

Summary
Expansions of microsatellite repeats are responsible for numerous hereditary diseases in humans, including myotonic dystrophy and Friedreich’s ataxia. While the length of an expandable repeat is the main factor determining disease inheritance, recent data point to genomic trans-modifiers that can impact the likelihood of expansions and disease progression. Detection of these modifiers may lead to understanding and treating repeat expansion diseases. Here we describe a method for the rapid, genome-wide identification of trans-modifiers for repeat expansion in a yeast experimental system. Using this method, we found that missense mutations in the endoribonuclease subunit (Ysh1) of the mRNA cleavage and polyadenylation complex dramatically increase the rate of (GAA)n repeat expansions, but only when they are actively transcribed. These expansions correlate with slower transcription elongation caused by the ysh1 mutation. These results reveal a previously unsuspected interplay between RNA processing and repeat-mediated genome instability, confirming the validity of our approach.
eTOC Blurb
McGinty, et al. developed a genetic screen in S. cerevisiae to identify genes promoting expansions of (GAA)n repeats. The authors uncovered the unexpected involvement of essential RNA-processing gene, YSH1. Mutation in YSH1 leads to slow transcription elongation, promoting DSBs, whose repair via HR cause repeat expansions.
Introduction
Expansions of DNA microsatellites are responsible for several dozens of hereditary diseases in humans, including fragile X syndrome (FXS), myotonic dystrophy (DM1 and DM2), Huntington’s disease (HD), Friedreich’s ataxia (FRDA), many spinocerebellar ataxias (SCA), the familial form of amyotrophic lateral sclerosis and frontotemporal dementia (ALS), and others (Lopez Castel et al., 2010; McMurray, 2010; Mirkin, 2007). The scale of expansions differs depending on the location of the DNA repeat: they are relatively small-scale when positioned in the protein-coding part of a gene, or very large-scale when in the non-coding parts of a gene, such as 5′- and 3′-UTRs, or introns (Mirkin, 2007). Repeat expansions readily occur during intergenerational transmissions in human pedigrees, which accounts for the phenomenon of genetic anticipation that is characteristic for these diseases. In some somatic tissues, repeats continue expanding throughout life, which affects age of onset and disease severity (Kovtun and McMurray, 2008).
It is generally believed that the length of an expandable repeat is the key factor determining disease inheritance and development. Significant amounts of data, however, point to the existence of trans-modifiers that can affect the likelihood of repeat expansions, and thus, disease progression. While most of these data came from studying repeat expansions in model experimental systems (Usdin et al., 2015), the idea is also supported by human genetics data (Morales et al., 2012).
Expansions of (CAG)n, (CGG)n, (GAA)n and (ATTCT)n repeats have been extensively studied in yeast experimental systems. These studies revealed that knocking out genes involved in DNA replication, repair, recombination and transcription machineries can strongly elevate or decrease the rate of repeat expansions in dividing cells (Kim and Mirkin, 2013). Studies of (CAG)n repeat expansions in a Drosophila system showed that repeat instability was decreased when a fly homolog of the nucleotide excision repair gene XPG, mus201 was mutated (Yu et al., 2011). Mice models for repeat expansions demonstrated the critical role of mismatch repair genes in promoting repeat expansions during both intergenerational transmission and in somatic cells (Kovtun and McMurray, 2001; McMurray, 2008; Savouret et al., 2003; Savouret et al., 2004). At the same time, mutations in the base excision repair machinery specifically prevented repeat expansions in somatic tissues (Kovtun et al., 2007). In a humanized mouse model of fragile X syndrome, the loss of the transcription-coupled DNA repair factor CSB led to a lower frequency of germ-line expansions and a reduction in the scale of somatic expansions (Zhao and Usdin, 2014). In cultured human cells, fork stabilizing proteins Claspin, Timeless, and Tipin were shown to counteract (CAG)n repeat expansions (Liu et al., 2012), while knockdown of the FANCJ protein resulted in the accumulation of DSBs and ectopic rearrangements at those repeats (Barthelemy et al., 2017). Finally, transcription-coupled repair was shown to trigger (CAG)n repeat contractions in human cells (Lin et al., 2010; Lin and Wilson, 2007).
Clinical genetics data, while more fragmentary and limited in scope, are generally in line with the conclusions of the model systems studies. In case of DM1, it was found that the rate of (CTG)n repeat expansions in the DMPK gene is a heritable trait in itself, pointing to the existence of trans-modifiers throughout the genome (Morales et al., 2012). More recently, a polymorphism in the MSH3 mismatch repair gene was specifically associated with the extent of somatic instability of (CTG)n repeats in the blood of DM1 patients (Morales et al., 2016). Single nucleotide polymorphisms (SNPs) in genes involved in DNA replication, repair and recombination have been associated with increased risk of repeat expansions in Huntington’s disease (HD) and spinocerebellar ataxia type 3 (SCA3) families (Genetic Modifiers of Huntington’s Disease, 2015; Martins et al., 2014). It was also suggested that differential expression levels for replication and repair genes in various parts of the HD patient brains might determine the extent of somatic instability in the corresponding brain regions (Mason et al., 2014).
There exists, however, a serious gap between the model systems and human genetics data. The former primarily describe the effect of gene knockouts, i.e. an all-or-none scenario, while the latter deal with SNPs, i.e. much more subtle changes in gene functioning. In this study, we attempted to fill this gap by conducting a genetic screening to detect trans-modifiers of repeat expansions in our yeast experimental system (Shah et al., 2012; Shishkin et al., 2009). The screening strategy involves mutagenesis and selection for repeat expansions, followed by whole-genome sequencing and identification of causal SNPs in the expansion process. Totally unexpectedly, this screening revealed mutations in YSH1, a gene central for RNA processing.
YSH1 encodes a component of the cleavage and polyadenylation specificity factor complex (CPSF or CPF), which in concert with cleavage stimulation factor (CstF or CFIA) and cleavage factor I (CFI or CFIB) cleaves mRNA transcript at poly(A) signals (Chan et al., 2011; Millevoi and Vagner, 2010). This subsequently allows both the addition of the poly-A tail to the 3′ end of the mRNA via Pap1 (poly-A polymerase), as well as the loading of Rat1 exonuclease to the 5′ end of the transcript, leading to the transcription termination (Porrua et al., 2016).
We found that in Ysh1 mutants that came from our screen, inefficient transcript cleavage is accompanied by slowed transcription elongation and accumulation of double stranded breaks within transcribed (GAA)n repeats followed by their expansions in a homologous recombination (HR)-dependent manner. These results reveal a totally unsuspected interplay between RNA processing and repeat-mediated genome instability, hence confirming the validity of our whole-genome screening approach. In the future, this approach can be used to identify trans-factors for large-scale expansions of other repeats, such as (CGG)n repeats responsible for fragile X syndrome or (CTG)n repeats responsible for myotonic dystrophy type 1.
RESULTS
Screen Design and Implementation
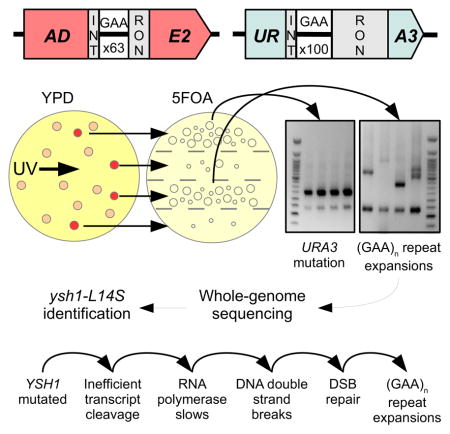
In yeast, large-scale repeat expansions are rare events as opposed to repeat contractions (Kim and Mirkin, 2013). To detect rare large-scale expansion events, we have developed an experimental system, in which expansions of the (GAA)100 repeat within an artificial intron of the URA3 gene (Fig. 1A) inhibits its splicing, resulting in yeast growth on 5-FOA-containing media (Shah et al., 2012; Shishkin et al., 2009). Crossing this reporter cassette into various yeast knockout libraries helped us to identify numerous genes involved in DNA replication, repair and transcription that affect repeat expansions (Zhang et al., 2012).
Fig. 1. Overview of screening method.

(A) Diagram of selectable ADE2 and URA3 cassettes. The ADE2 marker contains a short artificial intron with only 63 (GAA) repeats, while the URA3 marker contains a longer artificial intron with 100 (GAA) repeats. (B) Screening procedure: Cells are mutagenized and grown on complete (YPD) media. Colonies form, and those that turn red (ADE2 inactivation) are spread on sections of a plate containing the selective drug 5FOA. For each strain with a high number of 5FOA-resistant colonies (URA3 inactivation), four individual 5FOA colonies were tested via PCR for repeat length. C. Example PCRs for amplification of (GAA)n repeats in both cassettes. The URA3 (GAA)100 repeat consistently expands in strains containing genuine repeat expansion trans-modifiers (right), while remaining at wild-type length in strains containing off-target modifiers (left). The ADE2 (GAA)63 repeat does not appear to expand in any strains.
We were concerned, however, that gene knockouts are too blunt of a tool, particularly when it comes to essential genes, and, thus, wanted to assess the effect of subtler genetic changes on repeat expansions. To this end, we chose mild UV mutagenesis to induce point substitutions as opposed to gene deletions or gross-chromosomal rearrangements. While we expected this approach to generate point mutations in genes affecting repeat expansion in our system, we were acutely aware that it might also lead to the accumulation of mutations in the body of our reporter, or in other proteins involved in uracil biosynthesis. To minimize the latter prospect, we added another selectable cassette to make our screening a two-stage process. The second cassette, which contained the ADE2 reporter with a (GAA)100 repeat within its artificial intron, replaced the endogenous ADE2 gene on chromosome XV (Fig. 1A). Unexpectedly, however, the presence of even the starting length repeat within this intron completely inactivated the ADE2 gene, making yeast colonies red. Thus, we shortened the (GAA)n run down to 63 repeats (Fig. 1A) to keep the reporter active. Notably, the presence of 63 repeats in the ADE2 intron already decreased the reporter’s expression sufficiently that the resultant strain had a borderline ADE+ phenotype and pink colonies (see also below).
The strain carrying both selectable cassettes was irradiated with UV light followed by a two-step selection protocol (Fig. 1B): identification of red colonies (step 1 - ADE2 inactivation), which were then analyzed individually for 5-FOA-resistance (step 2 - URA3 inactivation). Nearly half of mutagenized red colonies gave rise to augmented papillae growth on 5-FOA-containing media. For roughly half of them, PCR analysis of those 5-FOA-resistant colonies revealed large-scale expansions in the URA3 cassette. Unexpectedly, however, repeat expansions in the ADE2 cassette were not detected in any of them (Fig. 1C). As shown later, the ADE2 inactivation is likely due to reduced expression of the ADE2 mRNA in these mutants. In summary, our screen revealed new genetic trans-modifiers that repress the reporter gene carrying a short (GAA)63 repeat, while simultaneously promoting expansions of longer (GAA)100 repeats.
Identification of mutations in the YSH1 gene
We conducted whole-genome sequencing of sixteen UV-mutagenized strains that simultaneously showed ADE2 inactivation and high rate of repeat expansions in the URA3 gene. In brief, genomic DNA was isolated from these strains, barcoded libraries were generated and sequenced using Illumina GAII with 100 bp Paired-End reads. This gave an average coverage of ~80x per strain. Reads were then aligned to the S288C reference genome using Bowtie (Langmead et al., 2009), and mutant variants were called using the SAMtools software (Li et al., 2009). This analysis revealed that our mutagenesis strategy resulted in the accumulation of ~10 mutations per yeast strain.
To assess which of these multiple mutations could potentially be causative, they were further analyzed using snpEFF (Cingolani et al., 2012) and PolyPhen2 (Adzhubei et al., 2013) tools. Remarkably, two out of sixteen sequenced strains contained missense mutations in the same essential gene, YSH1, which encodes a cleavage and polyadenylation factor subunit (Garas et al., 2008; Zhao et al., 1997). Furthermore, these mutations (ysh1-L439S and ysh1-L14S) affected highly conserved amino acids. The yeast L14S and L439S substitutions correspond to L17S and L427S in the human cleavage and polyadenylation factor CPSF-73 (Chan et al., 2011; Millevoi and Vagner, 2010). Both mutations are outside of the enzyme’s catalytic center (Fig. S4). The L14 residue appears to reside on the surface of the protein and could potentially affect the stability of the CPF complex, while the L439 residue resides internally, but not in the active site.
Since two independent mutational hits appeared in conserved parts of the YSH1 gene, we hypothesized that these mutations could be causative for the observed phenotype of increased repeat expansions and gene inactivation. To validate this hypothesis, we made two strains containing individual ysh1-L439S and ysh1-L14S mutations (see Methods) along with the two repeat-containing cassettes.
Characterization of the YSH1 mutant strains
We first looked at the growth characteristics of the strains with individual ysh1-L439S and ysh1-L14S mutations. These strains readily turned red, indicating that the ysh1 mutations are indeed responsible for inactivating the ADE2 cassette. Both mutants grew more slowly than the wildtype, and this slow growth was exacerbated at higher temperatures and rescued at lower temperatures. The ysh1-L14S mutant appeared to be the stronger of the two mutants in each test we conducted, and it had a clear-cut temperature-sensitive growth phenotype (Fig. 2). Consequently, this mutant was chosen for all further analyses.
Fig. 2. Mutant ysh1-L14S is temperature sensitive for growth.

WT and ysh1-L14S mutant strains were serially diluted and grown on complete media at three different temperatures. No difference in growth rate is observable at 15°C, which is below the optimal temperature for wild type yeast. At the optimal temperature of 30°C, the ysh1 mutant displays slightly reduced growth, best observable after 1 day of growth. Red pigment is observable after 3 days, due to inactivation of the ADE2 cassette. Incubation at 37°C severely slows the growth of the ysh1 mutant.
Ysh1 is the endonuclease responsible for cleavage of the nascent mRNA transcript during 3′ end processing (Mandel et al., 2006; Ryan et al., 2004). It has no other known enzymatic functions, though its presence in the CPF complex facilitates related processes, including polyadenylation and splicing (Chanfreau et al., 1996; Garas et al., 2008; Zhao et al., 1999a). Ysh1 mutants were shown to be defective in both cleavage and polyadenylation in vitro (Chanfreau et al., 1996; Garas et al., 2008; Zhao et al., 1999a). Therefore, we performed in vitro cleavage and polyadenylation assays for the wild-type and ysh1-L14S mutant as described (Zhao et al., 1999b). In brief, cell extracts from both strains were incubated with the full-length 32P-labeled GAL7-1 RNA in the presence of ATP, and the reaction products were separated on a denaturing polyacrylamide gel and visualized via phosphorimager (Fig. 3A). The ysh1-L14S mutation causes a strong decrease in the efficiency of RNA cleavage and polyadenylation at the non-permissive temperature. The individual steps of cleavage and poly(A) addition are also compromised in the mutant when uncoupled from each other (Fig. S1).
Fig. 3. The ysh1-L14S mutant is defective for mRNA 3′ end-processing and transcription elongation.

(A) In vitro 3′ end processing reaction. A precursor RNA is combined with cell extracts derived from WT or ysh-L14S mutant yeast, which were grown at 30°C and shifted to 37°C for 1.5 hours. The precursor RNA is shortened by the cleavage reaction, and then lengthened by the addition of the poly-A tail. (Positions indicated.) For ysh1-L14S mutants, less of the precursor RNA is converted to the polyadenylated form. Cleaved products are not observed, suggesting that the cleavage step is rate-limiting. See also Figure S1. (B) ysh1-L14S is sensitive to the transcription elongation inhibitor mycophenolic acid (MPA). WT and ysh1 L14S mutant strains were serially diluted and grown on synthetic media lacking uracil and containing the indicated MPA concentrations. Plates were incubated for 3 days at 30°C. ysh1-L14S strains display a pronounced growth inhibition under MPA treatment.
It was previously reported that mutants defective in the CF IA cleavage/polyadenylation factor are characterized by a slower rate of transcription elongation (Tous et al., 2011), but such effects from mutation of CPF, the complex in which Ysh1 resides, have not been reported. We were curious whether the same is true for the ysh1-L14S mutant. To address this question, we studied its sensitivity to mycophenolic acid (MPA), an inhibitor of inosine monophosphate dehydrogenase (IMPDH), which catalyzes the first committed step in GMP biosynthesis. Transcription elongation mutants are hypersensitive to the depletion of GTP pools in the presence of MPA (Desmoucelles et al., 2002). Fig. 3B shows that the ysh1-L14S strain is hypersensitive to MPA as compared to the wild-type strain.
Since inactivation of the ADE2 cassette in the ysh1-L14S mutant was not caused by the repeat expansions in its intron, we sought to determine to what extent 3′-end-processing defects of ysh1-L14S affect its expression. We first compared the steady-state levels of mRNA for the normal and split ADE2 gene in the wild-type and mutant strain using RT-qPCR. Owing to the concern that a polyadenylation mutant might affect any transcript used for normalization, we extracted DNA and RNA in parallel from an equal volume of cells, which allowed us to normalize RT-qPCR products to the total DNA. Fig. 4A shows that the presence of a repeat-bearing intron within the ADE2 gene decreased its expression 6-fold compared to the intron-less gene even in the wild-type strain. This result explains the border-line ADE+ phenotype in our starting strain used for mutagenesis. In the ysh1-L14S mutant, we observe an additional drop in the mRNA level in the selectable ADE2 cassette ranging from 2-fold at 30°C to 5-fold at 37°C.
Fig. 4. RNA analysis of ADE2 cassette transcripts.

Top panel: Diagram of the ADE2 gene and ADE2-(GAA)63 cassette, indicating the position of primer pairs used for RNA analysis. (A) Results of RT-qPCR using primer pair #29, which is specific to spliced mRNA in the split ADE2 cassette. Comparing the two versions of ADE2, the presence of the intron reduces mRNA expression in both the WT and ysh1-L14S mutant background. With the split ADE2 cassette, the ysh1-L14S mutant shows decreased levels of spliced ADE2 mRNA at both 30°C and 37°C. Reverse transcription was performed using oligo-dT primers. Error bars represent the SD of qPCR technical replicates. See also Figure S2. (B) Calculation of read-through transcription levels based on qRTPCR using primer pairs before (pair #31) and after the annotated poly-A site (pair #32). In both versions of the ADE2 gene, ysh1-L14S mutants show increased levels of read-through at 30°C, with a further increase at 37°C. Reverse transcription was performed using random hexamer primers. Error bars represent the SD of four qPCR technical replicates.
We then analyzed the usage of the main ADE2 poly(A) site in the wild type and mutant strain using RT-qPCR analysis with primers upstream or downstream from this site (Fig. 4B). Read-through of the poly(A) site is drastically increased in the ysh1-L14S mutant, reaching ~20-fold more than WT at 37°C. We conclude, therefore, that the ysh1-L14S mutant is also defective for mRNA 3′ end processing in vivo and cells with this mutation likely turn red due to decreased production of polyadenylated ADE2 mRNA.
In contrast to the ADE2 cassette, the ysh1-L14S mutation did not decrease the RNA level for the repeat-bearing URA3 cassette (Fig. S2A). While we do not know why the URA3 cassette behaves differently from the ADE2 cassette, our preliminary data are indicative of a peculiar interplay between slower transcription elongation (see Fig. 6A below) and higher splicing efficiency of the long repeat-containing intron in the URA3 cassette (Fig. S2), similarly to what was discussed in (Moehle et al., 2014). Whatever the reason, the lack of URA3 repression necessitated that 5-FOA-resistant clones originating in the ysh1-L14S mutant background arose as a result of expansions of the (GAA)100 repeat.
Fig. 6. Ysh1-L14S mutant exhibits slow transcription elongation, but expansions are not affected by R-loop processing enzymes.

(A) Diagram of the modified URA3-(GAA)100 cassette placed under control of the GAL1-10 promoter. This modified cassette was used to measure transcription elongation speed via RNA polymerase clearance assays. The ysh1-L14S mutant displays markedly slower elongation speed, especially downstream of the repeat tract, as indicated by a greater fraction of RNA Pol II remaining two minutes after glucose inhibition. Error bars represent standard error of two trials. Primer pairs are numbered by the midpoint of the PCR product, with respect to the ORF start position. See also Figure S3. (B) Knockout of RNaseH1 (rnh1Δ) and RNaseH2 (rnh201Δ) (left), which remove R-loops, or overexpression of RNaseH1 (right) do not affect expansions in a ysh1-L14S mutant background. Fluctuation assays were performed using the URA3 cassette located at ARS306, with cells grown at 30°C.
Effects of the ysh1-L14S mutation on (GAA)n repeat expansions
To study the effects of ysh1-L14S mutation on repeat instability, we first compared the expansion rates for the (GAA)100 repeat within the URA3 cassette (Fig. 5A) in the wild-type and mutant strain using the fluctuation test approach conducted as described previously (Shah et al., 2012). The results shown in Fig. 5B show that even at the semi-permissive temperature (30°C), ysh1-L14S mutation elevates the expansion rate ~4-fold, while in cells pre-grown at 37°C, it was up ~10-fold as compared to the WT.
Fig. 5. Mutation in YSH1 gene increases expansions of transcribed (GAA)n repeats.

(A) Selective system to assess large-scale (GAA)n repeat expansions in a transcribed setting. Repeats are placed within an artificial intron in the URA3 counterselectable marker. This distance is length constrained, with an expansion inhibiting splicing of the intron. Fluctuation tests were performed to determine the large-scale (GAA)n expansion rate for WT and ysh1-L14S mutant strains. (B) The ysh1-L14S mutant shows increased rates of repeat expansion, which increase further under temperature-sensitive conditions. (left side of graph). (C) Selective system to assess expansions in a non-transcribed setting. Repeats are placed between the galactose promoter and its upstream activating sequence. This distance is length constrained, with an expansion shutting off expression of the CAN1 marker. (D) The ysh1-L14S mutant shows no change in the rate of repeat expansion in the non-transcribed setting. Error bars represent 95% confidence intervals of two trials. * Significantly different from WT. # Significantly different from ysh1-L14S.
A mutation in the cleavage and polyadenylation factor complex likely affects expression of numerous yeast genes. We were concerned, therefore, whether its effect on repeat expansions could be mediated by a change in expression of a gene(s) involved in repeat expansions. If this were the case, one would expect ysh1-L14S to affect expansions of both transcribed and non-transcribed repeats to a similar extent.
To distinguish between these possibilities, we studied the influence of the ysh1-L14S mutation on expansions of (GAA)n repeats within a different selection cassette, in which they are located between the galactose promoter and its upstream activating sequence (UASGAL) (Fig. 5C), a region that is practically non-transcribed. Large-scale repeat expansions shut off transcription of the CAN1 reporter, which results in the appearance of canavanine-resistant colonies (Shah et al., 2014). Fig. 5D shows that, in contrast to transcribed repeats, ysh1-L14S mutation has no effect on the expansion of the non-transcribed (GAA)100 repeat at either at 30°C or 37°C. We conclude, therefore, that transcription is required for expansion of the repeat in the ysh1 mutant. Furthermore, the mutation is probably not affecting activity of a protein that directly represses repeat expansion. There remains the possibility that the mutation of Ysh1 affects the expression of a gene that promotes expansions solely within transcribed regions. However, the results below suggest a direct role for Ysh1.
Ysh1p plays a critical role in co-transcriptional 3′ end formation and in RNA polymerase II (RNAP II) transcription termination (Garas et al., 2008; Schaughency et al., 2014). In addition, the CPF factor in which Ysh1 resides is affiliated with actively transcribed chromatin (Kim et al., 2004) and the ysh1-L14S mutant is sensitive to the MPA inhibitor of elongation (Fig. 3B). These observations raise the possibility that transcription of the (GAA)n repeats might be important for expansion induced by the ysh1 mutation.
Given that ysh1-L14S mutation specifically elevates instability of transcribed DNA repeats, we next compared transcription elongation through the URA3 cassette in this mutant compared to the wildtype strain using an RNA polymerase clearance assay (Mason and Struhl, 2005). To this end, we replaced the URA3 promoter in our selectable cassette with the inducible GAL1-10 promoter. To analyze the transcription elongation rate, cells were grown in the presence of galactose, transcription was shut down by the addition of glucose, and RNAP II distribution along the body of the cassette was measured by ChIP. Fig. 6A shows the normalized (glucose/galactose) values for Pol II occupancy, i.e. the fraction of Pol II, which failed to clear the cassette following glucose repression. One can see that only 20% of RNA Pol II remains associated with promoter-distal parts of the URA3 cassette in the wild-type strain, which is indicative of a robust transcription elongation and efficient cassette clearance. In mutant cells, in contrast, the clearance rate appears to be much slower: up to 50% of all RNAP II remain bound to the cassette after glucose repression. Elongation defects were also observed in the ysh1-L14S mutant for the YLR454, GAL10 and GAL1 genes that do not have (GAA)n repeats (Fig. S3). We conclude, therefore, that transcription elongation rate is strongly decreased in the ysh1-L14S mutant.
Slow transcription elongation is known to stimulate R-loop formation at various sequences, including (GAA)n repeats (Butler and Napierala, 2015; Groh et al., 2014). It was foreseeable, therefore, that increased R-loop formation at (GAA)n repeats in the ysh1-L14S mutant could ultimately promote repeat expansions. RNase H is known to efficiently resolve R-loops by hydrolyzing their RNA component (Hamperl and Cimprich, 2014). Thus, to evaluate a possible role of R-loop formation in our mutant, we knocked out both RNase H1 and RNase H2 in the ysh1-L14S strain and measured the rate of (GAA)n repeat expansions in the URA3 selectable cassette described above (Fig. 5A). We found that double RNase H knockout has no effect on the rate of repeat expansions in the ysh1-L14S mutant (Fig. 6B). Our alternative approach was to overexpress RNase H1, which is known to counteract R-loop formation in vivo (Wahba et al., 2011). We first introduced the plasmid overexpressing human RNase H1 described in (Wahba et al., 2011) into our ysh1-L14S strain followed by measuring (GAA)n repeat instability. It appeared that RNase H1 overexpression had little if any effect on the repeat expansion rates. The caveat of these experiments, however, was that strains carrying the RNase H1-expressing plasmid appeared to be fairly sick. Thus, we used a different approach based on the regulation of RNase H1 expression under the control of the inducible MET25 promoter (Janke et al., 2004). To this end, the promoter of the endogenous RNH1 gene in our ysh1-L14S strain was replaced with the MET25 promoter as described in the Supplemental Experimental Methods. Fig. 6B shows that the rate of repeat expansions was quantitatively the same whether the expression of RNase H1 was low (in the presence of methionine) or high (in the absence of methionine). Altogether, we conclude that the elevated expansion rate of transcribed (GAA)n repeats in the ysh1 mutant is unlikely to be caused by R-loop formation.
We have previously shown that (GAA)n repeats cause chromosomal fragility in yeast (Kim et al., 2008). Compromised transcription elongation is also known to promote the formation of double-strand breaks (Dutta et al., 2011; Nudler, 2012). It is foreseeable, therefore, that slow transcription through the repeat in the ysh1-L14S mutant could result in the formation of double-strand breaks, ultimately resulting in expansions. To test this hypothesis, we moved our URA3 selectable cassette to the non-essential arm of chromosome V, centromere-proximal to the endogenous CAN1 marker gene (Chen and Kolodner, 1999). In this setting, breakage at the (GAA)n repeats could lead to a loss of the whole chromosomal arm containing both CAN1 and the URA3 reporters, - an event which is easily detectable on selective media containing canavanine and 5-FOA. Fig. 7A shows that the rate of arm loss is indeed significantly elevated in the ysh1-L14S mutant at 37°C. Thus, ysh1-L14S mutation indeed promotes breakage of the (GAA)n repeat.
Fig. 7. Ysh1-L14S mutation leads to double strand breaks, which may be processed by HR into repeat expansions.

(A) Selective system for chromosomal arm loss at (GAA)n repeats. The original URA3-(GAA)100 cassette was moved to the non-essential arm (marked in red) of chromosome V, just upstream of the endogenous CAN1 marker gene. An unrepaired double strand break at the repeats will confer resistance to both canavanine and 5FOA. Fluctuation assay shows an increase in the arm loss rate for the ysh1-L14S mutant, which becomes significant under temperature-sensitive conditions. Error bars represent 95% confidence intervals after two trials. (B) Knockout of RAD52 (left) reduces expansions in a ysh1-L14S mutant background, while knockout of RAD51 (right) does not affect expansions. Fluctuation assays were performed using the URA3 cassette located at ARS306, with cells grown at 30°C. * Significantly different from WT. # Significantly different from ysh1-L14S. C. Proposed chain of events leading to ysh1-L14S-driven repeat expansion.
In yeast, double-strand breaks are preferably repaired via homologous recombination (HR). Misalignment of the repetitive runs in the process of recombination could ultimately result in repeat expansions (Kim et al., 2017). Thus, we decided to assess the role of the key HR proteins, Rad51 and Rad52 (Symington, 2002) on repeat expansions in the ysh1-L14S genetic background. To this end, we compared repeat expansions between a double ysh1-L14S, rad52Δ mutant and a single rad52Δ mutant, as well a double ysh1-L14S, rad51Δ mutant and a single rad51Δ mutant. Since the ysh1-L14S, rad52Δ double mutant grew very slowly at 37°C, we were only able to generate reliable expansion data at the semi-permissive temperature (Fig. 7B). Clearly, knocking down Rad52 brings the rate of repeat expansions in L14S mutant down to the wild-type level. In contrast to rad52Δ, knocking out rad51Δ did not affect the rate of repeat expansions in the WT or ysh1-L14S genetic backgrounds (Fig. 7B). We believe, therefore, that a Rad51-independent sub-pathway of homologous recombination for DSB-repair might be responsible for the elevated rate of (GAA)n repeat expansions in the ysh1 mutant.
Discussion
Our screen revealed an unanticipated connection between RNA cleavage/polyadenylation and large-scale expansions of triplet DNA repeats in S. cerevisiae. The mechanisms responsible for this link are intriguing, as repeat expansions occur in the course of DNA, rather than RNA synthesis. That being said, there exists a substantial literature showing that transcription elevates triplet repeat instability. To give just a few examples: Transcription of (CAG)n repeats increased their instability in cultured human cells in a transcription-coupled repair dependent manner (Lin et al., 2009; Lin and Wilson, 2007). Changes in the chromatin structure during repeat transcription were also shown to promote expansions by making repeats more susceptible to inherent and external damage (Debacker et al., 2012; House et al., 2014; Shah et al., 2014; Yang and Freudenreich, 2010). Additionally, a number of studies implicated R-loops in triplet repeat instability. R-loops detected at triplet repeats (Groh and Gromak, 2014; Groh et al., 2014) were proposed to account for transcription-mediated repeat instability (Lin et al., 2010; Reddy et al., 2014; Reddy et al., 2011). Similarly, R-loop formation and transcription-coupled repair protein ERCC6/CSB were implicated in CGG repeat expansions in a mouse model of the fragile X-syndrome (Zhao and Usdin, 2014). None of these studies, however, investigated the role of co-transcriptional RNA processing.
In a separate development, recent genetic and molecular analyses began to identify RNA-binding proteins (RBPs) as important players in maintaining genome stability by preventing accumulation of harmful RNA/DNA hybrids and by regulating the DNA damage response (DDR) (Dutertre et al., 2014). In S. cerevisiae, seven essential subunits of the mRNA cleavage and polyadenylation machinery were implicated in DDR triggered by R loops (Stirling et al., 2012). Knockout of the TRF4 gene, encoding a non-canonical polyA-polymerase involved in RNA surveillance, gave rise to a transcription-associated recombination phenotype (Gavalda et al., 2013). Cleavage Factor I was shown to contribute to genome integrity by preventing replication hindrance (Gaillard and Aguilera, 2014). Similarly, S. pombe cleavage and polyadenylation factor Rna14 was implicated in the maintenance of genomic integrity (Sonkar et al., 2016). None of these studies, however, looked at triplet repeat expansions and/or fragility.
Contrary to the above examples, we found that RNA/DNA hybrids are not likely to be involved in elevated repeat instability in the ysh1-L14S mutant background (Fig. 6B). This difference may be due to the unique role that Ysh1 protein plays in RNA processing. Aguilera’s group has proposed that mutations in RNA binding proteins lead to their absence from the nascent RNA during transcription, which, in turn, allows this naked RNA to stably pair with its DNA template (Dominguez-Sanchez et al., 2011). We don’t think that mutations in Ysh1 protein would result in the presence of naked RNA during transcription, as other members of the CPSF complex are still expected to be bound to RNA. At the same time, we have demonstrated that mutations in the Ysh1 protein significantly slow down RNA polymerase progression (Fig. 6A), likely because it remains bound to the transcript, but cannot cleave efficiently. It was also demonstrated by others that transient depletion of Ysh1p triggers transcriptional pausing downstream of known polyadenylation sites (Schaughency et al., 2014).
Our working model combines the above observations with the data from this study. A mutation in the Ysh1 protein, which was isolated from our repeat expansion screen, cause defects in transcript cleavage and polyadenylation (Fig. 3). As this process occurs co-transcriptionally, we reasoned that the entire RNA Pol II elongating complex may slow or stall at potential poly(A) sites on the DNA template when Ysh1 is not efficient. In our mutants, transcription elongation is significantly slowed down across the whole URA3 cassette (Fig. 6A). Transcription stalling and backsliding is known to trigger the formation of double-stranded breaks in DNA, owing to their collisions with replication machinery or other mechanisms (Mirkin et al., 2006; Nudler, 2012). We do see elevated fragility of the (GAA)n run in the Ysh1 mutant, which is consistent with DSB formation. When homologous recombination machinery attempts to repair the broken DNA ends, repetitive DNA strands can align out of register, resulting in repeat expansions after the next round of replication (Fig. 7C). Supporting this reasoning, the increase in repeat expansions in the ysh1-L14S mutant fades when homologous recombination is completely shut down in the RAD52 knockout. At the same time, repeat expansions in the ysh1-L14S mutant were not diminished in the RAD51 knockout, indicating that a Rad51-independent sub-pathway of HR is either responsible for the expansions, or can compensate in the absence of canonical Rad51-dependent HR. One possibility is the involvement of the single-strand annealing pathway (SSA), which is known to act within repetitive regions and is not dependent on Rad51 protein (Downing et al., 2008). Another possibility is a Rad51-independent wing of the break-induced replication pathway (Ira and Haber, 2002).
While our studies were performed in S. cerevisiae, they may have implications for Friedrich’s ataxia in humans. An interesting repercussion from the transcription repeat breakage model is that expansions may pre-nucleate outside of the S-phase. This phenomenon might therefore shed light on how repeat expansions can occur in non-dividing neural and cardiac cells (McMurray, 2010). It would be of prime interest to investigate whether Friedreich’s ataxia patients carrying mutations in the YSH1 homolog CPSF-73 or other RNA processing genes might be at higher risk for repeat expansions, accounting for the variation in disease severity and age of onset between different individuals. Even in the absence of germline mutations in CPSF complex, transcription may already proceed more slowly through (GAA)n repeats (Krasilnikova et al., 2007). Transcriptional blocks at the (GAA)n repeat within the FRDA locus could become prominent in specific cell lineages or arise transiently to produce large-scale expansions in non-dividing cells. This can hint at a potential therapy, if it becomes possible to prevent RNA polymerase stalling at the repeat (Gottesfeld et al., 2013; Soragni et al., 2014). Reducing transcription pausing at (GAA)n repeats may both reduce DNA breakage and rescue the poorly expressed mutant allele of the FXN gene.
Experimental Procedures
Yeast strain construction
The list of our strains is presented in Table S1. See Supplemental Experimental Methods for further details.
Fluctuation assays
Fluctuation assays were performed as previously described (Shah et al., 2014; Shishkin et al., 2009). See Supplemental Experimental Methods for further details.
In Vitro 3′ End processing
Processing extracts were prepared as described (Zhao et al., 1999) using strains SMY732 and RMG89, which were grown at 30°C and then shifted to 37°C for 1.5h. Extracts were incubated with ATP and full-length or pre-cleaved 32P-labeled GAL7-1 RNA. Reaction products were run on a polyacrylimide urea gel and visualized via phosphoimager.
Quantitative RNA analysis
RNA levels were measured via qRT-PCR, employing a strategy wherein gDNA was extracted from an equal portion of the yeast culture used to extract RNA. See Supplemental Experimental Methods for further details.
RNA Pol II elongation assays
Assays were performed as previously described (Mason and Struhl, 2005). See Supplemental Experimental Methods for further details.
Supplementary Material
Highlights.
Genetic screen: UV mutagenesis → select for repeat expansions → genome sequencing
Point mutants in essential gene YSH1 increase the rate of GAA repeat expansions
YSH1 mutation → slow transcription elongation → DSB → repeat expansions
Acknowledgments
We thank Kartik Shah for sharing his experience with the CAN1 selectable cassette, Allen Su for sharing his experience with RNaseH1 overexpression and Doug Koshland for plasmids overexpressing RNase H1. Research in the lab of S.M.M. is supported by NIH grants R01GM60987 and P01GM105473 and generous contribution from the White family. Research in the lab of C.M. was supported by grant MCB-1244043 from NSF and an NIGMS Post-Baccalaureate Research Education Program Award 2R25GM066567. Anna Aksenova was supported by RFBR grant #15-04-08658 and research project in the Centre for Molecular and Cell Technologies (Research Park, Saint-Petersburg State University, Russia).
Footnotes
Author contributions
Conceptualization, R.J.M., A.Y.A., E.T.W. and S.M.M. Software, R.J.M. Investigation, R.J.M., F.P., A.Y.A, J.A.H., E.L.P. and E.T.W. Resources, A.A.S. Writing – Original Draft, R.J.M. and S.M.M. Writing – Review and Editing, R.J.M., C.M. and S.M.M. Visualization, R.J.M. and F.P. Supervision, D.E.H., C.M. and S.M.M. Funding acquisition, A.Y.A., S.M.M., C.M.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7(Unit7):20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelemy J, Hanenberg H, Leffak M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2016;44:6803–6816. doi: 10.1093/nar/gkw433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JS, Napierala M. Friedreich’s ataxia--a case of aberrant transcription termination? Transcription. 2015;6:33–36. doi: 10.1080/21541264.2015.1026538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Choi EA, Shi Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA. 2011;2:321–335. doi: 10.1002/wrna.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanfreau G, Noble SM, Guthrie C. Essential yeast protein with unexpected similarity to subunits of mammalian cleavage and polyadenylation specificity factor (CPSF) Science. 1996;274:1511–1514. doi: 10.1126/science.274.5292.1511. [DOI] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol. 2012;10:e1001257. doi: 10.1371/journal.pbio.1001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmoucelles C, Pinson B, Saint-Marc C, Daignan-Fornier B. Screening the yeast “disruptome” for mutants affecting resistance to the immunosuppressive drug, mycophenolic acid. J Biol Chem. 2002;277:27036–27044. doi: 10.1074/jbc.M111433200. [DOI] [PubMed] [Google Scholar]
- Dominguez-Sanchez MS, Barroso S, Gomez-Gonzalez B, Luna R, Aguilera A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011;7:e1002386. doi: 10.1371/journal.pgen.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing B, Morgan R, VanHulle K, Deem A, Malkova A. Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat Res. 2008;645:9–18. doi: 10.1016/j.mrfmmm.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre M, Lambert S, Carreira A, Amor-Gueret M, Vagner S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem Sci. 2014;39:141–149. doi: 10.1016/j.tibs.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard H, Aguilera A. Cleavage factor I links transcription termination to DNA damage response and genome integrity maintenance in Saccharomyces cerevisiae. PLoS Genet. 2014;10:e1004203. doi: 10.1371/journal.pgen.1004203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garas M, Dichtl B, Keller W. The role of the putative 3′ end processing endonuclease Ysh1p in mRNA and snoRNA synthesis. RNA. 2008;14:2671–2684. doi: 10.1261/rna.1293008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavalda S, Gallardo M, Luna R, Aguilera A. R-loop mediated transcription-associated recombination in trf4Delta mutants reveals new links between RNA surveillance and genome integrity. PLoS One. 2013;8:e65541. doi: 10.1371/journal.pone.0065541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington’s Disease C. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesfeld JM, Rusche JR, Pandolfo M. Increasing frataxin gene expression with histone deacetylase inhibitors as a therapeutic approach for Friedreich’s ataxia. J Neurochem. 2013;126(Suppl 1):147–154. doi: 10.1111/jnc.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Gromak N. Out of balance: R-loops in human disease. PLoS Genet. 2014;10:e1004630. doi: 10.1371/journal.pgen.1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Lufino MM, Wade-Martins R, Gromak N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014;10:e1004318. doi: 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 2014;19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House NC, Yang JH, Walsh SC, Moy JM, Freudenreich CH. NuA4 initiates dynamic histone H4 acetylation to promote high-fidelity sister chromatid recombination at postreplication gaps. Mol Cell. 2014;55:818–828. doi: 10.1016/j.molcel.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Haber JE. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol. 2002;22:6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, Lobachev KS. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 2008;27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Harris ST, Dinter T, Shah KA, Mirkin SM. The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol. 2017;24:55–60. doi: 10.1038/nsmb.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 2013;23:280–288. doi: 10.1016/j.gde.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ahn SH, Krogan NJ, Greenblatt JF, Buratowski S. Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J. 2004;23:354–364. doi: 10.1038/sj.emboj.7600053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet. 2001;27:407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18:198–213. doi: 10.1038/cr.2008.5. [DOI] [PubMed] [Google Scholar]
- Krasilnikova MM, Kireeva ML, Petrovic V, Knijnikova N, Kashlev M, Mirkin SM. Effects of Friedreich’s ataxia (GAA)n*(TTC)n repeats on RNA synthesis and stability. Nucleic Acids Res. 2007;35:1075–1084. doi: 10.1093/nar/gkl1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Dent SY, Wilson JH, Wells RD, Napierala M. R loops stimulate genetic instability of CTG.CAG repeats. Proc Natl Acad Sci U S A. 2010;107:692–697. doi: 10.1073/pnas.0909740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Hubert L, Jr, Wilson JH. Transcription destabilizes triplet repeats. Mol Carcinog. 2009;48:350–361. doi: 10.1002/mc.20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–6217. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Chen X, Gao Y, Lewis T, Barthelemy J, Leffak M. Altered replication in human cells promotes DMPK (CTG)(n). (CAG)(n) repeat instability. Mol Cell Biol. 2012;32:1618–1632. doi: 10.1128/MCB.06727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Castel A, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, Tong L. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature. 2006;444:953–956. doi: 10.1038/nature05363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins S, Pearson CE, Coutinho P, Provost S, Amorim A, Dube MP, Sequeiros J, Rouleau GA. Modifiers of (CAG)(n) instability in Machado-Joseph disease (MJD/SCA3) transmissions: an association study with DNA replication, repair and recombination genes. Hum Genet. 2014;133:1311–1318. doi: 10.1007/s00439-014-1467-8. [DOI] [PubMed] [Google Scholar]
- Mason AG, Tome S, Simard JP, Libby RT, Bammler TK, Beyer RP, Morton AJ, Pearson CE, La Spada AR. Expression levels of DNA replication and repair genes predict regional somatic repeat instability in the brain but are not altered by polyglutamine disease protein expression or age. Hum Mol Genet. 2014;23:1606–1618. doi: 10.1093/hmg/ddt551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, Struhl K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell. 2005;17:831–840. doi: 10.1016/j.molcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- McMurray CT. Hijacking of the mismatch repair system to cause CAG expansion and cell death in neurodegenerative disease. DNA Repair. 2008;7:1121–1134. doi: 10.1016/j.dnarep.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010;38:2757–2774. doi: 10.1093/nar/gkp1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Castro Roa D, Nudler E, Mirkin SM. Transcription regulatory elements are punctuation marks for DNA replication. Proc Natl Acad Sci USA. 2006;103:7276–7781. doi: 10.1073/pnas.0601127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Moehle EA, Braberg H, Krogan NJ, Guthrie C. Adventures in time and space: splicing efficiency and RNA polymerase II elongation rate. RNA Biol. 2014;11:313–319. doi: 10.4161/rna.28646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, Wilson RH, Adam B, del Valle G, Brian R, et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Human molecular genetics. 2012;21:3558–3567. doi: 10.1093/hmg/dds185. [DOI] [PubMed] [Google Scholar]
- Morales F, Vasquez M, Santamaria C, Cuenca P, Corrales E, Monckton DG. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst) 2016;40:57–66. doi: 10.1016/j.dnarep.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Nudler E. RNA polymerase backtracking in gene regulation and genome instability. Cell. 2012;149:1438–1445. doi: 10.1016/j.cell.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Boudvillain M, Libri D. Transcription Termination: Variations on Common Themes. Trends Genet. 2016;32:508–522. doi: 10.1016/j.tig.2016.05.007. [DOI] [PubMed] [Google Scholar]
- Reddy K, Schmidt MH, Geist JM, Thakkar NP, Panigrahi GB, Wang YH, Pearson CE. Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic Acids Res. 2014;42:10473–10487. doi: 10.1093/nar/gku658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2011;39:1749–1762. doi: 10.1093/nar/gkq935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K, Calvo O, Manley JL. Evidence that polyadenylation factor CPSF-73 is the mRNA 3′ processing endonuclease. RNA. 2004;10:565–573. doi: 10.1261/rna.5214404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, Junien C, Gourdon G. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 2003;22:2264–2273. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savouret C, Garcia-Cordier C, Megret J, te Riele H, Junien C, Gourdon G. MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Molecular and cellular biology. 2004;24:629–637. doi: 10.1128/MCB.24.2.629-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaughency P, Merran J, Corden JL. Genome-wide mapping of yeast RNA polymerase II termination. PLoS Genet. 2014;10:e1004632. doi: 10.1371/journal.pgen.1004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KA, McGinty RJ, Egorova VI, Mirkin SM. Coupling transcriptional state to large-scale repeat expansions in yeast. Cell Rep. 2014;9:1594–1602. doi: 10.1016/j.celrep.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KA, Shishkin AA, Voineagu I, Pavlov YI, Shcherbakova PV, Mirkin SM. Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2012;2:1088–1095. doi: 10.1016/j.celrep.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkar A, Yadav S, Ahmed S. Cleavage and polyadenylation factor, Rna14 is an essential protein required for the maintenance of genomic integrity in fission yeast Schizosaccharomyces pombe. Biochim Biophys Acta. 2016;1863:189–197. doi: 10.1016/j.bbamcr.2015.11.007. [DOI] [PubMed] [Google Scholar]
- Soragni E, Miao W, Iudicello M, Jacoby D, De Mercanti S, Clerico M, Longo F, Piga A, Ku S, Campau E, et al. Epigenetic therapy for Friedreich ataxia. Ann Neurol. 2014;76:489–508. doi: 10.1002/ana.24260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012;26:163–175. doi: 10.1101/gad.179721.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tous C, Rondon AG, Garcia-Rubio M, Gonzalez-Aguilera C, Luna R, Aguilera A. A novel assay identifies transcript elongation roles for the Nup84 complex and RNA processing factors. EMBO J. 2011;30:1953–1964. doi: 10.1038/emboj.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin K, House NC, Freudenreich CH. Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol. 2015;50:142–167. doi: 10.3109/10409238.2014.999192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica-Ross M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell. 2011;44:978–988. doi: 10.1016/j.molcel.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Freudenreich CH. The Rtt109 histone acetyltransferase facilitates error-free replication to prevent CAG/CTG repeat contractions. DNA Repair. 2010;9:414–420. doi: 10.1016/j.dnarep.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhu Y, Chen-Plotkin AS, Clay-Falcone D, McCluskey L, Elman L, Kalb RG, Trojanowski JQ, Lee VM, Van Deerlin VM, et al. PolyQ repeat expansions in ATXN2 associated with ALS are CAA interrupted repeats. PLoS One. 2011;6:e17951. doi: 10.1371/journal.pone.0017951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, Lobachev KS. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Molecular cell. 2012;48:254–265. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999a;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kessler M, Helmling S, O’Connor JP, Moore C. Pta1, a component of yeast CF II, is required for both cleavage and poly(A) addition of mRNA precursor. Mol Cell Biol. 1999b;19:7733–7740. doi: 10.1128/mcb.19.11.7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kessler MM, Moore CL. Cleavage factor II of Saccharomyces cerevisiae contains homologues to subunits of the mammalian Cleavage/ polyadenylation specificity factor and exhibits sequence-specific, ATP-dependent interaction with precursor RNA. J Biol Chem. 1997;272:10831–10838. doi: 10.1074/jbc.272.16.10831. [DOI] [PubMed] [Google Scholar]
- Zhao XN, Usdin K. Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum Mutat. 2014;35:341–349. doi: 10.1002/humu.22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.