

Abstract
Advances in single-cell technologies have highlighted the prevalence and biological significance of cellular heterogeneity. A critical question is how to design experiments that faithfully capture the true range of heterogeneity from samples of cellular populations. Here, we develop a data-driven approach, illustrated in the context of image data, that estimates sampling depth required for prospective investigations of single-cell heterogeneity from an existing collection of samples.
Cellular populations can exhibit widespread heterogeneity in morphology, signaling state and genotype. This heterogeneity can play a crucial role in normal tissue function as well as in disease progression and drug resistance1, 2. Advances in experimental and analytical technologies now allow individual cells to be probed and characterized3. A critical question is how to design experiments that faithfully capture the true range of heterogeneity from samples of cellular populations.
As studies of heterogeneity assess increasingly larger number of conditions, a practical consideration is to use sampling approaches that require fewer cells to represent each condition (Fig. 1a). For example, Tissue Microarrays (TMAs)4 are frequently used to overcome scarcity and non-renewability of patient tissue. By extracting small amounts of tissue (referred to as a “core”; Fig. 1b) from multiple tissue specimens and placing them on the same slide, TMAs have helped standardize tissue analysis, reduce cost and substantially improved efficiency and throughput5. Similarly, high-throughput, multi-well based screens capture each experimental condition with a small number of replicate wells. What determines the number of samples required within or across conditions to capture heterogeneity in single-cell studies (Fig. 1a)?
Figure 1. Sampling strategy to represent single-cell heterogeneity.
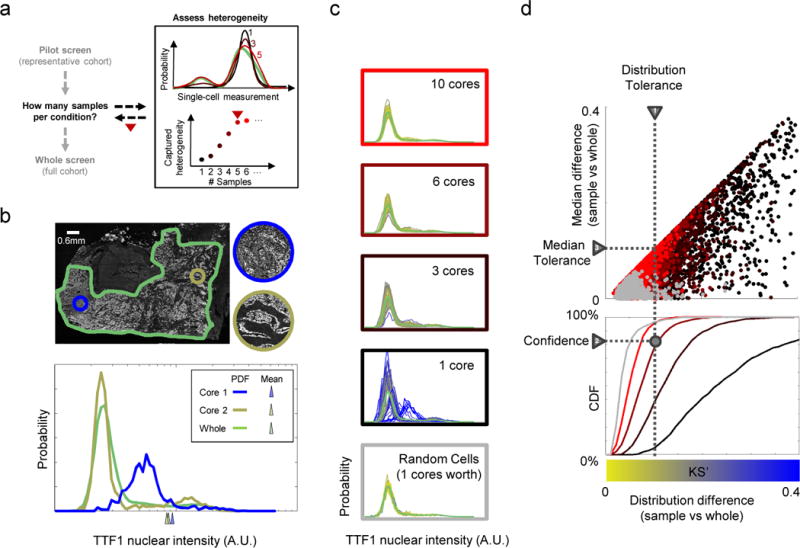
A) Overview of approach to determine how many samples (cores/replicates/draws) per condition are required for studies of heterogeneity. Top right: distributions of cellular phenotypes from different numbers of samples; bottom right: heterogeneity captured with different sample numbers. B) Samples that recover population averages may not capture heterogeneity. Papillary adenocarcinoma tumor (outlined in green) shows extensive heterogeneity in the staining of TTF1, but individual cores (circles) may not capture the full range of phenotypes. Bottom: distribution of TTF1 nuclear intensity (A.U.: arbitrary units, x-axis is on log scale) in the whole tissue and the cores. Cores and whole tissue have similar mean intensities (triangles below x-axis), yet differ greatly in their phenotypic distributions. C) Capturing whole-tissue heterogeneity depends on the number of cells sampled and the nature of the sampling. Plots: histograms of TTF1 distribution generated by repeated samplings of cells; colors based on agreement in distribution (blue or yellow show low or high (resp.) KS’ similarity) with whole tumor distribution (green curve). A single virtual core (~1000 cells on average) is unreliable, but spatially random draws with the same numbers of cells (bottom plot) captures heterogeneity as reliably as combining 10 virtual cores. D) Method for determining sample numbers needed to capture whole sample heterogeneity within a specified distribution tolerance (triangle “1”) at a desired level of confidence (triangle “2”). Upper scatter plot: comparison of whole tissue and samples generated as in B (point colors) based on their difference in distributions (x-axis: KS’ statistic) vs. medians (y-axis: deviation from the 50th percentile of the whole median). Differences in distributions places bounds on familiar quantities, such as differences in medians (triangle “1”; Methods). Bottom plot: confidence curves for achieving a desired KS’ tolerance as a function of sampling depth (number of cores) or type (core vs. random). This process allows rational user selection of the smallest number of samples (intersection of dotted lines) that capture whole specimen heterogeneity given desired tolerance and confidence levels. Given an existing library of specimens, confidence curves can be analyzed to estimate sampling depths of prospectively obtained samples for each choice of biomarker (e.g. Fig. 2C).
One might expect that the answer to this question depends on the degree of heterogeneity within a population (for example the central limit theorem would suggest that the number of samples required depends only on the standard deviation of the phenotypic distribution). However, individual samples may not reflect the heterogeneity of the whole population. In practice, the number of samples required may be dominated by non-homogeneity of the sampling. Experimentally, cells are not sampled independently, but rather in sub-sampled batches (e.g. cores or wells); cells within one batch may be strongly correlated and therefore not represent independent samplings of the population as a whole. For example, a single TMA core draws cells from the same region of the tissue. If tissue phenotypes vary on spatial scales larger than a core, then these sampled cells will be more phenotypically similar—and reveal far less about properties of the overall tissue—than a comparable number of cells sampled in a spatially random fashion (Fig. 1c, random vs. 1 core). Similarly, in cell culture, cells within a well may be more similar to one another than across replicate wells6 or areas within the same well may behave differently due to local cellular density variations7. Thus, the number of samples required to capture population heterogeneity depends on the phenotypic variation observed across samples drawn from the same experimental condition.
The question of how many samples to take has been studied in various contexts, including the number of TMA cores8, 9, the number of needle biopsies10 and so on. In many of these contexts, a consensus on appropriate sampling has emerged (e.g. three 0.6mm diameter TMA cores11). Crucially, these studies focus on recovering population-averaged properties, such as the mean or number of biomarker “positive” cells12–14 (although sometimes ability to recapture whole-population based correlations with biological/functional readouts is used8, 9). However, agreement in such bulk metrics does not ensure similar probability distributions of phenotypes (Fig. 1b bottom), and thus these approaches provide little guidance towards studying heterogeneity. Here, we develop an approach to determine the number of samples required to ensure that the probability distributions of cellular phenotypes in a sample (Fig. 1c, blue-yellow curves) matches that of the whole population (Fig. 1c, green curve). This allow us to not only ensure that similar phenotypes are present in similar proportions as the whole population (that is, phenotypic heterogeneity has been properly sampled) but also guarantees agreement in commonly used population quantifiers (including those of central tendency, variability) since these are derived from the probability distribution.
Our approach uses three steps to identify the number of samples needed to capture population heterogeneity (Fig. 1d). First, we designed a measure, referred to as a KS’ score, to quantify similarity between the phenotype distributions of samples to the whole. The KS’ score is designed to behave like the standard Kolmogorov-Smirnov (KS) test statistic15 for comparing phenotypic distributions, but has improved sensitivity (Online Methods, Supplementary Fig. 1, Supplementary Software) for detecting enrichment of extreme phenotype values. While we use the KS’ for our demonstrations, the basic approach outlined here is compatible with other measures of distribution similarity, such as KS, Anderson-Darling or Chi-Square test statistics. Second, we developed theoretical bounds to relate KS’ scores to familiar population-averaged metrics, such as the median or percentage of positive cells (Online Methods; diagonal line in Fig. 1d, top). Third, we developed a framework to estimate how many samples are required in future experiments to represent the phenotypic heterogeneity of the whole population (Fig. 1d, bottom).
In more detail, we start with a “representative” whole population of cells and a phenotype of interest. We then randomly “draw” a specified number of samples (e.g. TMA cores, wells, etc.) and compute the KS’ score to quantify the similarity of the phenotypic distribution of the pooled cellular population to that of the whole. To capture the inherent stochasticity of sampling, we repeat this process a large number of times (N=1000) to generate a distribution of KS’ scores for a given number of samples (Fig. 1d, top). As expected, if we repeat this procedure for increasingly larger number of samples, the distribution of KS’ scores will tend towards zero. This procedure provides a strategy to assign a confidence level (Fig. 1d, bottom, triangle “2”) that the whole and pooled distributions from a given number of samples will be close (in the sense of the KS’ score or a population-averaged score; Fig.1d, triangles “1” and “3”). Together, our approach provides a general and quantitative starting point for assessing the tradeoff between extracting more samples and obtaining better estimates of whole population heterogeneity.
We applied our approach to explore how sampling strategies could be designed for studies of heterogeneity in microscopy. The ability to capture simultaneously spatial context, morphology and biomarker expression of a whole population makes microscopy an ideal platform for studying heterogeneity. As with any assay, observed heterogeneity depends on the choice of assay readouts as well as non-biological (“technical”) variability introduced by the assay itself. Accordingly, here we sought to provide guidance on how sampling is affected by image generation, biomarkers, and cellular features.
First, we investigated the impact of the image generation processes—known to have strong quantitative effects on microscopy based measurements16, 17—in the context of designing TMAs to profile tissue heterogeneity. Here, the whole population was defined as the cells present in the imaged whole-tissue section and sampling was performed by computationally extracting cells within randomly placed TMA-sized core regions (~0.6mm; Online Methods). We applied our approach to a panel of 38 patient liver cancer specimens, of which 25 had two serial sections stained for the same antibody (YAP). Crucially, YAP staining was performed 5 months apart, and images were acquired using different microscopes. Despite these differences in the image generation process (Supplementary Fig. 2) we found broad agreement (Fig. 2a) in the number of TMA cores needed to capture the heterogeneity in YAP nuclear intensity (on the subset of 25 cases that had two consecutive sections stained for YAP). We found that these trends were robust for a wide range of KS’ threshold and confidence values (Supplementary Fig. 3). (We expect these trends to hold for other measures of distribution similarity, such as KS, though KS’ is particularly sensitive to identifying poorly represented tails in the distribution; Supplementary Fig. 4.) Thus, our approach allowed us to assert, in the context of this data set, that the number of samples determined by our method is largely a property of the specimen itself and is robust to changes in the image generation process.
Figure 2. The effect of experimental and analysis parameters on capturing heterogeneity.

A–B) Evaluation of the number of 0.6mm diameter cores needed for a panel of liver cancer specimens to capture the heterogeneity of individual whole-tissue images (with KS’ tolerance of 0.2 at 80% confidence) across different staining/imaging parameters and biomarkers. Point location: number of cores needed for the same specimen across different biomarker images; point size: number of specimens requiring the same numbers of cores. A) Comparison of serial sections stained for YAP and imaged 5 months apart using different microscopes (n = 25). Deviation from the diagonal represents the effect of imaging/staining variability. B) Comparison of the number of cores required by the most spatially heterogeneous (YAP) and homogenous (DAPI) biomarkers on the same section (n = 25). C) Tradeoffs between numbers of cores and sampling accuracy. Confidence curves (Online Methods; Fig. 1D bottom) were combined across the patient cohort to predict the proportion of patients whose heterogeneity will be captured (at a KS’ tolerance of 0.2) for different biomarkers and numbers of sampled cores (Imaging set I/II: YAP, n = 25/38; LKB1, n = 25/-; β-catenin, n = -/38; DAPI, n = 25/38). Dotted vertical line: 3 cores, the commonly accepted standard. D) Evaluation of the number of replicate wells needed in a high-content cell culture assay to capture heterogeneity of different cellular features. In each of 7 replicate 384-well plates, 215 single-cell image features (covering three imaged biomarkers) were extracted from 28 replicate wells. Features were divided (hand-curated) into feature types (color bar), including a sub-class of low contrast intensity features (Online Methods). For each feature, we calculated the number of wells required to ensure the distribution was close (KS’ < 0.05, 95% confidence) to the distribution from the full set of 28 wells. Error bars represent standard deviation across the 7 replicate plates, over which this analysis was independently repeated. Dashed horizontal line: 2 replicate wells, a common choice for high-throughput screens. The 8 rightmost features, denoted by *, all require >20 wells (the largest value tested) in at least one replicate plate.
Second, we investigated the impact of different biomarker choices on TMA design. We began by assessing the numbers of cores required to capture heterogeneity within a single patient specimen. By applying our analysis to biomarkers co-stained on the same tissue section, we found that YAP consistently requires more TMA cores than DAPI (Fig. 2b), LKB1 or β-catenin (Supplementary Fig. 5), perhaps reflecting intrinsic differences in the regulation of these markers. However, our results also revealed significant diversity across the patient cohort for the same biomarker: two cores seem adequate for some patients, while others required as many as 10. To understand the feasibility of using a single TMA design to study the heterogeneity of a large patient cohort, we examined how varying the number of cores affects the proportion of patients whose heterogeneity is well captured. Although all biomarkers require ~10 cores to capture the heterogeneity of every patient (Fig. 2c), the tradeoff between adding more cores vs. more patients being well represented is biomarker dependent. For example, TMA’s designed to sample phenotypic heterogeneity of LKB1 or β-catenin might poorly sample heterogeneity of YAP. Thus, our sampling strategies can be used to inform experimental design and biomarker selection of larger-scale studies of heterogeneity, as well as to compare heterogeneity within defined experimental conditions (e.g. patient-patient differences within one presumed clinical diagnosis).
Finally, we investigated the impact of cellular image feature choice in the context of designing high-throughput cell culture based experiments for profiling heterogeneity. In particular, how many replicate wells should be performed per condition to sample heterogeneity? We made use of A549 cells containing three, genetically encoded live-cell fluorescent markers to mark the nucleus, cytosol and a DNA-repair gene XRCC518. We analyzed seven 384-well imaging plates, each containing 28 replicate “control” wells. For each cell, we extracted 215 single cell features belonging to one of three feature classes (intensity, texture, and morphology; Supplementary Fig. 6). Cellular measurements were pooled across the 28 replicate control wells to define 215 whole-population feature distributions. We then used our approach to estimate how many replicate wells are required to recover the whole population distribution (Fig. 2d). The heterogeneity of some features, such as those relating to morphology, can be recovered with just one or two wells. In contrast, intensity features tend to be far more affected by well-to-well variation and require more wells to be sampled. Intensity features themselves are a highly diverse set: features quantifying biomarker intensities near background levels are particularly hard to sample (e.g. cytoplasmic levels of a biomarker that is largely localized to the nucleus) and thus require large numbers of wells. Nevertheless, these analyses predict that three replicate wells are sufficient to capture heterogeneity for many, but not all features. Thus, feature selection as well as biomarker selection plays a role in determining the number of samples (i.e. replicate wells or TMA cores) required for studies of heterogeneity.
Here, we provide a general approach for estimating how many samples are required to represent distributions of heterogeneous phenotypes, a question typically considered only in the context of population-averaged quantifiers. From a conceptual perspective, we highlight the importance of within-sample correlations in answering this question. These correlations can be complex and experiment or specimen specific, making its quantitative effects difficult to predict from first principles. We established a data-driven framework that quantifies mismatch in heterogeneity between sample and whole population, relates this mismatch to effects on familiar population average quantifiers, and allows the researcher to balance the tradeoff between number of samples and desired confidence for small heterogeneity mismatch.
Our approach makes use of representative single-cell data that capture the full range of phenotypic heterogeneity to estimate sampling depth of prospective studies. In many cases, such data may be readily available, though in other cases gathering such data may require upfront effort due to specimen size19 or rarity. Of course, it is not possible to determine via any analytical technique whether a set of specimens is truly representative. However, it is possible to ask whether fewer specimens would provide similar results; here, analysis of our data sets suggests that a smaller collection would have provided similar confidence estimates for numbers of samples taken (Supplementary Fig. 7). In practice, sample size may also be reduced by using assay-specific procedures to minimize non-biological effects (e.g. image correction) or by generating samples more efficiently (e.g. using stereology20 or H&E information). Beyond the contexts of immunofluorescence microscopy, highlighted in our case studies, our methodology also applies to other imaging modalities (e.g. IHC images, quantified by percent positive counts; Supplementary Fig. 8), to alternate sampling frameworks (e.g. choosing an appropriate placement of cores or designing “heterogeneity-TMAs”19 for samples too large to fit on a slide), as well as to other single-cell assay technologies. Taken together, our methodology provides a rational approach to the design of experiments targeting phenotypic heterogeneity.
ONLINE METHODS
Sample preparation, staining and imaging
1) Adenocarcinoma Tissue (Fig. 1)
Formalin-fixed paraffin-embedded (FFPE) human non-small cell lung cancer (papillary adenocarcinoma) tissue blocks were purchased from ILSBio, re-embedded and sliced in 5μm sections by the Molecular Pathology Core Facility at UTSW. Sudan Black B blocking was used to reduce auto-fluorescence. TTF1 staining was performed using polyclonal rabbit antibodies (Cat.# sc-13040, dilution 1:100 Santa Cruz Biotech). Digital images of stained tissue sections were obtained using a ScanScope Digital slide scanner at 20× (Aperio ePathology, Leica Biosystems).
2) Liver Cancer Tissue (Fig. 2a–c)
A collection of fresh frozen hepatocellular carcinoma (HCC, n=38) samples was used in the present study. HCC specimens were collected at the University of California, San Francisco (San Francisco, CA). Institutional Review Board approval was obtained and informed consent was obtained from all subjects. See Gordan et al., manuscript in preparation, for further details of this sample set. Immunofluorescence staining and imaging was performed at the Gladstone Institutes Histology and Light Microscopy core using fluorescently-labeled primary antibodies (all from Cell Signaling, Danvers, MA) to CTNNB1 (Mouse mAb L54E2, conjugated to Alexa Fluor® 555, 1:200, Cat#5612) YAP (Rabbit mAb D8H1X, conjugated to Alexa Fluor® 488, 1:200, Cat#14729) and LKB1 (Rabbit mAb, 1:250, Cat#13031, followed by goat anti-rabbit conjugated to Alexa Fluor® 633, 1:200). Staining and imaging for DAPI/YAP/LKB1 was first performed on 38 samples, and 5 months later serial sections for 25 of these samples were stained/imaged for DAPI/YAP/CTNNB1. Stitched images of entire sections were acquired at 20× (0.32μm per pixel) on automated slide scanners (BZ-X700; Keyence, Osaka, Japan; Aperio VERSA Digital Pathology Scanner, Leica Biosystems, and Axio Scan.Z1, Carl Zeiss Microscopy).
3) Lung Cancer Cell Lines (Fig. 2d)
We made use of a previously constructed18 adenocarcinoma cell line (A549) that was triply labeled for live cell reporters marking the nucleus, cytoplasm and XRCC5 (a nuclear-localized protein that functions in double-strand break repair). The A549 cells were cultured in RPMI1640 media containing 50 units/ml penicillin, and 50 μg/ml streptomycin (all from Life Technology, Inc.), and 10% FBS (Gemini BIO-PRODUCTS #100–106, Lot# A07F00G) at 37 °C, 5% CO2 and 100% humidity. Cells were grown in 10-cm culture plates for 72h, detached by trypsin, counted by TC10 automated cell counter (Bio-Rad Laboratories, Inc.) and seeded onto glass 384-well plates (ThermoFisher Scientific #164588) at a density of 1500 cells/well in 50uL media by the Matrix WellMate Liquid Dispenser (ThermoFisher Scientific). After 24 h at 37 °C, drugs were added using the Beckman Coulter BioMek FX liquid handler (Beckman Coulter, Inc.), and the plates were covered by BreathEasy sealing membranes (Sigma-Adrich, Inc.) and incubated at 37 °C for 48h. For the present analysis, only 28 replicate control wells in a plate that did not receive any drugs were used. Images were acquired using an IN Cell Analyzer 2000 epifluorescence microscope (GE) equipped with laser Autofocus and a Nikon 10×/0.45 Plan Apo objective lens. We used 1s exposure times and TexasRed, CFP and YFP emission filters, with 2×2 binning. All image acquisition was controlled by IN Cell Analyzer software (GE). One image was acquired per well. Images with obvious anomalies (e.g., out of focus, abnormal fluorescent patterns caused by dust, scratches on the plate) were discarded after manual inspection.
4) Immunohistochemistry (IHC) sample preparation and staining (Sup Fig. 8)
A FFPE human breast cancer specimen was obtained from UT Southwestern Shared Tissue resource. Ki67 immunostaining was performed using primary monoclonal rabbit anti- Ki67 antibody (Cat.#790-4286, clone 30-9, dilution 1:100, Ventana Medical Systems) on an automated BenchMark stainer (Ventana Medical Systems). Digital images of Ki67 stained tissue sections were obtained using a ScanScope Digital slide scanner at 20× (Aperio ePathology, Leica Biosystems)
Identification of regions of interest in tissue
We developed an image analysis approach to identify and exclude cells at the edge of the tissue (as these potentially display edge staining artifacts). Additionally, for the liver cancer data, staining artifacts and out of focus regions were automatically identified based on the lack of local intensity variations in these regions. For the adenocarcinoma (Fig. 1) and breast cancer (Supplementary Fig. 8) datasets analysis was performed on the non-edge tumor regions (pathologist-identified green curves in respective figures). For the liver cancer data (Fig. 2a–c), analysis was performed on the full tissue image excluding regions at the edge or displaying imaging artifacts.
Calculation of biomarker expression distributions
1) Tissue
The starting point is a tissue image with nuclear specific biomarker (DAPI levels for IF and deconvoluted haematoxylin intensity in IHC). A multilevel Otsu thresholding was applied to the nuclear image to identify nuclear pixels. A Laplacian of Gaussian filter, combined with an extended minima transform was applied to the nuclear biomarker image to identify well separated nuclei centers. These centers served as seeds for a watershed transform that was used to partition the nuclear pixels into distinct, spatially dis-connected, nuclear regions. Regions too large to be nuclei were successively divided, while those too small to reasonably be nuclei were dropped. Nuclei in regions identified as exhibiting artifactual staining or outside the region of interest (as identified above) were dropped. For each nucleus, the intensity of co-stained biomarkers (i.e. biomarkers stained on the same sections as the nuclear marker) was quantified by the mean intensity of the biomarker over pixels belonging to that nucleus. Given a region of interest (e.g. whole tumor or pooled cores), the nuclear distribution of a biomarker was calculated by pooling the nuclear intensities across all nuclei whose centroids fell within that region.
2) Cell Culture
Image background subtraction was performed using ImageJ’s Rolling Ball Background Subtraction algorithm21. Cells in an image were automatically identified using our in-house watershed based algorithm22 which identifies nuclear regions based on a nuclear marker and subsequently uses these nuclei as seeds for the identification of cell boundaries based on a cytoplasmic marker. For each cell, 215 different image features were calculated based on the intensities of the three biomarkers (H2B/cytoplasmic/XRCC5) in the pixels belonging to the cell. These features include: a) intensity features that are summaries of the intensities of the biomarkers in different cellular compartments (nucleus/cytoplasm/whole cell), b) texture features (Haralick/Zernike) that capture local biomarker intensity variations, or c) features that describe cellular morphology. See Supplementary Fig. 6 for a more detailed summary. Among the biomarkers, H2B and XRCC5 are expected to be localized within the nucleus, and any intensity features that included the cytoplasmic intensities of these biomarkers were classified as low contrast. The whole population distribution for a feature was calculated by pooling feature values for cells belonging to all 28 replicate wells in a plate, while sample distributions were generated by pooling (cells from) a subset of wells together.
Virtual sampling of cores in tissue
For each virtual sampling, N non-overlapping cores were placed randomly on the image such that 1) the entire core (circle of 0.6 mm diameter, between 600 and 1000 pixels in the 20× images analyzed here) was within the tissue and 2) the core was centered within the “good” tissue area (which was identified as described above). 3) More than 70% of the core area was covered by cells of interest. Cells whose centroids were within 0.6mm of the core center were considered as belonging to the core. Biomarker intensities of cells from all cores were pooled together to construct the core intensity distribution for each virtual sampling.
KS’: a new measure to compare phenotypic heterogeneity
A widely used measure to compare distributions is the Kolmogorov-Smirnov (KS) statistic15, which measures the maximum difference between two CDFs (Supplementary Fig. 1A bottom, double-sided arrows). More precisely, if C(I) and W(I) are the CDFs for the core and whole tissue, the corresponding KS score is given by . The KS statistic has several virtues, including that its value is insensitive to the nature of the distributions (i.e. normal, Poisson and so on), yet it can still detect changes to both the location and shape of the distributions.
However, a disadvantage of the KS statistic is that it tends to be less sensitive to changes near the tails of distributions23. This can be undesirable in contexts where subpopulations of the most- or least-stained cells have important biological meaning. The reason for the loss of sensitivity is that the KS statistic measures the largest difference between CDFs regardless of where this difference occurs. Yet, a large difference in CDFs is far rarer at the tails (differences between sampled and whole CDFs tend to zero at the tails; Supplementary Fig. 1C, top), and should be considered more significant than when the same difference occurs away from the tails. For our purposes, it is desirable to use a test that is more sensitive to changes across the whole range of intensities, including the tails.
We chose to modify the KS statistic to increase sensitivity at the intensity tails by normalizing differences of CDFs by the magnitude of expected deviations when sampling from the whole-tissue distribution W. Observed deviations near the tail would then become more apparent when divided by a small expected deviation, working in the same way as a z-score. Towards this end, we started with an explicit formula computed by Anderson and Darling (Supplementary Fig. 1C, bottom), , which computes the expected standard deviation, at each intensity value I, between the CDFs of the whole distribution W. Our desired normalized CDF difference between a core C(I) and whole tissue W(I) is then given by . Our modified KS statistic, which we refer to as the KS’ statistic, is then simply: . Thus, the KS’ allows us to compare the distributions of cores to that of whole tissue with particular sensitivity to extreme phenotypes. We note that the KS’ was designed as a measure of distribution similarity rather than a test statistic in the current work.
A particular strength of the KS’ is its ability to theoretical bound difference in various statistical quantifiers of the distribution. For example, if I50 is the median of the whole-specimen distribution (i.e. W(I50) = 0.5) then we have the relationship:
Since is the fraction of the sample population less than the whole-specimen median, it would be equal to 0.5 if the whole and sample medians matched. Thus, if the KS’ between the whole and its sample distributions is 0.1, the whole median must lie between the 40th and 60th percentile of the sample. (We note that in cases where C(I) is discontinuous exactly at I50, e.g. integrates one or more events at I50, then we interpret the right hand side of the inequality to be the smallest absolute difference of the range of values for the jump to 0.5.) Similar bounds can be established for the whole vs. sample mean, percentage positive cells, etc.
Determining the number of samples for a single whole specimen
The heterogeneity observed in a sample can be influenced by inhomogeneity introduced by sampling (i.e. the fact that not all cells within the whole have the same probability of belonging to the same sample). We assume here that a computational means has been established to simulate experimental sampling: in our case this was through construction of virtual cores of the same size as a true TMA core for tissue or selecting cells by well in cell culture.
Extract and pool the data for a given number N of samples, construct its distribution and calculate the corresponding KS’ score by comparing the sample distribution to the whole.
To model the inherent stochasticity of sampling, repeat 1) multiple times (we used 1000 repeats for our results) to construct a distribution (quantified by its CDF) of KS’ scores for N samples.
Repeat steps 1 & 2 while varying N over the experimentally reasonable range of samples. This will give us CDFs for the KS’ for different values of N (this is essentially Fig. 1d bottom, with the KS’ scores shifting to the left as more samples are extracted).
Determine the maximum value of KS’ for sampling to be considered “good”. This can be calibrated using the inequalities relating the KS’ to bounds on the median, mean, percentage positive, etc. For example, a KS’<0.1 means the median of the whole population will lie between the 40th and 60th percentile of the sample.
Determine the desired confidence level that sampling needs to be “good” (e.g. at least 80% of the times we generate samples we want the KS’<0.1).
By comparing the CDFs generated in Step 3, find the minimum value of N that the CDF at the KS’ tolerance exceeds the desired confidence level.
This procedure selects the minimum number of samples required to provide a desired level of confidence that the difference between sample and whole distributions is within a specified KS’ tolerance.
Design of experimental sampling using a pilot panel
The scenario we envisioned is to design a large experiment profiling heterogeneity based on whole-specimen data observed within a pilot set of specimens. In the paper (Fig. 2c), we illustrate the design of a TMA (potentially consisting of hundreds of patient tissue specimens) based on a pilot set of ~30 whole tissue liver cancer specimens. As with any statistical estimator, we treat the pilot set as representative of data for the large-scale experiment. The first step is to calculate the specimen’s KS’ CDF curves for each of the NPS pilot specimens across varying numbers of samples (as outlined above). For a pilot specimen i, let Ci(K,n) denote the probability of n pooled samples yielding a distribution that differs from the whole by KS’ score less than K. Then, as we average across specimen, the expected fraction of specimen for which of n pooled samples give a distribution that differs from the whole by KS’ score less than K is given by:
This process may be repeated for different biomarkers etc as demonstrated in Fig. 2c.
CODE AVAILABILITY
The MATLAB code used to generate the main figures is provided as supporting code. An R implementation of the KS’ is also included. The latest version of the code will be available at https://github.com/AltschulerWu-Lab/SamplingForHeterogeneity
DATA AVAILABILITY
The raw data used to generate Figure 1 is available at XXXX with DOI###. The raw data that support the findings of Fig. 2 are available from the corresponding author upon reasonable request. The processed source data used to generate the main figures is provided along with the manuscript as supporting information.
Supplementary Material
Acknowledgments
We would like to thank Meredith Calvert, Thea D. Tlsty and Philip B. Stark for helpful discussions. This work was supported by the NCI K08CA175143 (CEA), P01HL088594 (JSM), a Conquer Cancer Foundation Young Investigator Award supported by the Scopus Foundation (JDG), a gift from the Edmund Wattis Littlefield Foundation (RSW), NSF PHY-1545915 (SJA), Stand Up To Cancer (SJA), NCI R01 CA133253 (SJA), NCI RO1 CA185404 (LFW) and R01 CA184984 (LFW), and the Institute of Computational Health Sciences (ICHS) at UCSF (SJA and LFW).
Footnotes
AUTHOR CONTRIBUTIONS
S.R., S.J.A. and L.F.W. conceived of and designed the study. L.E.H, J.D.G., K.M.B. and A.K.W. performed the experiments and/or provided data. S.R., J.A., S.J.A., and L.F.W. developed the algorithms and S.R. performed the analysis. R.S.W. and A.K.W. contributed samples. The manuscript was written by S.R., S.J.A., and L.F.W. with contributions from C.E.A. and J.S.M.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol. 2013;8:277–302. doi: 10.1146/annurev-pathol-020712-163923. [DOI] [PubMed] [Google Scholar]
- 2.Altschuler SJ, Wu LF. Cellular heterogeneity: do differences make a difference? Cell. 2010;141:559–563. doi: 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan GC, et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017;18:84. doi: 10.1186/s13059-017-1218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wan WH, Fortuna MB, Furmanski P. A rapid and efficient method for testing immunohistochemical reactivity of monoclonal antibodies against multiple tissue samples simultaneously. Journal of immunological methods. 1987;103:121–129. doi: 10.1016/0022-1759(87)90249-3. [DOI] [PubMed] [Google Scholar]
- 5.Camp RL, Neumeister V, Rimm DL. A decade of tissue microarrays: progress in the discovery and validation of cancer biomarkers. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:5630–5637. doi: 10.1200/JCO.2008.17.3567. [DOI] [PubMed] [Google Scholar]
- 6.Bray MA, Carpenter A. In: Assay Guidance Manual. Sittampalam GS, et al., editors. Bethesda (MD): 2004. [Google Scholar]
- 7.Snijder B, et al. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature. 2009;461:520–523. doi: 10.1038/nature08282. [DOI] [PubMed] [Google Scholar]
- 8.Eckel-Passow JE, et al. Tissue microarrays: one size does not fit all. Diagnostic pathology. 2010;5:48. doi: 10.1186/1746-1596-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tennstedt P, et al. The impact of the number of cores on tissue microarray studies investigating prostate cancer biomarkers. International journal of oncology. 2012;40:261–268. doi: 10.3892/ijo.2011.1216. [DOI] [PubMed] [Google Scholar]
- 10.Jiang J, Colli J, El-Galley R. A simple method for estimating the optimum number of prostate biopsy cores needed to maintain high cancer detection rates while minimizing unnecessary biopsy sampling. J Endourol. 2010;24:143–147. doi: 10.1089/end.2009.0285. [DOI] [PubMed] [Google Scholar]
- 11.Rimm DL, et al. Cancer and Leukemia Group B Pathology Committee guidelines for tissue microarray construction representing multicenter prospective clinical trial tissues. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:2282–2290. doi: 10.1200/JCO.2010.33.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ping Y. Determining the optimal numbers of cores based on tissue microarray antibody assessment in non-small cell lung cancer. Journal of Cancer Science & Therapy. 2011 [Google Scholar]
- 13.Goethals L, et al. A new approach to the validation of tissue microarrays. The Journal of pathology. 2006;208:607–614. doi: 10.1002/path.1934. [DOI] [PubMed] [Google Scholar]
- 14.Khan AM, Yuan Y. Biopsy variability of lymphocytic infiltration in breast cancer subtypes and the ImmunoSkew score. Sci Rep. 2016;6:36231. doi: 10.1038/srep36231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massey FJ., Jr The Kolmogorov-Smirnov test for goodness of fit. Journal of the American statistical Association. 1951;46:68–78. [Google Scholar]
- 16.North AJ. Seeing is believing? A beginners’ guide to practical pitfalls in image acquisition. J Cell Biol. 2006;172:9–18. doi: 10.1083/jcb.200507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pawley J. The 39 steps: a cautionary tale of quantitative 3-D fluorescence microscopy. Biotechniques. 2000;28888:884–886. doi: 10.2144/00285bt01. [DOI] [PubMed] [Google Scholar]
- 18.Kang J, et al. Improving drug discovery with high-content phenotypic screens by systematic selection of reporter cell lines. Nat Biotechnol. 2016;34:70–77. doi: 10.1038/nbt.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minner S, et al. Marked heterogeneity of ERG expression in large primary prostate cancers. Mod Pathol. 2013;26:106–116. doi: 10.1038/modpathol.2012.130. [DOI] [PubMed] [Google Scholar]
- 20.Weibel ER, Hsia CC, Ochs M. How much is there really? Why stereology is essential in lung morphometry. J Appl Physiol (1985) 2007;102:459–467. doi: 10.1152/japplphysiol.00808.2006. [DOI] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loo LH, Wu LF, Altschuler SJ. Image-based multivariate profiling of drug responses from single cells. Nature Methods. 2007;4:445–453. doi: 10.1038/nmeth1032. [DOI] [PubMed] [Google Scholar]
- 23.Mason DM, Schuenemeyer JH. A modified Kolmogorov-Smirnov test sensitive to tail alternatives. The Annals of Statistics. 1983:933–946. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data used to generate Figure 1 is available at XXXX with DOI###. The raw data that support the findings of Fig. 2 are available from the corresponding author upon reasonable request. The processed source data used to generate the main figures is provided along with the manuscript as supporting information.