only 7.5% and 4.4%, respectively, that of the wild-type strain.
The C. militaris genome (147 × coverage) is predicted to have a total genome size of 32.2 Mb and over 9,684 genes [17]. In this study, 9,746 genes were detected, including 731 novel genes, which fulfilled the criterion for it being referred to as the C. militaris genome, suggesting that the genome size and gene number in C. militaris is similar to those of Metarhizium anisopliae (10,582 genes) [26] and Metarhizium acridum (9,849 genes) [27]. In addition, over 5,000 novel AS events of seven AS types were detected in each of the subcultured strains, indicating significantly higher rates of AS than in that observed in the wild-type strain. AS is an important mechanism for regulating gene expression and generating proteome diversity [28, 29]. Previous studies have shown that AS plays a decisive role in the generation of receptor diversity and regulation of growth and development [30]. Many genetic diseases have been closely linked to higher than normal rates of AS [31]. Therefore, we speculated the higher AS rates in filamentous fungi might be involved in the progression of strain degeneration.
Gene mutation [32], methylation modification [33], and DNA recombination can alter a strain’s genotype and can induce strain degeneration. In this study, we characterized and manually annotated the functions of four DEGs involved in methylation modification and in DNA recombination and repair. Methyltransferase type 11 (CCM_00251) and 6-O-methylguanine-DNA methyltransferase (CCM_02107) were significantly up-regulated in later generations and are crucial for genome stability, preventing mismatch during DNA replication and transcription [34, 35]. Two DNA repair genes, DNA excision repair protein (CCM_00249) and DNA double-strand break repair/VJ recombinationXRCC4 (CCM_04567), were also up-regulated in later generations.
Degenerated strains exhibit traits that are generally heritable, including significantly reduced sporulation, inability to form normal fruiting bodies, and significantly reduced secondary metabolites contents. Fruiting body and light-induced brown film formation areimportant characteristics that are altered in C. militaris strain degeneration [14]. Here, we analyzed the DEGs involved in fruiting body and light-induced brown film formation and found that the expression of α-1,3-mannosyltransferaseAlg3 (CCM_04231) was up-regulated while that of sexual development activator VeA (CCM_04531)was down-regulated. The expression of light-induced brown film formation-related genes phosducin I (CCM_05237) and phosducin II (CCM_09459) were significantly up-regulated beginning at the second generation.
Cordycepin, adenosine, polysaccharides, and ergosterol are the major bioactive constituents important in industrial cultivation [16, 36] speculated genes encoding ribonucleotide reductase, adenosine kinase, AMP deaminase, pyruvate kinase, 5’-nucleotidase among others were involved in the putative cordycepin metabolism pathway. This study showed that the expression of the genes encoding s-adenosylhomocysteine nucleosidase (CCM_02372) and oxidordeuctase (CCM_02761) was significantly down-regulated, and that of genes encoding adenine nucleotide alpha hydrolases like (CCM_07740) andadenine nucleotide alpha hydrolases like (CCM_08422) were significantly up-regulated during the degeneration progress. Ergosterol is one of the main components of the fungal cell membrane. The genes encoding dimethylallyl tryptophan synthase (CCM_04410), cytochrome P450 51 (CYP51, CCM_05535), cytochrome P450 61(CYP61, CCM_02962) were down-regulated, while that encoding lanosterol synthase (CCM_09526) was up-regulated during strain degeneration. Dimethylallyl tryptophan synthase is a key regulator of ergosterol synthesis [37, 38]. CYP51 and CYP61 are conserved members of P450 family and play important role in the biosynthesis of ergosterol [17]. qRT-PCR resultssuggested that the biosynthesis of ergosterol reduced with strain degenerations. C. militaris polysaccharidesare not only important immune regulators, but also exhibit antioxidant, antibacterial, antitumor, and antivirus activities in addition to other biological activities [1]. Several genes are related to the metabolism of C. militaris polysaccharide, including1,4-α-glucan branching enzyme(CCM_03369), α-N-acetylglucosaminidase (CCM_04090), mannan endo-1,6-α-mannosidase (CCM_05709), sorbitol dehydrogenase (CCM_06561), mannosidase MsdS (CCM_08733), and sorbitol utilization protein (CCM_09007)[39].
Toxic substances such as mycotoxins, active oxygen and other nonnutritional xenobiotic compounds, naturally exist in the environment and can be harmful to organisms [40]. Some scholars have speculated that accumulation of toxin and active oxygen molecules may cause fungal strain degeneration [14]. Here, the expression of several genes involved in detoxification was up-regulated during strain degeneration, including that encoding streptothricin-acetyltransferase (CCM_00152), which degrades streptothricin [41]; gamma-glutamyltranspeptidase (CCM_02065), which reduces glutathione [41]; MFS multidrug transporter (CCM_02974), which confers resistance to antibiotics [42]; glutathione S-transferase (CCM_03461), which processes pesticides, heavy metals, fluoride and eliminating reactive oxygen species (ROS) [43,44]; 30 kDa heat shock protein (CCM_06821), which is involved in stress response, signal transduction, and xenobiotic compounds metabolism [45]; and alcohol dehydrogenase (CCM_09345), which ameliorates the harmful effects of high concentration of ethanol [46]. However, expression of the gene encoding heavy metal tolerance protein (CCM_06182), which binds heavy metals ions, was down-regulated during strain degeneration [47]. In addition, levels of non-nutritional xenobiotic compounds increased during strain degeneration induced by subculture.
In contrast, expression of genes related to the metabolism of carbohydrate, lipid, protein, amino acid, nucleic acid, and nucleotide was significantly up-regulated during strain degeneration. These included the genes encoding mitochondrial hypoxia responsive protein (CCM_05400), mitochondrial co-chaperone GrpE (CCM_00863), glycoside hydrolase, family 2 (CCM_09220), trypsin-like serine protease (CCM_03489), metalloprotease 1 (CCM_07917), acetate transporter (CCM_07990), SGT1 and CS protein (CCM_09524), formyltetrahydrofolate deformylase (CCM_09394), nucleoside triphosphate hydrolases like (CCM_01902), and uracil phosphoribosyltransferase (CCM_07057).
The subcultured C. militaris strains degenerated beginning at the third generation, with incomplete fruiting body growth beginning at the fourth generation. Transcriptome analysis on the progress of C. militaris subculture degeneration showed that over 9,015 unigenes and 731 new genes were identified, and 35,323 alternative splicing (AS) events were detected. Compared with the first generation, the third generation (degenerated strain) included 2,498 differentially expressed genes (DEGs) including 1,729 up-regulated and 769 down-regulated genes. Validation of RNA-seq by qRT-PCR showed that the expression patterns of 51 DEGs were in accordance with the transcriptome data. In summary, our results indicated the mechanism of C. militaris strain degeneration is associated with gene involved in toxin biosynthesis, energy metabolism, DNA methylation, and chromosome remodeling.
Supporting information
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
Abbreviations
- C. militaris
Cordyceps militaris
- AS
alternative splicing
- DEGs
differentially expressed genes
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- qRT-PCR
quantitative real-time PCR
- YCC
the strain of C. militaris used in this study
- PDA
potato dextrose agar
- CmGAPDH
C. militaris housekeeping gene, glyceraldehyde-phosphate dehydrogenase
- CHM
correlation heat map
- MAT
mating-type
- CYP51
cytochrome P450 51
- CYP61
cytochrome P450 61
- ROS
reactive oxygen species
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was funded by the Special Fund for Agro-scientific Research in the Public Interest of China (No. 201403064).
References
- 1.Wu F, Yan H, Ma X, Jia J, Zhang G, Guo X, et al. Comparison of the structural characterization and biological activity of acidic polysaccharides from Cordyceps militaris cultured with different media. World J Microbiol Biotech. 2012; 28(5): 2029–2038. doi: 10.1007/s11274-012-1005-6 [DOI] [PubMed] [Google Scholar]
- 2.Leung PH, Zhao S, Ho KP, Wu JY. Chemical properties and antioxidant activity of exopolysaccharides from mycelial culture of Cordyceps sinensis fungus Cs-HK1. Food Chem. 2009;114(4): 1251–1256. doi: 10.1016/j.food -chem.2008.10.081 [Google Scholar]
- 3.Kuo YC, Tsai WJ, Wang JY, Chang SC, Lin CY, Shiao MS. Regulation of bronchoalveolar lavage fluids cell function by the immunomodulatory agents from Cordyceps sinensis. Life Sci. 2001;68(9): 1067–1082. doi: 10.1016/ S0024-3205(00)01011-0 [DOI] [PubMed] [Google Scholar]
- 4.Yu R, Yang W, Song L, Yan C, Zhang Z, Zhao Y. Structural characterization and antioxidant activity of a polysaccharide from the fruiting bodies of cultured Cordyceps militaris. Carbohydr Polym. 2007;70(4): 430–436. doi: 10.1016/ j.carbpol.2007.05.005 [Google Scholar]
- 5.Lin YW, Chiang BH. Anti-tumor activity of the fermentation broth of Cordyceps militaris cultured in the medium of Radix astragali. Process Biochem. 2008; 43(3): 244–250. doi: 10.1016/j.procbio.2007.11.020 [Google Scholar]
- 6.Park S, Yoo HS, Jin CY, Hong SH, Lee YW, Kim BW, et al. Induction of apoptosis and inhibition of telomerase activity in human lung carcinoma cells by the water extract of Cordyceps militaris. Food Chem Toxicol. 2009; 47: 1667–1675. doi: 10.1016/j.fct.2009.04.014 [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Wang LM, Jin C, Chen HJ, Li SH, Li SY, et al. Cordyceps militaris polysaccharide triggers apoptosis and G0/G1 cells arrest in cancer cells. J Asia Pac Entomol.2015; 18: 433–438. doi: 10.1016/j.aspen.2015.04.015 [Google Scholar]
- 8.Won SY, Park EH. Anti-inflammatory and related pharmacological activities of cultured mycelia and fruiting bodies of Cordyceps militaris. J Ethnopharmacol. 2005;96(3): 555–561. doi: 10.1016/j.jep.2004.10.009 [DOI] [PubMed] [Google Scholar]
- 9.Chiou WF, Chang PC, Chou CJ, Chen CF. Protein constituent contributes to the hypotensive and vasorelaxant activities of Cordyceps sinensis. Life Sci. 2000; 66(14): 1369–1376. doi: 10.1016/S0024-3205(00)00445-8 [DOI] [PubMed] [Google Scholar]
- 10.Li SP, Yang FQ, Tsim KWK. Quality control of Cordyceps sinensis, a valued traditional Chinese medicine. J Pharm Biomed Anal. 2006;41(5): 1571–1584. doi: 10.1016/j.jpba.2006.01.046 [DOI] [PubMed] [Google Scholar]
- 11.Jia G, Wang J, Zhao F, Bioengineering DO. Studies on artificial culture conditions of Cordyceps militaris high producing strain. Gansu Agri Sci Tech. 2016; 11: 32–35.CateGory Index: S567.35 [Google Scholar]
- 12.Lu Y, Xia Y, Luo F, Dong C, Wang C. Functional convergence and divergence of mating-type genes fulfilling in Cordyceps militaris. Fungal Genet Biol. 2016; 88:35–43. doi: 10.1016/j.fgb.2016.01.013 [DOI] [PubMed] [Google Scholar]
- 13.Chen A, Wang Y, Shao Y, Huang B.A novel technique for rejuvenation of degenerated caterpillar medicinal mushroom, Cordyceps militaris (Ascomycetes), a valued Traditional Chinese medicine. Int J Med Mushrooms.2017; 19(1): 87–91. doi: 10.1615/IntJMedMushrooms.v19.i1.90 [DOI] [PubMed] [Google Scholar]
- 14.Sun SJ, Deng CH, Zhang LY, Hu KH. Molecular analysis and biochemical characteristics of degenerated strains of Cordyceps militaris. Arch Microbiol. 2017; 199:939–944. doi: 10.1007/s00203-017-1359-0 [DOI] [PubMed] [Google Scholar]
- 15.Zhang GZ, Liang Y Improvement of fruiting body production in Cordyceps militaris by molecular assessment. Arch Microbiol. 2013; 195(8): 579–585. doi: 10.1007/s00203-013-0904-8 [DOI] [PubMed] [Google Scholar]
- 16.Yin Y, Yu G, Chen Y, Jiang S, Wang M, Jin Y, et al. Genome-wide transcriptome and proteome analysis on different developmental stages of Cordyceps militaris. Plos One. 2012; 7(12): e51853 doi: 10.1371/journal.pone.0051853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng P, Xia Y, Xiao G, Xiong C, Hu X, Zhang S, et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011;12(11): R116 doi: 10.1186/gb-2011-12-11-r116 PMCID: PMC3334602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu YH, Gui ZZ. Liquid fermentation with ventilation of Cordyceps militaris and its components analysis. J Food Sci Biotech. 2009;28(5): 699–704.CateGory Index: S567.35 [Google Scholar]
- 19.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B, Williams A. Lorian schaeffer and barbara wold mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5: 621–628. doi: 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 20.Hoon MJ, De L, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9): 1453–1454. doi: 10.1093/bioinformatics/bth078 [DOI] [PubMed] [Google Scholar]
- 21.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20(17): 3246–3248. doi: 10.1093/bioinformatics/bth349 [DOI] [PubMed] [Google Scholar]
- 22.Park JP, Kim SW, Hwang HJ, Yun JW. Optimization of submerged culture conditions for the mycelial growth and exo-biopolymer production by Cordyceps militaris. Lett Appl Microbiol. 2010;33(1): 76–81. doi: 10.1046/j. 1472-765X.2001.00950.x [DOI] [PubMed] [Google Scholar]
- 23.Han YF. Research progress about some problems on Cordyceps militaris. Microbiology. 2009; 32(9): 1423–1428. CateGory Index: S567.35 [Google Scholar]
- 24.Hu XC, Lv AJ, University JN. Discussion of the degenerate strains of Cordyceps militaris. Edi Fungi China. 2015;34: 1.–. CateGory Index: S567.35 [Google Scholar]
- 25.Yin J, Xin X, Weng Y, Li S, Jia J, Gui Z. Genotypic analysis of degenerative Cordyceps militaris cultured in the pupa of Bombyx mori. Entomol Res. Forthcoming 2017. [Google Scholar]
- 26.Ouyang L, Xu X, Freed S, Gao Y, Yu J, Wang S, et al. Cecropins from plutella xylostello and their interaction with petarhizium anisopliae. Plos One. 2015;10(11): e0142451 doi: 10.1371/journal.pone.0142451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He M, Hu J, Xia Y. Large scale expressed sequence tag (EST) analysis of Metarhizium acridum infecting Locusta migratoria reveals multiple strategies for fungal adaptation to the host cuticle. Curr Genet. 2012;58: 265–279. doi: 10.1007/s00294-012-0382-6 [DOI] [PubMed] [Google Scholar]
- 28.Maniatis T. Mechanisms of alternative pre-mRNA splicing. Science. 1991; 251(4989): 33–34. doi: 10.1126/science.1824726 [DOI] [PubMed] [Google Scholar]
- 29.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12): 1413–1415. doi: 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- 30.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010; 327(5968): 996–1000. doi: 10.1126/science.1184208 PMCID: PMC2913848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stamma S, Ben-Ari S, Rafalska I, Tanga YS, Zhang ZY, Toiber D, et al. Function of alternative splicing. Gene. 2013;514: 1–20. doi: 10.1016/j.gene.2012.07.083 [DOI] [PubMed] [Google Scholar]
- 32.Li M, Wu X, Li C, Feng B, Li Q. Molecular analysis of degeneration of artificial flanted Cordyceps militaris. Mycosystema. 2003;22(2): 277–282. CateGory Index: S567.3 [Google Scholar]
- 33.Wang YL, Wang ZX, Wang SB, Huang B. Genome-wide analysis of DNA methylation in the sexual stage of the insect pathogenic fungus Cordyceps militaris. Fungal Biol. 2015;119(12): 1246–1254. doi: 10.1016/j.funbio.2015.08.017 [DOI] [PubMed] [Google Scholar]
- 34.Holmberg N, Harker M, Gibbard CL, Wallace AD, Clayton JC, Rawlins S, et al. Sterol C-24 methyltransferase type 1 controls the flux of carbon into sterol biosynthesis in tobacco seed. Plant Physiol. 2002;130(1): 303–311. doi: 10.1104/pp.004226 PMCID: PMC166563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T. Promoter hypermethylation of the DNA repair gene O6-methylguanine-DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low-grade diffuse astrocytomas. Brain Pathol. 2010; 13(2): 176–184. doi: 10.1111/j.1750-3639.2003.tb00017.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lan D, Thu N, Lan P, Nha P, Tung B. Cordyceps militaris (L.) link: chemical bioactive compounds and pharmacological activities. J Pharm Nutrit Sci. 2016; 6: 153–159. doi: 10.6000/1927-5951.2016.06.04.4 [Google Scholar]
- 37.Črešnar B, Petrič Š. Cytochrome P450 enzymes in the fungal kingdom. Biochim Biophys Acta. 2011;1814(1): 29–35. doi: 10.1016/j.bbapap.2010.06.020 [DOI] [PubMed] [Google Scholar]
- 38.Liu M, Panaccione DG, Schardl CL. Phylogenetic analyses reveal monophyletic origin of the ergot alkaloid gene dmaW in fungi. Evol Bioinform. 2009;5(5):15–30. PMCID: PMC2747131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiong CM, Xia Y, Zheng P, Shi S, Wang C. TDMYC developmental stage-specific gene expression profiling for a medicinal fungus. Mycology. 2010;1(1): 25–66. doi: 10.1080/21501201003674581 [Google Scholar]
- 40.Ames BN, Profet M, Gold S. Nature's chemicals and synthetic chemicals: comparative toxicology. PNAS. 2013;87: 7782–7786. doi: 10.1073/pnas. 87.19.7782 PMCID: PMC54832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watkins JB, Klaunig JE, Smith HM, Cornwell P, Sanders RA. Streptozotocin-induced diabetes increases gamma-glutamyltranspeptidase activity but not expression in rat liver. J Food Chem Toxicol. 2015;12(4): 219–225. doi: 10.1002/(SICI)1099-0461(1998)12:4<219::AID-JBT4>3.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- 42.Zou P, Mchaourab H. Structure and dynamics of the MFS multidrug transporter EmrD Biophys J. 2012;102(3):60a doi: 10.1016/j.bpj.2011.11. 3594 [Google Scholar]
- 43.Aravindan V, Muthukumaravel S, Gunasekaran K. Interaction affinity of Delta and Epsilon class glutathione-s-transferases (GSTs) to bind with DDT for detoxification and conferring resistance in Anopheles gambiae, a malaria vector. J Vector Dis. 2014; 51(1):8–15. [PubMed] [Google Scholar]
- 44.Erban T, Stara J. Methodology for glutathione S-transferase purification and localization in two-dimensional gel electrophoresis performed on the pollen beetle, Meligethes aeneus (Coleoptera: Nitidulidae). J Asia Pac Entomol. 2014; 17(3): 369–373. doi: 10.1016/j.aspen.2014.02.010 [Google Scholar]
- 45.Behzadi E, Behzadi P, Sirmatel F. Identification of 30-kDa heat shock protein gene in Trichophyton rubrum. Mycoses. 2009;52(3): 234–238. doi: 10.1111/j.1439-0507.2008.01561.x [DOI] [PubMed] [Google Scholar]
- 46.Wall TL, Carr LG, Ehlers CL. Protective association of genetic variation in alcohol dehydrogenase with alcohol dependence in Native American Mission Indians. Am J Psych. 2003;160(1): 41–46. doi: 10.1176/appi.ajp.160.1.41 [DOI] [PubMed] [Google Scholar]
- 47.Chaturvedi N, Kajsik M, Forsythe S, Pandey PN. Protein sequences insight into heavy metal tolerance in Cronobacter sakazakii BAA-894 encoded by plasmid pESA3. Arch Microbiol. 2015;197(10): 1141–1149. doi: 10.1007/s00203-015-1147-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.