

Abstract
Alpha-Klotho (αKlotho) protein is encoded by the gene, Klotho, and functions as a coreceptor for endocrine fibroblast growth factor-23. The extracellular domain of αKlotho is cleaved by secretases and released into the circulation where it is called soluble αKlotho. Soluble αKlotho in the circulation starts to decline in chronic kidney disease (CKD) stage 2 and urinary αKlotho in even earlier CKD stage 1. Therefore soluble αKlotho is an early and sensitive marker of decline in kidney function. Preclinical data from numerous animal experiments support αKlotho deficiency as a pathogenic factor for CKD progression and extrarenal CKD complications including cardiac and vascular disease, hyperparathyroidism, and disturbed mineral metabolism. αKlotho deficiency induces cell senescence and renders cells susceptible to apoptosis induced by a variety of cellular insults including oxidative stress. αKlotho deficiency also leads to defective autophagy and angiogenesis and promotes fibrosis in the kidney and heart. Most importantly, prevention of αKlotho decline, upregulation of endogenous αKlotho production, or direct supplementation of soluble αKlotho are all associated with attenuation of renal fibrosis, retardation of CKD progression, improvement of mineral metabolism, amelioration of cardiac function and morphometry, and alleviation of vascular calcification in CKD. Therefore in rodents, αKlotho is not only a diagnostic and prognostic marker for CKD but the enhancement of endogenous or supplement of exogenous αKlotho are promising therapeutic strategies to prevent, retard, and decrease the comorbidity burden of CKD.
1. INTRODUCTION
The Klotho gene was discovered in 1997 when mice with silencing of this gene developed multiple organ dysfunction and failure with shortened life span resembling human premature aging (Kuro-o et al., 1997). The overexpression of the Klotho transgene with a ubiquitous promoter (Kurosu et al., 2005), viral-based transfer (Masuda, Chikuda, Suga, Kawaguchi, & Kuro-o, 2005), or direct parenteral administration (Chen, Kuro, et al., 2013) can extend mouse life span compared to normal mouse and rescue the most phenotypes observed in Klotho-deficient mouse (Kurosu et al., 2005). Two other paralogs βKlotho (Ito et al., 2000) and γKlotho (Ito, Fujimori, Hayashizaki, & Nabeshima, 2002) were identified, then Klotho gene was designated αKlotho to distinguish from the other two paralogs (Hu, Shiizaki, Kuro-o, & Moe, 2013).
αKlotho is highly expressed in the kidney, brain, and in lesser extent in other organs (Kato et al., 2000; Kuro-o et al., 1997). Human αKlotho is a single transmembrane 1012 amino acid 130 kDa protein encoded by human Klotho gene, while rodent αKlotho is a 1014 amino acid protein (Kuro-o et al., 1997; Matsumura et al., 1998; Shiraki-Iida et al., 1998; Tohyama et al., 2004). The extracellular domain of membrane αKlotho consisting of two repeat sequences (kl1 and kl2) can be shed by secretases and released into the circulation (Bloch et al., 2009; Chen, Podvin, Gillespie, Leeman, & Abraham, 2007; Chen, Tung, et al., 2014; Hu, Shi, Zhang, et al., 2015). This released extracellular domain of membrane αKlotho is referred as soluble or cleaved αKlotho. It is a main functional form in the circulation (Hu, Shi, Zhang, et al., 2015; Hu, Shi, Zhang, Pastor, et al., 2010; Hu, Shi, Zhang, Quinones, et al., 2010; Imura et al., 2004; Kurosu et al., 2005). Soluble αKlotho protein is also present in cerebrospinal fluid (Chen et al., 2015; Degaspari et al., 2015; Emami Aleagha et al., 2015; Imura et al., 2004; Semba et al., 2014) and urine of mammals (Akimoto et al., 2012; Hu, Shi, Zhang, Pastor, et al., 2010; Hu et al., 2011; Lau et al., 2012). Soluble αKlotho functions as a circulating substance exerting multiple systemic biological actions on distant organs and directly protects cells against a variety of insults including hypoxia, hyperoxia, oxidative stress, and cytotoxic medication and suppresses apoptosis (Cheng et al., 2015; Hu, Shi, Cho, et al., 2013; Panesso et al., 2014; Ravikumar et al., 2014; Sun et al., 2015; Wang et al., 2013).
Chronic kidney disease (CKD) is characterized by progressive deterioration of renal function with high risk of end-stage renal disease (ESRD) regardless of whether initial kidney insults have regressed or are continuously present (D’Hoore et al., 2015; Ferenbach & Bonventre, 2015; Rimes-Stigare et al., 2015; Venkatachalam, Weinberg, Kriz, & Bidani, 2015). As expected, CKD risk increases with age, and about half of the CKD stage ≥3 cases occurs in subjects >70 years old. CKD can be viewed as a state of accelerating aging (Kooman et al., 2013; Stenvinkel & Larsson, 2013). The relative risk for cardiovascular (CV) mortality of a 25- to 34-year-old dialysis patient is similar to a non-CKD patient of >75 years of age (Foley, Parfrey, & Sarnak, 1998). The similar phenotypes between αKlotho-deficient mice and CKD subjects also suggest a potential pathogenic role of αKlotho deficiency in CKD development and progression (Hu, Kuro-o, & Moe, 2012, 2013a, 2013b; Hu et al., 2011; Hu, Shiizaki, Kuro-o, et al., 2013; Shi et al., 2015).
In this chapter, we aim to summarize the current state of knowledge on αKlotho biology and pathophysiology in CKD, and provide a possible novel perspective on potential clinical applications of αKlotho in CKD.
2. CKD IS A STATE OF KLOTHO DEFICIENCY
2.1 The Kidney Is the Main Origin for Systemic αKlotho
Compared to the wide distribution of αKlotho mRNA in many organs and tissues, αKlotho protein expression is restricted to only a few tissues including the kidney, brain, heart, parathyroid gland, and testis (Kuro-o et al., 1997; Takeshita et al., 2004). αKlotho protein was also found in vascular endothelial cells and smooth muscle cells in humans and rodents (Fang et al., 2014; Jimbo et al., 2014; Lim et al., 2012; Ritter, Zhang, Delmez, Finch, & Slatopolsky, 2015), but this is still in debate because there is equally convincing evidence that do not support the presence of αKlotho protein in the vasculature (Hu, 2016; Lau et al., 2012; Lindberg et al., 2013; Mencke et al., 2015; Scialla et al., 2013). Therefore, whether αKlotho is expressed in the vasculature remains to be resolved. Among tissues expressing αKlotho protein, the kidney has the highest level. In mammalian kidney, αKlotho is prominently expressed in distal convoluted tubules (DCTs; Kato et al., 2000; Kuro-o et al., 1997), but is also unequivocally found in the proximal convoluted tubules although at lower levels compared to DCT (Hu, Shi, Zhang, Pastor, et al., 2010; Lim et al., 2015).
Despite the fact that the kidney is the organ expressing the highest levels of αKlotho protein, the confirmation that circulating αKlotho in serum mainly derived from the kidney under physiological conditions was demonstrated by Lindberg et al. (2014). The strongest evidence comes from mouse with renal tubule-specific partial deletion of αKlotho. This mouse line has reduced serum αKlotho levels and systemic features resembling the phenotype of global αKlotho deletion or the αKlotho hypomorphic mice, indicating that the kidney may be the principal organ mediating the systemic αKlotho effects (Lindberg et al., 2014). More direct evidence to support this notion is that αKlotho protein in the suprarenal vein is higher than that in infrarenal vein in rodents and humans (Hu, Shi, Zhang, et al., 2015) and circulating αKlotho levels were dramatically and quickly dropped in rodents that underwent bilateral nephrectomy (Hu, Shi, Zhang, et al., 2015) which strongly suggest that the kidney is the main source of αKlotho in the circulation under physiological conditions (Fig. 1). In living human kidney donors (Akimoto et al., 2013; Ponte et al., 2014), αKlotho was shown to drop after nephrectomy but it is difficult to distinguish this from the effect of surgery. However, under pathological renal conditions such as ESRD, circulating αKlotho is low rather absent, suggesting the possibility that αKlotho may come from extrarenal source(s), although its origin is not clear to date (Lau et al., 2012). Establishing extrarenal sources of αKlotho and characterizing how this can be upregulated when renal production fails are of paramount importance.
Fig. 1.
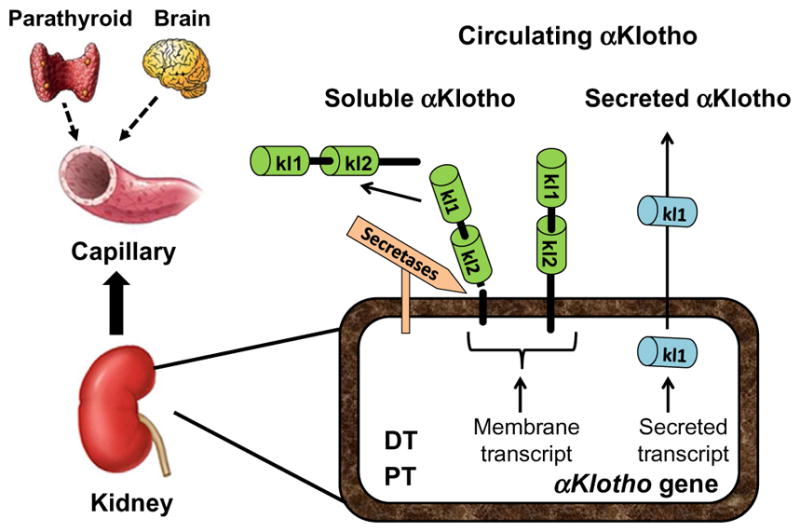
Source of circulatory αKlotho. αKlotho protein is expressed in a few organs, but the kidney is a main resource of circulating αKlotho under physiological conditions. The contribution of parathyroid gland and brain is not clear. Both renal proximal (PT) and distal tubules (DT) express membrane αKlotho protein and may also produce a secreted αKlotho protein which only contains kl1 domain and is directly secreted into the blood circulation. Extracellular domain of membrane αKlotho-containing kl1 and kl2 repeats is shed and cleaved by α and β-secretases, and released into the blood circulation.
2.2 Renal αKlotho Deficiency in CKD
With the renal source of circulating αKlotho established (Fig. 1), the next step is to understand why and how αKlotho is drastically reduced in kidney disease. As a general principle, if the organ of origin of an endocrine substance is diseased, it is logical to suspect that endocrine deficiency of that substance ensues. Therefore, it is not surprising to witness the reduction of αKlotho protein in the diseased kidney. However, whether the reduction of αKlotho is due to destruction of kidney, loss ability to produce/secrete αKlotho, or a maladaptive response, remains to be explored.
It has been shown that there is a significant reduction of renal αKlotho transcript and protein in the diseased kidney resulting from a wide variety of etiologies from glomerular and tubulointerstitial diseases, obstructive nephropathy, diabetic nephropathy, ischemic injury, subtotal nephrectomy, oxidative stress, chronic allograft rejection, and exposure to cisplatin, angiotensin II (Ang II), and calcineurin inhibitors in both humans and rodents (Hu et al., 2012; Hu et al., 2013a, 2013b; Hu, Shiizaki, Kuro-o, et al., 2013; Panesso et al., 2014; Sastre et al., 2013; Shi et al., 2015; Zhou, Li, Zhou, Tan, & Liu, 2013). The mechanisms underlying the relationship between renal αKlotho downregulation and kidney diseases therefore need to be further illustrated.
It is proposed that renal αKlotho deficiency in early stages of CKD may be attributed mainly to suppression of αKlotho expression rather than loss of viable renal tubules. Several intermediates are shown to be involved in the reduction of αKlotho expression: high serum phosphate (Hu, Kuro-o, & Moe, 2014), hypermethylation (Azuma et al., 2012; King, Rosene, & Abraham, 2012; Lee, Jeong, et al., 2010; Sun, Chang, & Wu, 2012; Young & Wu, 2012), and hyperdeacetylation (Moreno et al., 2011) in αKlotho gene promoter induced by inflammatory cytokines and the uremic toxin, indoxyl sulfate (Fig. 2). If these observations are correct, they provide an opportunity to reactivate αKlotho expression by modulation of these factors and thereby correcting circulating and renal αKlotho deficiency in early stages of CKD.
Fig. 2.
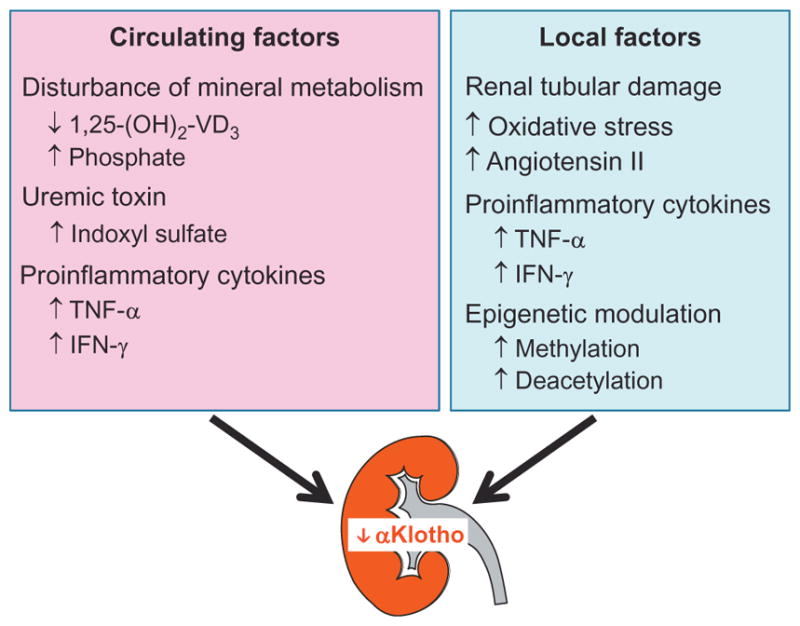
Circulating and local renal factors involved in the reduction of αKlotho expression in the kidney. In acute and chronic kidney disease, a variety of circulating factors including disturbed mineral metabolism, and accumulation of indoxyl sulfate and proinflammatory cytokines (left panel), can downregulate renal αKlotho expression. On the other hand, the elevation of reactive oxygen species, Ang II, and inflammatory cytokines in the diseased kidney can also downregulate renal αKlotho expression (right panel). Epigenetic modulation of αKlotho promoter via hypermethylation and deacetylation can reduce αKlotho expression and contribute to αKlotho deficiency in chronic kidney disease.
2.3 Circulating αKlotho Deficiency in CKD
αKlotho transcript and protein expression in diseased kidneys from humans and animals is clearly decreased. However, the relationship between renal αKlotho expression and serum and/or urinary αKlotho levels remains to be confirmed.
In a rodent model of CKD from uninephrectomy plus contralateral ischemia reperfusion, serum αKlotho concentration was remarkably decreased, and the degree of its reduction was similar in magnitude to that of decreased αKlotho protein in the kidneys and in the urine (Hu et al., 2011). Thus in rodents, CKD is a state of endocrine (circulating and urinary) αKlotho deficiency in addition to renal αKlotho deficiency (Hu et al., 2011).
In CKD patients, urinary αKlotho levels are significantly decreased at very early stages (stage 1) and sustainably reduced with progression of CKD (Hu et al., 2011), while the reduction of serum αKlotho starts later at stage 2 CKD (Barker et al., 2015). Moreover, human urinary αKlotho excretion is significantly decreased and the amount of urinary αKlotho decrease is directly correlated with estimated glomerular filtration rate (eGFR) decline (Yamazaki et al., 2010). These observations suggest that urinary soluble αKlotho may be a good biomarker for early CKD detection. A growing body of evidence has shown a reduction of circulating αKlotho in CKD and ESRD patients (Devaraj, Syed, Chien, & Jialal, 2012; Fliser, Seiler, Heine, & Ketteler, 2012; Pavik et al., 2013; Scholze et al., 2014; Shimamura et al., 2012; Zhou et al., 2013), therefore identifying the plausible mechanisms and clinical implications of serum and/or urinary αKlotho reduction in CKD/ESRD patients is of paramount importance.
3. αKlotho DEFICIENCY CONTRIBUTES TO CKD DEVELOPMENT AND PROGRESSION
αKlotho deficiency is not only an early biomarker of CKD but also a pathogenic intermediate for CKD development and progression, and extrarenal complications. Compared to wild-type mice, αKlotho-deficient mice have more severe kidney damage and faster progression to CKD with more fibrosis, and αKlotho overexpressors have milder kidney dysfunction and less fibrosis after exposure to renal insults including ischemic injury (Hu et al., 2011; Hu, Shi, Zhang, Quinones, et al., 2010; Shi et al., 2015), cisplatin (Panesso et al., 2014), Adriamycin (Zhou et al., 2013), and ureteric ligation (Sugiura et al., 2012; Zhou et al., 2013). αKlotho is a multifaceted protein. Different forms of αKlotho may be involved in different biological functions. Membrane αKlotho is confirmed to participate in maintenance of mineral homeostasis, while soluble αKlotho protein plays a more important and systemic role in cytoprotection, antifibrosis, and angiogenesis.
3.1 Increased Cell Senescence and Reduced Ability of Regeneration
Stem cells in most mammalian tissues participate in maintenance of tissue homeostasis and are involved in tissue repair or regeneration (Li & Clevers, 2010; Weissman, 2000). The dysfunction and depletion of stem cells and progenitor cells contribute to aging and aging-associated diseases including kidney disease. CKD can be a consequence of incomplete or failed tubule recovery after AKI (D’Hoore et al., 2015; Ferenbach & Bonventre, 2015; Kramann, Tanaka, & Humphreys, 2014; Polichnowski et al., 2014; Venkatachalam et al., 2015; Zhang et al., 2013). The repeated administration of bone marrow-derived mesenchymal stem cells improved renal function and histology, reduced blood pressure, and attenuated the infiltration of inflammatory cells on a remnant rat kidney (Lee, Lee, et al., 2010). More recently, evidence has shown human-induced pluripotent stem cells derived from any human somatic cell type after the introduction of reprogramming transcription factors contributing to kidney regeneration and improvement in kidney function (Schmitt, Susnik, & Melk, 2015).
The decrease in stem cell number is associated with an increase in progenitor cell senescence, a complicated process present not only in normal aging but also in pathophysiological states (Dmitrieva & Burg, 2007; Haruna et al., 2007; Jennings et al., 2007; Kailong et al., 2007; Nakano-Kurimoto et al., 2009; Yang et al., 2009; Yang & Fogo, 2010). Excessive senescence and subsequent stem cell deletion may decrease the ability of the kidney to defend against renal insults and impair regeneration (Schmitt et al., 2015).
αKlotho deficiency is associated with stem cell dysfunction and depletion which is part of normal aging (Bian, Neyra, Zhan, & Hu, 2015; Liu et al., 2007). αKlotho deficiency in CKD could enhance renal tubular and vascular cell senescence induced by oxidative stress, uremic toxin, and high phosphate (Carracedo et al., 2013; Clements, Chaber, Ledbetter, & Zuk, 2013; de Oliveira, 2006; Niwa & Shimizu, 2012; Small et al., 2012; Tsirpanlis, 2008; Verbeke, Van Biesen, & Vanholder, 2011; Yamada et al., 2015). Wnt signaling activity is significantly increased in tissues from kl/kl mice, which can be rescued by genetic αKlotho overexpression (Liu et al., 2007). Administration of exogenous Wnt stimulates Wnt signal transduction, and triggers or accelerates cell senescence both in vitro and in vivo. αKlotho appears to be a secreted Wnt antagonist and may utilize this mechanism to retard mammalian aging. Suppression of cell senescence may be one of many novel strategies for promotion of kidney regeneration after AKI and retardation of CKD progression (Camilli et al., 2011; Liu et al., 2007; Satoh et al., 2012; Zhou et al., 2013).
3.2 Defective Endothelial Function and Impaired Vasculogenesis
In CKD patients, there is endothelial dysfunction and impaired bone marrow-derived endothelial progenitor cells-mediated vascular regeneration and kidney repair ( Jie et al., 2010; Mohandas & Segal, 2010), both of which can contribute to progression of CKD and aging of the kidney (Chade et al., 2006; Mu et al., 2009; Reinders, Rabelink, & Briscoe, 2006; Taniyama & Morishita, 2010; Westerweel et al., 2007). Recent studies further indicated a potential causal link between vascular rarefaction and CKD progression. kl/kl mice do not only have abnormal vasodilatation due to abnormal endothelial function (Nakamura et al., 2002) and low blood flow after hind limb ischemia (Fukino et al., 2002) but also impaired angiogenesis and vasculogenesis (Shimada et al., 2004). The HMG coenzyme A reductase inhibitor, cerivastatin, increases αKlotho levels in cultured kidney cell lines (Narumiya et al., 2004) and mice (Yoon et al., 2012) and also restores impaired neovascularization in kl/kl mice (Shimada et al., 2004), but the causal relation between increased Klotho and restoration of vasculogenesis remains to be confirmed. The impaired vasculogenesis and angiogenesis might be attributable to downregulation of vascular endothelial growth factor (VEGF) in the aorta (Nakamura et al., 2002). Recent in vivo and in vitro studies showed that αKlotho is associated with VEGF receptor-2 (VEGFR-2) and the transient receptor potential canonical-1 (TRPC-1) Ca2+ channel to maintain endothelial integrity because in αKlotho-deficient endothelial cells, VEGF-mediated internalization of the VEGFR-2/TRPC-1 complex is impaired, and surface TRPC-1 expression increases which can be reversed by αKlotho protein (Kusaba et al., 2010). In addition, αKlotho mitigates the increased cell senescence and apoptosis triggered by oxidative stress in endothelial cells (Ikushima et al., 2006); and αKlotho also suppresses TNF-β-induced expression of intracellular adhesion molecule-1 and vascular cell adhesion molecule-1, attenuates NF-kappaB activation, and reverses the inhibition of eNOS phosphorylation by TNF-α. Thus αKlotho protein also protects vascular endothelium by inhibition of endothelial inflammation (Maekawa et al., 2009).
3.3 Promotion of Renal Fibrosis
Renal fibrosis is a histological hallmark of CKD and also believed to be a pathogenic intermediate for CKD progression (Ardura, Rayego-Mateos, Ramila, Ruiz-Ortega, & Esbrit, 2010; Iwano et al., 2002; Kalluri & Neilson, 2003; Liu, 2010; Zeisberg & Duffield, 2010; Zeisberg et al., 2003). kl/kl mice have more renal tubulointerstitial fibrosis (Sugiura et al., 2012) which is associated with upregulation of TGF-β in the kidneys. The renal fibrosis induced by unilateral ureteral obstruction (UUO) is accompanied by upregulation of TGF-β and fibronectin, and down-regulation of αKlotho mRNA and protein. These alterations are exaggerated in αKlotho-deficient UUO mice compared to WT UUO mice. Along the same line, more renal fibrosis was found in αKlotho-deficient mice injected with Adriamycin (Zhou et al., 2013). Soluble αKlotho alleviates renal fibrosis induced by UUO and suppresses expression of fibrosis markers and TGF-β1 target genes (eg, Snail, Twist), but does not reduce TGF-β1 expression in UUO kidney (Doi et al., 2011), suggesting that αKlotho suppresses renal fibrosis primarily through inhibiting TGF-β1 downstream signaling (Doi et al., 2011).
As discussed earlier, αKlotho is an antagonist for Wnt signaling and Wnt is associated with renal fibrosis. αKlotho’s suppression of renal fibrosis is conceivably attributable to inhibition of Wnt signal transduction. Mice with UUO and Adriamycin injection have high Wnt levels and β-catenin activity as well as myofibroblast activation which can be suppressed by administration of expression vector encoding the extracellular domain of αKlotho (Zhou et al., 2013).
When renal fibrosis and Wnt signaling are compared between αKlotho overexpression and αKlotho deficiency in UUO, Wnt signaling and tubulointerstitial fibrosis were attenuated in αKlotho-overexpressing compared to WT mice. In contrast, Wnt signaling and tubulointerstitial fibrosis were dramatically augmented in αKlotho heterozygous-deficient (kl/+) mice after UUO compared with WT mice. Interestingly, after transferring plasmid overexpressing αKlotho into skeletal muscle, kl/+ mutant mice had much lower Wnt signaling and extracellular matrix deposition. Therefore, αKlotho is a critical negative regulator of Wnt signaling and a suppressor of renal fibrosis in the obstructed kidney model (Satoh et al., 2012). In addition, αKlotho also promotes clearance of collagen I through upregulation of autophagy (Shi et al., 2015). Therefore, αKlotho suppresses fibrosis and enhances removal of collagen. Exogenous αKlotho administration may be a novel therapeutic agent for renal fibrosis.
4. αKlotho DEFICIENCY EXACERBATES DISORDERS OF MINERAL METABOLISM IN CKD
The fact that mice with complete αKlotho deficiency share similar features such as hyperphosphatemia, hyper-FGF23-temia, and high morbidity and mortality from CV disease than CKD subjects suggests that αKlotho deficiency may participate in CKD development (Hu et al., 2013a, 2013b; Hu, Shiizaki, Kuro-o, et al., 2013). Furthermore, αKlotho-deficient mice with AKI progress to CKD more rapidly and exhibit more severe vascular lesions (Hu et al., 2011; Shi et al., 2015) and uremic cardiac remodeling (Hu, Shi, Cho, et al., 2015), supporting the concept that αKlotho deficiency might be a pathogenic intermediate in CKD (Fig. 3). Given that disturbed mineral metabolism contributes to the high morbidity and mortality of CV disease in CKD (Davidovich, Davidovits, Peretz, Shapira, & Aframian, 2009; Fernandez-Martin et al., 2015; Kaisar, Isbel, & Johnson, 2007; Kestenbaum & Belozeroff, 2007; London, Marchais, Guerin, & Metivier, 2005; Obi, Hamano, & Isaka, 2015; Siomou & Stefanidis, 2012; van Ballegooijen, Rhee, Elmariah, de Boer, & Kestenbaum, 2016; Wesseling-Perry, 2015), a better understanding of the molecular mechanisms of how αKlotho deficiency dysregulates mineral metabolism will aid in the exploration of novel therapeutic strategies in CKD.
Fig. 3.

Proposed physiological role of αKlotho in mineral metabolism and pathophysiological consequences of αKlotho deficiency in CKD. In the setting of normal kidney function with normal αKlotho levels (left panel), αKlotho may suppress FGF23 production and release from the bone. But there is no direct evidence to prove it. αKlotho functions as coreceptor of FGFR to allow FGF23 to suppress PTH production and release from parathyroid gland. PTH stimulates and increases plasma levels of FGF23 and 1,25-(OH)2-vitamin D3. Increased 1,25-(OH)2-vitamin D3 further stimulates FGF23, and directly and indirectly suppresses PTH levels. Increased 1,25-(OH)2-vitamin D3 also stimulates αKlotho production in the kidney. Taken together, through several negative- or positive-feedback loops, αKlotho functions as both a phosphate and calcium regulatory hormone to directly or indirectly suppress PTH, 1,25-(OH)2-vitamin D3, and FGF23 production and release. αKlotho's action on the kidney is to prevent renal Pi retention and to prevent renal Ca loss. In CKD and ESRD (right panel), the network is deranged (red arrows). Renal αKlotho is decreased followed by decrease in plasma αKlotho. The downregulation of αKlotho increases FGF23 production via unknown mechanism, which in turn suppresses 1,25-(OH)2-vitamin D3 production in the kidney. Whether low plasma αKlotho renders parathyroid gland resistant to the suppressive effect of FGF23 on PTH production is not proven. However, decreased FGFR1/3 and αKlotho expression in the uremic parathyroid gland could make the gland resistant to FGF23, and triggers and/or promotes secondary hyperparathyroidism (SHPT). Low plasma Ca also participates in SHPT development. Hyperphosphatemia amplifies the high FGF23 and PTH levels, and low αKlotho levels in the blood. The high plasma PTH, Pi, and FGF23, and low plasma 1,25-(OH)2-vitamin D3 and αKlotho in concert contribute to the development of complications such as metabolic bone disease, SHPT, cardiomyopathy, and vascular calcification. Dash line: unproven putative roles of αKlotho. Ca, ion calcium; CKD, chronic kidney disease; ESRD, end-stage renal disease; FGFR, FGF receptor; Pi, phosphate; PTH, parathyroid hormone; SHPT, secondary hyperparathyroidism; 1,25-VD3, 1,25-(OH)2-vitamin D3.
4.1 Hyperphosphatemia
The role of αKlotho in phosphate homeostasis was recognized as soon as αKlotho was discovered because αKlotho-deficient mice have severe hyper-phosphatemia (Hu, Shi, Zhang, Pastor, et al., 2010; Kuro-o et al., 1997). This was further confirmed by the fact that there is low serum phosphate in αKlotho-overexpressing mice (Kurosu et al., 2005). A patient with homozygous missense mutation (H193R) in the αKLOTHO gene had severe calcinosis, dural and carotid artery calcifications, severe hyper-phosphatemia, hypercalcemia, and high serum 1,25-(OH)2-vitamin D3 and fibroblast growth factor (FGF23) (Ichikawa et al., 2007). This mutation conceivably destabilizes kl1 domain of αKlotho, thereby attenuating production of membrane-bound and soluble αKlotho protein (Ichikawa et al., 2007). Therefore, in one human, the manifestations are similar to those observed in αKlotho-deficient mice. Phosphate overload suppresses αKlotho expression in the kidney (Hu, Shi, Cho, et al., 2015; Shi et al., 2015; Fig. 2). Normal mice fed a high Pi diet have dramatically decreased αKlotho protein and mRNA in the kidney, while αKlotho hypomorphic mice fed low Pi diet can regain part of their Klotho expression (Hu, Shi, Cho, et al., 2015; Morishita et al., 2001; Shi et al., 2015).
Accumulating evidence showed that kidney disease is a status of αKlotho deficiency. Although the mechanism of reduced renal and circulating αKlotho is not understood, it is conceivable that αKlotho deficiency might be involved in the development of hyperphosphatemia, one of components of CKD–metabolic bone disease (CKD-MBD). αKlotho deficiency impairs phosphaturia (Hu, Shi, Zhang, Pastor, et al., 2010) and consequently accelerates Pi accumulation in CKD. The higher the level of serum Pi, the greater the degree of soft-tissue calcification, and the greater the risk of mortality (Kestenbaum et al., 2005). Higher serum Pi levels have been shown to be associated with high mortality in incident ESRD patients (Gutiérrez et al., 2008) and control of serum Pi may help decrease vascular calcification and suppress proliferation of parathyroid glands (Cannata-Andia & Rodriguez-Garcia, 2002; Isakova et al., 2009; Kestenbaum et al., 2005). αKlotho administration could be a novel strategy for the correction of hyperphosphatemia in CKD patients.
4.2 Increased FGF23 Levels
FGF23, a phosphatonin, is thought to be implicated in the systemic balance of phosphate maintained by the interaction of intestine, bone, and kidneys (Hu, Shiizaki, Kuro-o, et al., 2013) through interplay with αKlotho, parathyroid hormone (PTH), and 1,25-(OH)2-vitamin D3 (Bian, Xing, & Hu, 2014). One principal stimulus for FGF23 secretion is currently believed to be high serum phosphate caused by dietary phosphate load (Nishida et al., 2006). In CKD, there is an increase in FGF23 levels in parallel with the deterioration of renal function (Fliser et al., 2007) and the increase of serum phosphate and PTH (Ben-Dov et al., 2007; Nagano et al., 2006; Silver & Naveh-Many, 2010). High serum FGF23 in CKD may not only serve as a diagnostic biomarker of early CKD and predictor of CV disease and mortality in CKD/ESRD patients, but recently, it is proposed to be the necessary and sufficient contributor to uremic cardiomyopathy (Faul et al., 2011; Grabner et al., 2015) through activation of FGFR4 and independently from αKlotho. In contrast, recent data also showed that the relationship between FGF23 and cardiac remodeling depends on αKlotho, and the association of FGF23 with cardiac hypertrophy and fibrosis is only evident in the presence of αKlotho deficiency (Hu, Shi, Cho, et al., 2015).
High serum FGF23 levels antedate high serum levels of phosphate, suggesting a disrupted feedback loop resulting in very high levels of serum FGF23. αKlotho-deficient mice have very high serum levels of FGF23 further supporting that αKlotho might be a negative regulator of FGF23 regardless of the unknown precise mechanisms of how it suppresses FGF23 synthesis in bone (Fig. 3). Currently, there are no experimental data to show direct suppression of FGF23 by αKlotho in osteoblast or osteocytes in vitro. But other animal experiments have shown that extremely high circulating αKlotho with viral delivery can induce severe hypophosphatemia and increase blood FGF23 through stimulation of FGF23 production in the bone although the molecular mechanisms remain to be clarified (Smith et al., 2012). Because phosphate is a potent stimulus for FGF23 production in the bone, therefore, whether αKlotho directly or indirectly increases FGF23 remains to be explored. On the other hand, αKlotho-deficient mice have high levels of serum phosphate and conceivably high FGF23 results from high phosphate due to defective phosphate excretion induced by αKlotho deficiency (Hu, Shi, Zhang, Pastor, et al., 2010; Fig. 3).
4.3 Hypovitaminosis D
Low 1,25-(OH)2-vitamin D3 is a major component of disorders of mineral metabolism, and is conventionally attributed to cause bone disease and secondary hyperparathyroidism in CKD (Lips, 2001). However, hypervitaminosis D is present in αKlotho deficiency (Kuro-o et al., 1997) and removal of key components involved in vitamin D metabolism or function can rescue these phenotypes in αKlotho-deficient mice (Lanske & Razzaque, 2007; Ohnishi, Nakatani, Lanske, & Razzaque, 2009; Razzaque, 2012) suggesting that αKlotho is a suppressor of vitamin D signaling. If CKD is a state of αKlotho deficiency, why αKlotho deficiency does not raise the serum levels of 1,25-(OH)2-vitamin D3 in CKD? The increase in plasma FGF23 in CKD is thought to suppress 1α-hydroxylase in the kidney and initiate or accelerate vitamin D deficiency (Gutiérrez, 2010; Liu et al., 2006). Because 1,25-(OH)2-vitamin D3 induces αKlotho expression in the kidney (Tsujikawa, Kurotaki, Fujimori, Fukuda, & Nabeshima, 2003), it is plausible that low vitamin D levels in CKD may exacerbate renal αKlotho deficiency (Fig. 3).
4.4 Secondary Hyperparathyroidism
Secondary hyperparathyroidism is a common complication of CKD/ESRD and is induced by retention of phosphate as a result of reduced glomerular filtration. The “trade off” hypothesis formulated by Slatopolsky and Bricker has been used for several decades to explain the role of hyperphosphatemia in secondary hyperparathyroidism (Slatopolsky & Bricker, 1973). It was proposed that in the early stages of CKD, an increase in serum phosphate concentrations can be overcome by an increased rate of PTH release, which may be also a result of hypocalcemia. However, as CKD progresses to advanced stages or ESRD, hyperphosphatemia becomes sustained and PTH chronically elevated, suppressing the synthesis of 1α,25(OH)D3 in the kidney. The reduction in the synthesis of 1α,25(OH)D3 also results from hyperphosphatemia and reduced nephron mass. 1α,25(OH)D3 deficiency also contributes to an increase in PTH synthesis (Delmez & Slatopolsky, 1992).
Currently, secondary hyperparathyroidism is considered as part of the syndrome of CKD-metabolic bone disease (MBD; Galitzer, Ben-Dov, Silver, & Naveh-Many, 2010; Khan, 2007). Even mild increments in PTH levels are associated with an increased CV risk, regardless of the serum levels of calcium and phosphorus and whether vitamin D therapy is given, suggesting that decreasing PTH levels may improve mineral metabolism disorders (Floege et al., 2011; Panichi et al., 2010; Patel et al., 2011; Pontoriero, Cozzolino, Locatelli, & Brancaccio, 2010).
CKD patients have high blood FGF23 levels (Shimada et al., 2010) and low αKlotho and FGFR(s) in parathyroid gland (Canalejo et al., 2010; Krajisnik et al., 2010). In subjects with normal kidney function, FGF23 plays a crucial role, both as a phosphaturic factor (Gattineni & Baum, 2010; Goetz et al., 2010; Weber, Liu, Indridason, & Quarles, 2003) and as a calciotropic hormone to suppress 1,25-(OH)2-vitamin D production in the kidney (Liu et al., 2006) and PTH production in the parathyroid gland (Ben-Dov et al., 2007), and to increase renal calcium reabsorption through modulation of TRPV5 channel (Andrukhova et al., 2014). In contrast, in a CKD setting, FGF23 fails to inhibit PTH production probably due to downregulation of αKlotho and FGFR(s) in parathyroid gland (Fig. 3; Canalejo et al., 2010; Krajisnik et al., 2010). The mechanisms of downregulation of αKlotho and FGFR(s) in uremic parathyroid gland remain to be explored.
Renal and circulating αKlotho deficiency is associated with development and progression of CKD-MBD (Fahrleitner-Pammer et al., 2008; Hruska, Saab, Mathew, & Lund, 2007; Kalantar-Zadeh et al., 2010; Patel et al., 2011). Although whether αKlotho is present in the vasculature is still under debate, it has been shown that even early CKD-MBD may cause a reduction of vascular αKlotho (Fang et al., 2014), stimulate vascular osteoblastic transition, increase osteocytic secreted proteins, and consequently induce vascular calcification. Correction of αKlotho and maintenance of mineral homeostasis do not only benefit bone and mineral metabolism but also may attenuate CV disease and improve the quality of life of CKD/ESRD patients (Fernandez-Martin et al., 2015).
5. αKlotho DEFICIENCY IN CV DISEASE IN CKD
CKD confers significant CV morbidity and mortality (Go, Chertow, Fan, McCulloch, & Hsu, 2004; Gross & Ritz, 2008; Taddei, Nami, Bruno, Quatrini, & Nuti, 2011). A large number of CKD patients die from CV disease even before initiation of dialysis. The main clinical features of CV disease in CKD include uremic cardiomyopathy and vascular calcification.
5.1 Pathological Uremic Cardiomyopathy
Uremic cardiomyopathy or cardiomyopathy of advanced CKD, characterized by cardiac hypertrophy and fibrosis, is a major cause of CV disease, by causing congestive heart failure, cardiac dysrhythmias, and sudden cardiac death (Glassock, Pecoits-Filho, & Barberato, 2009; Go et al., 2004; Gross & Ritz, 2008; Taddei et al., 2011). There are traditional risk factors such as hypertension, coronary disease, atherosclerosis, anemia, and volume overload (Glassock et al., 2009; Gross & Ritz, 2008), and also CKD-specific factors such as hyperphosphatemia (Block, Hulbert-Shearon, Levin, & Port, 1998; Glassock et al., 2009; Gross & Ritz, 2008; Kestenbaum et al., 2005). Recent data have shown an association between elevated FGF23 levels and uremic cardiac remodeling (Faul et al., 2011; Gutiérrez et al., 2009). Soluble αKlotho deficiency may also be an intermediate mediator of the pathological cardiac remodeling observed in CKD (Hu, Shi, Cho, et al., 2015). Furthermore, αKlotho may protect the heart against stress-induced cardiac hypertrophy by inhibiting TRPC-6 channel-mediated abnormal Ca2+ signaling in the heart (Xie et al., 2012; Xie, Yoon, An, Kuro-o, & Huang, 2015) or against uremic solute indoxyl sulfate-induced myocardial hypertrophy probably by suppressing NADPH oxidase Nox2/Nox4-derived reactive oxygen species (ROS) production and its downstream signaling (Yang et al., 2015; Fig. 4).
Fig. 4.

Risk factors for uremic cardiomyopathy and proposed mechanisms of attenuation of pathological cardiac remodeling by αKlotho. αKlotho deficiency is a novel risk factor for uremic cardiomyopathy. Soluble αKlotho deficiency is an intermediate mediator of the pathological cardiac remodeling observed in CKD. Experimental αKlotho overexpression (Tg-Kl) suppressed phosphorylation of Smad2/3 and Erk, which are known to be involved in uremic cardiac fibrosis. Furthermore, αKlotho may protect the heart against stress-induced cardiac hypertrophy by inhibiting TRPC-6 channel-mediated abnormal Ca2+ signaling in the heart or against uremic solute indoxyl sulfate-induced myocardial hypertrophy probably by suppressing NADPH oxidase Nox2/Nox4-derived reactive oxygen species production and its downstream signaling. CHF, congestive heart failure; Erk, extracellular signal-regulated kinase; LVH, left ventricular hypertrophy; MAPK, mitogen-activated protein kinases; Nox, NADPH oxidase; SCD, sudden cardiac death; TRPC-6, transient receptor potential canonical-6.
5.1.1 αKlotho as a Modulator of Pathological Cardiac Remodeling
Uremic cardiomyopathy is a state of pathological cardiac remodeling characterized by left ventricular hypertrophy (LVH) and extensive fibrosis (Foley et al., 1995; Tyralla & Amann, 2003). Both primary genetic αKlotho deficiency (heterozygous αKlotho-deficient, kl/+ mice) and secondary αKlotho deficiency (from phosphate loading, aging, and CKD) triggered cardiac hypertrophy and fibrosis in mice, such that higher plasma phosphate and lower plasma αKlotho levels were associated with more severe cardiac hypertrophy and fibrosis (Hu, Shi, Cho, et al., 2015). Furthermore, higher plasma FGF23 levels were associated with more severe cardiac hypertrophy and fibrosis but only in the presence of moderate or low plasma αKlotho levels (Hu, Shi, Cho, et al., 2015). This suggests that FGF23 may not be cardiotoxic unless there is simultaneous αKlotho deficiency.
CKD models of secondary αKlotho deficiency included: (1) unilateral nephrectomy and contralateral ischemic–reperfusion injury followed by high-phosphate diet (2% phosphate) and (2) 5/6th nephrectomy. Both CKD models showed cardiac hypertrophy and left ventricular fibrosis. αKlotho levels in kidney tissue, plasma, and urine were decreased by high-phosphate diet starting at 6 months of age. When high-phosphate diet was given to older mice (12 months of age), additional reductions in plasma and kidney αKlotho were observed (Hu, Shi, Cho, et al., 2015). Cardiac hypertrophy and fibrosis were exaggerated in kl/+ mice and lessened in transgenic αKlotho-overexpressing mice (Tg-Kl) compared to WT mice and changes were more severe at age 15 months compared with 9 months. Aging exacerbated phosphate or αKlotho deficiency-induced pathological cardiac remodeling. Notably, αKlotho suppressed cardiac fibrosis triggered by high dietary phosphate. αKlotho overexpression (Tg-Kl) suppressed phosphorylation of Smad2/3 and extracellular signal-regulated kinase (Erk; Hu, Shi, Cho, et al., 2015), which are known to be involved in uremic cardiac fibrosis (Olson, Naugle, Zhang, Bomser, & Meszaros, 2005).
In vitro, αKlotho blocked TGF-β1- and angiotensin II (Ang II)-induced hypertrophy in cardiomyocytes (primary culture of neonatal rat) by inhibiting Smad2/3 phosphorylation. αKlotho also attenuated TGF-β1-, Ang II-, and high phosphate-induced upregulation of fibrosis markers in cultured cardiac fibroblasts by inhibiting Erk phosphorylation (Hu, Shi, Cho, et al., 2015).
Cardiac hypertrophy and fibrosis scores correlated negatively with plasma αKlotho levels and positively with plasma phosphate levels. In multivariable analysis, adjusting for plasma creatinine, FGF23, PTH, and 1,25-(OH)2-vitamin D3 levels, only plasma αKlotho and phosphorus levels were independent factors associated with pathological cardiac remodeling (Hu, Shi, Cho, et al., 2015).
5.1.2 αKlotho Protection Against Stress-Induced Cardiac Hypertrophy
Xie and coworkers reported that cardioprotection by αKlotho in normal mice is mediated by downregulation of TRPC-6 channels in the heart (Xie et al., 2012). In their experiments, deletion of TRPC-6 prevented stress-induced exaggerated cardiac remodeling in αKlotho-deficient mice (kl/+). In contrast, mice with heart-specific overexpression of TRPC-6 developed spontaneous cardiac hypertrophy and remodeling. Furthermore, αKlotho overexpression (Tg-Kl mice) ameliorated pathological cardiac remodeling and improved long-term survival (Xie et al., 2012). In addition, they proposed that soluble αKlotho inhibits TRPC-6 currents in cardiomyocytes by blocking phosphoinositide-3-kinase-dependent exocytosis of TRPC-6 channels (Xie et al., 2012).
Subsequently, the same investigators inferred that the decrease in soluble αKlotho in CKD is not only an important cause of uremic cardiomyopathy but independent of FGF23 and phosphotoxicity (Xie et al., 2015). They reported that αKlotho levels in αKlotho-deficient mice (kl/+) were about one half of those of WT mice, and they further decreased in kl/+ CKD mice to barely detectable levels (Xie et al., 2015). Heart weight-to-body weight ratio (a measure of cardiac hypertrophy) was significantly increased in both WT and kl/+ CKD mice, but the increase in kl/+ CKD mice was significantly more prominent than that in WT CKD mice. Similarly, the degree of fibrosis in kl/+ CKD was much more severe than that in WT CKD heart (Xie et al., 2015). WT CKD mice had ventricular hypertrophy, normal chamber size, and preserved contractility (diastolic dysfunction) in contrast to systolic dysfunction with dilated cardiomyopathy and impaired contractility in kl/+ CKD mice. Dietary phosphate restriction was successfully utilized to normalize serum phosphate and FGF23 levels in CKD mice (serum phosphate and FGF23 levels were similar in WT and kl/+ CKD mice compared with sham controls). Notably, dietary phosphate restriction did not significantly alter the pattern of cardiac hypertrophy in WT or kl/+ CKD mice (Xie et al., 2015). Moreover, viral-based deliver of αKlotho transgene significantly ameliorated cardiac hypertrophy and fibrosis in kl/+ CKD mice when compared with empty vector-injected mice. Functional TRPC-6-mediated currents were increased in cardiac myocytes isolated from CKD mice (vs sham), and the increase was more pronounced in kl/+ CKD vs WT CKD mice. Extracellular application of soluble αKlotho decreased these currents, confirming that αKlotho directly affects TRPC-6 functionality (Xie et al., 2015). The mechanisms of how the increase in TRPC-6 induces cardiomyopathy are not currently known.
5.1.3 αKlotho Protection Against Indoxyl Sulfate-Induced Myocardial Hypertrophy
Yang and colleges studied 86 patients with CKD and showed higher levels of indoxyl sulfate (Yang et al., 2015), a uremic solute derived from dietary protein and excreted by the kidney. Indoxyl sulfate accumulates with progressive loss of kidney function and can induce vascular endothelial cell dysfunction by enhancing oxidative stress (Tumur & Niwa, 2009; Tumur, Shimizu, Enomoto, Miyazaki, & Niwa, 2010). They showed a negative correlation between serum levels of indoxyl sulfate and αKlotho (r = −0.59, p<0.001). Importantly, serum levels of indoxyl sulfate and αKlotho were independently associated with LVH (Yang et al., 2015). This was further confirmed by experiments in normal mice in which intra-peritoneal injection of indoxyl sulfate for 8 weeks induced LVH, accompanied by substantial renal αKlotho downregulation. Notably, indoxyl sulfate-induced LVH was more severe in heterozygous αKlotho-deficient (kl/+) mice relative to WT mice, indicating that αKlotho deficiency may exacerbate indoxyl sulfate-mediated LVH (Yang et al., 2015) and αKlotho supplementation may be a strategy to counteract indoxyl sulfate-mediated LVH.
In vitro experiments showed that indoxyl sulfate induces cardiomyocyte hypertrophy through activation of Nox/ROS/MAPK (p38 and Erk1/2) signaling pathways and this activation can be attenuated by pretreatment with αKlotho protein, possibly through inhibition of ROS signaling. Indoxyl sulfate-induced cardiomyocyte hypertrophy is not mediated through TRPC-6 signaling pathway (Yang et al., 2015). Interestingly, the in vivo administration of exogenous αKlotho protein significantly alleviated the development of LVH in a mouse model of CKD-associated LVH characterized by high serum indoxyl sulfate levels, which confirmed in vitro findings (Yang et al., 2015). Moreover, indoxyl sulfate has been shown to suppress αKlotho deficiency in mice (Adijiang, Shimizu, Higuchi, Nishijima, & Niwa, 2011) and downregulate αKlotho expression in cultured cells (Shimizu et al., 2011; Sun et al., 2012). Therefore, indoxyl sulfate has a dual effect: induction of cardiomyocyte hypertrophy and induction of αKlotho deficiency to enhance cardiomyocyte hypertrophy.
5.2 Vascular Medial Calcification
Apart from traditional risk factors, the high CV morbidity and mortality in CKD have been linked to CKD-specific mechanisms of vascular calcification through modulation of the endothelium–vascular smooth muscle network (Hu et al., 2014; Vervloet, Adema, Larsson, & Massy, 2014). Calcium and phosphate play an important role in the initiation of osteochondrogenic changes of cellular elements in the arterial wall, and also in the final common pathway of alleged ectopic bone formation (Vervloet et al., 2014). Although the expression of αKlotho in the vasculature is highly controversial, there are data associating changes in circulating αKlotho levels with uremic vasculopathy (Hu et al., 2014; Vervloet et al., 2014). Blood vessels are composed of endothelial cells, mural cells (smooth muscle cells and pericytes), their shared basement membrane, and extracellular matrix. Vascular smooth muscle cells (VSMCs) and endothelial cells work synergistically for maintenance of the integrity of the vasculature (Heydarkhan-Hagvall et al., 2003; Fig. 5).
Fig. 5.

Proposed model of αKlotho as intermediate of endothelial cells (ECs)–vascular smooth muscle cells (VSMCs) cross talk. Left panel: Normal ECs–VSMCs cross talk. Normal ECs modulate VSMCs growth via release of growth factors (black line in left panel). NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostaglandin I2; VEGF, vascular endothelial growth factor. VEGF released from pericytes and/or VSMCs could also regulate endothelial cell. Right panel: In CKD, uremic toxins including high plasma Pi, damage ECs and induce release of growth factors, proinflammatory cytokines, and profibrotic factors, which exacerbate ECs injury and also induce VSMCs transition to osteoblast and promote vascular calcification in medial layer (red solid lines in right panel). Impaired ECs also directly contributes to vascular calcification through endo-osteoblast transition (green line in right panel). Whether dedifferentiated or damaged VSMCs could further modulate function of endothelial cells is speculative (red dash line in right panel). Central panel: αKlotho is a vascular protective protein. Whether resident aortic αKlotho protein in VSMCs functions in autocrine and/or paracrine mode to modulate VSMCs and/or ECs remains to be clarified (brown dash line in central panel). The mechanisms of how αKlotho is able to reach the VSMCs from the circulation and function as an endocrine factor remain to be defined (blue dash line in central panel). αKlotho, regardless of source, could increase NO production from ECs and NO consequently modulates VSMCs and ECs function in an autocrine mode (blue solid line in middle panel). αKlotho could protect EC from high phosphate and other uremic toxins and also attenuate oxidative stress and proinflammatory cytokines-induced cell senescence and apoptosis in VSMC (orange line in central panel). αKlotho also directly inhibits osteoblast transition induced by hyperphosphatemia and uremic milieu (orange line in central panel). Current experimental and clinical observations suggest that both ECs and VSMCs endothelium may be potential targets of soluble αKlotho to protect the vasculature from vascular calcification in CKD. ECs, endothelial cells; Pi, phosphorus; VSMCs, vascular smooth muscle cells.
5.2.1 αKlotho and Endothelium
Endothelial dysfunction is associated with CV morbidity and mortality in CKD (Ravani et al., 2005). Endothelial cells damaged by high phosphate or uremic solutes show increase in apoptosis, ROS, proinflammatory cytokines, profibrotic and proangiogenic growth factors, and impaired nitric oxide production (Carracedo et al., 2013; Di Marco et al., 2008). Vascular endothelium can be a source of osteoprogenitor cells in vascular calcification (Di Marco et al., 2008). Moreover, the endothelium could also stimulate VSMCs to initiate or participate in vascular calcification (Yao et al., 2013).
A functional vascular tone and low levels of oxidative stress are maintained by releasing nitric oxide, prostacyclin, and endothelin-1, and by controlling local angiotensin II activity. In addition, the endothelium also regulates vascular permeability, platelet and leukocyte adhesion and aggregation, and thrombosis (Sitia et al., 2010). Elevated asymmetric dimethylarginine, a known inhibitor of nitric oxide synthase, and consequent reduced nitric oxide production and reduced flow-mediated dilatation (FMD) of the vessels have been characterized in CKD (Schwedhelm & Boger, 2011; Yilmaz et al., 2006). High phosphate impairs FMD in experimental CKD and FMD is inversely related to serum phosphate level in humans (Shuto et al., 2009; Van et al., 2012). Similarly, oxidative stress and inflammation are implicated in the development of endothelial dysfunction in CKD (Recio-Mayoral, Banerjee, Streather, & Kaski, 2011).
Interestingly, circulating αKlotho regulates vasodilation through modulation of nitric oxide production in vascular endothelium (Nagai et al., 2000; Saito et al., 1998; Six et al., 2014; Yamagishi et al., 2001). In addition, treatment of cultured endothelial cells with αKlotho alleviates tumor necrosis factor α-mediated ROS activity, cell apoptosis, and induction of adhesion molecules (Carracedo et al., 2012; Maekawa et al., 2009; Yang et al., 2012). Importantly, αKlotho-deficient mice have increased VEGF-mediated calcium influx, downregulation of cadherin surface expression, increased apoptosis, and increased permeability (Kusaba et al., 2010). It is suggested that the Kl2 domain of αKlotho protein binds directly to VEGFR-2 and endothelial TRPC-1 Ca2+ channel and promotes their cointernalization and consequent reduction of cellular Ca2+ influx limiting the activity of Ca2+-dependent proteases that disrupt endothelial integrity (Kusaba et al., 2010). αKlotho protein is also capable of attenuating indoxyl sulfate-induced endothelial dysfunction, partly through inhibition of ROS/p38 mitogen-activated protein kinase and downstream nuclear factor-κB signaling pathways (Yang et al., 2012).
It is possible that αKlotho may act on the endothelium and induce a secondary effect via endothelial-VSMC crosstalk, or that VSMC-resident αKlotho regulates VSMC function in an autocrine mode or even endothelium in a paracrine mode (Fig. 5). The role of αKlotho protein in the disturbed endothelium–vascular smooth muscle network in CKD requires further investigation.
5.2.2 αKlotho and Vascular Smooth Muscle
Measurement of aortic αKlotho mRNA expression has not been consistent (Hu et al., 2014; Lim et al., 2012; Mencke et al., 2015; Navarro-Gonzalez et al., 2014; Ritter et al., 2015; Six et al., 2014). However, the association between αKlotho deficiency and medial vascular calcification has been well documented in hypomorphic αKlotho mice (Kuro-o et al., 1997), a phenotype rescued by transgenic overexpression, viral delivery of αKlotho, or recombinant αKlotho protein (Chen, Kuro, et al., 2013; Masuda et al., 2005; Shiraki-Iida et al., 2000). Furthermore, transgenic mice over-expressing αKlotho had significantly less vascular calcification after CKD induction in comparison to αKlotho-haploinsufficient mice with CKD that exhibited more severe vascular calcification (Hu et al., 2011).
The potential mechanisms underpinning the association between high serum phosphate and vascular calcification have been described in experimental models of CKD ( Jono et al., 2000; Lomashvili, Cobbs, Hennigar, Hardcastle, & O’Neill, 2004; Mathew et al., 2008). The beneficial effect of αKlotho on vascular calcification in CKD is thought to be a result of more than its effect on amelioration of renal dysfunction and hyperphosphatemia. In vitro experiments have shown that αKlotho suppresses type III Na+-dependent uptake of phosphate (Pit-1 and Pit-2 cotransporters) and mineralization induced by high phosphate in VSMCs (Hu et al., 2011). Runt-related transcription factor-2 (Runx2) expression, an early marker of ectopic osteogenesis, was decreased in the aortas of overexpressing αKlotho mice (Hu et al., 2011). The contractile phenotype of VSMCs was lost with exposure to high phosphate or resident αKlotho knockdown (Lim et al., 2012). Resident αKlotho knockdown in VSMCs accelerated the development of vascular calcification through Runx2 and myocardin-serum response factor-dependent pathway (Lim et al., 2012). Therefore, αKlotho may regulate VSMCs differentiation under CKD procalcific stressors (Hu et al., 2011; Lim et al., 2012). However, other experiments have resulted in conflicting evidence. One study did not show any effect of αKlotho protein on FGF23 and high phosphate-mediated vascular calcification in human or mouse VSMCs (Scialla et al., 2013). Another study performed in uremic rats showed that FGF23 augmented phosphate-induced aortic calcification in αKlotho-overexpressing but not naive VSMCs through Erk1/2 phosphorylation pathway ( Jimbo et al., 2014). The conflicting in vitro data may be, besides plausibility, a reflection of differences in VSMCs or αKlotho protein preparations utilized.
Vitamin D receptor agonists (eg, calcitriol or paracalcitol) were shown to increase serum and urine αKlotho levels and abate aortic calcification in CKD mice likely through modulation of osteopontin, an anticalcification factor in VSMCs (Lau et al., 2012). Importantly, no αKlotho mRNA expression was found in the aorta in these in vivo experiments (Lau et al., 2012). One independent study further confirmed that there is no membrane αKlotho expression in either healthy or uremic vessels in humans (Mencke et al., 2015). In contrast, Lim and colleagues showed that calcitriol effectively restored mRNA αKlotho expression in VSMCs (Lim et al., 2012). In a different experiment, αKlotho mRNA was detected in mouse aorta but specific deletion of αKlotho in mouse VSMCs did not induce vascular calcification (Lindberg et al., 2013) challenging the principal role of αKlotho in VSMCs. Moreover, αKlotho protein was found to be increased in atherosclerotic arteries (Donate-Correa et al., 2013) contradicting the suggested protective role of resident αKlotho in vasculature. The existence and role of resident αKlotho protein in the vasculature need to be clarified and further investigated (Table 1).
Table 1.
The Determination of Aortic αKlotho mRNA Expression Has Been Inconsistently Reproduced in Different Studies
| Aortic αKlotho mRNA Expression = Yes | Aortic αKlotho mRNA Expression = No |
|---|---|
| Klotho mRNA and protein is present in aorta and vascular smooth muscle cells Resident αKlotho knockdown induced vascular calcification through Runx2 and myocardin-serum response factor- dependent pathway Calcitriol effectively restored mRNA αKlotho expression in VSMCs (Lim et al., 2015, 2012) | No αKlotho mRNA expression was found in the aorta in in vivo experiments of aortic calcification in CKD (Lau et al., 2012) |
| αKlotho mRNA was detected in mouse aorta but specific deletion of αKlotho in mouse VSMCs did not induce vascular dysfunction and vascular calcification (Lindberg et al., 2013) | No membrane αKlotho expression in either healthy or uremic human vessels was found (Mencke et al., 2015) |
| αKlotho protein was found to be increased in atherosclerotic arteries (Donate-Correa et al., 2013) | |
| Human umbilical vein endothelial cells (HUVECs) express αKlotho. The decline in αKlotho preceded the manifestations of cell aging induced by repeated passage and those of senescence by TNF-α. The exogenous αKlotho administration prevented TNF-α-mediated senescence (Carracedo et al., 2012) |
In contrast to the inconclusive evidence of vascular αKlotho expression, the administration of exogenous recombinant αKlotho, αKlotho gene delivery, and increased endogenous circulating αKlotho significantly reduced vascular calcification and improved endothelial function, suggesting that soluble αKlotho may play a pivotal role in the protection of vasculature integrity as an endocrine factor (Chen, Kuro, et al., 2013; Lau et al., 2012; Masuda et al., 2005; Saito et al., 2000; Utsugi et al., 2000; Fig. 5).
It was shown that αKlotho protein attenuates endothelial cell damage from high phosphate and oxidative stress, and inhibits osteogenic transformation of VSMCs induced by high phosphate. The administration of exogenous αKlotho or modulators of αKlotho expression may represent novel therapies for the management of vascular calcification in CKD patients. Studies of endothelial cells or VSMCs in isolation may not fully represent the vascular system to dissect out the role of the individual players. Coculture of endothelial cells and VSMCs may be a viable intermittent system to further elucidate the role of αKlotho in the vasculature under normal and pathological conditions (Hu et al., 2014).
6. αKlotho DEFICIENCY AS A BIOMARKER OF CKD
CKD is a global public health problem that affects over 20 million people in the United States (Snyder, Foley, & Collins, 2009). The major complications in this population are progression to ESRD and CV morbidity and mortality (Eckardt et al., 2013; Hemmelgarn et al., 2010; Levey et al., 2011).
There has been intense search for highly sensitive (diagnostic value) or highly specific (treatment effect value) biomarkers of CKD onset and/or prognosis of progression. An ideal prognostic CKD biomarker should be able to predict CKD onset and progression, characterize the severity of CKD stage, display similar reliability across multiple species (particularly humans), and be accessible in readily available body fluids or tissues.
In the following sections, novel functional (detecting primarily loss of kidney function) and injury biomarkers (with or without loss of kidney function) of CKD and the potential role of FGF23 and αKlotho as early diagnostic and prognostic biomarkers of CKD will be discussed.
6.1 Functional Biomarkers in Human CKD
New filtration markers such as β-trace protein (BTP), β2 microglobulin (β2M), and cystatin C were associated with mortality risk in a representative sample of 6445 US adults from the Third National Health and Nutrition Examination Survey (Foster et al., 2013). The highest quintile for cystatin C, BTP, and β2M were associated with increased all-cause mortality risk, whereas the association was weaker for serum creatinine-based eGFR (Foster et al., 2013). A >50% decline in serum creatinine-based eGFR is an established surrogate marker for ESRD in clinical trials but a >30% decline in kidney function assessed using novel filtration markers (cystatin C and β2M) has been strongly associated with ESRD (Rebholz, Grams, Matsushita, Selvin, & Coresh, 2015).
6.2 Injury Biomarkers in Human CKD
The assessment of the plasma proteome through mass spectrometry analysis identified three fragments of high-molecular-weight kininogen associated with early progressive renal function decline in microalbuminuric patients with type 1 diabetes (Merchant et al., 2013). The performance of urine neutrophil gelatinase-associated lipocalin (NGAL) was analyzed in a cohort of 3386 patients with CKD in the Chronic Renal Insufficiency Cohort (CRIC) study. Urine NGAL was independently associated with 50% decreased eGFR or incident ESRD development over a mean follow-up of 3.2 years. However, it did not improve prediction models for CKD progression (Liu et al., 2013). More recently, urine NGAL has been independently associated with ischemic atherosclerotic events but not heart failure events or death (Fufaa et al., 2015). In a cohort of 124 patients with type 1 diabetes and proteinuria, serum kidney injury molecule-1 (KIM-1) levels at baseline strongly predicted rate of eGFR loss and risk of ESRD during 5–15 years of follow-up, after adjustment for baseline urinary albumin-to-creatinine ratio, eGFR, and hemoglobin A1C (Sabbisetti et al., 2014). Similarly, urinary NGAL and liver fatty acid-binding protein (L-FABP) were independently associated with incident ESRD and mortality but did not meaningfully improved clinical prediction models of CKD progression in a cohort of 260 Pima Indians with median follow-up of 14 years (Fufaa et al., 2015).
6.3 FGF23 in Human CKD
Current evidence favors a direct pathogenic role for dysregulated FGF23-α Klotho in CKD-related adverse outcomes, in particular CV disease. The relationship between baseline serum intact FGF23 and incident ESRD was evaluated in 13,448 Atherosclerosis Risk in Communities (ARIC) study participants during a median follow-up of 19 years. After adjustment for demographics, baseline eGFR, and traditional CKD risk factors, the highest FGF23 quintile (>54.6 pg/mL) compared with the lowest quintile (<32.0 pg/mL) was associated with risk of developing ESRD (Rebholz, Grams, Coresh, et al., 2015). Similarly, elevated FGF23 has been proposed as an early indicator of kidney injury or CKD progression (Fliser et al., 2007; Wolf, 2012). Elevated serum FGF23 has been independently associated with LVH and a causal inference through Klotho-independent activation of FGF receptor-dependent activation of the calcineurin–NFAT signaling pathway in rat cardiomyocytes has been proposed (Faul et al., 2011). Increased FGF23 was independently associated with mortality among incident hemo-dialysis patients (Gutiérrez et al., 2008) and has been linked to mortality and higher risk of ESRD or CV disease in patients with CKD during a median follow-up of 3.5 years (Faul et al., 2011; Ix et al., 2012). In contrast, FGF23 was not associated with arterial calcification in 1501 patients from the CRIC study (Scialla et al., 2013).
6.4 The Role of αKlotho as a Marker of Adverse Outcomes in Human CKD
CKD is a state of αKlotho deficiency in multiple tissues. αKlotho mRNA levels in parathyroid gland declined in parallel with decreasing eGFR over CKD stages (Krajisnik et al., 2010). Similarly, αKlotho mRNA expression in kidney tissue was greatly reduced and positively and significantly correlated with eGFR in CKD patients (Asai et al., 2012,; Koh et al., 2001). In a larger sample of 236 CKD patients with available kidney biopsies, αKlotho mRNA levels were significantly and positively correlated with eGFR (p<0.001) in multiple regression analysis including CKD-MBD parameters (Sakan et al., 2014). Most importantly, αKlotho mRNA in the kidney was the only independent contributing factor to serum αKlotho across all strata of CKD patients. Renal αKlotho was significantly correlated with serum calcium, serum phosphorus, 1,25-(OH)2-vitamin D3, FGF23, and intact PTH (Sakan et al., 2014). Correspondingly, circulating serum αKlotho levels were progressively lower with each CKD stage when compared to healthy controls (Pavik et al., 2013). This was also observed in kidney transplant recipients vs healthy controls (Sawires, Essam, Morgan, & Mahmoud, 2015). Furthermore, adjusted mean serum αKlotho decrease was 3.2 pg/mL for each 1 mL/min eGFR decrease in adult CKD patients (Pavik et al., 2013). A positive correlation between αKlotho levels (serum and urine) and eGFR has been further characterized in adult CKD patients (Akimoto et al., 2012; Hu et al., 2011; Kim et al., 2013; Kitagawa et al., 2013; Ozeki et al., 2014), although only 24-h urine αKlotho but not serum αKlotho has been shown to be independently associated with eGFR change (Akimoto et al., 2012). However, questions about the stability of αKlotho in the urine have emerged (Adema, Vervloet, Blankenstein, & Heijboer, 2015). Therefore, a standardized urine protocol is required. A similar positive correlation between plasma αKlotho levels and eGFR was shown in CKD children without kidney transplant (Wan et al., 2013). Moreover, serum αKlotho levels in children on chronic peritoneal dialysis were significantly lower (~41%) than healthy controls (Cano et al., 2014). The progressive decline of serum αKlotho in adults with early stages of CKD (eg, stage 2) has been subsequently demonstrated, suggesting that the decrease in serum αKlotho may antecede high FGF23, PTH, and hyperphosphatemia (Barker et al., 2015; Kim et al., 2013; Rotondi et al., 2015; Shimamura et al., 2012; Table 2).
Table 2.
Clinical Studies Demonstrating the Implication of Kidney and Circulating αKlotho in Different Strata of Chronic Kidney Disease and Complications of Chronic Kidney Disease
| Study | Design | Sample | Exclusion | Measurement | Observations | Comments |
|---|---|---|---|---|---|---|
| Koh et al. (2001) | Cross sectional | 10 CKD and 15 controls | None | Kidney tissue kl mRNA expression | αKlotho expression was greatly reduced in CKD vs controls | Controls included healthy parts of resected kidneys with renal cell carcinoma or angiomyolipoma |
| Asai et al. (2012) | Cross sectional | 31 pts with DN, 31 IgAN, and 7 MCD | None | Kidney tissue kl mRNA expression | Kidney kl mRNA expression levels were positively and significantly correlated with eGFR (r = 0.35, p = 0.003) | Different slopes depending on CKD cause were observed, particularly in patients with eGFR >60 |
| Pavik et al. (2013) | Cross sectional | 87 CKD pts (stages 1–5) and 21 controls | PKD, kidney transplantation | Serum αKlotho | Adjusted mean αKlotho decrease was 3.2 pg/mL for each 1 mL/min eGFR decrease | Age and eGFR were independently associated with αKlotho |
| Wan et al. (2013) | Cohort | 154 children with CKD (1–5, 28 on dialysis, 44 posttransplant) | None | Predictor: Plasma αKlotho Outcome: eGFR Follow-up: 1 year | αKlotho levels decreased with decreasing eGFR in CKD children without kidney transplant | No independent association between plasma αKlotho and eGFR. Decreased αKlotho was associated with increased FGF23 and PTH levels in CKD children without kidney transplant |
| Kitagawa et al. (2013) | Cross sectional | 114 CKD pts | Patients with established atherosclerotic complications (CAD, CHF, PVOD) or those treated with vitamin D or phosphate binders | Serum αKlotho | αKlotho was a significant determinant of arterial stiffness (adjusted OR per 100 pg/mL increase 0.60, 95% CI 0.39–0.98, p = 0.008) | Positive correlation between serum αKlotho and eGFR. Arterial stiffness was determined by ankle– brachial pulse wave velocity |
| Kim et al. (2013)a | Cohort | 243 pts with CKD (stages 1–5), post hoc analysis | RRT, organ transplantation, heart failure, cirrhosis, malignancy, pregnancy, acute coronary syndrome, or ischemic stroke within 3 months prior to the study, progressive CKD within 3 months prior to the study | Predictor: Serum αKlotho Outcome: composite of doubling SCr, ESRD, or death Follow-up: median 29.7 months | αKlotho level independently predicted the composite outcome after multivariate adjustment (adjusted HR per 10 pg/mL increase 0.96, 95% CI 0.94–0.98, p<0.001). If serum αKlotho was ≤396.3 pg/mL, 35.2% reached the composite outcome vs 15.7% if >396.3 pg/mL (p = 0.03) | αKlotho levels were lower at more advanced CKD stages (p for trend <0.001) and correlated positively with eGFR and negatively with FGF23 and phosphate level. In multivariate linear regression analysis, αKlotho was independently associated with eGFR (p<0.001) |
| Akimoto et al. (2012) | Cross sectional | 131 CKD pts (stages 1–5) | RRT | Serum and urine αKlotho | Positive correlation between serum and urine αKlotho and eGFR | 24-h urine αKlotho was independently associated with eGFR |
| Sakan et al. (2014) | Cross sectional | 236 CKD pts (stages 1–5), 43 on dialysis | None | Kidney tissue kl mRNA expression and serum αKlotho | Kidney kl mRNA levels were significantly and positively correlated with eGFR (p<0.001) in multiple regression analysis including CKD-MBD parameters | Kidney kl mRNA was the only independent contributing factor to serum αKlotho across all strata of CKD patients (p<0.001) |
| Seiler et al. (2014) | Cohort | 444 CKD pts (stages 2–4) | None | Predictor: plasma αKlotho Outcome: (1) composite of atherosclerotic event or death or (2) time until ADHF admission or death Follow-up: median of 2.6 years | Plasma αKlotho levels (highest vs lowest tertile) did not predict atherosclerotic events/death (HR 0.75, 95% CI 0.43–1.30, p = 0.30) or ADHF/death | Events were adjudicated by two independent nephrologists |
| Ozeki et al. (2014) | Cross sectional | 185 CKD pts (stages 1–5) | RRT | Serum αKlotho | eGFR was an independent predictor of serum αKlotho levels in multivariate linear regression analysis, specifically when eGFR was <60 | Serum αKlotho was not significantly correlated with serum calcium or phosphate |
| Cano et al. (2014) | Cohort | 31 children on chronic PD and 45 healthy controls | None | Predictor: CKD-MBD parameters Outcome: serum αKlotho Follow-up: 1 year | Baseline αKlotho levels were lower than controls (132±58 vs 320±119 pg/mL, p<0.001) and remained virtually unchanged throughout the observation period | αKlotho levels did not correlated with FGF23 and phosphorus levels |
| Park et al. (2015) | Cross sectional | 24 HTN pts (12 with RVH and 12 EH) and 12 HV controls | eGFR<30, uncontrolled BP, diabetes, recent CV event (within 6 months), pregnancy, kidney transplant | Serum αKlotho | Serum αKlotho was significantly reduced in HTN pts vs HV controls after adjustment by eGFR | eGFR correlated directly with serum αKlotho levels |
| Sawires et al. (2015) | Cross sectional | 40 CKD pts (stages 2–5), 44 pts with ESRD on HD, 40 kidney transplant recipients, and 40 healthy controls | Marked hypocalcemia, dysfunctional HD access, combined organ transplantation, surgical parathyroidectomy, sarcoidosis | Serum αKlotho | Using multivariate regression analysis, only serum calcium was an independent predictor of serum αKlotho in all groups with kidney disease | There was an inverse significant correlation between serum calcium and αKlotho |
| Shroff et al. (2016) | Cohort | ESCAPE cohort post hoc analysis, 167 children with CKD on ACEI | None | Predictor: ACEI therapy Outcome: serum αKlotho Follow-up: 8 months | ACEI therapy significantly increased serum αKlotho levels without any associated changes in serum calcium or phosphate | αKlotho levels did not correlate with eGFR at baseline (r = 0.02, p = 0.82) or at 8-month follow-up (r = 0.06, p = 0.45) |
Highlighted as an important observational study.
6.4.1 α lotho and CV Disease in CKD
A cross-sectional study of 114 CKD patients (mean eGFR 48±29 mL/min/1.73 m2) revealed that serum αKlotho was a significant marker of arterial stiffness measured by ankle–brachial pulse waive velocity (Kitagawa et al., 2013). In contrast, a large cohort study of 444 patients with CKD stages 2–4 showed that plasma αKlotho levels (highest vs lowest tertile) did not predict atherosclerotic events or death at 2.6 years follow-up. Serum αKlotho was also significantly reduced in hypertensive (essential and renovascular) patients with mild CKD when compared to healthy controls, even after adjustment by eGFR (Park et al., 2015). The proposed cross talk between the renin–angiotensin–aldosterone system and the vitamin D–FGF23–α Klotho pathways supports the concept that modulation of one system can have positive effects on the other (de Borst, Vervloet, ter Wee, & Navis, 2011). In this context, a post hoc analysis of the ESCAPE trial in children with CKD (all received fixed dose of ramipril 6 mg/m2 per day) showed that 25(OH)D ≥50 nmol/L was associated with greater preservation of renal function. Interestingly, ACEI therapy significantly increased serum αKlotho levels without any associated changes in serum calcium or phosphate (Shroff et al., 2016; Table 2).
6.4.2 αKlotho and Progressive CKD
The most conclusive evidence thus far about the role of αKlotho as a predictor of adverse outcomes in CKD is based on a post hoc cohort study of 243 adult patients with CKD. In this study, serum αKlotho levels independently predicted the composite outcome of doubling SCr, ESRD, or death after multivariable adjustment. If serum αKlotho was ≤396.3 pg/mL, 35.2% reached the composite outcome vs 15.7% if >396.3 pg/mL (adjusted HR 2.03, 95% CI 1.07–3.85, p = 0.03). The areas under the curve (a measure of discrimination, that is, the ability of αKlotho to correctly classify those with and without the outcome) for 1/serum αKlotho to predicate the composite outcome (doubling SCr, ESRD, or death) were 0.81, 0.78, and 0.72 at 12, 24, and 36 months (Kim et al., 2013; Table 2). Further studies are needed to corroborate these findings.
Taken together, CKD is a state of αKlotho deficiency and dysfunctional vitamin D–FGF23–αKlotho pathways. Plasma αKlotho level positively correlates with eGFR and negatively with SCr and FGF23 and is a promising biomarker for the prediction of adverse outcomes (eg, CKD progression, CV morbidity, and death) in CKD patients. Therefore, αKlotho may represent a novel therapy for CKD patients that needs to be further investigated.
7. αKlotho AS A PROMISING TREATMENT STRATEGY FOR CKD
The kidney is confirmed as the major site that contributes to circulating αKlotho (Hu, Shi, Zhang, et al., 2015; Lindberg et al., 2014) and dysfunctional or decreased number of αKlotho-producing cells contribute to αKlotho deficiency leading to accelerated aging (Hu, Shi, Zhang, et al., 2015; Lindberg et al., 2014). Therefore, αKlotho deficiency may not only be a pathogenic intermediate for accelerating CKD progression but also a main promoter of complications such as secondary hyperparathyroidism and CV disease in CKD. Conceivably, any therapy that restores or stimulates endogenous αKlotho or administration of exogenous αKlotho might provide a novel treatment strategy in CKD.
7.1 Epigenetic regulation of αKlotho Expression
αKlotho deficiency in the kidney of hypomorphic αKlotho-deficient mice was thought to result from interruption of the promoter of αKlotho gene by the exogenous transgene (Kuro-o et al., 1997). But recent data do not entirely support this notion because there was aberrant αKlotho promoter methylation in kl/kl mice. The in vitro study showed that αKlotho gene promoter methylation reduced promoter activity by 30–40%, whereas DNA demethylating agents increased αKlotho expression 1.5- to 3.0-fold (Azuma et al., 2012). Similarly, uremic toxins—indoxyl sulfate or p-cresyl sulfate—induced hypermethylation of the αKlotho gene, and decreased αKlotho expression in renal tubules and kidney cell line, which can be reversed by demethylation of the αKlotho gene. Therefore, hypermethylation may be one of the mechanisms of αKlotho gene expression inhibition in CKD (Chen, Zhang, et al., 2013; Sun et al., 2012; Young & Wu, 2012). In addition, promoter histone acetylation was also proposed as a possible mechanism for αKlotho silencing in many types of cancer cell lines (Rubinek et al., 2012; Xie et al., 2013). Furthermore, TNF and TNF-like weak inducer of apoptosis (TWEAK)-induced downregulation of αKlotho expression in the kidney and kidney cell lines can be blunted by inhibition of histone deacetylase (Moreno et al., 2011). Therefore, demethylating agents and deacetylase inhibitors may be agents to reactivate αKlotho expression in the kidney and consequently increase circulating αKlotho.
7.2 Reactivation of Endogenous αKlotho Expression Independently of Epigenetics
To date, several categories of drugs in the market including peroxisome proliferator-activated receptors-gamma (PPAR-γ) agonists (Chen, Cheng, Ku, & Lin, 2014; Yang et al., 2009; Zhang et al., 2008; Zhang & Zheng, 2008), angiotensin II type I receptor antagonists (Karalliedde, Maltese, Hill, Viberti, & Gnudi, 2013; Yoon et al., 2011; Zhou et al., 2010), 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase inhibitors (statin; Narumiya et al., 2004), and vitamin D active derivatives (de Borst et al., 2011; Forster et al., 2011; Lau et al., 2012; Lim et al., 2012; Ritter et al., 2015) have been shown to upregulate αKlotho expression in vivo and in vitro. The effect of upregulating αKlotho is definitely not associated with their well-identified original pharmacological targets, and pharmacological mechanisms remain to be explored.
7.3 Administration of Soluble αKlotho Protein
αKlotho gene delivery is shown to effectively rescue many phenotypes observed in αKlotho-deficient mice (Shiraki-Iida et al., 2000), attenuating the progression of hypertension and kidney damage in spontaneous hypertensive rats (Wang & Sun, 2009, 2014), improving kidney function in acute kidney injury (Sugiura et al., 2005), ameliorating angiotensin II-induced kidney injury (Mitani et al., 2002), improving endothelial function (Saito et al., 2000), and protecting from uremic cardiomyopathy (Xie et al., 2012, 2015). Although gene therapy is effective in animal studies, its safety is still questionable and human clinical application is not in the proximity. There are only few clinical trials testing gene therapy in specific human diseases including genetic diseases (Williams, 2014) and some types of cancers (Heller & Heller, 2015).
In contrast, administration of exogenous αKlotho protein is more direct, safer, and an easier modality to restore endocrine αKlotho deficiency. Animal studies have already provided prove-of-concept encouraging data that soluble αKlotho protein administration is safe and effective (Hu, Shi, Zhang, Quinones, et al., 2010; Shi et al., 2015). Soluble αKlotho protein attenuates kidney damage and preserves kidney function in an ischemia-reperfusion injury model causing acute kidney injury, which is a state of acute αKlotho deficiency (Hu, Shi, Zhang, Quinones, et al., 2010). Furthermore, αKlotho protein inhibited renal fibrosis in a UUO kidney injury model (Doi et al., 2011), and effectively extended the life span of homozygous αKlotho-deficient mice, ameliorating premature aging-related phenotypes (Chen, Kuro, et al., 2013). Although there are no clinical data showing αKlotho protein administration to CKD patients, the preclinical data clearly support the therapeutic potential of soluble αKlotho protein in CKD patients.
8. CONCLUSION AND FUTURE DIRECTIONS
Although progress has been made in understanding that αKlotho is not only an aging suppressor involved in longevity and aging, but also a renoprotective factor which has a central impact on renal physiology and renal pathophysiology, the utility of αKlotho in clinical practice is still under development (Table 3). First, αKlotho could serve as an early and sensitive biomarker of CKD, although its specificity and its prognostic value require further exploration in humans. Similarly, whether urine or serum αKlotho is a better biomarker remains to be confirmed. Second, exogenous αKlotho supplementation may represent a novel therapy to retard or block progressive CKD, post-AKI transition to CKD, as well as preventing and reversing CV complications associated with renal disease as shown in animal studies. The therapeutic efficacy of αKlotho in kidney disease has been unequivocally demonstrated in animal models. One needs to validate the efficacy of αKlotho therapy in different stages of kidney disease.
Table 3.
Potential Applications of αKlotho in Chronic Kidney Disease
| Biomarker | Therapeutic Agent | ||
|---|---|---|---|
| Diagnostic | Early detection of CKD | Prevention |
|
| Prognostic | Prediction of progression to ESRD | Treatment |
|
CKD, chronic kidney disease; CV, cardiovascular event; ESRD, end-stage renal disease.
Although the effects of αKlotho protein on the kidney will invariably be pleiotropic, how αKlotho exerts its renoprotection is largely inconclusive. High levels of FGF23 have been found to be associated with high mortality and morbidity in CKD (Arnlov et al., 2013; Desjardins et al., 2012; Faul et al., 2011; Fliser et al., 2007; Grabner et al., 2015; Greenhill, 2011; Guo & Yuan, 2015; Hanks, Casazza, Judd, Jenny, & Gutierrez, 2015; Hasegawa et al., 2010; Krupp & Madhivanan, 2014; Mencke et al., 2015; Mirza, Larsson, Melhus, Lind, & Larsson, 2009; Razzaque, 2009a, 2009b; Rotondi et al., 2015; Sawires et al., 2015; Silswal et al., 2014; Silver, Rodriguez, & Slatopolsky, 2012; Sinha et al., 2015; Wolf, 2010; Wright et al., 2014; Zhang, Yan, Zhu, & Ni, 2015; Zhang, Yang, et al., 2015). Given that administration of exogenous αKlotho has a favorable effect on CKD animals in terms of improvement of renal function, better maintenance of phosphate homeostasis, and attenuation of vascular calcification and cardiac hypertrophy, whether synergistic utilization of FGF23 antagonist or inhibitor and αKlotho can enhance αKlotho therapeutic efficacy needs to be tested.
Some therapeutic modalities including ACE inhibition, HMG-CoA reductase inhibition, vitamin D derivatives, and antioxidants could sustain or increase endogenous αKlotho expression. It is conceivable that a multi-targeted regimen including αKlotho protein, FGF23 antagonist, and endogenous αKlotho inducers may enhance the beneficial effect of αKlotho on retardation of CKD progression and prevention or reduction of CV morbidity and mortality in CKD. The immediate challenge is to test whether human CKD resembles the rodent counterpart and if so, how to more efficiently increase αKlotho levels in patients with CKD, either by stimulating endogenous αKlotho or by administering recombinant αKlotho. Finally, αKlotho has shown to be effective in experimental CKD models such as renal ablation (Xie et al., 2015), hypertensive kidney damage (Tang et al., 2011; Wang & Sun, 2009), UUO (Doi et al., 2011; Guan et al., 2014; Satoh et al., 2012; Sugiura et al., 2012), but few experimental data showed its effect on CKD secondary to diabetes or other glomerular diseases which account for large part of the CKD population. More animal studies and clinical observational studies are required to consider the administration of exogenous αKlotho protein a potential novel therapeutic strategy for CKD.
Acknowledgments
The authors appreciate Dr. Orson Moe for helpful interactions in the editing of this chapter. The research work in MC Hu’s research laboratory is in part supported by the NIH (R01-DK091392, R01-DK092461), the George M. O’Brien Kidney Research Center (P30-DK-07938), the Charles and Jane Pak Foundation, and the Pak Center Innovative Research Support. J.A.N. is in part supported by the Ben J. Lipps Research Fellowship Program of American Society of Nephrology Foundation for Kidney Research and the Truelson Fellowship Fund at the Charles and Jane Pak Center of Mineral Metabolism and Clinical Research.
References
- Adema AY, Vervloet MG, Blankenstein MA, Heijboer AC. α-Klotho is unstable in human urine. Kidney International. 2015;88(6):1442–1444. doi: 10.1038/ki.2015.238. [DOI] [PubMed] [Google Scholar]
- Adijiang A, Shimizu H, Higuchi Y, Nishijima F, Niwa T. Indoxyl sulfate reduces klotho expression and promotes senescence in the kidneys of hypertensive rats. Journal of Renal Nutrition. 2011;21(1):105–109. doi: 10.1053/j.jrn.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Kimura T, Watanabe Y, Ishikawa N, Iwazu Y, Saito O, et al. The impact of nephrectomy and renal transplantation on serum levels of soluble Klotho protein. Transplantation Proceedings. 2013;45(1):134–136. doi: 10.1016/j.transproceed.2012.07.150. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Yoshizawa H, Watanabe Y, Numata A, Yamazaki T, Takeshima E, et al. Characteristics of urinary and serum soluble Klotho protein in patients with different degrees of chronic kidney disease. BMC Nephrology. 2012;13:155. doi: 10.1186/1471-2369-13-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R, et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. The EMBO Journal. 2014;33(3):229–246. doi: 10.1002/embj.201284188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardura JA, Rayego-Mateos S, Ramila D, Ruiz-Ortega M, Esbrit P. Parathyroid hormone-related protein promotes epithelial-mesenchymal transition. Journal of the American Society of Nephrology. 2010;21(2):237–248. doi: 10.1681/ASN.2009050462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnlov J, Carlsson AC, Sundstrom J, Ingelsson E, Larsson A, Lind L, et al. Serum FGF23 and risk of cardiovascular events in relation to mineral metabolism and cardiovascular pathology. Clinical Journal of the American Society of Nephrology. 2013;8(5):781–786. doi: 10.2215/CJN.09570912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai O, Nakatani K, Tanaka T, Sakan H, Imura A, Yoshimoto S, et al. Decreased renal alpha-Klotho expression in early diabetic nephropathy in humans and mice and its possible role in urinary calcium excretion. Kidney International. 2012;81(6):539–547. doi: 10.1038/ki.2011.423. [DOI] [PubMed] [Google Scholar]
- Azuma M, Koyama D, Kikuchi J, Yoshizawa H, Thasinas D, Shiizaki K, et al. Promoter methylation confers kidney-specific expression of the Klotho gene. The FASEB Journal. 2012;26(10):4264–4274. doi: 10.1096/fj.12-211631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker SL, Pastor J, Carranza D, Quinones H, Griffith C, Goetz R, et al. The demonstration of alphaKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrology, Dialysis, Transplantation. 2015;30(2):223–233. doi: 10.1093/ndt/gfu291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. The Journal of Clinical Investigation. 2007;117(12):4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian A, Neyra JA, Zhan M, Hu MC. Klotho, stem cells, and aging. Clinical Interventions in Aging. 2015;10:1233–1243. doi: 10.2147/CIA.S84978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian A, Xing C, Hu MC. Alpha Klotho and phosphate homeostasis. Journal of Endocrinological Investigation. 2014;37(11):1121–1126. doi: 10.1007/s40618-014-0158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, et al. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Letters. 2009;583(19):3221–3224. doi: 10.1016/j.febslet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium × phosphate product with mortality risk in chronic hemodialysis patients: A national study. American Journal of Kidney Diseases. 1998;31(4):607–617. doi: 10.1053/ajkd.1998.v31.pm9531176. [DOI] [PubMed] [Google Scholar]
- Camilli TC, Xu M, O’Connell MP, Chien B, Frank BP, Subaran S, et al. Loss of Klotho during melanoma progression leads to increased filamin cleavage, increased Wnt5A expression, and enhanced melanoma cell motility. Pigment Cell & Melanoma Research. 2011;24(1):175–186. doi: 10.1111/j.1755-148X.2010.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalejo R, Canalejo A, Martinez-Moreno JM, Rodriguez-Ortiz ME, Estepa JC, Mendoza FJ, et al. FGF23 fails to inhibit uremic parathyroid glands. Journal of the American Society of Nephrology. 2010;21(7):1125–1135. doi: 10.1681/ASN.2009040427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannata-Andia JB, Rodriguez-Garcia M. Hyperphosphataemia as a cardiovascular risk factor—How to manage the problem. Nephrology, Dialysis, Transplantation. 2002;17(Suppl 11):16–19. doi: 10.1093/ndt/17.suppl_11.16. [DOI] [PubMed] [Google Scholar]
- Cano FJ, Freundlich M, Ceballos ML, Rojo AP, Azocar MA, Delgado IO, et al. Longitudinal FGF23 and Klotho axis characterization in children treated with chronic peritoneal dialysis. Clinical Kidney Journal. 2014;7(5):457–463. doi: 10.1093/ckj/sfu074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo J, Buendia P, Merino A, Madueno JA, Peralbo E, Ortiz A, et al. Klotho modulates the stress response in human senescent endothelial cells. Mechanisms of Ageing and Development. 2012;133(11–12):647–654. doi: 10.1016/j.mad.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Carracedo J, Buendia P, Merino A, Soriano S, Esquivias E, Martin-Malo A, et al. Cellular senescence determines endothelial cell damage induced by uremia. Experimental Gerontology. 2013;48(8):766–773. doi: 10.1016/j.exger.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. The FASEB Journal. 2006;20(10):1706–1708. doi: 10.1096/fj.05-5680fje. [DOI] [PubMed] [Google Scholar]
- Chen LJ, Cheng MF, Ku PM, Lin JW. Rosiglitazone increases cerebral klotho expression to reverse baroreflex in type 1-like diabetic rats. BioMed Research International. 2014;2014:309151. doi: 10.1155/2014/309151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TH, Kuro OM, Chen CH, Sue YM, Chen YC, Wu HH, et al. The secreted Klotho protein restores phosphate retention and suppresses accelerated aging in Klotho mutant mice. European Journal of Pharmacology. 2013;698(1–3):67–73. doi: 10.1016/j.ejphar.2012.09.032. [DOI] [PubMed] [Google Scholar]
- Chen CD, Li H, Liang J, Hixson K, Zeldich E, Abraham CR. The anti-aging and tumor suppressor protein klotho enhances differentiation of a human oligodendrocytic hybrid cell line. Journal of Molecular Neuroscience. 2015;55(1):76–90. doi: 10.1007/s12031-014-0336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(50):19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Tung TY, Liang J, Zeldich E, Tucker Zhou TB, Turk BE, et al. Identification of cleavage sites leading to the shed form of the anti-aging protein klotho. Biochemistry. 2014;53(34):5579–5587. doi: 10.1021/bi500409n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang X, Zhang H, Lin J, Zhang C, Wu Q, et al. Elevated Klotho promoter methylation is associated with severity of chronic kidney disease. PloS One. 2013;8(11):e79856. doi: 10.1371/journal.pone.0079856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MF, Chen LJ, Niu HS, Yang TT, Lin KC, Cheng JT. Signals mediating Klotho-induced neuroprotection in hippocampal neuronal cells. Acta Neurobiologiae Experimentalis (Wars) 2015;75(1):60–71. [PubMed] [Google Scholar]
- Clements ME, Chaber CJ, Ledbetter SR, Zuk A. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PloS One. 2013;8(8):e70464. doi: 10.1371/journal.pone.0070464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich E, Davidovits M, Peretz B, Shapira J, Aframian DJ. The correlation between dental calculus and disturbed mineral metabolism in paediatric patients with chronic kidney disease. Nephrology, Dialysis, Transplantation. 2009;24(8):2439–2445. doi: 10.1093/ndt/gfp101. [DOI] [PubMed] [Google Scholar]
- de Borst MH, Vervloet MG, ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. Journal of the American Society of Nephrology. 2011;22(9):1603–1609. doi: 10.1681/ASN.2010121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira RM. Klotho RNAi induces premature senescence of human cells via a p53/p21 dependent pathway. FEBS Letters. 2006;580(24):5753–5758. doi: 10.1016/j.febslet.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Degaspari S, Tzanno-Martins CB, Fujihara CK, Zatz R, Branco-Martins JP, Viel TA, et al. Altered KLOTHO and NF-kappaB-TNF-alpha signaling are correlated with nephrectomy-induced cognitive impairment in rats. PloS One. 2015;10(5):e0125271. doi: 10.1371/journal.pone.0125271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmez JA, Slatopolsky E. Hyperphosphatemia: Its consequences and treatment in patients with chronic renal disease. American Journal of Kidney Diseases. 1992;19:303–317. doi: 10.1016/s0272-6386(12)80446-x. [DOI] [PubMed] [Google Scholar]
- Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, Choukroun G, et al. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporosis International. 2012;23(7):2017–2025. doi: 10.1007/s00198-011-1838-0. [DOI] [PubMed] [Google Scholar]
- Devaraj S, Syed B, Chien A, Jialal I. Validation of an immunoassay for soluble Klotho protein: Decreased levels in diabetes and increased levels in chronic kidney disease. American Journal of Clinical Pathology. 2012;137(3):479–485. doi: 10.1309/AJCPGPMAF7SFRBO4. [DOI] [PubMed] [Google Scholar]
- D’Hoore E, Neirynck N, Schepers E, Vanholder R, Verbeke F, Van Thielen M, et al. Chronic kidney disease progression is mainly associated with non-recovery of acute kidney injury. Journal of Nephrology. 2015;28(6):709–716. doi: 10.1007/s40620-015-0181-5. [DOI] [PubMed] [Google Scholar]
- Di Marco GS, Hausberg M, Hillebrand U, Rustemeyer P, Wittkowski W, Lang D, et al. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. American Journal of Physiology. Renal Physiology. 2008;294(6):F1381–F1387. doi: 10.1152/ajprenal.00003.2008. [DOI] [PubMed] [Google Scholar]
- Dmitrieva NI, Burg MB. High NaCl promotes cellular senescence. Cell Cycle. 2007;6(24):3108–3113. doi: 10.4161/cc.6.24.5084. [DOI] [PubMed] [Google Scholar]
- Doi S, Yorioka N, Doi S, Zou Y, Togao O, Pastor JV, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. The Journal of Biological Chemistry. 2011;286(10):8655–8665. doi: 10.1074/jbc.M110.174037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donate-Correa J, Mora-Fernandez C, Martinez-Sanz R, Muros-de-Fuentes M, Perez H, Meneses-Perez B, et al. Expression of FGF23/KLOTHO system in human vascular tissue. International Journal of Cardiology. 2013;165(1):179–183. doi: 10.1016/j.ijcard.2011.08.850. [DOI] [PubMed] [Google Scholar]
- Eckardt KU, Coresh J, Devuyst O, Johnson RJ, Kottgen A, Levey AS, et al. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet. 2013;382(9887):158–169. doi: 10.1016/S0140-6736(13)60439-0. [DOI] [PubMed] [Google Scholar]
- Emami Aleagha MS, Siroos B, Ahmadi M, Balood M, Palangi A, Haghighi AN, et al. Decreased concentration of Klotho in the cerebrospinal fluid of patients with relapsing-remitting multiple sclerosis. Journal of Neuroimmunology. 2015;281:5–8. doi: 10.1016/j.jneuroim.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Fahrleitner-Pammer A, Herberth J, Browning SR, Obermayer-Pietsch B, Wirnsberger G, Holzer H, et al. Bone markers predict cardiovascular events in chronic kidney disease. Journal of Bone and Mineral Research. 2008;23(11):1850–1858. doi: 10.1359/jbmr.080610. [DOI] [PubMed] [Google Scholar]
- Fang Y, Ginsberg C, Sugatani T, Monier-Faugere MC, Malluche H, Hruska KA. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney International. 2014;85(1):142–150. doi: 10.1038/ki.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. The Journal of Clinical Investigation. 2011;121(11):4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nature Reviews. Nephrology. 2015;11(5):264–276. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martin JL, Martinez-Camblor P, Dionisi MP, Floege J, Ketteler M, London G, et al. Improvement of mineral and bone metabolism markers is associated with better survival in haemodialysis patients: The COSMOS study. Nephrology, Dialysis, Transplantation. 2015;30(9):1542–1551. doi: 10.1093/ndt/gfv099. [DOI] [PubMed] [Google Scholar]
- Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The Mild to Moderate Kidney Disease (MMKD) Study. Journal of the American Society of Nephrology. 2007;18(9):2600–2608. doi: 10.1681/ASN.2006080936. [DOI] [PubMed] [Google Scholar]
- Fliser D, Seiler S, Heine GH, Ketteler M. Measurement of serum soluble Klotho levels in CKD 5D patients: Useful tool or dispensable biomarker? Nephrology, Dialysis, Transplantation. 2012;27(5):1702–1703. doi: 10.1093/ndt/gfs076. [DOI] [PubMed] [Google Scholar]
- Floege J, Kim J, Ireland E, Chazot C, Drueke T, de Francisco A, et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrology, Dialysis, Transplantation. 2011;26(6):1948–1955. doi: 10.1093/ndt/gfq219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, et al. Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney International. 1995;47(1):186–192. doi: 10.1038/ki.1995.22. [DOI] [PubMed] [Google Scholar]
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. American Journal of Kidney Diseases. 1998;32(5 Suppl 3):S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- Forster RE, Jurutka PW, Hsieh JC, Haussler CA, Lowmiller CL, Kaneko I, et al. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochemical and Biophysical Research Communications. 2011;414(3):557–562. doi: 10.1016/j.bbrc.2011.09.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MC, Inker LA, Levey AS, Selvin E, Eckfeldt J, Juraschek SP, et al. Novel filtration markers as predictors of all-cause and cardiovascular mortality in US adults. American Journal of Kidney Diseases. 2013;62(1):42–51. doi: 10.1053/j.ajkd.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fufaa GD, Weil EJ, Nelson RG, Hanson RL, Bonventre JV, Sabbisetti V, et al. Association of urinary KIM-1, L-FABP, NAG and NGAL with incident end-stage renal disease and mortality in American Indians with type 2 diabetes mellitus. Diabetologia. 2015;58(1):188–198. doi: 10.1007/s00125-014-3389-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukino K, Suzuki T, Saito Y, Shindo T, Amaki T, Kurabayashi M, et al. Regulation of angiogenesis by the aging suppressor gene klotho. Biochemical and Biophysical Research Communications. 2002;293(1):332–337. doi: 10.1016/S0006-291X(02)00216-4. [DOI] [PubMed] [Google Scholar]
- Galitzer H, Ben-Dov IZ, Silver J, Naveh-Many T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney International. 2010;77(3):211–218. doi: 10.1038/ki.2009.464. [DOI] [PubMed] [Google Scholar]
- Gattineni J, Baum M. Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): Implications for disorders of phosphate metabolism. Pediatric Nephrology. 2010;25(4):591–601. doi: 10.1007/s00467-009-1273-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassock RJ, Pecoits-Filho R, Barberato SH. Left ventricular mass in chronic kidney disease and ESRD. Clinical Journal of the American Society of Nephrology. 2009;4(Suppl 1):S79–S91. doi: 10.2215/CJN.04860709. [DOI] [PubMed] [Google Scholar]
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. The New England Journal of Medicine. 2004;351(13):1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):407–412. doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metabolism. 2015;22(6):1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill C. Risk factors: Levels of FGF23 predict outcomes in advanced CKD. Nature Reviews. Nephrology. 2011;7(11):606. doi: 10.1038/nrneph.2011.130. [DOI] [PubMed] [Google Scholar]
- Gross ML, Ritz E. Hypertrophy and fibrosis in the cardiomyopathy of uremia—Beyond coronary heart disease. Seminars in Dialysis. 2008;21(4):308–318. doi: 10.1111/j.1525-139X.2008.00454.x. [DOI] [PubMed] [Google Scholar]
- Guan X, Nie L, He T, Yang K, Xiao T, Wang S, et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. The Journal of Pathology. 2014;234(4):560–572. doi: 10.1002/path.4420. [DOI] [PubMed] [Google Scholar]
- Guo YC, Yuan Q. Fibroblast growth factor 23 and bone mineralisation. International Journal of Oral Science. 2015;7(1):8–13. doi: 10.1038/ijos.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez OM. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: Updating the “trade-off” hypothesis. Clinical Journal of the American Society of Nephrology. 2010;5(9):1710–1716. doi: 10.2215/CJN.02640310. [DOI] [PubMed] [Google Scholar]
- Gutiérrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119(19):2545–2552. doi: 10.1161/CIRCULATIONAHA.108.844506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. The New England Journal of Medicine. 2008;359(6):584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks LJ, Casazza K, Judd SE, Jenny NS, Gutierrez OM. Associations of fibroblast growth factor-23 with markers of inflammation, insulin resistance and obesity in adults. PloS One. 2015;10(3):e0122885. doi: 10.1371/journal.pone.0122885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruna Y, Kashihara N, Satoh M, Tomita N, Namikoshi T, Sasaki T, et al. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(7):2331–2336. doi: 10.1073/pnas.0611079104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney International. 2010;78(10):975–980. doi: 10.1038/ki.2010.313. [DOI] [PubMed] [Google Scholar]
- Heller R, Heller LC. Gene electrotransfer clinical trials. Advances in Genetics. 2015;89:235–262. doi: 10.1016/bs.adgen.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Hemmelgarn BR, Manns BJ, Lloyd A, James MT, Klarenbach S, Quinn RR, et al. Relation between kidney function, proteinuria, and adverse outcomes. JAMA. 2010;303(5):423–429. doi: 10.1001/jama.2010.39. [DOI] [PubMed] [Google Scholar]
- Heydarkhan-Hagvall S, Helenius G, Johansson BR, Li JY, Mattsson E, Risberg B. Co-culture of endothelial cells and smooth muscle cells affects gene expression of angiogenic factors. Journal of Cellular Biochemistry. 2003;89(6):1250–1259. doi: 10.1002/jcb.10583. [DOI] [PubMed] [Google Scholar]
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Seminars in Dialysis. 2007;20(4):309–315. doi: 10.1111/j.1525-139X.2007.00300.x. [DOI] [PubMed] [Google Scholar]
- Hu MC. Klotho connects intermedin1–53 to suppression of vascular calcification in chronic kidney disease. Kidney International. 2016;89(3):534–537. doi: 10.1016/j.kint.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Kuro-o M, Moe OW. Secreted klotho and chronic kidney disease. Advances in Experimental Medicine and Biology. 2012;728:126–157. doi: 10.1007/978-1-4614-0887-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Kuro-o M, Moe OW. Klotho and chronic kidney disease. Contributions to Nephrology. 2013a;180:47–63. doi: 10.1159/000346778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Kuro-o M, Moe OW. Renal and extrarenal actions of Klotho. Seminars in Nephrology. 2013b;33(2):118–129. doi: 10.1016/j.semnephrol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Kuro-o M, Moe OW. alphaKlotho and vascular calcification: An evolving paradigm. Current Opinion in Nephrology and Hypertension. 2014;23(4):331–339. doi: 10.1097/01.mnh.0000447024.97464.a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. Journal of the American Society of Nephrology. 2015a;26(6):1290–1302. doi: 10.1681/ASN.2014050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shi M, Cho HJ, Zhang J, Pavlenco A, Liu S, et al. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney International. 2013;84(3):468–481. doi: 10.1038/ki.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, et al. Renal production, uptake, and handling of circulating αKlotho. Journal of the American Society of Nephrology. 2015b;27(1):79–90. doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. The FASEB Journal. 2010a;24(9):3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shi M, Zhang J, Quinones H, Griffith C, Kuro-o M, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. Journal of the American Society of Nephrology. 2011;22(1):124–136. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shi M, Zhang J, Quinones H, Kuro OM, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney International. 2010b;78(12):1240–1251. doi: 10.1038/ki.2010.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annual Review of Physiology. 2013;75:503–533. doi: 10.1146/annurev-physiol-030212-183727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. The Journal of Clinical Investigation. 2007;117(9):2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima M, Rakugi H, Ishikawa K, Maekawa Y, Yamamoto K, Ohta J, et al. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochemical and Biophysical Research Communications. 2006;339(3):827–832. doi: 10.1016/j.bbrc.2005.11.094. [DOI] [PubMed] [Google Scholar]
- Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, et al. Secreted Klotho protein in sera and CSF: Implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Letters. 2004;565(1–3):143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- Isakova T, Gutierrez OM, Chang Y, Shah A, Tamez H, Smith K, et al. Phosphorus binders and survival on hemodialysis. Journal of the American Society of Nephrology. 2009;20(2):388–396. doi: 10.1681/ASN.2008060609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Fujimori T, Hayashizaki Y, Nabeshima Y. Identification of a novel mouse membrane-bound family 1 glycosidase-like protein, which carries an atypical active site structure. Biochimica et Biophysica Acta. 2002;1576(3):341–345. doi: 10.1016/s0167-4781(02)00281-6. [DOI] [PubMed] [Google Scholar]
- Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, et al. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mechanisms of Development. 2000;98(1–2):115–119. doi: 10.1016/s0925-4773(00)00439-1. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. The Journal of Clinical Investigation. 2002;110(3):341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ix JH, Katz R, Kestenbaum BR, de Boer IH, Chonchol M, Mukamal KJ, et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study) Journal of the American College of Cardiology. 2012;60(3):200–207. doi: 10.1016/j.jacc.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings P, Koppelstaetter C, Aydin S, Abberger T, Wolf AM, Mayer G, et al. Cyclosporine A induces senescence in renal tubular epithelial cells. American Journal of Physiology Renal Physiology. 2007;293(3):F831–F838. doi: 10.1152/ajprenal.00005.2007. [DOI] [PubMed] [Google Scholar]
- Jie KE, Zaikova MA, Bergevoet MW, Westerweel PE, Rastmanesh M, Blankestijn PJ, et al. Progenitor cells and vascular function are impaired in patients with chronic kidney disease. Nephrology, Dialysis, Transplantation. 2010;25(6):1875–1882. doi: 10.1093/ndt/gfp749. [DOI] [PubMed] [Google Scholar]
- Jimbo R, Kawakami-Mori F, Mu S, Hirohama D, Majtan B, Shimizu Y, et al. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney International. 2014;85(5):1103–1111. doi: 10.1038/ki.2013.332. [DOI] [PubMed] [Google Scholar]
- Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circulation Research. 2000;87(7):E10–E17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- Kailong L, Du X, Yani H, Lin Z, Jvrong Y, Ruihua S, et al. P53-Rb signaling pathway is involved in tubular cell senescence in renal ischemia/reperfusion injury. Biocell. 2007;31(2):213–223. [PubMed] [Google Scholar]
- Kaisar M, Isbel N, Johnson DW. Cardiovascular disease in patients with chronic kidney disease. A clinical review. Minerva Urologica e Nefrologica. 2007;59(3):281–297. [PubMed] [Google Scholar]
- Kalantar-Zadeh K, Shah A, Duong U, Hechter RC, Dukkipati R, Kovesdy CP. Kidney bone disease and mortality in CKD: Revisiting the role of vitamin D, calcimimetics, alkaline phosphatase, and minerals. Kidney International. Supplement. 2010;117:S10–S21. doi: 10.1038/ki.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. The Journal of Clinical Investigation. 2003;112(12):1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalliedde J, Maltese G, Hill B, Viberti G, Gnudi L. Effect of renin-angiotensin system blockade on soluble Klotho in patients with type 2 diabetes, systolic hypertension, and albuminuria. Clinical Journal of the American Society of Nephrology. 2013;8(11):1899–1905. doi: 10.2215/CJN.02700313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, et al. Establishment of the anti-Klotho monoclonal antibodies and detection of Klotho protein in kidneys. Biochemical and Biophysical Research Communications. 2000;267(2):597–602. doi: 10.1006/bbrc.1999.2009. [DOI] [PubMed] [Google Scholar]
- Kestenbaum B, Belozeroff V. Mineral metabolism disturbances in patients with chronic kidney disease. European Journal of Clinical Investigation. 2007;37(8):607–622. doi: 10.1111/j.1365-2362.2007.01840.x. [DOI] [PubMed] [Google Scholar]
- Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. Journal of the American Society of Nephrology. 2005;16(2):520–528. doi: 10.1681/ASN.2004070602. [DOI] [PubMed] [Google Scholar]
- Khan S. Vitamin D deficiency and secondary hyperparathyroidism among patients with chronic kidney disease. The American Journal of the Medical Sciences. 2007;333(4):201–207. doi: 10.1097/MAJ.0b013e31803bb129. [DOI] [PubMed] [Google Scholar]
- Kim HR, Nam BY, Kim DW, Kang MW, Han JH, Lee MJ, et al. Circulating alpha-klotho levels in CKD and relationship to progression. American Journal of Kidney Diseases. 2013;61(6):899–909. doi: 10.1053/j.ajkd.2013.01.024. [DOI] [PubMed] [Google Scholar]
- King GD, Rosene DL, Abraham CR. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age (Dordrecht, Netherlands) 2012;34(6):1405–1419. doi: 10.1007/s11357-011-9315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Sugiyama H, Morinaga H, Inoue T, Takiue K, Ogawa A, et al. A decreased level of serum soluble Klotho is an independent biomarker associated with arterial stiffness in patients with chronic kidney disease. PloS One. 2013;8(2):e56695. doi: 10.1371/journal.pone.0056695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochemical and Biophysical Research Communications. 2001;280(4):1015–1020. doi: 10.1006/bbrc.2000.4226. [DOI] [PubMed] [Google Scholar]
- Kooman JP, Broers NJ, Usvyat L, Thijssen S, van der Sande FM, Cornelis T, et al. Out of control: Accelerated aging in uremia. Nephrology, Dialysis, Transplantation. 2013;28(1):48–54. doi: 10.1093/ndt/gfs451. [DOI] [PubMed] [Google Scholar]
- Krajisnik T, Olauson H, Mirza MA, Hellman P, Akerstrom G, Westin G, et al. Parathyroid Klotho and FGF-receptor 1 expression decline with renal function in hyperparathyroid patients with chronic kidney disease and kidney transplant recipients. Kidney International. 2010;78(10):1024–1032. doi: 10.1038/ki.2010.260. [DOI] [PubMed] [Google Scholar]
- Kramann R, Tanaka M, Humphreys BD. Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. Journal of the American Society of Nephrology. 2014;25(9):1924–1931. doi: 10.1681/ASN.2013101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp K, Madhivanan P. FGF23 and risk of all-cause mortality and cardiovascular events: A meta-analysis of prospective cohort studies. International Journal of Cardiology. 2014;176(3):1341–1342. doi: 10.1016/j.ijcard.2014.07.142. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309(5742):1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba T, Okigaki M, Matui A, Murakami M, Ishikawa K, Kimura T, et al. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(45):19308–19313. doi: 10.1073/pnas.1008544107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske B, Razzaque MS. Vitamin D and aging: Old concepts and new insights. The Journal of Nutritional Biochemistry. 2007;18(12):771–777. doi: 10.1016/j.jnutbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney International. 2012;82(12):1261–1270. doi: 10.1038/ki.2012.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Jeong DJ, Kim J, Lee S, Park JH, Chang B, et al. The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Molecular Cancer. 2010;9:109. doi: 10.1186/1476-4598-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Lee SH, Moon JY, Park JY, Lee D, Lim SJ, et al. Repeated administration of bone marrow-derived mesenchymal stem cells improved the protective effects on a remnant kidney model. Renal Failure. 2010;32(7):840–848. doi: 10.3109/0886022X.2010.494803. [DOI] [PubMed] [Google Scholar]
- Levey AS, de Jong PE, Coresh J, El Nahas M, Astor BC, Matsushita K, et al. The definition, classification, and prognosis of chronic kidney disease: A KDIGO Controversies Conference report. Kidney International. 2011;80(1):17–28. doi: 10.1038/ki.2010.483. [DOI] [PubMed] [Google Scholar]
- Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327(5965):542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K, Groen A, Molostvov G, Lu T, Lilley KS, Snead D, et al. α-Klotho expression in human tissues. The Journal of Clinical Endocrinology and Metabolism. 2015;100(10):E1308–E1318. doi: 10.1210/jc.2015-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125(18):2243–2255. doi: 10.1161/CIRCULATIONAHA.111.053405. [DOI] [PubMed] [Google Scholar]
- Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Ostman Wernerson A, et al. The kidney is the principal organ mediating klotho effects. Journal of the American Society of Nephrology. 2014;25(10):2169–2175. doi: 10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg K, Olauson H, Amin R, Ponnusamy A, Goetz R, Taylor RF, et al. Arterial klotho expression and FGF23 effects on vascular calcification and function. PloS One. 2013;8(4):e60658. doi: 10.1371/journal.pone.0060658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: Consequences for bone loss and fractures and therapeutic implications. Endocrine Reviews. 2001;22(4):477–501. doi: 10.1210/edrv.22.4.0437. [DOI] [PubMed] [Google Scholar]
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. Journal of the American Society of Nephrology. 2010;21(2):212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317(5839):803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. Journal of the American Society of Nephrology. 2006;17(5):1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- Liu KD, Yang W, Anderson AH, Feldman HI, Demirjian S, Hamano T, et al. Urine neutrophil gelatinase-associated lipocalin levels do not improve risk prediction of progressive chronic kidney disease. Kidney International. 2013;83(5):909–914. doi: 10.1038/ki.2012.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI, O’Neill WC. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. Journal of the American Society of Nephrology. 2004;15(6):1392–1401. doi: 10.1097/01.asn.0000128955.83129.9c. [DOI] [PubMed] [Google Scholar]
- London GM, Marchais SJ, Guerin AP, Metivier F. Arteriosclerosis, vascular calcifications and cardiovascular disease in uremia. Current Opinion in Nephrology and Hypertension. 2005;14(6):525–531. doi: 10.1097/01.mnh.0000168336.67499.c0. [DOI] [PubMed] [Google Scholar]
- Maekawa Y, Ishikawa K, Yasuda O, Oguro R, Hanasaki H, Kida I, et al. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine. 2009;35(3):341–346. doi: 10.1007/s12020-009-9181-3. [DOI] [PubMed] [Google Scholar]
- Masuda H, Chikuda H, Suga T, Kawaguchi H, Kuro-o M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mechanisms of Ageing and Development. 2005;126(12):1274–1283. doi: 10.1016/j.mad.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mathew S, Tustison KS, Sugatani T, Chaudhary LR, Rifas L, Hruska KA. The mechanism of phosphorus as a cardiovascular risk factor in CKD. Journal of the American Society of Nephrology. 2008;19(6):1092–1105. doi: 10.1681/ASN.2007070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochemical and Biophysical Research Communications. 1998;242(3):626–630. doi: 10.1006/bbrc.1997.8019. [DOI] [PubMed] [Google Scholar]
- Mencke R, Harms G, Mirkovic K, Struik J, van Ark J, van Loon E, et al. Membrane-bound Klotho is not expressed endogenously in healthy or uremic human vascular tissue. Cardiovascular Research. 2015;108(2):220–231. doi: 10.1093/cvr/cvv187. [DOI] [PubMed] [Google Scholar]
- Merchant ML, Niewczas MA, Ficociello LH, Lukenbill JA, Wilkey DW, Li M, et al. Plasma kininogen and kininogen fragments are biomarkers of progressive renal decline in type 1 diabetes. Kidney International. 2013;83(6):1177–1184. doi: 10.1038/ki.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis. 2009;207(2):546–551. doi: 10.1016/j.atherosclerosis.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Mitani H, Ishizaka N, Aizawa T, Ohno M, Usui S, Suzuki T, et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension. 2002;39(4):838–843. doi: 10.1161/01.hyp.0000013734.33441.ea. [DOI] [PubMed] [Google Scholar]
- Mohandas R, Segal MS. Endothelial progenitor cells and endothelial vesicles—What is the significance for patients with chronic kidney disease? Blood Purification. 2010;29(2):158–162. doi: 10.1159/000245643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Izquierdo MC, Sanchez-Nino MD, Suarez-Alvarez B, Lopez-Larrea C, Jakubowski A, et al. The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. Journal of the American Society of Nephrology. 2011;22(7):1315–1325. doi: 10.1681/ASN.2010101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita K, Shirai A, Kubota M, Katakura Y, Nabeshima Y, Takeshige K, et al. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. The Journal of Nutrition. 2001;131(12):3182–3188. doi: 10.1093/jn/131.12.3182. [DOI] [PubMed] [Google Scholar]
- Mu W, Long DA, Ouyang X, Agarwal A, Cruz PE, Roncal CA, et al. Angiostatin overexpression is associated with an improvement in chronic kidney injury by an anti-inflammatory mechanism. American Journal of Physiology. Renal Physiology. 2009;296(1):F145–F152. doi: 10.1152/ajprenal.90430.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai R, Saito Y, Ohyama Y, Aizawa H, Suga T, Nakamura T, et al. Endothelial dysfunction in the klotho mouse and downregulation of klotho gene expression in various animal models of vascular and metabolic diseases. Cellular and Molecular Life Sciences. 2000;57(5):738–746. doi: 10.1007/s000180050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano N, Miyata S, Abe M, Kobayashi N, Wakita S, Yamashita T, et al. Effect of manipulating serum phosphorus with phosphate binder on circulating PTH and FGF23 in renal failure rats. Kidney International. 2006;69(3):531–537. doi: 10.1038/sj.ki.5000020. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Saito Y, Ohyama Y, Masuda H, Sumino H, Kuro-o M, et al. Production of nitric oxide, but not prostacyclin, is reduced in klotho mice. Japanese Journal of Pharmacology. 2002;89(2):149–156. doi: 10.1254/jjp.89.149. [DOI] [PubMed] [Google Scholar]
- Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. American Journal of Physiology. Heart and Circulatory Physiology. 2009;297(5):H1673–H1684. doi: 10.1152/ajpheart.00455.2009. [DOI] [PubMed] [Google Scholar]
- Narumiya H, Sasaki S, Kuwahara N, Irie H, Kusaba T, Kameyama H, et al. HMG-CoA reductase inhibitors up-regulate anti-aging klotho mRNA via RhoA inactivation in IMCD3 cells. Cardiovascular Research. 2004;64(2):331–336. doi: 10.1016/j.cardiores.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Navarro-Gonzalez JF, Donate-Correa J, Muros de Fuentes M, Perez-Hernandez H, Martinez-Sanz R, Mora-Fernandez C. Reduced Klotho is associated with the presence and severity of coronary artery disease. Heart. 2014;100(1):34–40. doi: 10.1136/heartjnl-2013-304746. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney International. 2006;70(12):2141–2147. doi: 10.1038/sj.ki.5002000. [DOI] [PubMed] [Google Scholar]
- Niwa T, Shimizu H. Indoxyl sulfate induces nephrovascular senescence. Journal of Renal Nutrition. 2012;22(1):102–106. doi: 10.1053/j.jrn.2011.10.032. [DOI] [PubMed] [Google Scholar]
- Obi Y, Hamano T, Isaka Y. Prevalence and prognostic implications of vitamin D deficiency in chronic kidney disease. Disease Markers. 2015;2015:868961. doi: 10.1155/2015/868961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney International. 2009;75(11):1166–1172. doi: 10.1038/ki.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ER, Naugle JE, Zhang X, Bomser JA, Meszaros JG. Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. American Journal of Physiology. Heart and Circulatory Physiology. 2005;288(3):H1131–H1138. doi: 10.1152/ajpheart.00763.2004. [DOI] [PubMed] [Google Scholar]
- Ozeki M, Fujita S, Kizawa S, Morita H, Sohmiya K, Hoshiga M, et al. Association of serum levels of FGF23 and alpha-Klotho with glomerular filtration rate and proteinuria among cardiac patients. BMC Nephrology. 2014;15:147. doi: 10.1186/1471-2369-15-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panesso MC, Shi M, Cho HJ, Paek J, Ye J, Moe OW, et al. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney International. 2014;85(4):855–870. doi: 10.1038/ki.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panichi V, Bigazzi R, Paoletti S, Mantuano E, Beati S, Marchetti V, et al. Impact of calcium, phosphate, PTH abnormalities and management on mortality in hemodialysis: Results from the RISCAVID study. Journal of Nephrology. 2010;23(5):556–562. [PubMed] [Google Scholar]
- Park MY, Herrmann SM, Saad A, Eirin A, Tang H, Lerman A, et al. Biomarkers of kidney injury and klotho in patients with atherosclerotic renovascular disease. Clinical Journal of the American Society of Nephrology. 2015;10(3):443–451. doi: 10.2215/CJN.07290714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Barron JL, Mirzazedeh M, Gallagher H, Hyer S, Cantor T, et al. Changes in bone mineral parameters, vitamin D metabolites, and PTH measurements with varying chronic kidney disease stages. Journal of Bone and Mineral Metabolism. 2011;29(1):71–79. doi: 10.1007/s00774-010-0192-1. [DOI] [PubMed] [Google Scholar]
- Pavik I, Jaeger P, Ebner L, Wagner CA, Petzold K, Spichtig D, et al. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: A sequence suggested from a cross-sectional study. Nephrology, Dialysis, Transplantation. 2013;28(2):352–359. doi: 10.1093/ndt/gfs460. [DOI] [PubMed] [Google Scholar]
- Polichnowski AJ, Lan R, Geng H, Griffin KA, Venkatachalam MA, Bidani AK. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. Journal of the American Society of Nephrology. 2014;25(7):1496–1507. doi: 10.1681/ASN.2013040359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte B, Trombetti A, Hadaya K, Ernandez T, Fumeaux D, Iselin C, et al. Acute and long term mineral metabolism adaptation in living kidney donors: A prospective study. Bone. 2014;62:36–42. doi: 10.1016/j.bone.2014.01.020. [DOI] [PubMed] [Google Scholar]
- Pontoriero G, Cozzolino M, Locatelli F, Brancaccio D. CKD patients: The dilemma of serum PTH levels. Nephron. Clinical Practice. 2010;116(4):c263–c268. doi: 10.1159/000318787. [DOI] [PubMed] [Google Scholar]
- Ravani P, Tripepi G, Malberti F, Testa S, Mallamaci F, Zoccali C. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: A competing risks modeling approach. Journal of the American Society of Nephrology. 2005;16(8):2449–2455. doi: 10.1681/ASN.2005010076. [DOI] [PubMed] [Google Scholar]
- Ravikumar P, Ye J, Zhang J, Pinch SN, Hu MC, Kuro-o M, et al. alpha-Klotho protects against oxidative damage in pulmonary epithelia. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2014;307(7):L566–L575. doi: 10.1152/ajplung.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: Is Klotho an essential player? American Journal of Physiology. Renal Physiology. 2009a;296(3):F470–F476. doi: 10.1152/ajprenal.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrology, Dialysis, Transplantation. 2009b;24(1):4–7. doi: 10.1093/ndt/gfn620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS. FGF23, klotho and vitamin D interactions: What have we learned from in vivo mouse genetics studies? Advances in Experimental Medicine and Biology. 2012;728:84–91. doi: 10.1007/978-1-4614-0887-1_5. [DOI] [PubMed] [Google Scholar]
- Rebholz CM, Grams ME, Coresh J, Selvin E, Inker LA, Levey AS, et al. Serum fibroblast growth factor-23 is associated with incident kidney disease. Journal of the American Society of Nephrology. 2015a;26(1):192–200. doi: 10.1681/ASN.2014020218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebholz CM, Grams ME, Matsushita K, Selvin E, Coresh J. Change in novel filtration markers and risk of ESRD. American Journal of Kidney Diseases. 2015b;66(1):47–54. doi: 10.1053/j.ajkd.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio-Mayoral A, Banerjee D, Streather C, Kaski JC. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease—A cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis. 2011;216(2):446–451. doi: 10.1016/j.atherosclerosis.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Reinders ME, Rabelink TJ, Briscoe DM. Angiogenesis and endothelial cell repair in renal disease and allograft rejection. Journal of the American Society of Nephrology. 2006;17(4):932–942. doi: 10.1681/ASN.2005121250. [DOI] [PubMed] [Google Scholar]
- Rimes-Stigare C, Frumento P, Bottai M, Martensson J, Martling CR, Walther SM, et al. Evolution of chronic renal impairment and long-term mortality after de novo acute kidney injury in the critically ill; a Swedish multi-centre cohort study. Critical Care. 2015;19:221. doi: 10.1186/s13054-015-0920-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter CS, Zhang S, Delmez J, Finch JL, Slatopolsky E. Differential expression and regulation of Klotho by paricalcitol in the kidney, parathyroid, and aorta of uremic rats. Kidney International. 2015;87(6):1141–1152. doi: 10.1038/ki.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotondi S, Pasquali M, Tartaglione L, Muci ML, Mandanici G, Leonangeli C, et al. Soluble alpha-Klotho serum levels in chronic kidney disease. International Journal of Endocrinology. 2015;2015:872193. doi: 10.1155/2015/872193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinek T, Shulman M, Israeli S, Bose S, Avraham A, Zundelevich A, et al. Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Research and Treatment. 2012;133(2):649–657. doi: 10.1007/s10549-011-1824-4. [DOI] [PubMed] [Google Scholar]
- Sabbisetti VS, Waikar SS, Antoine DJ, Smiles A, Wang C, Ravisankar A, et al. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. Journal of the American Society of Nephrology. 2014;25(10):2177–2186. doi: 10.1681/ASN.2013070758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Nakamura T, Ohyama Y, Suzuki T, Iida A, Shiraki-Iida T, et al. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochemical and Biophysical Research Communications. 2000;276(2):767–772. doi: 10.1006/bbrc.2000.3470. [DOI] [PubMed] [Google Scholar]
- Saito Y, Yamagishi T, Nakamura T, Ohyama Y, Aizawa H, Suga T, et al. Klotho protein protects against endothelial dysfunction. Biochemical and Biophysical Research Communications. 1998;248(2):324–329. doi: 10.1006/bbrc.1998.8943. [DOI] [PubMed] [Google Scholar]
- Sakan H, Nakatani K, Asai O, Imura A, Tanaka T, Yoshimoto S, et al. Reduced renal alpha-Klotho expression in CKD patients and its effect on renal phosphate handling and vitamin D metabolism. PloS One. 2014;9(1):e86301. doi: 10.1371/journal.pone.0086301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre C, Rubio-Navarro A, Buendía I, Gómez-Guerrero C, Blanco J, Mas S, et al. Hyperlipidemia-associated renal damage decreases Klotho expression in kidneys from ApoE knockout mice. PloS One. 2013;8(12):e83713. doi: 10.1371/journal.pone.0083713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. American Journal of Physiology. Renal Physiology. 2012;303(12):F1641–F1651. doi: 10.1152/ajprenal.00460.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawires HK, Essam RM, Morgan MF, Mahmoud RA. Serum klotho: Relation to fibroblast growth factor-23 and other regulators of phosphate metabolism in children with chronic kidney disease. Nephron. 2015;129(4):293–299. doi: 10.1159/000377633. [DOI] [PubMed] [Google Scholar]
- Schmitt R, Susnik N, Melk A. Molecular aspects of renal senescence. Current Opinion in Organ Transplantation. 2015;20(4):412–416. doi: 10.1097/MOT.0000000000000214. [DOI] [PubMed] [Google Scholar]
- Scholze A, Liu Y, Pedersen L, Xia S, Roth HJ, Hocher B, et al. Soluble alpha-klotho and its relation to kidney function and fibroblast growth factor-23. The Journal of Clinical Endocrinology and Metabolism. 2014;99(5):E855–E861. doi: 10.1210/jc.2013-4171. [DOI] [PubMed] [Google Scholar]
- Schwedhelm E, Boger RH. The role of asymmetric and symmetric dimethylarginines in renal disease. Nature Reviews. Nephrology. 2011;7(5):275–285. doi: 10.1038/nrneph.2011.31. [DOI] [PubMed] [Google Scholar]
- Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney International. 2013;83(6):1159–1168. doi: 10.1038/ki.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler S, Rogacev KS, Roth HJ, Shafein P, Emrich I, Neuhaus S, et al. Associations of FGF-23 and sKlotho with cardiovascular outcomes among patients with CKD stages 2–4. Clinical Journal of the American Society of Nephrology. 2014;9(6):1049–1058. doi: 10.2215/CJN.07870713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba RD, Moghekar AR, Hu J, Sun K, Turner R, Ferrucci L, et al. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neuroscience Letters. 2014;558:37–40. doi: 10.1016/j.neulet.2013.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, et al. αKlotho mitigates progression of acute kidney injury to chronic kidney disease through activation of autophagy. Journal of the American Society of Nephrology. 2015 doi: 10.1681/ASN.2015060613. pii: ASN.2015060613 Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Takeshita Y, Murohara T, Sasaki K, Egami K, Shintani S, et al. Angiogenesis and vasculogenesis are impaired in the precocious-aging klotho mouse. Circulation. 2004;110(9):1148–1155. doi: 10.1161/01.CIR.0000139854.74847.99. [DOI] [PubMed] [Google Scholar]
- Shimada T, Urakawa I, Isakova T, Yamazaki Y, Epstein M, Wesseling-Perry K, et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. The Journal of Clinical Endocrinology and Metabolism. 2010;95(2):578–585. doi: 10.1210/jc.2009-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura Y, Hamada K, Inoue K, Ogata K, Ishihara M, Kagawa T, et al. Serum levels of soluble secreted alpha-Klotho are decreased in the early stages of chronic kidney disease, making it a probable novel biomarker for early diagnosis. Clinical and Experimental Nephrology. 2012;16(5):722–729. doi: 10.1007/s10157-012-0621-7. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Bolati D, Adijiang A, Adelibieke Y, Muteliefu G, Enomoto A, et al. Indoxyl sulfate downregulates renal expression of Klotho through production of ROS and activation of nuclear factor-κB. American Journal of Nephrology. 2011;33(4):319–324. doi: 10.1159/000324885. [DOI] [PubMed] [Google Scholar]
- Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, et al. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Letters. 1998;424(1–2):6–10. doi: 10.1016/s0014-5793(98)00127-6. [DOI] [PubMed] [Google Scholar]
- Shiraki-Iida T, Iida A, Nabeshima Y, Anazawa H, Nishikawa S, Noda M, et al. Improvement of multiple pathophysiological phenotypes of klotho (kl/kl) mice by adenovirus-mediated expression of the klotho gene. The Journal of Gene Medicine. 2000;2(4):233–242. doi: 10.1002/1521-2254(200007/08)2:4<233::AID-JGM110>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Shroff R, Aitkenhead H, Costa N, Trivelli A, Litwin M, Picca S, et al. Normal 25-hydroxyvitamin D levels are associated with less proteinuria and attenuate renal failure progression in children with CKD. Journal of the American Society of Nephrology. 2016;27(1):314–322. doi: 10.1681/ASN.2014090947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto E, Taketani Y, Tanaka R, Harada N, Isshiki M, Sato M, et al. Dietary phosphorus acutely impairs endothelial function. Journal of the American Society of Nephrology. 2009;20(7):1504–1512. doi: 10.1681/ASN.2008101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silswal N, Touchberry CD, Daniel DR, McCarthy DL, Zhang S, Andresen J, et al. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. American Journal of Physiology. Endocrinology and Metabolism. 2014;307(5):E426–E436. doi: 10.1152/ajpendo.00264.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Naveh-Many T. FGF23 and the parathyroid glands. Pediatric Nephrology. 2010;25(11):2241–2245. doi: 10.1007/s00467-010-1565-3. [DOI] [PubMed] [Google Scholar]
- Silver J, Rodriguez M, Slatopolsky E. FGF23 and PTH—Double agents at the heart of CKD. Nephrology, Dialysis, Transplantation. 2012;27(5):1715–1720. doi: 10.1093/ndt/gfs050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha MD, Turner C, Booth CJ, Waller S, Rasmussen P, Goldsmith DJ, et al. Relationship of FGF23 to indexed left ventricular mass in children with non-dialysis stages of chronic kidney disease. Pediatric Nephrology. 2015;30(10):1843–1852. doi: 10.1007/s00467-015-3125-3. [DOI] [PubMed] [Google Scholar]
- Siomou E, Stefanidis CJ. FGF-23 in children with CKD: A new player in the development of CKD-mineral and bone disorder. Nephrology, Dialysis, Transplantation. 2012;27(12):4259–4262. doi: 10.1093/ndt/gfs315. [DOI] [PubMed] [Google Scholar]
- Sitia S, Tomasoni L, Atzeni F, Ambrosio G, Cordiano C, Catapano A, et al. From endothelial dysfunction to atherosclerosis. Autoimmunity Reviews. 2010;9(12):830–834. doi: 10.1016/j.autrev.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Six I, Okazaki H, Gross P, Cagnard J, Boudot C, Maizel J, et al. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PloS One. 2014;9(4):e93423. doi: 10.1371/journal.pone.0093423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatopolsky E, Bricker NS. The role of phosphorus restrict ion in the prevention of secondary hyperparathyroidism in chronic renal disease. Kidney International. 1973;4:141–146. doi: 10.1038/ki.1973.92. [DOI] [PubMed] [Google Scholar]
- Small DM, Bennett NC, Roy S, Gabrielli BG, Johnson DW, Gobe GC. Oxidative stress and cell senescence combine to cause maximal renal tubular epithelial cell dysfunction and loss in an in vitro model of kidney disease. Nephron. Experimental Nephrology. 2012;122(3–4):123–130. doi: 10.1159/000350726. [DOI] [PubMed] [Google Scholar]
- Smith RC, O’Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, et al. Circulating αKlotho influences phosphate handling by controlling FGF23 production. The Journal of Clinical Investigation. 2012;122(12):4710–4715. doi: 10.1172/JCI64986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JJ, Foley RN, Collins AJ. Prevalence of CKD in the United States: A sensitivity analysis using the National Health and Nutrition Examination Survey (NHANES) 1999–2004. American Journal of Kidney Diseases. 2009;53(2):218–228. doi: 10.1053/j.ajkd.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvinkel P, Larsson TE. Chronic kidney disease: A clinical model of premature aging. American Journal of Kidney Diseases. 2013;62(2):339–351. doi: 10.1053/j.ajkd.2012.11.051. [DOI] [PubMed] [Google Scholar]
- Sugiura H, Yoshida T, Shiohira S, Kohei J, Mitobe M, Kurosu H, et al. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. American Journal of Physiology. Renal Physiology. 2012;302(10):F1252–F1264. doi: 10.1152/ajprenal.00294.2011. [DOI] [PubMed] [Google Scholar]
- Sugiura H, Yoshida T, Tsuchiya K, Mitobe M, Nishimura S, Shirota S, et al. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrology, Dialysis, Transplantation. 2005;20(12):2636–2645. doi: 10.1093/ndt/gfi165. [DOI] [PubMed] [Google Scholar]
- Sun CY, Chang SC, Wu MS. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney International. 2012;81(7):640–650. doi: 10.1038/ki.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Cheng B, Sun PG, Wu XH, Wu QQ, He P. RTEF-1 protects against oxidative damage induced by H2O2 in human umbilical vein endothelial cells through Klotho activation. Experimental Biology and Medicine (Maywood, NJ) 2015;240(12):1606–1613. doi: 10.1177/1535370215587914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei S, Nami R, Bruno RM, Quatrini I, Nuti R. Hypertension, left ventricular hypertrophy and chronic kidney disease. Heart Failure Reviews. 2011;16(6):615–620. doi: 10.1007/s10741-010-9197-z. [DOI] [PubMed] [Google Scholar]
- Takeshita K, Fujimori T, Kurotaki Y, Honjo H, Tsujikawa H, Yasui K, et al. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109(14):1776–1782. doi: 10.1161/01.CIR.0000124224.48962.32. [DOI] [PubMed] [Google Scholar]
- Tang R, Zhou QL, Ao X, Peng WS, Veeraragoo P, Tang TF. Fosinopril and losartan regulate klotho gene and nicotinamide adenine dinucleotide phosphate oxidase expression in kidneys of spontaneously hypertensive rats. Kidney & Blood Pressure Research. 2011;34(5):350–357. doi: 10.1159/000326806. [DOI] [PubMed] [Google Scholar]
- Taniyama Y, Morishita R. Does therapeutic angiogenesis overcome CKD? Hypertension Research. 2010;33(2):114–115. doi: 10.1038/hr.2009.208. [DOI] [PubMed] [Google Scholar]
- Tohyama O, Imura A, Iwano A, Freund JN, Henrissat B, Fujimori T, et al. Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid beta-glucuronides. The Journal of Biological Chemistry. 2004;279(11):9777–9784. doi: 10.1074/jbc.M312392200. [DOI] [PubMed] [Google Scholar]
- Tsirpanlis G. Cellular senescence, cardiovascular risk, and CKD: A review of established and hypothetical interconnections. American Journal of Kidney Diseases. 2008;51(1):131–144. doi: 10.1053/j.ajkd.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Molecular Endocrinology. 2003;17(12):2393–2403. doi: 10.1210/me.2003-0048. [DOI] [PubMed] [Google Scholar]
- Tumur Z, Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. American Journal of Nephrology. 2009;29(6):551–557. doi: 10.1159/000191468. [DOI] [PubMed] [Google Scholar]
- Tumur Z, Shimizu H, Enomoto A, Miyazaki H, Niwa T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. American Journal of Nephrology. 2010;31(5):435–441. doi: 10.1159/000299798. [DOI] [PubMed] [Google Scholar]
- Tyralla K, Amann K. Morphology of the heart and arteries in renal failure. Kidney International. Supplement. 2003;84:S80–S83. doi: 10.1046/j.1523-1755.63.s84.1.x. [DOI] [PubMed] [Google Scholar]
- Utsugi T, Ohno T, Ohyama Y, Uchiyama T, Saito Y, Matsumura Y, et al. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism. 2000;49(9):1118–1123. doi: 10.1053/meta.2000.8606. [DOI] [PubMed] [Google Scholar]
- van Ballegooijen AJ, Rhee EP, Elmariah S, de Boer IH, Kestenbaum B. Renal clearance of mineral metabolism biomarkers. Journal of the American Society of Nephrology. 2016;27(2):392–397. doi: 10.1681/ASN.2014121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van TV, Watari E, Taketani Y, Kitamura T, Shiota A, Tanaka T, et al. Dietary phosphate restriction ameliorates endothelial dysfunction in adenine-induced kidney disease rats. Journal of Clinical Biochemistry and Nutrition. 2012;51(1):27–32. doi: 10.3164/jcbn.11-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. Journal of the American Society of Nephrology. 2015;26(8):1765–1776. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke F, Van Biesen W, Vanholder R. The role of collagen metabolism in CKD-associated arterial senescence: Underestimated and underappreciated. Nephrology, Dialysis, Transplantation. 2011;26(9):2726–2728. doi: 10.1093/ndt/gfr421. [DOI] [PubMed] [Google Scholar]
- Vervloet MG, Adema AY, Larsson TE, Massy ZA. The role of klotho on vascular calcification and endothelial function in chronic kidney disease. Seminars in Nephrology. 2014;34(6):578–585. doi: 10.1016/j.semnephrol.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Wan M, Smith C, Shah V, Gullet A, Wells D, Rees L, et al. Fibroblast growth factor 23 and soluble klotho in children with chronic kidney disease. Nephrology, Dialysis, Transplantation. 2013;28(1):153–161. doi: 10.1093/ndt/gfs411. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen L, Huang G, He D, He J, Xu W, et al. Klotho sensitizes human lung cancer cell line to cisplatin via PI3k/Akt pathway. PloS One. 2013;8(2):e57391. doi: 10.1371/journal.pone.0057391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54(4):810–817. doi: 10.1161/HYPERTENSIONAHA.109.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sun Z. Antiaging gene Klotho regulates endothelin-1 levels and endothelin receptor subtype B expression in kidneys of spontaneously hypertensive rats. Journal of Hypertension. 2014;32(8):1629–1636. doi: 10.1097/HJH.0000000000000233. [DOI] [PubMed] [Google Scholar]
- Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. Journal of Bone and Mineral Research. 2003;18(7):1227–1234. doi: 10.1359/jbmr.2003.18.7.1227. [DOI] [PubMed] [Google Scholar]
- Weissman IL. Stem cells: Units of development, units of regeneration, and units in evolution. Cell. 2000;100(1):157–168. doi: 10.1016/s0092-8674(00)81692-x. [DOI] [PubMed] [Google Scholar]
- Wesseling-Perry K. Defective skeletal mineralization in pediatric CKD. Current Osteoporosis Reports. 2015;13(2):98–105. doi: 10.1007/s11914-015-0253-4. [DOI] [PubMed] [Google Scholar]
- Westerweel PE, Hoefer IE, Blankestijn PJ, de Bree P, Groeneveld D, van Oostrom O, et al. End-stage renal disease causes an imbalance between endothelial and smooth muscle progenitor cells. American Journal of Physiology. Renal Physiology. 2007;292(4):F1132–F1140. doi: 10.1152/ajprenal.00163.2006. [DOI] [PubMed] [Google Scholar]
- Williams DA. Curing genetic disease with gene therapy. Transactions of the American Clinical and Climatological Association. 2014;125:122–128. discussion 8–9. [PMC free article] [PubMed] [Google Scholar]
- Wolf M. Forging forward with 10 burning questions on FGF23 in kidney disease. Journal of the American Society of Nephrology. 2010;21(9):1427–1435. doi: 10.1681/ASN.2009121293. [DOI] [PubMed] [Google Scholar]
- Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney International. 2012;82(7):737–747. doi: 10.1038/ki.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CB, Dong C, Stark M, Silverberg S, Rundek T, Elkind MS, et al. Plasma FGF23 and the risk of stroke: The Northern Manhattan Study (NOMAS) Neurology. 2014;82(19):1700–1706. doi: 10.1212/WNL.0000000000000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nature Communications. 2012;3:1238. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. Journal of the American Society of Nephrology. 2015;26(5):1150–1160. doi: 10.1681/ASN.2014040325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B, Zhou J, Yuan L, Ren F, Liu DC, Li Q, et al. Epigenetic silencing of Klotho expression correlates with poor prognosis of human hepatocellular carcinoma. Human Pathology. 2013;44(5):795–801. doi: 10.1016/j.humpath.2012.07.023. [DOI] [PubMed] [Google Scholar]
- Yamada S, Tatsumoto N, Tokumoto M, Noguchi H, Ooboshi H, Kitazono T, et al. Phosphate binders prevent phosphate-induced cellular senescence of vascular smooth muscle cells and vascular calcification in a modified, adenine-based uremic rat model. Calcified Tissue International. 2015;96(4):347–358. doi: 10.1007/s00223-014-9929-5. [DOI] [PubMed] [Google Scholar]
- Yamagishi T, Saito Y, Nakamura T, Takeda S, Kanai H, Sumino H, et al. Troglitazone improves endothelial function and augments renal klotho mRNA expression in Otsuka Long-Evans Tokushima Fatty (OLETF) rats with multiple atherogenic risk factors. Hypertension Research. 2001;24(6):705–709. doi: 10.1291/hypres.24.705. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: Age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochemical and Biophysical Research Communications. 2010;398(3):513–518. doi: 10.1016/j.bbrc.2010.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HC, Deleuze S, Zuo Y, Potthoff SA, Ma LJ, Fogo AB. The PPARgamma agonist pioglitazone ameliorates aging-related progressive renal injury. Journal of the American Society of Nephrology. 2009;20(11):2380–2388. doi: 10.1681/ASN.2008111138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Fogo AB. Cell senescence in the aging kidney. Journal of the American Society of Nephrology. 2010;21(9):1436–1439. doi: 10.1681/ASN.2010020205. [DOI] [PubMed] [Google Scholar]
- Yang K, Nie L, Huang Y, Zhang J, Xiao T, Guan X, et al. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicology Letters. 2012;215(2):77–83. doi: 10.1016/j.toxlet.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Yang K, Wang C, Nie L, Zhao X, Gu J, Guan X, et al. Klotho protects against indoxyl sulphate-induced myocardial hypertrophy. Journal of the American Society of Nephrology. 2015;26(10):2434–2446. doi: 10.1681/ASN.2014060543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circulation Research. 2013;113(5):495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz MI, Saglam M, Caglar K, Cakir E, Sonmez A, Ozgurtas T, et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. American Journal of Kidney Diseases. 2006;47(1):42–50. doi: 10.1053/j.ajkd.2005.09.029. [DOI] [PubMed] [Google Scholar]
- Yoon HE, Ghee JY, Piao S, Song JH, Han DH, Kim S, et al. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrology, Dialysis, Transplantation. 2011;26(3):800–813. doi: 10.1093/ndt/gfq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HE, Lim SW, Piao SG, Song JH, Kim J, Yang CW. Statin upregulates the expression of klotho, an anti-aging gene, in experimental cyclosporine nephropathy. Nephron Experimental Nephrology. 2012;120(4):e123–e133. doi: 10.1159/000342117. [DOI] [PubMed] [Google Scholar]
- Young GH, Wu VC. KLOTHO methylation is linked to uremic toxins and chronic kidney disease. Kidney International. 2012;81(7):611–612. doi: 10.1038/ki.2011.461. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Duffield JS. Resolved: EMT produces fibroblasts in the kidney. Journal of the American Society of Nephrology. 2010;21(8):1247–1253. doi: 10.1681/ASN.2010060616. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature Medicine. 2003;9(7):964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li Y, Fan Y, Wu J, Zhao B, Guan Y, et al. Klotho is a target gene of PPAR-gamma. Kidney International. 2008;74(6):732–739. doi: 10.1038/ki.2008.244. [DOI] [PubMed] [Google Scholar]
- Zhang M, Yan J, Zhu M, Ni Z. Fibroblast growth factor 23 predicts coronary calcification and poor prognosis in patients with chronic kidney disease stages 3–5D. Annals of Clinical and Laboratory Science. 2015;45(1):17–22. [PubMed] [Google Scholar]
- Zhang LN, Yang G, Cheng C, Shen C, Cui YY, Zhang J, et al. Plasma FGF23 levels and heart rate variability in patients with stage 5 CKD. Osteoporosis International. 2015;26(1):395–405. doi: 10.1007/s00198-014-2862-7. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang Y, Tang Y, Zhao Y, Cao Y, Su B, et al. Recovery from AKI following multiple wasp stings: A case series. Clinical Journal of the American Society of Nephrology. 2013;8(11):1850–1856. doi: 10.2215/CJN.12081112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zheng F. PPAR-gamma and aging: One link through klotho? Kidney International. 2008;74(6):702–704. doi: 10.1038/ki.2008.382. [DOI] [PubMed] [Google Scholar]
- Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. Journal of the American Society of Nephrology. 2013;24(5):771–785. doi: 10.1681/ASN.2012080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lin S, Tang R, Veeraragoo P, Peng W, Wu R. Role of fosinopril and valsartan on Klotho gene expression induced by angiotensin II in rat renal tubular epithelial cells. Kidney & Blood Pressure Research. 2010;33(3):186–192. doi: 10.1159/000316703. [DOI] [PubMed] [Google Scholar]