

Abstract
Inflammation is a critical factor in early atherosclerosis and its progression to myocardial infarction. The search for valid surrogate markers of arterial vascular inflammation led to the increasing use of positron emission tomography (PET)–computed tomography. Indeed, vascular inflammation is associated with future risk of MI, and can be modulated with short-term therapies, such as statins, that mitigate cardiovascular risk. However, to better understand vascular inflammation and its mechanisms, we recently convened a panel of world experts in immunology, human translational research, and PET vascular imaging. This contemporary review first strives to understand the diverse roles of immune cells implicated in atherogenesis. Next, we describe human chronic inflammatory disease models that can help elucidate the pathophysiology of vascular inflammation. Finally, we review PET-based imaging techniques to characterize the vessel wall in vivo.
Keywords: cardiovascular imaging, monocytes, neutrophils, T cells
INTRODUCTION
Cardiovascular disease (CVD) remains the leading cause of death worldwide, highlighting the need to elucidate its pathogenesis. Once considered a passive biological process, CVD is now recognized as an active, immune-driven process that may begin in childhood (1). Current research into the natural history of atheroma development has implicated many immune cells, including phagocytes, lymphocytes, dendritic cells, and neutrophils (2). Because these cells play a major role in initiating plaque development and complication, leukocytes are promising targets for acute and chronic atherosclerosis therapy. However, the complexity of the immune system and its role as a defensive force against infection requires novel tools to precisely identify and treat only the inflammatory cells or processes that promote atherosclerosis. Biomedical engineering, specifically in human imaging, offers unique possibilities for diagnosing and treating atherosclerotic plaque inflammation before cardiovascular (CV) events. Thus, interfacing novel engineering to enhance human imaging with immunology will be essential to accelerate advances in management of this disease.
In this review, we begin with an overview of the emerging understanding of CVD as a systemic inflammatory disorder relating to monocytes, macrophages, neutrophils, and T cells. We then discuss specific human conditions with increased CVD risk to study the natural history of atherosclerosis, including human immunodeficiency virus (HIV), systemic lupus erythematosus (SLE), and psoriasis as human models of vascular disease initiation and progression. Finally, we review current applications of positron emission tomography (PET) imaging and emerging PET tracer agents used in vascular characterization.
IMMUNOLOGY OF INFLAMMATION AS IT PERTAINS TO THE VESSEL WALL
MONOCYTES AND MACROPHAGES
The most numerous cells in atherosclerotic plaques are macrophages (3), leukocytes that are central to innate immunity. In atherosclerosis, macrophage accumulation commences as bone marrow–derived, Ly6Chigh monocytes, infiltrate the lesion. These Ly6Chigh inflammatory monocytes exit the bone marrow in a C-C motif chemokine receptor 2 (CCR2)–dependent manner, accumulate in the vessel wall, and differentiate into macrophages, which are sustained through self-renewal (4). Notably, as atherosclerosis progresses, local macrophage proliferation, rather than monocyte recruitment, becomes more important in lesion growth.
In addition to the accumulation of monocytes in atherosclerotic lesions, these innate immune cells contribute to the biological response following a myocardial infarction (MI). Monocytes are both destructive and protective, in that they give rise to infarct rupture and contribute to infarct healing. However, an overabundance of monocytes can interfere with healing, resulting in heart failure. In an acute MI, an oversupply of monocytes to the aorta is rapid, concomitant with a reduction in C-X-C motif chemokine ligand 12 (CXCL12) expression in the bone marrow. Diminished CXCL12 expression enables myeloid cells and their progenitors to exit the bone marrow and take up residence in the spleen, where they trigger extramedullary hematopoiesis (5). Additionally, differentiated leukocytes, especially monocytes and neutrophils, take up residence in end-organ tissues, giving rise to plaques and inflammation. The vascular sympathetic innervation (i.e., nerve fibers travelling along the aorta and arterioles) plays a role in the increased emergency supply of leukocytes. In the periphery, sympathetic innervation activates endothelial cells on the luminal surface of atherosclerotic plaques, increasing adhesion molecule expression, which augments leukocyte recruitment (6). To directly investigate the role of the vascular sympathetic innervation system in atherogenesis, the role of stress in monocyte proliferation is a topic of ongoing investigation. Stress elevates noradrenaline levels in the bone marrow and activates bone marrow stem cells (7). Hematopoietic stem and progenitor cell proliferation is significantly enhanced, CXCL12 is lowered, and monocytes enter the systemic circulation in increased numbers. Collectively, these mechanisms suggest a multiorgan communication system that activates the bone marrow through the sympathetic innervation system, increasing hematopoietic stem and progenitor cell proliferation and thus enhancing leukocytosis. Therefore, current studies aim to unravel the systemic mechanisms that control the production, recruitment, differentiation, and proliferation of monocytes and their descendent macrophages in atherosclerosis, to determine how these processes can be balanced to exert the most benefit, and to elucidate specific control points in atherogenesis.
NEUTROPHILS
Neutrophils are increasingly recognized in the initiation of atherosclerotic plaque development. Neutrophils are the initial immune cell to infiltrate inflammatory sites produced by injury or infection. Although the antimicrobial action of neutrophils is indispensable to combat infection, their mechanisms of action yield significant tissue damage and toxic debris. A newly-identified defense mechanism is the ability of neutrophils to form neutrophil extracellular traps (NETs)(8); however, NET formation, or “NETosis,” remains poorly understood. Currently, 2 distinct mechanisms have been described, suicidal and vital NETosis. During suicidal NETosis, the internal membranes of the neutrophils dissolve, followed by cell lysis and decondensed chromatin release (8). During an infectious process, neutrophils directly secrete nuclear contents from the cell without killing the neutrophils (9). Although many questions remain unanswered regarding NETosis, it is evident that NETs themselves are proinflammatory, induce endothelial and tissue damage, and are highly prothrombotic (Figure 1) (10). Additionally, NETs provide a communication platform between neutrophils and macrophages, priming macrophages to produce pro-interleukin (IL)1β, culminating in atherosclerotic plaque destabilization (11).
Figure 1. NETosis Induces Vascular Damage.

Neutrophil extracellular traps (NETs) in the time line of deep vein thrombosis (DVT): a model. (A) DVT is initiated by local hypoxia and activation of endothelial cells (ECs) as a result of flow restriction/disturbances. Activated endothelium releases ultralarge von Willebrand factor (ULVWF) and P-selectin from Weibel-Palade bodies (WPB), which mediate platelet and neutrophil adhesion. Activated platelets recruit tissue factor (TF)–containing microparticles that enhance thrombin generation in the growing thrombus. (B) Activated platelets and endothelium or other stimulus induce NET formation in adherent neutrophils. NETs provide an additional scaffold for platelet and red blood cell (RBC) adhesion, promote fibrin formation, and exacerbate platelet and endothelial activation. (C) Plasmin, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), and deoxyribonuclease (DNase) mediate thrombolysis by degrading fibrin, ULVWF, and DNA, respectively. Monocytes/macrophages (Mø) release an additional source of DNase and generate plasmin and promote restoration of blood flow (10).
NET formation is a peptidylarginine deiminase-4 (PAD4)-dependent mechanism induced by extracellular stimuli via microbes, activated platelets, cytokines, antibodies to neutrophils, hypoxia, and cholesterol crystals. Upon activation, PAD4 converts arginine residues on histones to citrulline, triggering chromatin decondensation and NET release (12). Consequently, PAD4 is expelled in conjunction with a milieu of toxic proteins, resulting in citrullination of surrounding proteins, altering their functional properties and producing neoantigens, which may play a role in autoimmune diseases (13). In a murine model of MI, ischemia of the heart induces NETosis. Furthermore, PAD4−/−, a murine model incapable of NETosis, displays smaller infarct size and has significantly better heart function, demonstrated by an elevated ejection fraction subsequent to an ischemic event (14). In aged or diabetic mice, neutrophils are primed for NETosis, producing excessive thrombosis and inflammation (15). Myocardial NET deposition delays wound healing, leading to fibrosis in the cardiac pressure-overload model. Intriguingly, spontaneous fibrosis of organs produced by aging is greatly reduced in PAD4−/− mice. Functionally, PAD4−/− hearts are comparable to young murine hearts, and their systolic and diastolic function does not decline with age (15). Thus, there is recent interest in further study of the specific roles of neutrophils, NETosis, and the PAD4 pathway in atherosclerosis.
T CELLS
When immune effector mechanisms are active within the blood vessel wall or endothelial surface, vascular dysfunction, thrombosis, and ischemia usually result. T cells play important roles in the promotion and regulation of inflammation in atherosclerotic lesions. Human arteries have interferon-γ (IFN-γ)– and IL-17–producing CD4+ T cells in atherosclerotic lesions, and mouse studies have shown that IFN-γ–producing Th1 cells promote atheroma development (16). Consistent with the findings that T cells are involved in plaque inflammation, murine models show that the B7-CD28 T-cell costimulatory pathway is involved in promoting proatherogenic T-cell responses, as well as atheroprotective regulatory T-cell responses (17). Importantly, murine models indicate that inhibitory members of the B7-CD28 family, in particular PD-L1 (B7-H1) expressed on antigen-presenting cells, endothelium, and the cells of various tissues, and its receptor on T-cells, PD-1, are important in suppressing T-cell–driven inflammation in arteriosclerosis and myocytes (18). For example, mice lacking PD-L1 or PD-1 display a marked increase in CD4+ and CD8+–mediated inflammation in arterial lesions. Mice lacking PD-1 or PD-L1, or treated with PD-1 blockade, are more susceptible to T-cell–mediated myocardial injury. Furthermore, IFNγ-induced up-regulation of PD-L1 by heart endothelial cells in vitro or in vivo protects against CD8+ cytotoxic T lymphocyte–mediated damage (19).
Mouse studies demonstrating the protective roles of the PD-1 pathway in arteries and heart highlight the possibility of increasing CV risks by targeting PD-1 or PD-L1 in cancer immunotherapy. In fact, many cases of acute severe lymphocytic myocarditis are now being reported in the context of checkpoint blockade cancer immunotherapy, including patients treated with anti–PD-1 (20). Histopathological analyses of tissues from some of these cases reveal up-regulation of endothelial HLA-DR (Human Leukocyte Antigen–antigen D Related), as well as myocyte and endothelial PD-L1 associated with T-cell infiltrates, consistent with an IFN-mediated effect.
Notably, CVD initiation and progression involves biological activity from multiple immune cells, both innate and adaptive. Future studies deciphering the interplay among these immune cells in CVD are critical for developing therapies targeting CVD initiation in order to reduce clinical CVD outcomes, and ultimately decrease CVD prevalence.
HUMAN DISEASE MODELS OF IMAGING TO STUDY VASCULAR INFLAMMATION
Currently, 2 large ongoing CV trials in patients with prior MI are testing if treatment of inflammation will reduce a second CV event: CIRT (Cardiovascular Inflammation Reduction Trial), and inhibition of IL-1B in CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcomes Study). There is an initial report that the CANTOS trial met the primary endpoint for a reduction in recurrent major adverse CV events. These trials will provide critical data on whether inhibition of nonspecific T-cell inflammation (methotrexate) or inflammasome activation (canakinumab) reduces further CV events in high-risk patients. As the results of these trials become available, emerging data from human chronic diseases associated with high CV risk and systemic inflammation provide models to understand vascular disease initiation and progression. Indeed, 18F-fluorodeoxyglucose (FDG)-PET CT has been used to characterize vascular inflammation (VI) in HIV, psoriasis, and SLE (Figure 2).
Figure 2. Vascular Images of Chronically Inflamed Human Models.

Representative 18F-FDG-PET/CT imaging of the aorta in a healthy volunteer (A), compared with the aortas of patients with human immunodeficiency virus (B), psoriasis (C), and systemic lupus erythematosus (D). CT = computed tomography; FDG = fluorodeoxyglucose; PET = positron emission tomography.
HUMAN IMMUNODEFICIENCY VIRUS
HIV treatment has become very effective over the last decade; however, the rate of MI, stroke, and sudden cardiac death remains elevated 50% to 100% in HIV (21). Although dyslipidemia is more prevalent in HIV, traditional risks, including dyslipidemia, hypertension, and diabetes only account for 25% of the excess risk. Using coronary CT angiography (CCTA), a study demonstrated a high prevalence of noncalcified plaque with high-risk morphological features, associated with increased immune activation indexes (22) and inflammation, imaged by FDG-PET remains elevated after effective antiretroviral therapy (23).
Among HIV patients, coronary plaques are often inflamed and noncalcified (24), and patients exhibit increased myocardial fibrosis imaged by cardiac magnetic resonance (CMR) (25). Imaging with FDG has demonstrated that HIV-infected subjects with an undetectable viral load have an increased aortic target-to-background ratio compared with healthy controls and subjects with known CVD, indicating significant arterial inflammation, even in the context of immune restoration and viral suppression (Figure 3) (26). Together, CCTA and FDG-PET have helped to identify the unique pathophysiology of arterial inflammation and plaque in HIV, proven to be a readout for the efficacy of anti-inflammatory strategies now being targeted in HIV, including newer statins. Statins effectively decrease noncalcified plaque as well as low-density lipoprotein in patients with HIV (27). Based on these data, a large trial of 6,500 patients with HIV and a low-to-moderate risk of CVD is now underway to test whether pitavastatin can prevent CV events by reducing low-density lipoprotein and concomitantly improve inflammatory pathways of immune activation (The Randomized Trial to Prevent Vascular Events in HIV [REPRIEVE]: NCT02344290).
Figure 3. NaF Uptake in the Coronary Arteries.
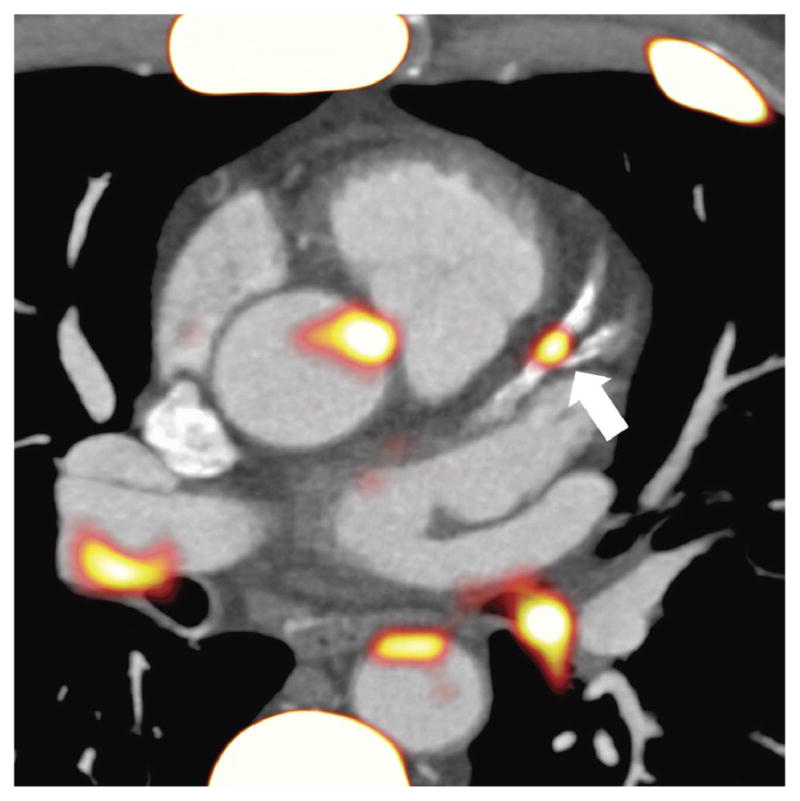
A discrete focus of fluorine 18 sodium fluoride (18F-NaF) uptake overlying an otherwise heavily calcified left anterior descending artery, suggesting a locus of active calcification with potentially increased vulnerability for rupture.
PSORIASIS
Psoriasis is a chronic T helper 1 cell (Th1) T helper 17 cell (Th17) inflammatory disease that affects more than 125 million people worldwide and about 2% to 4% of the adult population, with the most common type manifesting as plaque psoriasis. The pathophysiology of the disease is localized skin inflammation, epidermal hyperproliferation, up-regulated T-cell and neutrophil activation, and increases in C-reactive protein, serum amyloid A, and intercellular adhesion molecular 1 (ICAM-1) (28). Consistent with chronically inflamed pathologies, large population-based epidemiological studies suggest that patients with psoriasis, particularly moderate to severe, have an increased risk of MI, stroke, and CV mortality independent of traditional risk factors (29–31). Notably, studies demonstrate strong associations of psoriasis and CVD with increasing severity of skin disease. Patients with moderate to severe psoriasis experience premature mortality, dying approximately 5 years younger than their nonpsoriatic counterparts in adjusted analysis (32). Over 10-year periods, patients with moderate to severe psoriasis have about 6% excess risk of developing a major CV event, beyond the conventional Framingham risk score calculation (33). Interestingly, translational studies have demonstrated dramatic up-regulation of genes known to adversely affect CV risk in the skin lesions of psoriasis. Furthermore, transgenic mouse models of psoriasis in which the genetic construct is confined to the skin demonstrate aortic inflammation and thrombosis (34). Similarly, patients with psoriasis have an increased amount of aortic FDG-PET activity that is positively correlated with skin disease severity (35).
There are a series of randomized, placebo-controlled clinical trials evaluating the impact of treatments such as adalimumab compared to phototherapy (NCT01553058), ustekinumab (NCT02187172), and secukinumab (NCT02690701) on aortic inflammation (measured by FDG-PET) and CV biomarkers in psoriasis. These trials will provide greater insight into the ability of anti-inflammatory therapies to improve the risk of CVD.
SYSTEMIC LUPUS ERYTHEMATOSUS
Patients with SLE have a heightened risk of developing atherosclerosis, including MI and stroke (36), with young women having up to a 50-fold increased risk of developing vascular events (37). Furthermore, even SLE patients with no prior CVD events have subclinical vascular dysfunction, endothelial dysfunction, arterial stiffness, myocardial perfusion abnormalities, carotid plaque, and coronary calcification (36) not fully explained by Framingham risk score or by medications used to treat SLE (36).
Altered innate immune responses characteristic of SLE contribute to the development of lupus vasculopathy and atherosclerotic plaque formation (38). Type I IFNs, a group of cytokines elevated in many patients with SLE, particularly during periods of flare, have pleiotropic deleterious roles in the vasculature (39). Type I IFNs independently associated with endothelial function, carotid plaque, and severity of coronary calcification in patients without a history of CVD (40). Specifically, these cytokines promote an imbalance of enhanced endothelial cell damage and decreased vascular repair that may promote initiation of vasculopathy, and contribute to the development of foam cell formation and platelet activation. SLE is also characterized by the presence of a distinct subset of pathogenic neutrophils, low-density granulocytes, which have an enhanced capacity to form NETs (38,39). NETs can amplify inflammatory responses in the plaque and other tissues, including inflammasome activation in macrophages and induction of type I IFN synthesis by plasmacytoid dendritic cells (38,41).
Two pathways are of potential interest in targeting vascular risk in SLE: the inhibition of type 1 IFNs and the blockage of NETosis through PAD-4 inhibition. Type 1 IFNs signal through the JAK/STAT pathway, which is inhibited by tofacitinib. Tofacitinib ameliorated vascular dysfunction in a murine lupus model (42), and is being tested in humans in an ongoing clinical trial (NCT02535689). Inhibition of pathways implicated in NET formation using chemical inhibitors of PAD-4 leads to amelioration of endothelial dysfunction, a prothrombotic phenotype, and plaque formation in murine models of lupus and atherosclerosis (41). Overall, SLE represents a unique model for understanding the role of the innate immune system in the development of vascular disease, and may allow for the characterization of novel therapeutic targets in subsets of patients.
PET IMAGING OF THE ARTERY WALL
Currently, FDG-PET imaging is a cost-effective clinical standard of care for diagnosis, staging, and monitoring the response of many malignancies, and its role is significantly enhanced by the introduction of PET-CT and PET-CMR. Imaging human inflammation models with PET allows investigations to be conducted without expensive animal models, a step towards clinical translation.
PET imaging of vascular inflammation (VI), using FDG was first described more than 15 years ago (43,44), and has become important in atherosclerosis research. FDG accumulates within living cells in proportion to their glycolytic rates (45). Several tissues have particularly high glycolytic rates and hence tend to accumulate FDG, including tumors, brain tissue, myocytes, and inflammatory cells. Phagocytes have particularly high glycolytic rates, especially after proinflammatory activation (46), leading to high FDG retention. A potential advantage of PET imaging in addition to other available anatomic imaging techniques (e.g., CT, CMR, and ultrasound) is through the ability to administer miniscule concentrations of a substance that can be targeted for a specific molecular process. PET images can be interpreted both qualitatively and quantitatively to evaluate early phases of disease or short-term changes in VI related to treatment that may not have concordant changes in morphology. Several studies show that FDG uptake, when measured in the arterial wall in vivo, reflects the level of macrophage accumulation within the atheroma (47). Atherosclerotic lesions show higher VI by FDG and tend to experience greater subsequent progression (48). Moreover, the FDG measurement of VI may provide an independent index of the risk of subsequent CV events (48–50), which is the subject of larger ongoing prospective studies (51) to evaluate the standardization of ideal imaging procedures (52,53) and subsequent clinical application.
FDG imaging of VI is increasingly used to evaluate therapeutic compounds targeting atherosclerosis. The best therapies studied in this regard are statins, which show a reduction in the arterial FDG signal consistent with their impact on CV events in randomized clinical trials (54,55). However, a lack of reduction in arterial FDG uptake associates with a parallel failure to reduce CV events for a number of experimental treatments (56–58). Prospective outcome data and response to novel therapies using FDG in vascular diseases are limited. However, given the general concordance between VI imaging and CVD outcomes, relatively small and swift FDG imaging studies may facilitate drug discovery.
Although FDG imaging adequately addresses certain pathophysiological and treatment-related questions, the specificity for inflammation with this agent is not clearly defined due to the variable affinity for glucose of all cells in the body. Additionally, high FDG activity in the blood pool and tissues near the vessel wall complicates quantification. Thus, there are opportunities for other imaging agents for atherosclerosis, such as vascular calcification imaged with 18F-sodium fluoride (NaF). Originally Food and Drug Administration–approved for bone imaging in oncology, NaF-PET is an emerging technique reputed to capture a different aspect of the development of atheroma compared with FDG (59), related to calcification/microcalcification within an atherosclerotic plaque (Figure 3). In patients with MI and stable angina, Joshi et al. (59) demonstrated conspicuous uptake of NaF in plaque, with histological evidence of macrophage infiltration, calcification, necrosis, and apoptosis, as well as high-risk plaque features imaged by intravascular ultrasound. Advantages of NaF over FDG include rapid background blood and tissue clearance and absence of activity in the myocardium, improving the potential for imaging pathological NaF uptake in the coronary arteries (53). As with FDG, elements of the cell/process-specificity of NaF in atherosclerosis have been described, and imaging technique standardization is an active realm of research.
In parallel with further FDG and NaF work, investigation continues with other PET, CT, and CMR techniques to study varied mechanisms and applications for CVD imaging. An abbreviated list of PET agents currently under investigation is shown in Table 1. Agents from the list that already have FDA approval for human imaging of nonvascular processes include gallium 68 (68Ga)-DOTA-octreotate and 18F-florbetapen. 68Ga-DOTA-octreotate has high affinity for somatostatin receptors that are present in high concentration on inflammatory leukocytes (60). Its low background activity and high signal make it a promising candidate for plaque imaging, and its feasibility in humans has been demonstrated in comparison to FDG (60). 18F-florbetapen is a PET agent that is used clinically to characterize β-amyloid plaque in neurodegenerative processes, such as Alzheimer’s disease. Bucerius et al. (61 demonstrated that higher carotid artery uptake of 18F-florbetapen is associated with male sex, independent of central nervous system uptake, which potentially implicates the presence of higher concentrations of inflammatory β-amyloid within the vessel wall. Further investigations of the performance characteristics of these new agents are ongoing.
Table 1.
A Summary of Agents and Their Potential Mechanisms of Uptake Applicable to VI Imaging
| Agent | Potential Mechanism of Uptake |
|---|---|
| 18F-FLT (62) | Structural analogue of thymidine, images DNA synthesis within atheroma |
| 11C-PK11195 (63) | Affinity for translocator protein (TSPO), upregulated on inflammatory cells |
| 18F-A85380 (64) | Binds arterial nicotinic acetylcholine receptors (nAChRs), possibly related to vascular damage |
| 18F-choline (65) | Images increased cell wall synthesis within atheroma |
| 68Ga-DOTA-octreotate (60) | Affinity for somatostatin receptors, which are highly expressed on macrophages |
| 64Cu-ATSM (66) | Trapped within cells in hypoxic state |
| 18F-MISO (67) | Trapped within cells in hypoxic state |
| 68Ga-NOTA-RGD (68) | Images neoangiogenesis as a result of hypoxia or chronic inflammation |
| 64Cu-DOTA-CANF (69) | Images angioneogenesis via natriuretic peptide receptor affinity |
| 18F-FDG | A glucose analogue imaging increased metabolic rate in the presence of inflammation and hypoxia |
| 18F-sodium fluoride (59) | Images active calcification as a result of necrosis or inflammation |
| 68Ga-CXCR4 (70) | Images CXCR4 receptor expressed by inflammatory cells |
| 18F-florbetapen (61) | Imaging β-amyloid plaque as a component of inflammation |
11C-PK11195 = carbon 111-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide; 18F = fluorine 18; 64Cu = copper 64; 68Ga = gallium 68; A85380 = 3-([2S]-Azetidinylmethoxy)pyridine dihydrochloride; ATSM = diacetyl-bis(N-methylthiosemicarbazone; CXCR4 = C-X-C chemokine receptor type 4; DOTA-CANF = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid atrial natriuretic factor; FDG = fluorodeoxyglucose; FLT = fluorothymidine; MISO = fluoromisonidazole; NOTA-RGD = 1,4,7-triazacyclononane-N, N′, N″-triacetic acid arginine-glycine-aspartate.
In summary, with a new understanding of immunological processes applied to the established feasibility of promising PET agents for imaging atherosclerosis in humans, we now can administer these techniques in newly-identified human disease models to better understand atherogenesis. Acknowledging the great potential for pathology-specific and quantitative information that PET imaging has for atherosclerosis, an outcome-based multidisciplinary approach for further investigation, integrating biology, engineering, imaging physics, and economics, will continue to help us understand how these techniques ultimately fit into clinical care and translational research.
CONCLUSIONS
Inflammation has emerged as a critical factor in early atherosclerosis. An understanding of the immune cells involved in the initiation and progression of vascular inflammation may facilitate the identification of important pathways to target for future CVD therapeutics. Furthermore, the use of PET imaging to measure vascular inflammation may improve our ability to identify the most promising therapies to take to phase III clinical trials. As such, advances in immunology and vascular imaging have the potential to accelerate discovery of new treatments to reduce the burden of CVD.
Central Illustration. Progression of Vascular Inflammation in Human Inflammatory Models.

A gross illustration of the aortic arch has been taken in cross-section to magnify the vessel wall. Neutrophil activation due to systemic inflammation leads to NETosis and may initiate damage to the endothelium. Monocytes and T cells then infiltrate the lesion. Monocytes differentiate into macrophages, where they proliferate to sustain their population. Macrophages within the vessel wall have high glycolytic rates and take up the 18F-fluorodeoxyglucose (FDG) tracer, which is detectable by FDG positron emission tomography in human models of inflammation. NETosis = neutrophil extracellular trap activation and release. IFN = interferon; IL = interleukin
ABBREVIATIONS AND ACRONYMS
- CMR
cardiac magnetic resonance
- CT
computed tomography
- CV
cardiovascular
- CVD
cardiovascular disease
- CXCL12
C-X-C motif chemokine ligand 12
- FDG
18F-fluorodeoxyglucose
- HIV
human immunodeficiency virus
- IFN
interfero
- IL
interleukin
- MI
myocardial infarction
- NaF
18F sodium fluoride
- NET
neutrophil extracellular trap
- PAD4
peptidylarginine deiminase 2
- PET
positron emission tomography
- SLE
systemic lupus erythematosus
- VI
vascular inflammation
Footnotes
Disclosures: Dr. Nestle is an employee of Sanofi. Dr. Gelfand has: served as a consultant for Coherus (DSMB), Dermira, Janssen Biologics, Merck (DSMB), Novartis Corp, Regeneron, Dr Reddy’s labs, Sanofi, and Pfizer Inc.; received honoraria; and received research grants (to the Trustees of the University of Pennsylvania) from Abbvie, Janssen, Novartis Corp, Regeneron, Sanofi, Celgene, and Pfizer Inc.; and received payment for continuing medical education work related to psoriasis that was supported indirectly by Lilly and Abbvie. Dr. Gelfand coholds a patent of resiquimod for treatment of cutaneous T cell lymphoma. Dr. Grinspoon has served as a consultant for and received honoraria from Navidea Pharmaceuticals and Theratechnologies, and has received research grants (to the MGH) from Navidea Pharmaceuticals, KOWA Pharmaceuticals, Gilead Sciences, and Theratechnologies. Dr. Ridker has received investigator-initiated research support from the NHLBI, Pfizer, AstraZenenca, Novartis, and Kowa, and is listed as a coinventor on patents held by the Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease and diabetes that have been licensed to AstraZeneca and Siemens. Dr. Tawakol has served as a consultant for Actelion Pharmaceuticals and has received research grants (to the MGH) from Actelion and Genentech. Dr. Mehta is a full-time U.S. Government employee, and has received research grants from Abbvie, Janssen, Novartis Corp, and Celgene. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–97. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–72. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–9. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sager HB, Dutta P, Dahlman JE, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8:342ra80. doi: 10.1126/scitranslmed.aaf1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754–8. doi: 10.1038/nm.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 9.Yipp BG, Petri B, Salina D, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–93. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1777–83. doi: 10.1161/ATVBAHA.111.242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–13. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis. 2014;73:483–91. doi: 10.1136/annrheumdis-2013-203844. [DOI] [PubMed] [Google Scholar]
- 14.Savchenko AS, Borissoff JI, Martinod K, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123:141–8. doi: 10.1182/blood-2013-07-514992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinod K, Witsch T, Erpenbeck L, et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med. 2017;214:439–58. doi: 10.1084/jem.20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014;9:73–102. doi: 10.1146/annurev-pathol-020712-163936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscleros Thrombos Vasc Biol. 2015;35:280–7. doi: 10.1161/ATVBAHA.114.303568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotsman I, Sharpe AH, Lichtman AH. T-cell costimulation and coinhibition in atherosclerosis. Circ Res. 2008;103:1220–31. doi: 10.1161/CIRCRESAHA.108.182428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lichtman AH. The heart of the matter: protection of the myocardium from T cells. J Autoimmun. 2013;45:90–6. doi: 10.1016/j.jaut.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–55. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab. 2007;92:2506–12. doi: 10.1210/jc.2006-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanni MV, Abbara S, Lo J, et al. Increased coronary atherosclerotic plaque vulnerability by coronary computed tomography angiography in HIV-infected men. AIDS. 2013;27:1263–72. doi: 10.1097/QAD.0b013e32835eca9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zanni MV, Toribio M, Robbins GK, et al. Effects of antiretroviral therapy on immune function and arterial inflammation in treatment-naive patients with human immunodeficiency virus infection. JAMA Cardiol. 2016;1:474–80. doi: 10.1001/jamacardio.2016.0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo J, Abbara S, Shturman L, et al. Increased prevalence of subclinical coronary atherosclerosis detected by coronary computed tomography angiography in HIV-infected men. AIDS. 2010;24:243–53. doi: 10.1097/QAD.0b013e328333ea9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiara DK, Liu CY, Raman F, et al. Abnormal myocardial function is related to myocardial steatosis and diffuse myocardial fibrosis in HIV-infected adults. J Infect Dis. 2015;212:1544–51. doi: 10.1093/infdis/jiv274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian S, Tawakol A, Burdo TH, et al. Arterial inflammation in patients with HIV. JAMA. 2012;308:379–86. doi: 10.1001/jama.2012.6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo J, Lu MT, Ihenachor EJ, et al. Effects of statin therapy on coronary artery plaque volume and high-risk plaque morphology in HIV-infected patients with subclinical atherosclerosis: a randomised, double-blind, placebo-controlled trial. Lancet HIV. 2015;2:e52–63. doi: 10.1016/S2352-3018(14)00032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–55. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelfand JM, Dommasch ED, Shin DB, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol. 2009;129:2411–8. doi: 10.1038/jid.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735–41. doi: 10.1001/jama.296.14.1735. [DOI] [PubMed] [Google Scholar]
- 31.Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. 2010;31:1000–6. doi: 10.1093/eurheartj/ehp567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeung H, Takeshita J, Mehta NN, et al. Psoriasis severity and the prevalence of major medical comorbidity: a population-based study. JAMA Dermatol. 2013;149:1173–9. doi: 10.1001/jamadermatol.2013.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta NN, Yu Y, Pinnelas R, et al. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med. 2011;124:775, e1–6. doi: 10.1016/j.amjmed.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Gao H, Loyd CM, et al. Chronic skin-specific inflammation promotes vascular inflammation and thrombosis. J Invest Dermatol. 2012;132:2067–75. doi: 10.1038/jid.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003–2004. J Am Acad Dermatol. 2009;60:218–24. doi: 10.1016/j.jaad.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
- 37.Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997;145:408–15. doi: 10.1093/oxfordjournals.aje.a009122. [DOI] [PubMed] [Google Scholar]
- 38.Villanueva E, Yalavarthi S, Berthier CC, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–52. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denny MF, Yalavarthi S, Zhao W, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010;184:3284–97. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Somers EC, Zhao W, Lewis EE, et al. Type I interferons are associated with subclinical markers of cardiovascular disease in a cohort of systemic lupus erythematosus patients. PloS One. 2012;7:e37000. doi: 10.1371/journal.pone.0037000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knight JS, Luo W, O’Dell AA, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. 2014;114:947–56. doi: 10.1161/CIRCRESAHA.114.303312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furumoto Y, Smith CK, Blanco L, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. 2017;69:148–60. doi: 10.1002/art.39818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yun M, Yeh D, Araujo LI, Jang S, Newberg A, Alavi A. F-18 FDG uptake in the large arteries: a new observation. Clin Nucl Med. 2001;26:314–9. doi: 10.1097/00003072-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Rudd JH, Warburton EA, Fryer TD, et al. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation. 2002;105:2708–11. doi: 10.1161/01.cir.0000020548.60110.76. [DOI] [PubMed] [Google Scholar]
- 45.Joseph P, Tawakol A. Imaging atherosclerosis with positron emission tomography. Eur Heart J. 2016;37:2974–80. doi: 10.1093/eurheartj/ehw147. [DOI] [PubMed] [Google Scholar]
- 46.Tawakol A, Singh P, Mojena M, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc Biol. 2015;35:1463–71. doi: 10.1161/ATVBAHA.115.305551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taqueti VR, Di Carli MF, Jerosch-Herold M, et al. Increased microvascularization and vessel permeability associate with active inflammation in human atheromata. Circ Cardiovasc Imaging. 2014;7:920–9. doi: 10.1161/CIRCIMAGING.114.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdelbaky A, Corsini E, Figueroa AL, et al. Early aortic valve inflammation precedes calcification: a longitudinal FDG-PET/CT study. Atherosclerosis. 2015;238:165–72. doi: 10.1016/j.atherosclerosis.2014.11.026. [DOI] [PubMed] [Google Scholar]
- 49.Rominger A, Saam T, Wolpers S, et al. 18F-FDG PET/CT identifies patients at risk for future vascular events in an otherwise asymptomatic cohort with neoplastic disease. J Nucl Med. 2009;50:1611–20. doi: 10.2967/jnumed.109.065151. [DOI] [PubMed] [Google Scholar]
- 50.Figueroa AL, Abdelbaky A, Truong QA, et al. Measurement of arterial activity on routine FDG PET/CT images improves prediction of risk of future CV events. J Am Coll Cardiol Img. 2013;6:1250–9. doi: 10.1016/j.jcmg.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Mehta NN, Torigian DA, Gelfand JM, Saboury B, Alavi A. Quantification of atherosclerotic plaque activity and vascular inflammation using [18-F] fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT) J Vis Exp. 2012:e3777. doi: 10.3791/3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huet P, Burg S, Le Guludec D, Hyafil F, Buvat I. Variability and uncertainty of 18F-FDG PET imaging protocols for assessing inflammation in atherosclerosis: suggestions for improvement. J Nucl Med. 2015;56:552–9. doi: 10.2967/jnumed.114.142596. [DOI] [PubMed] [Google Scholar]
- 53.Bucerius J, Hyafil F, Verberne HJ, et al. Cardiovascular Committee of the European Association of Nuclear Medicine (EANM) Position paper of the Cardiovascular Committee of the European Association of Nuclear Medicine (EANM) on PET imaging of atherosclerosis. Eur J Nucl Med Mol Imaging. 2016;43:780–92. doi: 10.1007/s00259-015-3259-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tawakol A, Fayad ZA, Mogg R, et al. Intensification of statin therapy results in a rapid reduction in atherosclerotic inflammation: results of a multicenter fluorodeoxyglucose-positron emission tomography/computed tomography feasibility study. J Am Coll Cardiol. 2013;62:909–17. doi: 10.1016/j.jacc.2013.04.066. [DOI] [PubMed] [Google Scholar]
- 55.Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fayad ZA, Mani V, Woodward M, et al. dal-PLAQUE Investigators. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging (dal-PLAQUE): a randomised clinical trial. Lancet. 2011;378:1547–59. doi: 10.1016/S0140-6736(11)61383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tawakol A, Singh P, Rudd JH, et al. Effect of treatment for 12 weeks with rilapladib, a lipoprotein-associated phospholipase A2 inhibitor, on arterial inflammation as assessed with 18F-fluorodeoxyglucose-positron emission tomography imaging. J Am Coll Cardiol. 2014;63:86–8. doi: 10.1016/j.jacc.2013.07.050. [DOI] [PubMed] [Google Scholar]
- 58.O’Donoghue ML, Glaser R, Cavender MA, et al. LATITUDE-TIMI 60 Investigators. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA. 2016;315:1591–9. doi: 10.1001/jama.2016.3609. [DOI] [PubMed] [Google Scholar]
- 59.Joshi NV, Vesey AT, Williams MC, et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: a prospective clinical trial. Lancet. 2014;383:705–13. doi: 10.1016/S0140-6736(13)61754-7. [DOI] [PubMed] [Google Scholar]
- 60.Tarkin JM, Joshi FR, Evans NR, et al. Detection of atherosclerotic inflammation by 68Ga-DOTATATE PET compared to [18F]FDG PET Imaging. J Am Coll Cardiol. 2017;69:1774–91. doi: 10.1016/j.jacc.2017.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bucerius J, Barthel H, Tiepolt S, et al. Feasibility of in vivo 18F-florbetaben PET/MR imaging of human carotid amyloid-β. Eur J Nucl Med Mol Imaging. 2017;44:1119–28. doi: 10.1007/s00259-017-3651-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ye YX, Calcagno C, Binderup T, et al. Imaging macrophage and hematopoietic progenitor proliferation in atherosclerosis. Circ Res. 2015;117:835–45. doi: 10.1161/CIRCRESAHA.115.307024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pugliese F, Gaemperli O, Kinderlerer AR, et al. Imaging of vascular inflammation with [11C]-PK11195 and positron emission tomography/computed tomography angiography. J Am Coll Cardiol. 2010;56:653–61. doi: 10.1016/j.jacc.2010.02.063. [DOI] [PubMed] [Google Scholar]
- 64.Bucerius J, Manka C, Schmaljohann J, et al. Feasibility of [18F]-2-Fluoro-A85380-PET imaging of human vascular nicotinic acetylcholine receptors in vivo. J Am Coll Cardiol Img. 2012;5:528–36. doi: 10.1016/j.jcmg.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vöö S, Kwee RM, Sluimer JC, et al. Imaging intraplaque inflammation in carotid atherosclerosis with 18F-fluorocholine positron emission tomography-computed tomography: prospective study on vulnerable atheroma with immunohistochemical validation. Circ Cardiovasc Imaging. 2016;9:e00447. doi: 10.1161/CIRCIMAGING.115.004467. [DOI] [PubMed] [Google Scholar]
- 66.Nie X, Laforest R, Elvington A, et al. PET/MRI of hypoxic atherosclerosis using 64Cu-ATSM in a rabbit model. J Nucl Med. 2016;57:2006–11. doi: 10.2967/jnumed.116.172544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joshi FR, Manavaki R, Fryer TD, et al. Vascular imaging with 18F-fluorodeoxyglucose positron emission tomography is influenced by hypoxia. J Am Coll Cardiol. 2017;69:1873–4. doi: 10.1016/j.jacc.2017.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paeng JC, Lee YS, Lee JS, et al. Feasibility and kinetic characteristics of 68Ga-NOTA-RGD PET for in vivo atherosclerosis imaging. Ann Nucl Med. 2013;27:847–54. doi: 10.1007/s12149-013-0757-x. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Pressly ED, Abendschein DR, et al. Targeting angiogenesis using a C-type atrial natriuretic factor-conjugated nanoprobe and PET. J Nucl Med. 2011;52:1956–63. doi: 10.2967/jnumed.111.089581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hyafil F, Pelisek J, Laitinen I, et al. Imaging the cytokine receptor CXCR4 in atherosclerotic plaques with the radiotracer 68Ga-pentixafor for PET. J Nucl Med. 2017;58:499–506. doi: 10.2967/jnumed.116.179663. [DOI] [PubMed] [Google Scholar]