

Abstract
Diabetic retinopathy (DR) is characterized by early loss of retinal capillary pericytes and microvascular dysfunction. We recently showed that pro-oxidative stress and pro-apoptotic thioredoxin interacting protein (TXNIP) is significantly up-regulated in rat retinas in experimental diabetes and mediates inflammation and apoptosis. Therefore, we hypothesize here that TXNIP up-regulation in pericyte plays a causative role in oxidative stress and apoptosis under sustained high glucose exposure in culture. We maintained a rat retinal capillary pericyte cell line (TR-rPCT1) for 5 days under low glucose (LG, 5.5 mM) or high glucose (HG, 25 mM) with or without anti-oxidant N-acetylcysteine (5 mM, NAC), Azaseine (2 μM, AzaS), an inhibitor of TXNIP, and TXNIP siRNA (siTXNIP3, 20 nM). The results show that HG increases TXNIP expression in TR-rPCT1, which correlates positively with ROS generation, protein S-nitrosylation, and pro-apoptotic caspase-3 activation. Furthermore, pericyte apoptosis is demonstrated by DNA fragmentation (alkaline comet assay) and a reduction in MTT survival assay. Treatment of TR-rPCT1 with NAC or an inhibition of TXNIP by AzaS or siTXNIP3 each reduces HG-induced ROS, caspase-3 activation and DNA damage demonstrating that TXNIP up-regulation under chronic hyperglycemia is critically involved in cellular oxidative stress, DNA damage and retinal pericyte apoptosis. Thus, TXNIP represents a novel gene and drug target to prevent pericyte loss and progression of DR.
Keywords: Hyperglycemia, TXNIP, Oxidative stress, Mitochondrial dysfunction, Chromatin break, Pericyte apoptosis, Diabetic retinopathy
Introduction
Diabetic retinopathy (DR) is the most common disease of diabetes leading to visual deficiency and blindness among working adults in the US and around the globe [1]. About one-third of adult diabetic patients both with type 1 and type 2 diabetes will develop one form of proliferative or non-proliferative DR [1,2]. Hyperglycemia-associated oxidative/nitrosative stress, chronic low grade inflammation and pre-mature cell death are considered to play critical roles in the pathogenesis of diabetic ocular complications [3–5]. Early features of DR include capillary basement membrane thickening, pericyte loss, and blood retinal barrier (BRB) breakdown, which ultimately progress to proliferative DR manifested by capillary neovascularization and blindness [6,7]. Capillary endothelial cells and pericytes share a common basal lamina and influence each other for maintaining vascular tone and proper functioning of the endothelial cell–cell junction and regulate nutrient transport across the BRB for neuro-retinal homeostasis and visual function [8,9]. However, under chronic hyperglycemia and diabetes, pericyte apoptosis occurs early, which leads to formation of ghost and acellular capillaries and leaky vessels [10]. Nonetheless, the cellular and molecular mechanisms of retinal capillary pericyte apoptosis in diabetes are not fully understood yet.
We have recently shown that pro-oxidative stress and pro-apoptotic protein, thioredoxin interacting protein (TXNIP), which binds to thioredoxin (Trx) and inhibits its oxidant scavenging and thiol (cysteine) reducing capacity [11,12], is highly induced in the diabetic retina in vivo as well as in vitro in retinal capillary endothelial cells and Muller glia [13–15]. Trx is a 12-kDa protein, which scavenges ROS and maintains protein cysteine sulfahydryl groups by its thiol redox-active sulfides. Trx1 is present both in the cytosol and nucleus while Trx2 is present in the mitochondria. Trx together with Trx reductase 1 (cytosol/nuclear) and Trx reductase 2 (mitochondria) and NADPH maintain the cellular reducing environment. The other redox regulating system is the GSH/glutaredoxin system [16,17]. Trx activity is however required for reduction of oxidized glutathione reductase, which converts GSSG to GSH. Trx denitrosylates a broad spectrum of S-nitrosylated proteins [16,17]. While a reversible physiological level of protein-SNO is important for maintaining cellular function and cell viability [18]; a sustained up-regulation of TXNIP in chronic hyperglycemia can lead to excessive ROS/RNS accumulation, abnormal protein SNO and pre-mature cell death.
Recently, TXNIP has been shown to be involved in NLRP3 inflammasome assembly and caspase 1 activation and pro-IL-1β expression and maturation [15,19–23] and endoplasmic reticular (ER) stress induced cell death [24,25]. Nonetheless, a critical role of TXNIP in oxidative stress and pericyte demise under sustained high glucose exposure has not been investigated so far. We hypothesize that high glucose induces TXNIP up-regulation in pericyte in culture and causes mitochondrial bioenergetic imbalance, oxidative/nitrosative stress, chromatin instability/DNA damage and apoptosis. Indeed, our studies show first evidence that high glucose increases TXNIP expression, ROS/RNS stress, chromatin breakage, and pericyte apoptosis. Our findings thus implicate a novel role of TXNIP in early pericyte loss and BRB dysfunction in DR and suggest that TXNIP is an important therapeutic target for preventing vascular lesions of blinding ocular complications in diabetes.
Materials and methods
Materials
Tissue culture media, serum and antibiotics were purchased from Invitrogen (Carlsbad, CA). Antibodies for TXNIP were obtained from MBL (cat# K0205-3, Woburn, MA). Anti-S-nitroso-cysteine (SNO-Cys, cat# N5411) antibody was from Sigma-Aldrich (St. Louis, MO). Histone H3 lysine 9 trimethyl antibody (cat# 5327) and total H3 (cat# 9717) were purchased from Cell Signaling Technologies (cat# 5327, Danvers, MA). The LC3B antibody kit for autophagy (cat# L10382), ROS detection reagent CM-H2DCFDA (cat# C6827) and ATP (cat# A22066) assay kits were from Molecular Probes (Invitrogen). Electrophoresis Mobility Shift Assay (EMSA) kit for Transcription factors – p53 (cat# GS-0033), GADD153 (also known as CHOP), and sXBP1 (custom-synthesis) – binding to respective consensus DNA sequences was purchased from Signosis Inc. (Sunnyvale, CA). Fluorescent-labeled secondary antibodies anti-rabbit and anti-mouse were obtained from Molecular Probes while that of the anti-goat antibodies were purchased from Abcam (Cambridge, MA). Active Caspase-3 Red SR FLIVOTM in vivo Apoptosis Kit (cat# 982) was obtained from Immunochemistry Technologies (Bloomington, MN). Pre-designed primers for rat TXNIP, Bim, Puma, Bcl2 and actin were purchased from Qiagen (SABiosciences). Similarly, control and TXNIP siRNAs were also obtained from Qiagen (SABiosciences). TRIZOL for RNA isolation was from Invitrogen while First strand cDNA synthesis kit and SYBR green reagents were purchased from Biorad, CA.
Retinal pericyte culture
A conditionally immortalized rat retinal capillary pericytes (TR-rPCT1) was used in this study similar to those described for rat retina capillary endothelial cells (TR-iBRB2) [26]. These cells were characterized to express pericyte markers such as rat intercellular adhesion molecule-1, platelet-derived growth factor-receptor beta, angiopoietin-1, and osteopontin, suggesting a relevant cell type to study pericyte function in vitro [26–29]. As mentioned above, we have used TR-iBRB2, a rat retinal endothelial cell line derived from the same animal as TR-rPCT1 in our previous studies [13–15] to investigate TXNIP function under HG in culture. Therefore, we chose this rat retinal pericyte in the present study as well. TR-rPCT1 cells are temperature sensitive and they were propagated at 33 °C in medium containing low glucose DMEM/F-12 (4:1 ratio), 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin in collagen 1 coated plates similar to those described for TR-iBRB2 [13,14]. After ~70–80 confluence, TR-PCT1 cells were cultured at 37 °C at low serum (0.2% FBS) medium for 24 h to slow down cell growth and proliferation. Subsequently, TR-rPCT1 cells were maintained at 0.2% serum either at low glucose (5.5 mM, LG) or high glucose (25 mM, HG) for 5 days. Media were changed every 48 h except for the last day of the culture, which is at 24 h [15]. When azaserine (AzaS, 2 μM), N-acetylcysteine (NAC, 5 mM) or siRNAs (scrRNA or siTXNIP3, 20 nM) were present, they were included in the final 2 days of HG treatment before terminating the experiment. Transfection of scrRNA and siTXNIP3, which significantly reduces TXNIP protein, was performed as previously described using the HiFect transfection reagent from Qiagen [15].
Real-time quantitative PCR
Total RNAs were isolated by TRIZOL method and first-strand cDNAs [from 1 μg RNA] were synthesized in 20 μl volume using the Bio-Rad iScript cDNA synthesis kit [13–15]. Messenger RNA expression was analyzed by real-time quantitative PCR using the Bio-Rad Chromo 4 detection system and SYBR Green PCR Master Mix from Bio-Rad or Applied Biosystems (Foster City, CA). The real time PCR reaction mixture contained 1 × SYBR Green PCR Master Mix, 400 nM forward and reverse primers, and 2 μl cDNA in a final volume of 25 μl. The PCR cycling was programmed as 95 °C for 15 s, 55 °C for 30 s and 72 °C for 30 s 40 cycles followed by the construction of a melting curve through increasing the temperature from 60 °C to 95 °C at a ramp rate of 2% for 20 min. The real time PCR samples were evaluated using a single predominant peak as a quality control. Ct values were used to calculate the relative expression level of mRNAs that were normalized to actin.
SDS-PAGE and Western blotting
Proteins were extracted in RIPA buffer containing protease inhibitors and their concentrations were determined using a Coomassie Plus (Bradford) Assay Reagent from Pierce (Product 23238) with BSA as the standard. Absorbance of blue dye of the Coomassie–protein complex was measured at 595 nm using a Gemini Microplate reader (Molecular Devices, Sunnyvale, CA). Thirty micrograms of protein extracts were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDA-PAGE) and Western blot analysis of proteins were performed as described previously [14,15]. Primary antibodies were used at 1:1000 dilutions while secondary antibodies were used at 1:3000 dilutions. ECL used to detect the immunoreactive bands using a Cell Bioscience FluorChem E System (Santa Clara, CA).
Immunohistochemistry (IHC)
Cells were grown in four-chambered tissue culture glass slides (NUNC, Naperville, IL) and exposed to HG for 5 days as described [15]. Cells were fixed with freshly prepared paraformaldehyde (4%) for 2 h in ice or 4 °C overnight, washed 10 min each with PB (three times) and blocked with 5% horse serum in PB for 1 h at room temperature. Following a 30 min wash with PBS, cells were incubated with primary antibodies (1:100 dilutions) overnight at 4 °C in a humidified chamber. After washing with PB, cells were further incubated with corresponding secondary antibodies conjugated with Alexa Fluor 488 or Alexa 594 at 1:500 dilutions for 1 h at 37 °C in a darkened humidified chamber. DAPI (Blue) was used to counter stain the nuclei. The images were captured by an OLYMPUS BX 51 fluorescence microscope, which is fitted with a triple DAPI/FITC/TRITC cube, a DP70 digital camera and image acquisition software. Similar magnification (40 ×) and exposure time were maintained throughout for comparing images unless otherwise mentioned.
Determination of cell viability
MTT assay for cell viability was performed in 24 or 48 well plates and with 0.5 mg/ml MTT (cat# V-13154, Invitrogen) in each well as previously described [12,15]. TR-rPCT1 cells (1 × 104 cells) were grown to 70% confluence and serum-starved overnight and treated with HG for 5 days. MTT was added for 3 h at the end of the experiment, the media was removed and cells were kept in 100 μl of DMSO for 10 min. The resulting color was diluted with 500 μl of distilled H2O and detected at 570 nm using a Gemini Microplate reader (Molecular Devices, Sunnyvale, CA).
Intracellular reactive oxygen species (ROS) measurement
The formation of intracellular ROS in TR-rPCT1 cells was detected as previously described [13,15] by using the fluorescent probe, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA). This dye can enter living cells by passive diffusion and it is non-fluorescent until the acetate group is cleaved off by intracellular esterase and oxidation occurs within the cell. Approximately 1 × 105 cells/ml were cultured in 24 well plates, serum-starved overnight and glucose were added for the specified time period. Then, CM-H2DCFDA (10 μM) was incubated for 60 min at 37 °C. The medium with the dye was aspirated (to remove the extracellular dye), washed with PBS (3 ×), then the PBS is added to cells. The fluorescence was measured in a Gemini Fluorescent Microplate Reader (Molecular Devices) with the bottom read scanning mode at 480 nm excitation and emission at 530 nm.
DNA fragmentation detection
To measure DNA damage under chronic hyperglycemia in TR-rPCT1 cells, we used (i) IHC to detect chromatin fragmentation and condensation using DAPI staining of the nucleus as described above in IHC section and (ii) single cell gel electrophoresis (SCGE) or the alkaline comet assay using the OxiSelectTM Comet Assay kit (cat# STA-350) from Cell Biolabs, Inc (San Diago, CA). The SCGE or Comet assay is a useful method to measure DNA damage in individual cells under an electrophoretic field. For this, TR-rPCT1 cells were cultured in 6 well plates and maintained in LG or HG conditions for 3 or 5 days. Cells were scrapped off and resuspended in cold-PBS (without Mg2+ and Ca2+) at 1 × 105 cells/ml. Cell suspension (10 μl) and 90 μl of Comet Agarose (melted at 90 °C and maintained at 37 °C) were mixed and immediately pipette (75 μl) on the OxiSelectTM Comet Slide. The slides were kept at 4 °C for 15 min in the dark. The slides were then carefully immersed (while maintaining horizontal position to prevent agarose slipping off the slide) in the pre-chilled lysis buffer in a small container (~25 ml) for 30 min at 4 °C in the dark. The slides were then transferred to an electrophoresis chamber and filled with cold Alkaline Electrophoresis Solution (300 mM NaOH, 1 mM EDTA, pH 13). Electrophoresis was run at 20 V (1 V/cm for 20 cm apart chamber electrodes) for 25 min. After completing the electrophoresis, the agarose slide was carefully transferred to a container and immerged in pre-chilled sterile H2O for 2 min, aspirated and repeated two times. Then the slides were emerged again in 70% alcohol for 5 min and air dried for 30 min. After the agarose is completely dried, 100 μl of a diluted Vista Green DNA Dye was added and kept for 15 min at room temperature. The slide is then view under OLYMPUS BX 51 fluorescence microscope for green fluorescence. The DNA fragmentation is determined by the presence of a Head and a Comet Tail ((displacement of nuclear DNA (head) to a resulting DNA streaking (tail) due to breakage)).
Measurement of mitochondrial membrane potential
We used a MitoPT™-JC1 Assay Kit (cat# 924, Immunohistochemistry Technologies, Bloomington, MN) to detect mitochondrial membrane depolarization in TR-rPCT1 5 days after HG exposure according to Manufacturer’s instructions. As a member of the carbocyanine family of potentiometric dyes, 5,5′, 6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine I-/Cl- salt, commonly known as JC-1, penetrates cell and accumulates within mitochondria as a orange-red aggregate when excited at 490 nm. However, upon mitochondrial membrane potential collapses in apoptotic or metabolically stressed cells, the MitoPT™ JC-1 reagent will no longer concentrate within the mitochondria. Instead, it exits mitochondria and disperses throughout the cell in a monomeric form, which emits a green fluorescence at 490 nm excitation. Therefore, non-apoptotic cells exhibit as orange-red stained mitochondria while apoptotic cells with mitochondrial membrane potential depolarization stains green. TR-rPCT1 cells (~to 1 × 105 cells/ml) were cultured in four well slide chambers for 5 days at low serum medium with LG or HG. Cell were then incubated with a JC-1 working solution (5 μM) for 30 min at 37 °C in the dark and then washed with the wash buffer (provided in the kit). The slides were lightly covered with a cover slip and fluorescence was detected in OLYMPUS BX 51 fluorescence microscope, fitted with triple DAPI/FITC/TRITC cubes and a DP70 digital camera.
ATP assay
Cellular ATP concentration was determined in cell supernatants with an ATP biolumescence assay kit as described previously [15]. Briefly, ATP was released using the boiling method from TR-rPCT1 cells maintained in 24 well plates under LG or HG for 5 days. A volume of 250 μl TE buffer (50 mM Tris–Cl, pH 7.4, 4 mM EDTA) was added to the wells and scrapped off. Cells were immediately transferred to a boiling water bath for 4 min. Samples were then put on ice and briefly sonicated and centrifuged. Relative fluorescence units (RLUs) were detected in a Luminometer (Promega, Madison, WI).
Autophagy determination
Autophagy is a survival mechanism for long-lived and fully differentiated cells under starvation and oxidative stress to remove aggregated proteins and damaged organelles including mitochondria via a lysosomal degradation pathway and recycling of cell contents. The microtubule light chain LC3B protein is a late marker of autophagy. LC3BI, an inactive protein, is converted to its active form LC3BII during autophagic punctae formation (vesicular autophagolysosomes usually observed in the peri-nuclear region). Therefore, we use the LC3B antibody kit to measure LC3B punctae to determine HG induction of autophagy in TR-rPCT1 cells on IHC with a rabbit anti-LC3B antibody (Invitrogen, cat# L10382) as previously described [15]. In addition, the conversion of LC3BI to LC3BII (LC3B activation) was determined on WB by the presence of a faster moving LC3BII band in gels.
Gel-shift assay
The gel-shift or electrophoretic mobility-shift assay (EMSA) provides a simple and rapid method for detecting DNA-binding activity of proteins. We investigated p53, GADD153 and sXBP1 activities of TR-rPCT1 nuclear protein extracts using commercially available (p53) and custom-made (GADD153 and sXBP1) gel-shift assay kits from Signosis (Sunnyvale, CA) and according to manufacturer’s instructions [15,30]. After incubation at 16 °C for 30 min with biotin-labeled probes and nuclear extracts (5.0 μg protein) in a PCR machine, the protein–DNA complexes were subjected to 6.5% non-denaturing polyacrylamide gels prepared with TBE gel formulation. The gel was run at 100 V for 1 h until the Bromophenol blue dye front moved three quarters down the gel, using 0.5 × TBE running buffer. For competitive assays, excess cold probes were added to the reaction. Streptavin-HRP and ECL were used to detect and capture the reactive bands using a Cell Bioscience FluorChem E System (Santa Clara, CA).
Statistical analysis
Results have been expressed as means +/−SE. Student’s t-test was used to compare differences between the control and treated samples. Multiple mean comparisons were performed by one-way ANOVA followed by Bonferroni test [15,30]. A preset p value of <0.05 was considered statistically significant.
Results
High glucose increases TXNIP expression, oxidative stress and apoptosis of retinal pericytes in culture
A critical role of TXNIP in retinal pericyte apoptosis under chronic hyperglycemia, which is a hallmark of early diabetic retinopathy, is currently unknown. Therefore, we investigated up-regulation of TXNIP in TR-rPCT1 under sustained HG exposure in culture and whether TXNIP expression correlates with cellular oxidative and nitrosative stresses. Initially, we performed a time-depended effect of HG on ROS and RNS generation and DNA damage at 3 and 5 days. We observed that up to 3 days of HG exposure, no DNA damage/cell death occurs in retinal pericytes although ROS and RNS levels increase (Fig. 1S). Therefore, we maintained retinal pericytes for 5 days by changing the medium for every 48 h and once at 24 h in the last day, and further experiments were performed at this duration of HG exposure.
As shown in Fig. 1A and B, HG (25 mM) exposure of TR-rPCT1 for 5 days increases TXNIP mRNA expression and IHC staining of TXNIP protein, respectively, when compared with LG (5.5 mM). In agreement, TXNIP protein is also enhanced in HG compared to LG (Fig. 1C). Nonetheless, TXNIP protein level is higher than its mRNA. This result might be due to TXNIP message stabilization under prolonged HG exposure as recently described [24,25] although we need further investigation. TXNIP up-regulation is associated with significant increases in ROS generation (p<0.05) as revealed by enhanced CM-H2DCFDA fluorescence emission (Fig. 2A). Addition of an equimolar mannitol to HG (osmolar effect) does not increase ROS level suggesting that intracellular glucose metabolism is indeed linked to the observed ROS production. In addition, we also observed an increase in protein S-nitrosylation (SNO) in retinal pericyte under HG, which is revealed by greater intensity in IHC staining of thiol-nitrosylated proteins by an anti-SNO antibody, which is a marker of cellular reactive nitrogen species (RNS) stress (Fig. 2B). These results show that HG increases TXNIP expression in pericytes and induces ROS/RNS stress.
Fig. 1.

HG induces TXNIP expression and ROS/RNS generation in retinal pericytes. (a) Quantitative RT-PCR for TXNIP. HG increases TXNIP mRNA level significantly in retinal pericytes at day 5 (1.45+/−0.14; n=4; p=0.01) when compared with LG. (b) IHC of TXNIP in pericytes cultured under HG for 5 days shows enhanced TXNIP staining than in LG (arrows). A representative of n=3 is shown. (c) WB of TXNIP in TR-rPCT1. There is also a significant increase (p<0.05) in TXNIP protein in HG at day 5 (2.58+/−0.77; n=4) when compared with LG.
Fig. 2.

HG induces pericyte ROS/RNS levels and apoptosis in retinal pericytes. (a) Reactive oxygen species (ROS) detected by CM-H2DCFDA. ROS level is significantly increased by HG at day 5 (p<0.05) compared to LG. On the other hand, a 20 mM mannitol plus 5.5 mM LG, an equimolar osmotic effect of HG, does not increase ROS suggesting a specific effect of excess glucose metabolism on ROS production in pericytes. (b) IHC of protein thiol (S) nitrosylation using an anti-S-nitroso-cysteine (SNO-Cys) antibody shows enhanced S-nitrosylation of proteins in pericytes under HG (arrows). A representative of n=3 is shown. (c) Cell viability assay by MTT. HG significantly decreases MTT activity (p<0.0002) when compared with LG. Mannitol has no significant effect on MTT activity in pericytes. (d) IHC of active caspase-3: Cells were cultured in 4 well slide chambers with HG or LG for 5 days. Red SR FLIVO, which is cell membrane permeable and binds to active caspase-3, was added for 30 min, washed and fixed in 4% paraformaldehyde. DAPI was used to stain nuclei. HG activates caspase-3 in retinal pericytes as revealed by enhanced staining of SR FLIVO (arrows). A representative of n=3 is shown.
Next, we determined cell viability using MTT assay, which measures the succinate dehydrogenase activity of mitochondrial electron transport chain (etc) complex II is also reduced (p<0.0001) vs. LG (Fig. 2C), indicating mitochondrial dysfunction and pericyte cell death. In agreement, the IHC staining of activated capspase-3 in pericytes is enhanced by HG compared with LG (Fig. 2D). The results suggest that exposure of retinal pericytes to chronic high glucose induces mitochondrial dysfunction and apoptosis, and correlates positively with TXNIP up-regulation and oxidative stress in these cells (Fig. 1).
We further examined whether HG induces mitochondrial membrane depolarization in retinal pericytes and causes cell death since damaged mitochondria generates excess ROS. As shown in Fig. S2A, there is an increase in green fluorescence accumulation in the cytosol (monomeric JC-1) outside mitochondria as detected by IHC, which suggests a mitochondrial membrane potential collapse and metabolically stressed pericyte. The mitochondrial damage in pericyte is also accompanied by a significant decrease in ATP content after treatment with HG (p<0.0002 vs. LG) as shown in Fig. S2B.
High glucose induces nuclear DNA break and chromatin condensation in retinal pericytes
The mechanism(s) of cell death under chronic hyperglycemia and oxidative stress are complex but it may involve mitochondria oxidative stress-induced caspase 3 activation (as shown above) and nuclear chromatin break [31,32]. Here, we observe, on IHC staining of nuclear DNA with DAPI, that there is an increase in chromatin fragmentation and condensation under HG thereby indicating cells undergoing apoptosis (Fig. 3A). In addition, chromatin breakage in HG treated pericytes is also associated with TXNIP staining in these cells (Fig. 3A). Chromatin damage is absent in LG treated pericytes. We further demonstrate chromatin break by HG in pericytes using a different method of alkaline single cell gel electrophoresis (comet assay). In this assay, the intake nuclear DNA is seen as circular spot (unbroken genomic DNA) under LG while treatment with HG causes formation of a comet tail indicating chromatin break (Fig. 3B). Furthermore, there is a centrally located and condensed nucleoid DNA in HG treated pericytes. As shown in Fig. 3C, Western blot analysis indicates that heterochromatin marker, histone H3 lysine 9 trimethylation (H3K9Me3), is increased by HG at day 5. It is not enhanced at day 3 of HG. This result is in agreement with the observation that although ROS and RNS generation occur at day 3 of HG treatment in pericytes, there is no DNA breakage at day 3 in comet assay (Fig. S1).
Fig. 3.

HG induces DNA breakage and chromatin condensation in retinal pericytes. (a) DAPI staining (IHC) of DNA break and chromatin condensation in pericytes by HG maintained for 5 days (arrows). Note that TXNIP staining (red) colocalizes with broken chromatin. (b) A SINGLE CELL ALKALINE COMET Assay for DNA break also shows DNA tailing in HG suggesting chromatin breakage. A central chromatin nucleoid is also present in HG condition indicating chromatin condensation. The tail portion and chromatin nucleoid are absent in LG indicating intact chromatin. A representative of n=3 is shown. (c) Western Blot for histone H3 lysine 9 trimethylation (H3K9Me3), a heterochromatin mark for chromatin condensation, and total H3. HG treatment of TR-rPCT1 for 5 days leads to increases in H3K9Me3 but not at day 3. A representative of n=3 is shown.
Anti-oxidant N-acetylcysteine and azaserine reduce high glucose induced ROS generation and DNA breakage in retinal pericytes
We have previously shown that several effects of chronic hyperglycemia on TXNIP expression and oxidative stress are mediated by excess glucose metabolic flux through the hexosamine biosynthesis pathway (HBP) [13,14]. Therefore, in this study, we also examined if AzaS, an inhibitor of the HBP and TXNIP, and anti-oxidant NAC prevent HG induced ROS generation and DNA damage. First, we treated pericytes with NAC (5 mM) during the final 2 days of 5 days HG exposure in pericytes, and examined if HG induced ROS generation in pericytes is decreased. As shown in Fig. 4A, NAC significantly reduces ROS levels induced by HG (p<0.001) to the level of LG. Similarly, HG-induced DNA breakage in pericytes is prevented by NAC, both in DAPI staining (Fig. 4B) and comet assay (Fig. 4C). Second, we treated pericytes with AzaS (2 μM) for 2 days similar to NAC above and show that HG induced ROS generation in pericytes is significantly decreased (Fig. 5A). In addition, MTT activity is also increased by AzaS under HG (p<0.001; n=8, Fig. 5B), suggesting cell viability. The observed actions of AzaS in reducing HG-induced ROS and enhanced MTT activity in pericytes correlate well with decreases in chromatin breakage in DAPI staining of nuclear DNA by AzaS (Fig. 5C). These results suggest that TXNIP and the HBP, which mediates TXNIP expression and cellular oxidative stress, may play a causal role in pericyte oxidative stress and apoptosis.
Fig. 4.

N-acetylcysteine prevents HG-induced ROS generation and DNA damage in retinal pericytes. (a) ROS generation. N-acetylcysteine (NAC, 5 mM) was added during the last 2 days of treatment while HG was present for 5 days. NAC significantly reduces HG-induced ROS generation in TR-rPCT1 (p<0.001; HG vs. HG+NAC). (b) IHC of DNA break and chromatin condensation (DAPI staining). HG induces TXNIP and chromatin breakage (middle panel, arrows). TXNIP up regulation and chromatin breakage by HG are reduced by NAC (right panel), which is more of less comparable to LG (left panel). A representative of n=3 is shown. (c) NAC also reduces COMET tailing by DNA break in HG treated pericytes suggesting a role for ROS in chromatin instability and breakage. A representative of n=3 is shown.
Fig. 5.

Azaserine prevents HG-induced ROS generation, maintains MTT activity, and reduces DNA break in retinal pericytes. (a) ROS generation. Azaserine (AzaS, 2 μM) was added during the last 2 days of treatment while HG was present for 5 days. ROS generation was determined by CM-H2DCFDA. AzaS has not effect on ROS level in LG but prevents the HG-induced ROS generation in TR-rPCT1. (b) MTT activity. In the presence of AzaS, a reduction in the MTT activity by HG is reversed in retinal pericytes. In fact there is a significant increase (p<0.001) in MTT activity in HG+AzaS. (c) IHC of DNA break and chromatin condensation. HG induces chromatin breakage (middle panel, arrows), which is reduced by AzaS (right panel), which is more of less comparable to LG (left panel). A representative of n=3 is shown.
TXNIP knock down by siRNA reduces high glucose-induced ROS and DNA break in retinal pericytes
We further determine the extent to which TXNIP critically regulates ROS/RNS generation, DNA damage and pericyte apoptosis in retinal pericytes under chronic hyperglycemia. For this, we used a transient transfection method to knock down TXNIP by siRNA when compared with a scramble RNA. HG induces ROS generation in pericytes transfected with the scrRNA while ROS level in siTXNIP3 transfected cells is significantly reduced (p<0.01) to levels observed in LG (Fig. 6A). Furthermore, siTXNIP3 reduces TXNIP expression and chromatin breakage as shown by TXNIP and DAPI staining (Fig. 6B). In addition, DNA tailing by broken chromatins in pericytes under HG is attenuated by siTXNIP3 transfection as seen in the alkaline comet assay (Fig. 6C). Finally, caspase-3 activation by HG in scrRNA treated pericytes is absent in siTXNIP3 treated cells (Fig. 3S), suggesting that TXNIP may play an important role in caspase-3 activation, DNA damage and retinal pericyte loss under chronic hyperglycemia and diabetes.
Fig. 6.

TXNIP siRNA prevents HG-induced ROS generation and DNA damage in retinal pericytes. Scramble (scr)RNA or siTXNIP3 were transiently transfected in pericytes for the last 2 days of 5 days HG treatment. (a) ROS determination by CM-H2DCFDA. HG is able to increase ROS level in scrRNA-treated pericytes (p<0.06 vs. LG+scrRNA). However, after siTXNIP3 transfection, ROS level is significantly reduced (p<0.01; scrRNA+HG vs. siTXNIP3+HG). (b) IHC. SrcRNA transfection has no effect on TXNIP and DNA integrity in LG (left panel). However, HG increases TXNIP staining and DNA damage/breakage (middle panel, arrows) in scrRNA transfected pericytes. On the other hand, siTXNIP3 reduces both TXNIP staining and DNA breakage in pericytes by HG, which is more or less comparable to the level of LG+scrRNA (right panel). (c) Comet assay further shows DNA tailing by HG in scrRNA treated TR-rPCT1 suggesting chromatin breakage. However, with siTXNIP3 transfection, the DNA tailing are absent indicating that TXNIP is involved in chromatin breakage under chronic hyperglycemia (middle panel vs. right panel). A representative of n=3 is shown.
High glucose does not increase autophagic response in retinal pericytes but activates pro-apoptotic GADD153
We have recently shown that, under chronic hyperglycemia (HG, 5 days), Muller cells induce an autophagic/mitophagic response to remove damaged mitochondria and maintain cell viability [15]. We therefore reasoned that pericyte apoptosis occurs in HG due to a weak cellular defense response against oxidative/nitrosative stress. To test this notion, we measure whether autophagy is or is not activated in retinal pericytes under HG exposure. We do not observe a significant increase in the number and size of LC3B punctae in IHC nor the conversion of LC3BI (inactive form) to LC3BII (active form) on Western blots (Fig. 7A and B, respectively), a marker for autophagy induction. Furthermore, we do not find a change in the activity of DNA damage repair protein p53 as measured by EMSA (Fig. 7C). Nonetheless, the DNA binding activity of DNA-damage and ER-stress inducible pro-apoptotic transcription factor GADD153/CHOP is enhanced in EMSA (Fig. 7D), suggesting that under HG GADD153 may be involved in pericyte apoptosis. Surprisingly, we do not observe an induction of pro-apoptotic genes such as Bim and PUMA, which are considered to be targets of GADD153 and p53, respectively (Fig. S4A) or an increase in ER-stress response protein sXBP1 activity (Fig. S4B).
Fig. 7.

HG does not increase autophagic response in retinal pericytes but induces GADD153 DNA binding activity. (a) IHC of LC3B antibody. The number and size of LC3B punctae (red dots) were marginally increased in IHC. (b) However, on Western blots LC3BII levels, the cleaved and active form, are not significantly different between HG and LG. β-Actin was used for normalization. A representative of n=3 is shown for each experiment. ((c)–(d)) EMSA for DNA damage repair protein p53 and pro-apotptotic GADD153. EMSA shows that the DNA binding activity of (c) p53 is unaltered by HG while that of (d) GADD153 is enhanced. The specificity of the reaction is indicated by a competitive inhibition with excess cold probe and a lack of protein–DNA band shift by mannitol. Lane 1. Probe alone; Lane 2. LG; Lane 3. HG; Lane 4. Mannitol; and Lane 5. HG+cold probe (n=3–4 for each experiment).
Discussion
In summation, we provide first evidence that high glucose up regulates TXNIP expression in retinal pericytes in culture and induces cellular oxidative/nitrosative stress, mitochondrial dysfunction, DNA damage and apoptosis. Treatment with anti-oxidant NAC or with azaserine, which inhibits TXNIP [14] or TXNIP knock down itself by siRNA prevents pericyte oxidative stress, DNA damage and apoptosis. These results provide important mechanistic insights into how pericyte loss and vascular dysfunction may occur in early stages of DR. We also find that retinal pericyte apoptosis under hyperglycemic exposure seen in this study differs from our recent report in Muller cells under similar duration of hyperglycemic exposure [15]. In that study, we showed that high glucose causes cellular oxidative stress in Muller cells but maintains cell viability via induction of host innate defense and survival mechanisms including autophagy/mitophagy and anti-oxidant capacity [15]. Furthermore, Busik et al. [33] also demonstrated that retinal endothelial cells are able to sustain viability under chronic hyperglycemia in culture and that endothelial cell death is mediated by cytokines secreted by neighboring cells in the retina. Therefore, a reason for pericyte ROS/RNS stress and apoptosis under HG observed in this study may be due to accumulation of damaged mitochondria, which generate ROS and are inefficient in energy (ATP) production. We suggest further that this might lead to activation of an intrinsic cell death signal and cell-autonomous apoptosis. In support of this, we show that mitochondrial damage, oxidative stress, caspase-3 activation and DNA break occur in pericytes under HG, however, these effects are prevented by anti-oxidant NAC treatment as well as TXNIP knock down by siRNA.
Mitochondria are critical for cellular bioenergetics, metabolism and cell death signals. This is supported by our observation that MTT activity, which measures the reductase activity of mitochondrial complex II enzyme succinate dehydrogenase, is reduced in pericytes by HG, and ATP level is reduced while caspase-3 is activated. In contrast, Muller cells maintain MTT succinate dehydrogenase activity and viability under HG exposure [15]. Succinate dehydrogenase participates in mitochondrial glucose metabolism both in tricarboxylic acid (TCA) cycle and OxPhos at electron transport chain (ETC) complex II, which uses FADH2 as an electron donor to complex III and converts succinate to fumarate at TCA cycle to generate FADH2 [34,35]. Complex II proteins are exclusively synthesized by nuclear genes and are imported into mitochondria as unfolded proteins for assembly. Other ETC proteins at complexes I, III–V are synthesized by both mitochondrial and nuclear genes [36] and they may also be affected in pericytes by HG and oxidative/nitrosative stress. Accumulation of oxidatively stressed and depolarized mitochondria in the cell will therefore result in protein misfolding and disassembly of ETC protein complexes leading to low ATP synthesis and bioenergetic imbalance [37]. It appears that pericytes depend on mitochondrial OxPhos for cellular bioenergetics, i.e., ATP generation (this study) while Muller cells maintain both cytosolic glycolysis and mitochondrial OxPhos [15,38]. In agreement with this, Trudeau et al. [39] also demonstrated that mitochondrial fragmentation occurs in retinal pericytes exposed to HG and that fragmentation also causes low oxygen consumption and DNA nick. An injury to the mitochondrion therefore will have a profound effect on pericyte energy level and cell functioning [40–42]. Pericytes are like renal mesangial and vascular smooth muscle cells that are contractile, and we have previously shown that mesangial cells like pericytes also undergo oxidative stress and apoptosis under chronic hyperglycemia and TXNIP up regulation [43,44]. Further indepth studies and a comparison of bioenergetic requirement between retinal pericyte and Muller cell, as well as with other retinal cells, will be important to determine cell type specific hyperglycemic injury in DR pathogenesis.
TXNIP is present in the cytosol, nucleus and mitochondria; therefore, TXNIP up regulation by HG in pericytes could increase both the cytosolic/nuclear and mitochondrial ROS and RNS levels. ROS (H2O2) and RNS (NO) are signaling molecules at low level however they are toxic at higher concentrations. Mitochondrial OxPhos is the major source of ROS in the cell while membrane bound and cytosolic NADPH oxidase and xanthine oxidase also contribute to ROS generation. In the absence of an efficient autophagy/mitophagy response, which exclusively removes depolarized and fragmented mitochondria, and a weak DNA damage repair mechanism (Fig. 7), pericytes will accumulate damaged mitochondria that can enhance cellular ROS/RNS stress, protein misfolding, and DNA damage. We propose that p53, which is activated following DNA damage and plays a critical role in DNA repair, is not activated while pro-apoptotic GADD153/CHOP is activated will promote cell death processes in pericyte [45–47]. Interestingly, we did not observe activation of GADD153 down-stream target genes that are involved in apoptosis such as Bim and PUMA (Fig. S4). Neither the pro-survival protein Bcl2 nor ER-stress induced spliced form of XBP1 that are important for survival and anti-oxidant gene induction are activated in pericytes. In line with this observation, other investigators have reported an induction of ER stress in retinal pericytes by glucose deprivation, not by hyperglycemia, [48] and with intermittent hyperglycemia [45]. Therefore, signaling mechanisms of pericyte apoptosis under chronic hyperglycemia and TXNIP up regulation remains to be resolved. Nonetheless, we could consider that TXNIP-mediated oxidative stress and mitochondrial cell death signals are involved in pericyte demise since caspase-3 activation and chromatin breakage occur under chronic hyperglycemia (this study). Several other studies also have shown a role for TXNIP in mitochondrial TXNIP-Trx2-ASK1 axis in pancreatic beta cell death [49] and NMDA mediated excitotoxicity in the retina [50,51] and glaucoma [52].
One of the consequences of sustained hyperglycemia and TXNIP up regulation in diabetes will be thiol redox imbalance due to Trx inactivation, ROS/RNS stress, and aberrant protein-S-nitrosylation (SNO). We have recently published that diabetes up regulates both TXNIP and iNOS in the retina [15]. NO is a diffusible molecule, which is also constitutively produced by endothelial eNOS and neuronal nNOS [52,53]. However, induction of iNOS with TXNIP in retinal pericytes as well as in Muller glia/microglia and endothelial cells in diabetes [15,54,55] will exacerbate excessive cell redox imbalance and protein-SNO. This can occur because iNOS/NO will increase protein SNO as well as tyrosine nitration while TXNIP by trapping Trx1 (cytosol/nuclear) and Trx2 (mitochondria) will mitigate against denitrosylation of protein-SNO. Pathologically, thiol S-nitrosylation of certain proteins may be initially protective, such as the SNO modification of caspase-3, which prevents its release from the mitochondrion [16,17,56]; however, thiol modification of pro-inflammatory proteins such as Cox-2-SNO [57], HIF-1α-SNO [58] and glycolytic and apoptotic GAPDH-SNO [59,60] will have deleterious effects in chronic hyperglycemia and cellular stress. These observations are in concert with our hypothesis that TXNIP’s effect on pericyte apoptosis in early diabetic retinopathy may involve S-nitrosylation of critical enzymes and signaling proteins via a TXNIP-mediated Trx inhibition.
Conclusion
We demonstrate a critical function of TXNIP in pericyte apoptosis under chronic high glucose in culture. The molecular events may involve ROS/RNS stress, mitochondrial membrane depolarization, bioenergetic imbalance, chromatin damage, and premature pericyte demise in the diabetic retina. We summarize results of this study in Fig. 8. Our working hypothesis is that anti-oxidant therapies targeting mitochondria and suppression of TXNIP up-regulation in diabetic retinopathy may have potentially beneficial effects in preventing ocular complications. In support of this hypothesis, we have previously shown that TXNIP knock down by intravitreal delivery of siRNA in rats in vivo ameliorates early molecular abnormalities of diabetes in the retina, which include aberrant extracellular matrix gene expression, inflammation, gliosis, and retinal cell death [14,15]. Furthermore, other investigators have recently shown that TXNIP knock out mouse protects the myocardium from ischemia-reperfusion injury [61], endotoxemia insult [62], and ER-stress mediated beta cell apoptosis [24,25]. Together, our current data showing that TXNIP is critical for retinal pericyte oxidative stress, DNA damage and apoptosis under chronic hyperglycemia further support the notion that TXNIP is a novel therapeutic target to prevent or treat DR. Further in vivo studies using TXNIP knockout mouse and/or TXNIP siRNA knock down approaches in animal models of diabetes are warranted to elucidate the role of TXNIP in inflammation, pericyte loss and acellular capillary formation in DR.
Fig. 8.
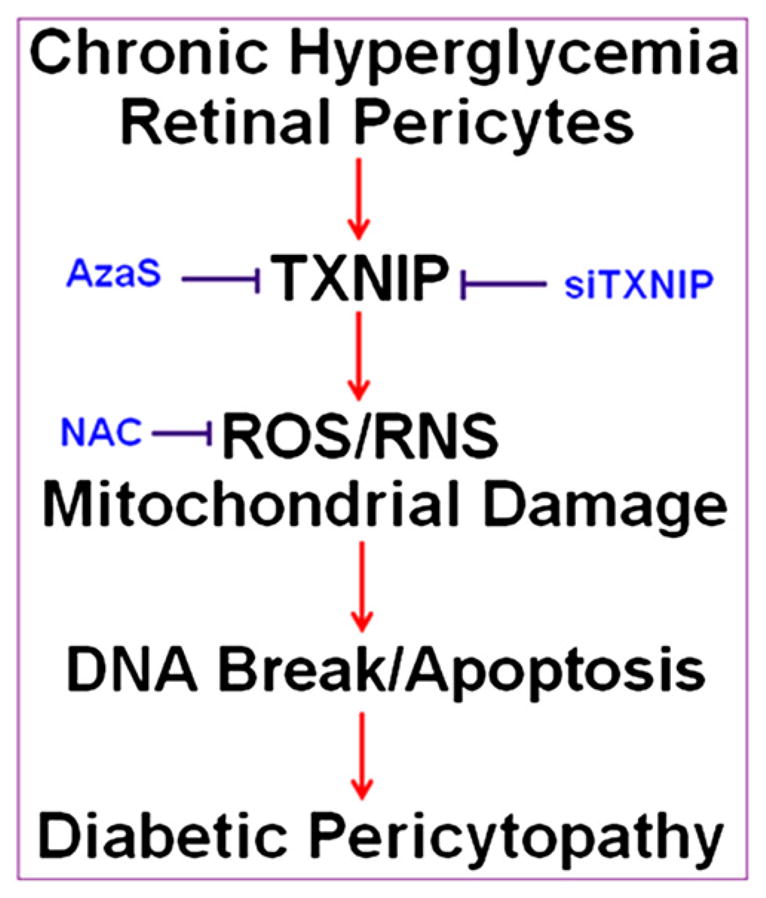
Summary: A potential role of TXNIP in hyperglycemia-induced ROS/RNS stress, DNA damage and pericyte demise (Pericytopathy) in DR. Chronic hyperglycemia-induced TXNIP up-regulation leads to cellular ROS/RNS stress, mitochondrial membrane depolarization, bioenergetic imbalance (low ATP), chromatin (DNA) fragmentation and pericyte apoptosis. Retinal pericytes appear to have a weaker anti-oxidant and mitophagic response to cellular stress to scavenge ROS and remove depolarized mitochondria, which leak ROS and are inefficient in ATP production. In addition, the DNA damage repair mechanism (p53 activation) is not evoked that may result in chromatin breakage and early demise of pericytes in DR. Anti-oxidant treatment such as NAC and a blockade of TXNIP via an inhibition of the HBP may represent potential therapeutic approaches to ameliorate DR pathogenesis.
Supplementary Material
Acknowledgments
Research grants from Mid-West Eye Bank, Michigan, and Bridge Funds from the Department of Anatomy/Cell Biology and School of Medicine to Dr. Singh are also acknowledged. Research funding to the Department of Anatomy and Cell Biology for Core facilities by grant P30 EY04068 from the National Eye Institute, the National Institutes of Health is also acknowledged. Supports from Research to Prevent Blindness to the Department of Ophthalmology are also acknowledged. We thank Dr. Kwaku D. Nantwi at the Department of Anatomy and Cell Biology, Wayne state University, Detroit, MI, for reading the manuscript critically. Parts of this study were presented at the Annual Meeting of ARVO at Ft. Lauderdale, FL, June 6–10, 2012.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.yexcr.2013.01.012.
References
- 1.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010;376:124–136. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 2.Robinson R, Barathi VA, Chaurasia SS, Wong TY, Kern TS. Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech. 2012;5:444–456. doi: 10.1242/dmm.009597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–358. doi: 10.1016/j.preteyeres.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frey T, Antonetti DA. Alterations to the blood-retinal barrier in diabetes: cytokines and reactive oxygen species. Antioxid Redox Signal. 2011;15:1271–1284. doi: 10.1089/ars.2011.3906. [DOI] [PubMed] [Google Scholar]
- 5.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–1306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy S, Nasser S, Yee Y, Graves DT, Roy SS. A long-term siRNA strategy regulates fibronectin overexpression and improves vascular lesions in retinas of diabetic rats. Mol Vis. 2011;17:3166–3174. [PMC free article] [PubMed] [Google Scholar]
- 7.Hammes HP, Feng Y, Pfister F, Brownlee M. Diabetic retinopathy: targeting vasoregression. Diabetes. 2011;60:9–16. doi: 10.2337/db10-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puro DG. Retinovascular physiology and pathophysiology: new experimental approach/new insights. Prog Retin Eye Res. 2012;31:258–270. doi: 10.1016/j.preteyeres.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1156–1163. doi: 10.1167/iovs.10-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem. 2004;279:30369–30374. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- 12.Forrester MT, Seth D, Hausladen A, Eyler CE, Foster MW, Matsumoto A, Benhar M, Marshall HE, Stamler JS. Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J Biol Chem. 2009;284:36160–36166. doi: 10.1074/jbc.M109.057729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol. 2009;221:262–272. doi: 10.1002/jcp.21852. [DOI] [PubMed] [Google Scholar]
- 14.Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010;1:65. doi: 10.1038/cddis.2010.42. http://dx.doi.org/10.1038/cddis.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devi TS, Lee I, Hüttemann M, Kumar A, Nantwi KD, Singh LP. Txnip links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res. 2012:438238. doi: 10.1155/2012/438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu C, Parrott AM, Fu C, Liu T, Marino SM, Gladyshev VN, Jain MR, Baykal AT, Li Q, Oka S, Sadoshima J, Beuve A, Simmons WJ, Li H. Thioredoxin 1-mediated post-translational modifications: reduction, transnitrosylation, denitrosylation, and related proteomics methodologies. Antioxid Redox Signal. 2011;15:2565–2604. doi: 10.1089/ars.2010.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barglow KT, Knutson CG, Wishnok JS, Tannenbaum SR, Marletta MA. Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc Nat Acad Sci USA. 2011;108:E600–E606. doi: 10.1073/pnas.1110736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 20.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nuñez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LA. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AF, Joosten LA, Tack CJ, Netea MG. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1β transcription in human adipose tissue. Diabetes. 2011;60:517–524. doi: 10.2337/db10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trueblood KE, Mohr S, Dubyak GR. Purinergic regulation of high glucose-induced caspase-1 activation in the rMC-1 rat retinal Muller cell line. Am J Physiol Cell Physiol. 2011;301:C1213–C1223. doi: 10.1152/ajpcell.00265.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sbai O, Devi TS, Melone MA, Feron F, Khrestchatisky M, Singh LP, Perrone L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J Cell Sci. 2010;123:4332–4339. doi: 10.1242/jcs.074674. [DOI] [PubMed] [Google Scholar]
- 24.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome andpromote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, Greiner D, Kaufman RJ, Bortell R, Urano F. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondo T, Hosoya K, Hori S, Tomi M, Ohtsuki S, Takanaga H, Nakashima E, Iizasa H, Asashima T, Ueda M, Obinata M, Terasaki T. Establishment of conditionally immortalized rat retinal pericyte cell lines (TR-rPCT) and their application in a co-culture system using retinal capillary endothelial cell line (TR-iBRB2) Cell Struct Funct. 2003;28:145–153. doi: 10.1247/csf.28.145. [DOI] [PubMed] [Google Scholar]
- 27.Kador PF, Randazzo J, Blessing K, Makita J, Zhang P, Yu K, Hosoya K, Terasaki T. Polyol formation in cell lines of rat retinal capillary pericytes and endothelial cells (TR-rPCT and TR-iBRB) J Ocul Pharmacol Ther. 2009;25:299–308. doi: 10.1089/jop.2008.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adachi T, Yasuda H, Aida K, Kamiya T, Hara H, Hosoya K, Terasaki T, Ikeda T. Regulation of extracellular-superoxide dismutase in rat retina pericytes. Redox Rep. 2010;15:250–258. doi: 10.1179/135100010X12826446921662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makita J, Hosoya K, Zhang P, Kador PF. Response of rat retinal capillary pericytes and endothelial cells to glucose. J Ocul Pharmacol Ther. 2011;27:7–15. doi: 10.1089/jop.2010.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh LP, Devi TS, Nantwi KD. Theophylline regulates inflammatory and neurotrophic factor signals in functional recovery after C2-hemisection in adult rats. Exp Neurol. 2012;238:79–88. doi: 10.1016/j.expneurol.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sen N, Snyder SH. Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc Nat Acad Sci USA. 2011;108:20178–20183. doi: 10.1073/pnas.1117820108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57:1952–1965. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ackrell BA. Cytopathies involving mitochondrial complex II. Mol Aspects Med. 2002;23:369–384. doi: 10.1016/s0098-2997(02)00012-2. [DOI] [PubMed] [Google Scholar]
- 35.Sandhir R, Sood A, Mehrotra A, Kamboj SS. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener Dis. 2012;9:145–157. doi: 10.1159/000334273. [DOI] [PubMed] [Google Scholar]
- 36.Morán M, Moreno-Lastres D, Marín-Buera L, Arenas J, Martín MA, Ugalde C. Mitochondrial respiratory chain dysfunction: implications in neurodegeneration. Free Radical Biol Med. 2012;53:595–609. doi: 10.1016/j.freeradbiomed.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2012 Mar;14 doi: 10.1016/j.bbamcr.2012.02.019. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabezas R, El-Bachá RS, González J, Barreto GE. Mitochondrial functions in astrocytes: neuroprotective implications from oxidative damage by rotenone. Neurosci Res. 2012 Aug 10; doi: 10.1016/j.neures.2012.07.008. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Trudeau K, Molina AJ, Roy S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Invest Ophthalmol Vis Sci. 2011;52:8657–8664. doi: 10.1167/iovs.11-7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jelenik T, Roden M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid Redox Signal. 2012 Sep 3; doi: 10.1089/ars.2012.4910. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res. 2011;36:1069–1077. doi: 10.3109/02713683.2011.607536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest Ophthalmol Vis Sci. 2011;52:8739–8746. doi: 10.1167/iovs.11-8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devi TS, Singh LP, Hosoya KI, Terasaki T. GSK-3β/CREB axis mediates IGF-1-induced ECM/adhesion molecule expression, cell cycle progression and monolayer permeability in retinal capillary endothelial cells: implications for diabetic retinopathy. Biochim Biophys Acta Mol Basis Dis. 2011;1812:1080–1088. doi: 10.1016/j.bbadis.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Cheng DW, Jiang Y, Shalev A, Kowluru R, Crook ED, Singh LP. An analysis of high glucose and glucosamine-induced gene expression and oxidative stress in renal mesangial cells. Arch Physiol Biochem. 2006;112:189–218. doi: 10.1080/13813450601093518. [DOI] [PubMed] [Google Scholar]
- 45.Zhong Y, Wang JJ, Zhang SX. Intermittent but not constant high glucose induces ER stress and inflammation in human retinal pericytes. Adv Exp Med Biol. 2012;723:285–292. doi: 10.1007/978-1-4614-0631-0_37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jauhiainen A, Thomsen C, Strömbom L, Grundevik P, Andersson C, Danielsson A, Andersson MK, Nerman O, Rörkvist L, Ståhlberg A, Åman P. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS One. 2012;7:e33208. doi: 10.1371/journal.pone.0033208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Awai M, Koga T, Inomata Y, Oyadomari S, Gotoh T, Mori M, Tanihara H. NMDA-induced retinal injury is mediated by an endoplasmic reticulum stress-related protein, CHOP/GADD153. J Neurochem. 2006;96:43–52. doi: 10.1111/j.1471-4159.2005.03502.x. [DOI] [PubMed] [Google Scholar]
- 48.Ikesugi K, Mulhern ML, Madson CJ, Hosoya K, Terasaki T, Kador PF, Shinohara T. Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr Eye Res. 2006;31:947–953. doi: 10.1080/02713680600966785. [DOI] [PubMed] [Google Scholar]
- 49.Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem. 2010;285:3997–4005. doi: 10.1074/jbc.M109.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al-Gayyar MM, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Thioredoxin interacting protein is a novel mediator of retinal inflammation and neurotoxicity. Br J Pharmacol. 2011;164:170–180. doi: 10.1111/j.1476-5381.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Munemasa Y, Kwong JM, Kim SH, Ahn JH, Caprioli J, Piri N. Thioredoxins 1 and 2 protect retinal ganglion cells from pharmacologically induced oxidative stress, optic nerve transection and ocular hypertension. Adv Exp Med Biol. 2010;664:355–363. doi: 10.1007/978-1-4419-1399-9_41. [DOI] [PubMed] [Google Scholar]
- 52.Li Q, Verma A, Han PY, Nakagawa T, Johnson RJ, Grant MB, Campbell-Thompson M, Jarajapu YP, Lei B, Hauswirth WW. Diabetic eNOS-knockout mice develop accelerated retinopathy. Invest Ophthalmol Vis Sci. 2010;51:5240–5246. doi: 10.1167/iovs.09-5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giove TJ, Deshpande MM, Gagen CS, Eldred WD. Increased neuronal nitric oxide synthase activity in retinal neurons in early diabetic retinopathy. Mol Vis. 2009;15:2249–2258. [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra A, Newman EA. Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia. 2010;58:1996–2004. doi: 10.1002/glia.21068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng L, Du Y, Miller C, Gubitosi-Klug RA, Ball S, Berkowitz BA, Kern TS. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia. 2007;50:1987–1996. doi: 10.1007/s00125-007-0734-9. [DOI] [PubMed] [Google Scholar]
- 56.Lai YC, Pan KT, Chang GF, Hsu CH, Khoo KH, Hung CH, Jiang YJ, Ho FM, Meng TC. Nitrite-mediated S-nitrosylation of caspase-3 prevents hypoxia-induced endothelial barrier dysfunction. Circ Res. 2011;109:1375–1386. doi: 10.1161/CIRCRESAHA.111.256479. [DOI] [PubMed] [Google Scholar]
- 57.Tian J, Kim SF, Hester L, Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Nat Acad Sci USA. 2008;105:10537–10540. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, Vujaskovic Z, Dewhirst MW, Li CY. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007;13:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sen N, Snyder SH. Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc Nat Acad Sci USA. 2011;108:20178–20183. doi: 10.1073/pnas.1117820108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG, Lee RT. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. 2012;122:267–279. doi: 10.1172/JCI44927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oka S, Liu W, Yoshihara E, Ahsan MK, Ramos DA, Son A, Okuyama H, Zhang L, Masutani H, Nakamura H, Yodoi J. Thioredoxin binding protein-2 mediates metabolic adaptation in response to lipopolysaccharide in vivo. Crit Care Med. 2010;38:2345–2351. doi: 10.1097/CCM.0b013e3181f85b2a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.