

SUMMARY
Localized extracellular matrix (ECM) remodeling is thought to stabilize the cleavage furrow and maintain cell shape during cytokinesis [1–14]. This remodeling is spatiotemporally coordinated with a cytoskeletal structure pertaining to a kingdom of life, for example, the FtsZ ring in bacteria [15], the phragmoplast in plants [16], and the actomyosin ring in fungi and animals [17, 18]. While the cytoskeletal structures have been analyzed extensively, the mechanisms of ECM remodeling remain poorly understood. In the budding yeast Saccharomyces cerevisiae, the ECM remodeling refers to sequential formations of the primary and secondary septa that are catalyzed by chitin synthase-II (Chs2) and chitin synthase-III (the catalytic subunit Chs3 and its activator Chs4), respectively [18, 19]. Surprisingly, both Chs2 and Chs3 are delivered to the division site at the onset of cytokinesis [6, 20]. What keeps Chs3 inactive until secondary septum formation remains unknown. Here, we show that Hof1 binds to the Sel1-like repeats (SLR) of Chs4 via its F-BAR domain, and inhibits Chs3-mediated chitin synthesis during cytokinesis. In addition, Hof1 is required for rapid accumulation as well as efficient removal of Chs4 at the division site. This study uncovers a mechanism by which Hof1 controls timely activation of Chs3 during cytokinesis, and defines a novel interaction and function for the conserved F-BAR domain and SLR that are otherwise known for their abilities to bind membrane lipids [21, 22] and scaffold protein complex formation [23].
Keywords: cytokinesis, F-BAR, Sel1-like repeats (SLR), Hof1, Chs4, Chs3, Chs2, chitin, septum formation, budding yeast
eTOC Blurb
ECM remodeling during cytokinesis occurs in a spatiotemporally controlled manner. Oh et al. report that the F-BAR protein Hof1 interacts with the SLR protein Chs4 to ensure that secondary septum formation occurs after actomyosin ring constriction and primary septum formation during cytokinesis in budding yeast.
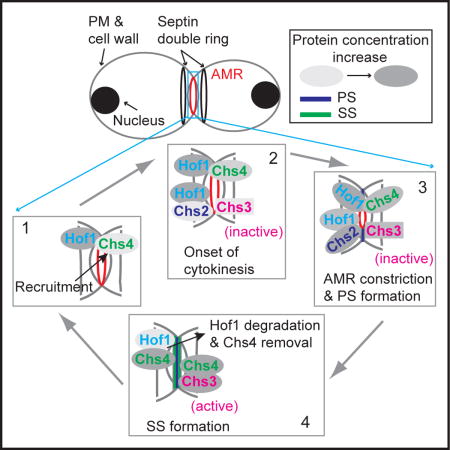
RESULTS AND DISCUSSION
Hof1 binds directly to the SLR of Chs4 via its F-BAR domain
Chs3, the catalytic subunit of chitin synthase-III (CSIII), and its activator Chs4 first localize to the incipient bud site and start catalyzing the formation of a chitin ring at the mother side of the bud neck [1, 24–26]. Both Chs3 and Chs4 disappear from the bud neck around G2/M and then re-localize to the neck during cytokinesis to drive secondary septum (SS) formation [20, 27]. Chs3 and Chs4 are delivered to and removed from the division site independently of each other [25]. However, it remains unclear how Chs4 regulates Chs3 activity at the plasma membrane (PM) and how their localizations at the division site are controlled.
The disappearance of Chs3 from the bud neck during G2/M coincided with the arrival of the F-BAR protein Hof1 at the same location (Figures S1A and S1B; Movie S1A). Hof1, a conserved cytokinetic protein [28, 29], is one of the very few proteins in S. cerevisiae (less than 10 out of ~6000 in total) that display asymmetric localization to the mother side of the bud neck before cytokinesis [30]. Both Chs3 and Hof1 also localized to the division site during cytokinesis and cell separation, and the surge and fall of Hof1 at the division site preceded that of Chs3 (Figures S1C and S1D; Movies S1B and S1C for a side view and an en-face view, respectively). While Hof1 is involved in coupling actomyosin ring (AMR) to primary septum (PS) formation during cytokinesis [29, 31], its role in G2/M remained unknown. These observations prompted us to consider the possibility that Hof1 might regulate the localization and/or activity of Chs3 and Chs4 during G2/M as well as during cytokinesis and cell separation. This hypothesis predicts that: 1) Hof1 might interact with Chs3 and/or Chs4, 2) Hof1 might control Chs3 activity, and 3) Hof1 might control the localization dynamics of Chs3 and/or Chs4 at the bud neck. To test these predictions, we first examined for possible interactions between Hof1 and Chs3, Chs4 as well as the “adaptor” protein Bni4, which links Chs4 to the septin hourglass at the bud neck before cytokinesis [24]. We found by two-hybrid analysis that Hof1 interacted with Chs4-C693S (the Cysteine in the prenylation motif, CVIM, was mutated to allow the assessment of protein-protein interactions in the nucleus by the two-hybrid system) and Chs4 (1–610), which lacks the prenylation site, but not with the wild-type (WT) protein (Figures 1A and 1B). In addition, Hof1 failed to interact with Chs3, Chs3 (1–700), and Bni4 (65–730; also known as Bni4E). These two-hybrid constructs have been used to discover the interactions between Chs3, Chs4, and Bni4 before [24]. Further analysis indicated that the N-terminal half of Hof1 (Hof1-N-term; 1–340), which consists of the F-BAR domain (1–275) and the CC2 region (300–333) [31, 32], was sufficient for interaction with Chs4 (Figures 1A and 1B).
Figure 1. Hof1 interacts with Chs4 in vitro and in vivo.
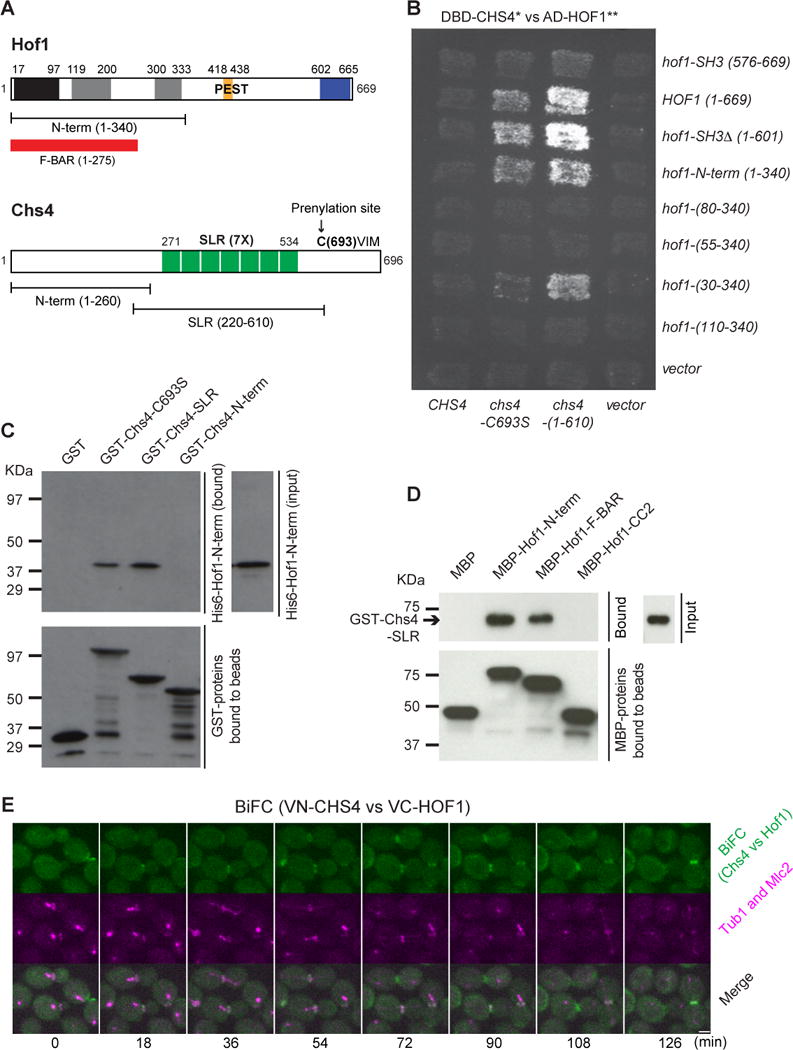
(A) Domains and motifs of Hof1 and Chs4. PCH, Pombe Cdc15 homology; CC1 and CC2, coiled-coil regions 1 and 2; PEST, proline-glutamic acid-serine-threonine-rich sequence that acts as a signal for protein degradation; SH3, Src homology 3 domain; SLR, Sel1-like repeats. See also Figure S1 and Movie S1.
(B) Hof1-N-term interacts with Chs4. Full-length and various truncation alleles of HOF1 were examined pair-wise for interactions with different alleles of CHS4 by two-hybrid analysis.
(C) Hof1-N-term binds directly to the SLR of Chs4 in vitro. Purified GST-Chs4 fragments, including the full-length Chs4-C693S, SLR (220–610), and Chs4-N-terrm (1–260) were examined for interactions with His6-Hof1-N-term in vitro.
(D) Hof1-F-BAR binds directly to the SLR of Chs4 in vitro. Purified MBP-Hof1 fragments (Hof1-N-term, Hof1-F-BAR, and Hof1-CC2) were examined for interactions with GST-Chs4-SLR in vitro.
(E) Hof1 and Chs4 interact at the bud neck strongly during cytokinesis. Strain YEF7980 (VN-CHS4, VC-HOF1, mRuby2-TUB1, MLC2-mApple) (See also Table S1) was grown to exponential phase in SC medium at 25°C and the BiFC signal was then analyzed during the cell cycle by spinning-disk microscopy (See also Movie S2). Scale bar = 2 μm.
Next, we demonstrated by in vitro binding assay that Hof1-N-term bound directly to the evolutionarily conserved Sel1-like repeats (Chs4-SLR; 220–610) of Chs4, but not its N-terminal fragment (Chs4-N-term; 1–260) (Figures 1A and 1C). Furthermore, we found that the F-BAR domain, not the CC2 region, of Hof1-N-term interacted directly with Chs4-SLR (Figure 1D). Hof1-F-BAR is known to form dimers and bind non-selectively to a number of phospholipids in vitro with no apparent tubulation activity in vivo [33].
We then employed the bimolecular complementation assay (BiFC) [34] to test whether Hof1 interacts with Chs4 in vivo, and if so, when and where the interaction occurs during the cell cycle. For this purpose, the N- and C-terminal fragments of an YFP (Venus) molecule were attached to the N-termini of Chs4 and Hof1, respectively, in two haploid yeast strains of opposite mating types. Time-lapse analysis of the resulting diploid cells showed that the interaction between Hof1 and Chs4 started at the bud neck in cells with a medium-sized bud (Figure 1E, cell 1, 18 min; Movie S2; approximately in G2/M as indicated by the mRuby2-Tub1-labeled short spindle near the bud neck in the mother compartment [35]) and became stronger shortly before and during cytokinesis (Cell 1, 72–90 min; as indicated by the elongated spindle and the Mlc2-mApple-labeled constricting AMR [36]), and then at both mother and daughter sides of the bud neck during cell separation (Cell 1, 108 min; as indicated by the disappearance of the AMR). Thus, Hof1 and Chs4 interact in vivo in a spatiotemporally controlled fashion that is consistent with their localization profiles during the cell cycle.
Together, these data indicate that Hof1 and Chs4 bind to each other directly through conserved domains, and also demonstrate that the F-BAR domain is not only a lipid-binding module, but also capable of interacting with other proteins. This analysis has also led to the identification of a novel binding partner for the SLR of Chs4.
Deletion of HOF1 causes an increase in Chs3-dependent chitin synthesis at the division site
The Hof1-Chs4 interaction compelled us to examine the role of Hof1 in Chs3-mediated chitin synthesis. Yeast mutants requiring a higher level of chitin for survival are known to be sensitive to Calcofluor White (CW), a fluorescent dye that binds specifically to chitin in fungal cell walls [37]. We found that WT and bni4Δ cells grew well on YPD plates containing 25 μg/ml CW (Figure 2A). As expected, chs3Δ and chs4Δ cells, which are deficient in chitin synthesis [24, 37], were resistant to CW. In contrast, deletion of CYK3, which encodes a protein involved in PS formation during cytokinesis [38, 39], is known to cause an increase in Chs3-mediated chitin synthesis [37]. Not surprisingly, cyk3Δ cells failed to grow on the CW plate. Similarly, hof1Δ cells also failed to grow on the same CW plate, suggesting that chitin might be increased in hof1Δ cells. Because deletion of HOF1 causes temperature-sensitive defects in growth and cytokinesis [28, 29], we measured the chitin content in WT and different mutant strains at both the permissive (25°) and non-permissive (37°C) temperatures by a quantitative colorimetric assay [37] (Figure 2B). Consistent with the plate assay, the chitin level was significantly increased in hof1Δ cells in comparison to WT cells at both 25°C (p = 0.00098) and 37°C (p = 0.02163).
Figure 2. Chs3-dependent chitin synthesis at the bud neck is increased in hof1Δ cells.
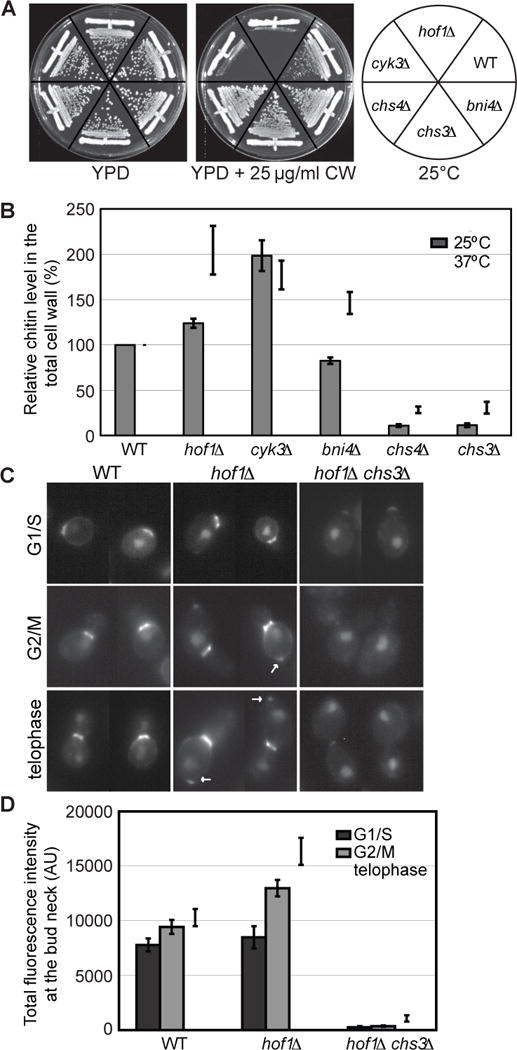
(A) hof1Δ cells are sensitive to Calcofluor White (CW). Strains YEF473A (wild-type, WT), YEF4600 (hof1Δ), YEF2368 (cyk3Δ), YEF4559 (chs3Δ), YEF2197 (chs4Δ), and YEF2769 (bni4Δ) (See also Table S1) were streaked out on an YPD plate and an YPD plate containing 25 μg/ml CW and incubated at 25°C for 3 days before documentation.
(B) Chitin level in the cell wall of hof1Δ cells is increased. Chitin levels in the total cell walls of the strains listed in (A), which were grown in YM-1 medium at 25°C or 37°C, were measured as described in STAR METHODS. Data are averaged from 9 and 4 independent experiments for the 25°C and 37°C samples, respectively. Error bars represent SEM (standard error of the mean).
(C–D) Chitin level at the bud neck of hof1Δ cells is increased from G2/M to telophase in a Chs3-dependent manner. Chitin at the bud neck of WT (YEF473A), hof1Δ (YEF4600), and hof1Δ chs3Δ (YEF2757) (See also Table S1) cells was visualized by CW staining (C) and then quantified as described in STAR METHODS (D). Cell cycle stages were estimated based on nuclear staining by DAPI. The number of cells used for quantification was: WT, n=34, 26, and 27; hof1Δ, n=12, 24, and 28; hof1Δ chs3Δ, n=10, 17, and 13 for G1/S, G2/M, and telophase, respectively. Error bars represent SEM (standard error of the mean).
To determine when and where Hof1 acts to regulate chitin synthesis during the cell cycle, we stained the WT and hof1Δ cells with CW and measured their relative chitin levels at the bud neck during different phases of the cell cycle (Figures 2C and 2D). Nuclear position in the cell (as indicated by DNA staining) as well as bud size were used to gauge cell cycle progression (Figure 2C). We found that in G1/S cells (DNA in the mother compartment, but not near the bud neck), the chitin level was similar in both WT and hof1Δ cells (p = 0.42225) (Figure 2D). However, in G2/M (DNA in the mother compartment, and close to the bud neck), hof1Δ cells had significantly more chitin at the bud neck than WT cells did (p = 0.00017). The difference was even more striking in telophase cells (p = 3.3401E-07) (DNA in both the mother and daughter compartments). Thus, deletion of HOF1 causes an increase in chitin synthesis at the bud neck during G2/M as well as during cytokinesis.
A tiny patch of chitin was frequently observed at the distal pole of the mother compartment in hof1Δ cells (Figure 2C, arrows). The underlying mechanism remains unknown. Nonetheless, this observation suggests that Hof1 plays a fine-tuning role in restricting chitin synthase activity to the bud neck under normal growth conditions. We also found that deletion of CHS3 abolished all the ectopic chitin at the mother pole and nearly all the chitin at the bud neck in hof1Δ cells (Figures 2C and 2D), except a faint chitin ring in some telophase cells (Figure 2C, arrowhead), which presumably represents the PS that is catalyzed by the chitin synthase-II (CSII) Chs2 during cytokinesis. Together, these data suggest that Hof1 inhibits Chs3-mediated chitin synthesis during the cell cycle, especially, during cytokinesis.
The F-BAR domain of Hof1 competes with the catalytic region of Chs3 for binding to the SLR of Chs4
Previous two-hybrid analysis suggests that Chs4-SLR interacts with Chs3, and this interaction is critical for Chs3 activity [40]. Based on this observation and our findings described above, we hypothesize that direct binding of Hof1-F-BAR to Chs4-SLR prevents Chs3-Chs4 interaction, thus, inhibiting Chs3-dependent chitin synthesis at the bud neck during G2/M and cytokinesis. To test this possibility, we first examined whether the catalytic region of Chs3 binds directly to Chs4-SLR using in vitro binding assays with recombinant proteins purified from E. coli. The middle region of Chs3 (Chs3-MID, 477–1028) is predicted to be cytosolic and flanked by transmembrane domains. This region is known to contain residues (991–999) that are essential for Chs3 activity [41]. We found that His6-Chs3-MID bound directly to MBP-Chs4-SLR (Figure 3A). As expected, His6-Hof1-N-term also bound to MBP-Chs4-SLR (Figure 3A). Due to the toxicity caused by the expression of His6-Hof1-F-BAR in E. coli, this fragment could not be obtained for the binding assay. However, with different affinity tags, we already showed that both Hof1-N-term and Hof1-F-BAR bound well to Chs4-SLR whereas Hof1-CC2 did not (Figure 1D). Thus, both the F-BAR domain of Hof1 and the catalytic region of Chs3 interact directly with the SLR of Chs4.
Figure 3. Hof1-F-BAR is a competitive inhibitor of Chs3-Chs4 interaction.
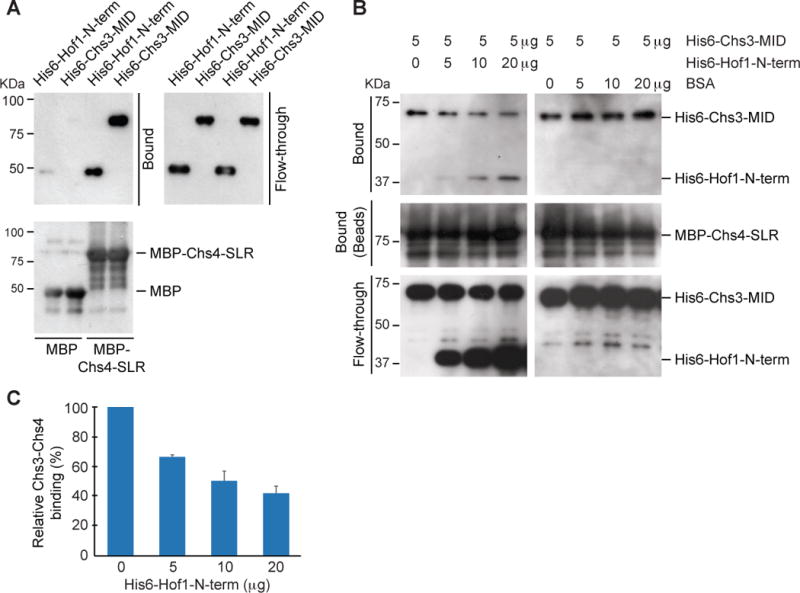
(A) The catalytic region of Chs3 binds directly to the SLR of Chs4. Purified MBP-Chs4-SLR, along with the control MBP, was tested for interactions with His6-Hof1-N-term and His6-Chs3-MID in an in vitro binding assay as described in STAR METHODS.
(B) Hof1-F-BAR inhibits the interaction between Chs3-MID and Chs4-SLR. MBP-Chs4-SLR bound to amylase beads was incubated with 5 μg of His6-Chs3-MID and different amounts of His6-Hof1-N-term or BSA as indicated above. Proteins associated with the beads (Bound) or in the supernatant (flow-through) were then probed by Western-blot analysis using appropriate antibodies.
(C) Quantification of the binding data in (B). The relative Chs3-Chs4 binding is calculated as the Chs3 band intensity subtracted by background in the Western blot at a given concentration of Hof1-N-term divided by the background-subtracted Chs3 band intensity in the absence of Hof1-N-term. The average and the standard deviation were calculated from two independent experiments.
Next, we examined whether Hof1-N-term could compete with Chs3 for binding to Chs4. As the amount of His6-Hof1-N-term in the reactions increased, the binding of His6-Chs3-MID to MBP-Chs4-SLR decreased (Figures 3B and 3C). Together, these data support our hypothesis that Hof1 inhibits Chs3-dependent chitin synthesis at the bud neck by binding to the SLR of Chs4 via its F-BAR domain.
Hof1 governs the localization dynamics of Chs4 and Chs3 during cytokinesis
To determine when and where the Hof1-Chs4 regulatory mechanism functions during the cell cycle, we imaged WT and hof1Δ cells carrying Chs4-GFP and Spc42-mCherry (RFP) by time-lapse microscopy. Spc42 is a component of the spindle pole body (equivalent of centrosome in animal cells) [42] that serves as a cell-cycle marker here. We found that the retention of Chs4 at the bud neck was increased in hof1Δ cells, mildly during G2/M (p = 0.0440) and dramatically during cytokinesis (p < 0.0001) (Figures 4A and S2; Movie S3). Interestingly, Chs4 accumulation at the bud neck appeared to be biphasic in WT cells, with a rapid phase followed by a slow phase (Figure 4B). Strikingly, only the rapid phase depended on Hof1 (Figure 4B). The peak accumulation of Chs4 at the bud neck was followed by its quick removal in WT cells, and this process was also attenuated in hof1Δ cells (Figures 4A and 4B). Thus, Hof1 is required for the rapid accumulation and removal of Chs4 at the division site during cytokinesis.
Figure 4. Hof1 governs the localization dynamics of Chs4 and Chs3 during cytokinesis.
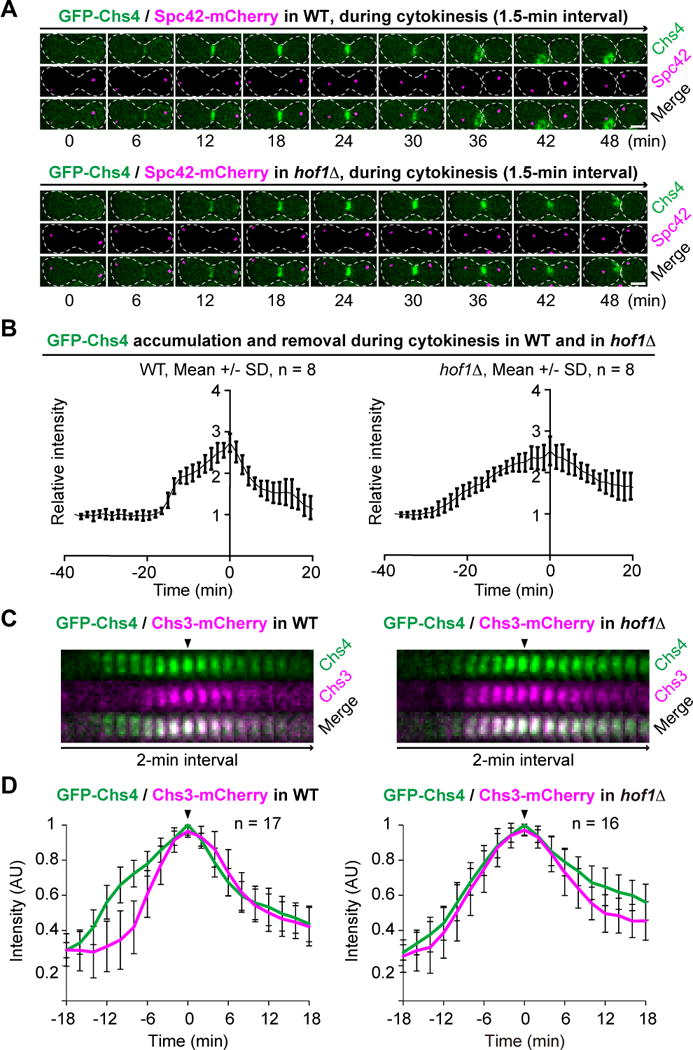
(A) The duration of Chs4 at the bud neck is lengthened in hof1Δ cells. Cells of WT (YEF5678) and hof1Δ (YEF5694) strains carrying Chs4-GFP and Spc42-mCherry (See also Table S1) were grown in SC-His media at 25°C and then imaged by spinning-disk microscopy (See also Movie S3). Scale bar = 2 μm.
(B) Hof1 is required for the rapid accumulation and removal of Chs4 at the bud neck. Imaging data from (A) were analyzed for the localization kinetics. See also Figure S2.
(C) Chs3 precociously localizes to the bud neck in hof1Δ cells during cytokinesis. Cells of WT (YEF7965) and hof1Δ (YEF7966) strains carrying Chs4-GFP and Chs3-mCherry (See also Table S1) were grown in SC-His media at 25°C and then imaged by spinning-disk microscopy to determine their relative timing of localization during cytokinesis (See also Movie S4A) and subsequent budding event (Movie S4B). See also Figure S3.
(D) Localization kinetics of Chs4 and Chs3 during cytokinesis. Imaging data from (C) were analyzed to generate the plots.
The Hof1-Chs4 regulatory mechanism most likely acts to ensure that SS formation occurs after PS formation. To explore this possibility, we first compared the localization kinetics of Chs2 and Chs3 in WT cells. Consistent with the hypothesis, Chs2 localization to the bud neck peaked ~ 4 min before that of Chs3 (Figure S3). Surprisingly, both proteins were delivered to the bud neck simultaneously at the onset of cytokinesis (Figure S3). What keeps Chs3 inactive until SS formation was unclear.
To determine whether and how Hof1-Chs4 interaction controls the timely activation of Chs3 during cytokinesis, we followed Chs4-GFP and Chs3-mCherry localization in WT and hof1Δ cells by time-lapse microscopy. As expected, Chs4 localized to the bud neck via a biphasic mechanism in WT cells, and peaked at ~18 min after its initial arrival; in contrast, Chs3 began to arrive at the bud neck ~6 min after Chs4, but peaked at the exactly the same time as Chs4 did (Figures 4C and 4D, arrowhead; Movie S4A). This temporal separation between Chs4 and Chs3 occurred only during cytokinesis, as both proteins localized simultaneously to the incipient bud site and the mother side of the bud neck during bud emergence and bud growth (Movie S4B). Remarkably, Chs3 and Chs4 displayed nearly identical kinetics of accumulation, but differed in the kinetics of removal in hof1Δ cells (Figures 4C and 4D, arrowhead; Movie S4A). Importantly, the absence of Hof1 did not delay the initial localization of Chs4, but changed its kinetics of accumulation, and also caused precocious localization of Chs3 to the bud neck. Consistent with a role in Chs4 accumulation during cytokinesis, localization of Hof1 to the bud neck preceded that of Chs4 (Figures S1C, S1D, and 4D; Chs3 served as the reference point for determining the relative timing of Hof1 and Chs4 localizations). Consistent with a role in the relief of Chs4 inhibition, Hof1 level began to drop before Chs4 and Chs3 reached their peak at the bud neck (Figures S1D and 4D). Strikingly, at ~ 4 min after its initial drop, Hof1 re-surged to form a “small peak” at ~ 2 min after the peaking of Chs4 and Chs3 at the bud neck (Figures S1D and 4D). This small peak of Hof1 might be involved in the endocytic removal of Chs4. Taken together, these data suggest that Hof1 is required for preventing precocious interaction between Chs4 and Chs3 or untimely activation of Chs3 at the PM during cytokinesis as well as efficient removal of Chs4 after cytokinesis.
ECM remodeling at the division site is an important problem that has been underappreciated in the field of cytokinesis. In this study, we have defined a molecular mechanism that ensures timely execution of specific ECM-remodeling events during cytokinesis. Specifically, our findings provide an answer to the question of how CSIII is kept inactive until SS formation in WT cells. The delivery of both Chs2 and Chs3 to the bud neck occurs simultaneously at the onset of cytokinesis, although they peak at different times. What keeps Chs3 inactive during PS formation was unknown. Hof1 associates with the AMR at the onset of cytokinesis via a C-terminal region [31, 43] and couples AMR constriction to PS formation during cytokinesis [31]. Here, we show that Hof1 interacts with the SLR of Chs4 via its N-terminal F-BAR domain. This interaction prevents Chs3 from binding to Chs4, which explains the delayed accumulation and activation of Chs3 at the bud neck, as Chs4 is required for stabilized localization of Chs3 at the PM [25]. After AMR constriction and PS formation, Hof1 is progressively degraded, which is mediated by its PEST sequence and the F-box protein Grr1 [29, 44]. This degradation leads to the relief of Chs4 from Hof1 inhibition. The relieved Chs4 associates with the PM via its lipid-modified tail [24, 25], stabilizes and activates Chs3 at the PM to promote SS formation. Thus, the Hof1-Chs4 interaction controls timely recruitment and activation of Chs3 at the bud neck during cytokinesis. This conclusion predicts that deletion of HOF1 would cause precocious as well as enhanced SS formation, which was indeed observed in hof1Δ cells by electron microscopy [43]. The precocious SS deposition might contribute to a failure to effectively cleave the PS, leading to a delay in cell separation. Besides the role for rapid accumulation and inhibition of Chs4 at the bud neck, Hof1 is also required for efficient removal of Chs4 after cytokinesis. Hof1 could target Chs4 for ubiquitylation-dependent proteolysis independently of Chs3, which presumably ensures that Chs3 is kept inactive once it leaves the PM after cytokinesis. This endocytic role of Hof1 is likely mediated by an interaction between its C-terminal SH3 domain and verprolin (Vrp1) [45]. Thus, Hof1 controls Chs3 activity via two sequential mechanisms, first controlling Chs4 accumulation during cytokinesis, and then controlling its endocytic removal after cytokinesis.
This study also defines a novel interaction between and function of a F-BAR domain and SLR. All F-BAR domains form crescent dimers that bind to negatively charged phospholipids. Unlike Hof1 in budding yeast or Cdc15 in the fission yeast S. pombe [33, 46], many F-BAR domain-containing proteins can generate membrane curvatures by forming helical filaments via lateral as well as end-to-end associations [21]. The F-BAR proteins usually link cellular membranes to the actin cytoskeleton and/or cellular signaling via other domains such as the SH3 and GTPase-activating domains (GAPs) for Rho GTPases and function in a number of cellular processes such as endocytosis, cell migration, and cytokinesis [22, 46]. Very few protein partners of F-BAR domains have been identified and characterized, with the exceptions that the F-BAR of Cdc15 in fission yeast interacts with the formin Cdc12 to fine-tune AMR assembly [47], and that the F-BAR of PSTPIP1 in mammalian cells interacts with pyrin to regulate inflammatory responses [48]. SLR proteins generally act as adaptors for macromolecular complex formation in controlling various cellular responses [23]. For example, Hrd3 in budding yeast and its counterparts in C. elegans (Sel1) and mammalian cells (Sel1 and Sel1L) are involved in ER-associated protein degradation [23]. The transmembrane SLR protein PodJ in the bacterium Caulobacter crescentus binds to cell wall component via its C-terminus and scaffolds protein complex formation to affect organelle development at the flagellar pole of the swarmer cell [23]. Here, we show that the F-BAR of Hof1 binds directly to the SLR of Chs4 to regulate chitin synthesis during cytokinesis, which reveals a novel interaction and function for two highly conserved protein domains.
STAR METHODS
KEY RESOURECES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erfei Bi (ebi@mail.med.upenn.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The budding yeast Saccharomyces cerevisiae strains used in this study are listed in Table S1. All strains except those (Y860 and Y1026) used for two-hybrid analysis are isogenic to the wild-type YEF473 [49].
METHOD DETAILS
Yeast media and culture conditions
Standard culture media were used [50]. The YPD medium consists of 1% yeast extract (Becton, Dickson, and Co., Sparks, MD), 2% Peptone (Becton, Dickson, and Co.), and 2% dextrose (Fisher Scientific, Fair Lawn, NJ). The synthetic complete (SC) medium consists of 0.67% yeast nitrogen base without amino acids (Becton, Dickson, and Co.), 0.0086% of each of the 20 amino acids except 0.0171% leucine (Fisher Scientific), 0.0021% adenine (Fisher Scientific), 0.0009% 4-aminobenzoic acid (Fisher Scientific), 0.0086% uracil (Sigma, St. Louis, MO), 0.0086% myo-inositol (Sigma), and 2% dextrose (Fisher Scientific). The SC-Dropout medium is the same as SC, except that specific amino acid(s) were omitted. The YM-1, a buffered rich liquid medium [51], consists of 0.5% yeast extract (Becton, Dickson, and Co.), 1% Peptone (Becton, Dickson, and Co.), 0.7% yeast nitrogen base without amino acids (Becton, Dickson, and Co.), 0.6% sodium hydroxide (Fisher Scientific), 1% succinic acid (Fisher Scientific), and 2% dextrose (Fisher Scientific). All yeast strains were grown at 25°C, unless otherwise indicated.
Constructions of plasmids and strains
The parent vectors for two-hybrid analyses were the DNA-binding-domain (DBD) plasmid pEG202 (2μ, HIS3) and the activation-domain (AD) plasmid pJG4-5 (2μ, TRP1) [52]. The DBD-fusion plasmids pEG202-CHS4, pEG202-Chs4-C693S, pEG202-Chs4-(1–610), and pEG202-Chs3-(1–700) and the AD-fusion plasmids pJG4-5-Chs3-(1–700) and pJG4-5-Bni4pE-(65–730) were described previously [24] (supplied by John Pringle at Stanford University, Stanford, CA). The chs4-C693S allele has cysteine to serine change in the CAAX box in the C-terminus and the chs4-(1–610) allele encodes amino acids 1–610 and is missing the CAAX box. The AD-HOF1* plasmids, except pJG4-5-HOF1-SH3(576–669) (supplied by Charlie Boone at the University of Toronto, Toronto, Canada), were constructed by PCR-amplifying full-length and different fragments of HOF1 and then gap-repairing into NcoI-digested pJG4-5 plasmid. All these AD-HOF* constructs were confirmed by sequencing. Plasmids for in vitro protein-interaction assays were constructed as follows. BamHI-XhoI-digested DNA fragments encoding chs4-C693S, chs4-SLR (220–610), and chs4-N-term (1–260) were subcloned from pEG202 plasmids into the corresponding sites of pGEX-5X-1 (GE Healthcare Life Sciences, Buckinghamshire, UK) to generate plasmids expressing GST-fusion proteins. DNA fragments encoding HOF1-N-term or Chs3-MID (477–1028) were PCR-amplified, digested with BamHI and SalI (sites included in the primers), and then cloned into BamHI/SalI-digested pCOLA-Duet-1 (EMD Biosciences, Darmstadt, Germany) to generate plasmids encoding His6-tagged proteins. Plasmids pMAL-C2-Hof1-N-term (1–340), -Hof1-F-BAR (1–275), and -Hof1-CC2 (276–340) expressing MBP-fusions were described previously [31]. Plasmid pMAL-C2-chs4-SLR (220–610) was constructed by cloning a BamHI and SalI (both sites introduced in PCR primers)–digested CHS4 fragment into the corresponding sites of pMAL-C2 (New England BioLabs, Ipswich, MA). The PCR condition for a 100 μl reaction is: 2 μl PfuUltra-II fusion HS DNA polymerase (Agilent Technologies, Santa Clara, CA), 200 ng genomic DNA from wild-type yeast strain YEF473A (see Table S1), 0.2 μM for each of the forward and reverse primers (see Table S2), PfuUltra-II Hotstart PCR Master Mix (0.25 mM for each dNTP, 2 mM MgCl2) (Agilent Technologies) and distilled water (to fill to 100 μl), 30 cycles of 94°C for 30 sec, 54°C for 30 sec, and 72°C for 15 sec per kb fragment. Plasmid pRS306-GFP-Chs4Δ610–696 (pJL68) (integrative, URA3) was kindly supplied by Dr. Kelly Tatchell (Louisiana State University Health Science Center. Shreveport, LA) [26]. This plasmid was digested with NheI and then integrated at the CHS4 locus to generate the yeast strains carrying GFP-CHS4. Plasmid bWL737 containing mRuby2-TUB1 (kindly supplied by Dr. Wei-Lih Lee at the University of Massachusetts, Amherst, MA) [35] was digested with BsaBI and integrated at the TUB1 locus to yield the strain YEF7980 for BiFC imaging (see below). The strains carrying C-terminal tagging of Hof1, Mlc2, Chs2, and Chs3 with GFP or RFP (mApple or mCherry) were constructed following the standard PCR-based approach [53] [54]. The PCR condition for a 50 μl reaction is: 1 μl Universe High-Fidelity Hot Start DNA Polymerase (Biotool, Houston, TX), 0.20 mM for each dNTP, 2 mM MgCl2, 100 ng tagging vector DNA [53] [54], 0.4 μM for each of the defined forward and reverse primers [53, 54], and distilled water (to fill to 50 μl), 95°C for 30 sec for initial denaturation, 30 cycles of 95°C for 15 sec, 55°C for 15 sec, and 72°C for 90 sec, followed by 72°C for 5 min for the final extension.
Two-hybrid interactions
Strain Y1026 carrying various DBD plasmids was mated to strain Y860 carrying various AD plasmids. Diploids were selected on SC-His-Trp plates, replica-plated to SC-His-Trp-Ade plates containing 1% raffinose plus 2% galactose (to induce production of the fusion proteins), and incubated at 30°C for 4 days to detect interactions.
In vitro protein-binding assays
To purify His6-tagged proteins [31], E. coli strain BL21 (Invitrogen, Carlsbad, CA) was transformed with pCOLA-Duet-1-based plasmids (see above), grown to exponential phase at 37°C for 4 h, and induced with 1 mM IPTG for 3 h at 23°C. Cells were centrifuged and resuspened with Ni-NTA lysis buffer (300 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl (pH 8.0), 20 mM imidazole, 10 mM β-mercaptoethanol, 0.1% NP-40) containing a cocktail of protease inhibitors (Sigma). Cells were then sonicated six times with 40 amplitude (QSonica Q55, Newtown, CT) for 15 sec with 1-min interval on ice. The protein extracts were centrifuged at 13,500 rpm for 20 minutes at 4°C. The supernatant was mixed with Ni-NTA beads (Qiagen, MA) that had been freshly washed three times with Ni-NTA lysis buffer. After rocking for 2 hours at 4°C, the beads were centrifuged at 3,000 rpm for 10 sec, then washed three times with Ni-NTA buffer. His6-tagged proteins were then eluted four times with freshly prepared elution buffer (300 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl (pH 8.0), 300 mM imidazole, 10 mM β-mercaptoethanol, 0.1% NP-40).
To express MBP- or GST-tagged proteins [55], E. coli strain BL21 (Invitrogen) was transformed with pMAL-C2- or pGEX-5X-based plasmids, grown in 250 ml LB medium containing 100 μg/ml ampicillin at 37°C to OD600 ≤ 1.0, and induced with 1 mM IPTG for 3 h at 24°C. Cells were then collected by centrifugation (6,000 rpm, 10 min at 4°C), washed twice with ice cold water, resuspended into 3 ml cell lysis buffer (CLB) [the CLB for MBP-tagged proteins: 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.1% NP-40, 10mM-β Me and 2× complete protein inhibitor cocktail (Sigma); the CLB for GST tagged protein: 1x PBS, 0.1% NP-40, 10 mM β-mecaptoethanol and 2× complete protein inhibitor cocktail (Sigma)]. Protein extracts were obtained by sonicating cells for 7 × 15 sec with 15 sec interval on ice, centrifuged at 18,500 rpm for 20 min at 4°C. To purify MBP- or GST-tagged proteins, protein extracts incubated with prewashed amylose beads (New England Biolabs, Inc.) or glutathione conjugated Sepharose 4B beads (GE Healthcare), rocked for 1 h at 4°C. Beads were then washed five times each with 1.0 ml corresponding CLB buffer. MBP-tagged proteins were resuspended in 1.0 ml CLB. The GST-tagged proteins were eluted with 250 μl of 20 mM reduced glutathione in 50 mM TrisHCl (pH 8.0) and 10 mM β-mecaptoethanol four times at 4°C. The concentrations of the purified recombinant proteins were estimated by comparing the sample proteins to bovine serum albumin (BSA) of known concentrations by SDS-PAGE analysis, followed by staining the gel with simplelyblue™ (Invitrogen).
To test in vitro binding between His6- and GST-tagged proteins, 20 μg of His6-tagged protein was mixed with 10 μg of GST (as negative control) or GST-tagged protein that was still bound to the glutathione beads (400 μl total volume) and rocked for 1 h at 4°C. The beads were washed five times with freshly prepared CLB for GST fusion proteins (see above) and resuspended in 50 μl 2x SDS-PAGE sample buffer, and proteins were analyzed by SDS-PAGE (10% gel) and Western blotting using monoclonal mouse anti-His (GE Healthcare Life Sciences, Buckinghamshire, UK), and anti-GST (Abcam, Cambridge, MA) primary antibodies and an HRP-conjugated rabbit anti-mouse-IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA).
To test in vitro binding between MBP- and GST-tagged proteins, approximately 40 μg MBP-tagged proteins that were still bound on beads were added to different 1.5 ml Eppendorf tubes, the amounts of beads were normalized by adding more pre-washed amylose beads, and then incubated with 500 μl of 5% BSA at 4°C for 1 h to block nonspecific binding by the beads. After a brief centrifugation to pellet the beads and remove the supernatant, the beads were mixed with 10 μg GST-tagged proteins (GST alone as the negative control). The CLB for MBP-tagged proteins without the protease inhibitors was added to the reaction to make the final volume of 500 μl. The binding reaction was incubated with rotation at 4°C for 1 h, and then spun down to pellet the beads, which were then washed three times each with 500 μl CLB for MBP-tagged proteins at 4°C. The final protein complexes were eluted by adding 100 μl 2× SDS sample buffer and boiled for 5 min, resolved by SDS-PAGE and analyzed by Western blotting. Monoclonal mouse anti-MBP (Sigma-Aldrich, St. Louis, MO) and anti-GST (Abcam) primary antibodies and the Fast Western Blot Kit (Supersignal West Pico, Mouse, Thermo Scientific) were used for Western blotting following the manufacturer’s instructions.
Bimolecular fluorescence complementation (BiFC) assay
Yeast strains used for the BiFC assay [56] were constructed by a PCR-based approach [34]. A PCR fragment containing the TRP1∷pCET1 promoter and the N-terminal fragment of Venus (VN) was directly inserted in-frame before the START codon of CHS4 at its chromosomal locus in YEF473A (MATa), generating the TRP1∷pCET1-VN-CHS4 strain. Similarly, the His3MX6∷pCET1-VC-HOF1 strain was derived from YEF473B (MATα). These strains were mated and diploids were selected on SC-His-Trp plates. The diploid cells were imaged using the Nikon spinning-disk confocal imaging system as described below.
Chitin measurements
Chitin assay
Chitin assay: to determine the chitin context in the total cell walls of different strains (data presented in Figure 2B), we used a quantitative colorimetric assay as described previously [37], with slight modification. Yeast cells were grown in YM-1 with 2% glucose for 48 hours at 23°C to stationary phase. The optical density (OD) of the culture was measured at a wavelength of 600 nm. The culture was diluted approximately 1:100 into duplicates of 5 ml of fresh YM-1 medium, trying to get the same amount of starting cells in each duplicate. The cultures were grown for 22–24 hours at 23°C or 37°C. A total of 3 ml of each culture was centrifuged into a pre-weighed 1.5 ml tube at 15,000 rpm for 2 min. The tubes were then placed in a 37°C incubator for 48–96 hours to dry the pellets. The tubes with the pellets were weighed again and the weights subtracted the initial weight of the empty tube to yield the dry weight of the cell pellet. 1 ml 6% KOH was added to the cell pellets, which were then heated to 80°C for 90 min with occasional mixing. The alkaline insoluble material was pelleted at 15,000 rpm for 20 min, and neutralized with 1ml PBS for 10–20 min with occasional mixing. The mixture was centrifuged at 15,000 rpm for 20 min and the supernatant was discarded. 200 μl of McIlvaine’s Buffer (0.2 M Na2HPO4/0.1 M citric acid, pH 6.0) was added to the pellets, which were stored at −20°C until ready to process for chitin measurements. Samples were thawed and digested with 10 μl of Serratia marcescens chitinase (0.004 g freshly dissolved in 1 ml cold 200 mM potassium phosphate buffer, pH 6.0, with 2 mM CaCl2; Sigma-Aldrich, St. Louis, MO) 18–20 hours on shaking 23°C platform. 10 μl of supernatant was mixed with 10 μl of 0.27 M sodium borate (pH 9.0) in a 0.2 ml PCR tube, heated in a thermocycler to 99.9°C for about 60 sec, mixed gently, and incubated at 99.9°C for 10 min. Immediately after cooling to room temperature, 100 μl of freshly diluted DMAB solution (Ehrlich’s reagent, consisting of 10 g of p-dimethylaminobenzaldehyde in 12.5 ml of concentrated HCl and 87.5 ml of glacial acetic acid, diluted 1:10 with glacial acetic acid) was added to samples, and incubated at 37°C for 20 min. The absorbance at 585 nm was immediately recorded. Standard curves were prepared from stocks of 0.2 to 2.0 mM N-acetylglucosamine (GlcNAc). The levels of chitin were normalized, expressed as GlcNAc concentration, to the dry weight of the sample.
Calcofluor White and DNA staining
Calcofluor White and DNA staining: to determine the relative chitin levels at the bud neck in different strains (data presented in Figure 2C), cells were grown to exponential phase in liquid YM-1 medium at 25°C. Formaldehyde was added to a 5-ml culture to the final concentration of 3.7% to fix the cells at 25°C for 1 hour. Cells were pelleted in an Eppendorf tube by centrifugation and washed once with 1.0 ml distilled water. Cells were then resuspended in 1.0 ml of 0.1 mg/ml Calcofluor White (CW) (Sigma) for chitin staining at 25°C for 5 min [57]. After staining, cells were washed three times each with 1.0 ml distilled water. Cell pellet was then resuspended in VECTASHIELD Antifade Mounting Medium with DAPI (Vector laboratories, Burlingame, CA) to stain DNA.
Imaging and data analysis
For imaging CW-stained cells (Figure 2C), a computer-controlled Eclipse 800 microscope (Nikon, Tokyo, Japan) and a high-resolution charge-coupled device camera (model C4742-95; Hamamatsu Photonics, Bridgewater, NJ) were used. Cells were divided into different categories based on the following criteria: if the long axis of the bud was smaller than 50% of that of the mother compartment, the cell was designated “G1/S”. If the long axis of the bud was larger than 50% of that of the mother compartment and the nucleus was located in the vicinity of bud-neck region, the cell designated “G2/M”. If the long axis of the bud was larger than 50% of that of the mother compartment and the nuclei were located at the cellular poles, the cell designated “telophase”. Image-based quantification of chitin at the bud neck was performed using Fiji [58]. A region of interest (ROI) was set to cover the chitin signal at the bud-neck region. After subtraction of background signal, the total intensity in the ROI was used for quantitative analysis and statistical test.
For imaging cells carrying GFP-Chs4 and Spc42-mCherry (Figure 4A), cells were grown at 23°C to exponential phase in SC-His media, and then concentrated by centrifugation, and spotted on top of a 2% agarose pad containing SC-His media on a slide. The cover glass was sealed with nail polish along its edges. Images were acquired at 23°C on a spinning-disk confocal microscope equipped with a Yokogawa CSU 10 scan head combined with an Olympus IX 71 microscope and an Olympus 100× objective (1.4 NA, Plan S-Apo oil immersion). Acquisition and hardware were controlled by MetaMorph version 7.7 (Molecular Devices, Downingtown, PA). A Hamamatsu ImagEM EMCCD camera (model C9100-13, Bridgewater, NJ) was used for capture. Diode lasers for excitation (488 nm for GFP and 561 nm for mCherry/RFP) were housed in a launch constructed by Spectral Applied Research (Richmond Hill, Ontario). Images were taken every 1.5 min with a z-stack consisting of 14 × 0.4-μm steps. Integrated intensity of GFP-Chs4 at the bud-neck region during the cell cycle was measured using Fiji/ImageJ, as described previously [59].
All other imaging experiments presented in this study were performed as described previously [60], with slight modification. In brief, cells were cultured to exponential phase at 25°C in SC or SC-His medium, and placed to poly-lysine-coated glass-bottom dish, followed by embedding with agarose-containing SC or SC-His medium. Images were acquired at 24°C by a Nikon spinning-disk confocal microscope (model Eclipse Ti-U, Tokyo, Japan) with a Nikon 100×/1.49NA oil objective (model CFI Apo TIRF 100×), combined with a Yokogawa confocal scanner unit (model CSU-X1, Tokyo, Japan). A Photometrics QuantEM EMCCD camera (model 512SC, Tucson, AZ, USA) was used for capture. Solid-state lasers for excitation (488 nm for GFP and 561 nm for RFP) were housed in a launch constructed by Spectral Applied Research (model ILE-400, Richmond Hill, Ontario, Canada). The imaging system was controlled by MetaMorph version 7.8.10.0 (Molecular Devices, Downingtown, PA, USA). Images were taken every 2 or 3 min with z-stacks ranging from 7×1 μm or 8×1 μm. A sum projection was created with NIH ImageJ (1.51j) [61]. For quantification of fluorescence intensities, the integrated density at the bud-neck region was calculated by subtracting the fluorescence intensity in background area from the total intensity in an ImageJ-drawn polygon covering the neck region. Data analyses were performed with Microsoft Excel.
QUANTITATION AND STATISTICAL ANALYSIS
For the statistical analyses on chitin levels (related to Figures 2B and 2D), one-tailed unpaired t-test (assuming unequal variances) was performed. P-values are described in the main text. For the statistical analysis of Chs4 duration in WT and hof1Δ cells (related to Figure S2), unpaired t-test was performed using Prism Version 5 (GraphPad Software, La Jolla, CA). “n” refers to the number of cells analyzed unless indicated otherwise.
Supplementary Material
Movie S1. Localization of Hof1 in relation to Chs3 during the cell cycle. Related to Figure 1.
Movie S2. Interaction of Hof1 and Chs4 during the cell cycle as detected by BiFC. Related to Figure 1E.
Movie S3. Localization of Chs4 in relation to Spc42 during the cell cycle in WT and hof1Δ cells. Related to Figure 4A.
Movie S4. Localization of Chs4 in relation to Chs3 during the cell cycle in WT and hof1Δ cells. Related toFigure 4C.
Highlights.
Hof1 and Chs4 interact via the conserved F-BAR domain and Sel1-like repeats
Hof1 and Chs3 competes for binding to the SLR of Chs4
Hof1 regulates the localization dynamics of Chs4 at the division site
Hof1 regulates Chs3-mediated chitin synthesis during cytokinesis
Acknowledgments
We thank Andrea Stout for assistance with imaging, John Pringle, Won-Ki Huh, Kelly Tatchell, Wei-Lih Lee, and Charlie Boone for plasmids and strains, and the members of the Bi laboratory for stimulating discussions. This work was supported by NIH grant GM115420 to E.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Y.O. and X.D. designed and performed the in vitro protein-binding experiments. J.H.S. performed the two-hybrid analysis, chitin assays, and BiFC experiment. H.O. and C.W. performed imaging experiments and data analysis. S.O. and D.Y. performed chitin staining and analysis of chitin levels at the bud neck during the cell cycle. E.B. supervised the entire project and was the primary writer for the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures, one table, and four movies and can be found with this article online at
References
- 1.Shaw JA, Mol PC, Bowers B, Silverman SJ, Valdivieso MH, Duran A, Cabib E. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J Cell Biol. 1991;114:111–123. doi: 10.1083/jcb.114.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J, Wang H, McCollum D, Balasubramanian MK. Drc1p/Cps1p, a 1,3-beta-glucan synthase subunit, is essential for division septum assembly in Schizosaccharomyces pombe. Genetics. 1999;153:1193–1203. doi: 10.1093/genetics/153.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tammi R, Tammi M. Correlations between hyaluronan and epidermal proliferation as studied by [3H]glucosamine and [3H]thymidine incorporations and staining of hyaluronan on mitotic keratinocytes. Exp Cell Res. 1991;195:524–527. doi: 10.1016/0014-4827(91)90405-j. [DOI] [PubMed] [Google Scholar]
- 4.Mizuguchi S, Uyama T, Kitagawa H, Nomura KH, Dejima K, Gengyo-Ando K, Mitani S, Sugahara K, Nomura K. Chondroitin proteoglycans are involved in cell division of Caenorhabditis elegans. Nature (London) 2003;423:443–448. doi: 10.1038/nature01635. [DOI] [PubMed] [Google Scholar]
- 5.Evanko SP, Parks WT, Wight TN. Intracellular hyaluronan in arterial smooth muscle cells: association with microtubules, RHAMM, and the mitotic spindle. J Histochem Cytochem. 2004;52:1525–1535. doi: 10.1369/jhc.4A6356.2004. [DOI] [PubMed] [Google Scholar]
- 6.VerPlank L, Li R. Cell cycle-regulated trafficking of Chs2 controls actomyosin ring stability during cytokinesis. Mol Biol Cell. 2005;16:2529–2543. doi: 10.1091/mbc.E04-12-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang G, Kashimshetty R, Ng KE, Tan HB, Yeong FM. Exit from mitosis triggers Chs2p transport from the endoplasmic reticulum to mother-daughter neck via the secretory pathway in budding yeast. J Cell Biol. 2006;174:207–220. doi: 10.1083/jcb.200604094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neto H, Collins LL, Gould GW. Vesicle trafficking and membrane remodelling in cytokinesis. Biochem J. 2011;437:13–24. doi: 10.1042/BJ20110153. [DOI] [PubMed] [Google Scholar]
- 9.Izumikawa T, Kanagawa N, Watamoto Y, Okada M, Saeki M, Sakano M, Sugahara K, Sugihara K, Asano M, Kitagawa H. Impairment of embryonic cell division and glycosaminoglycan biosynthesis in glucuronyltransferase-I-deficient mice. J Biol Chem. 2010;285:12190–12196. doi: 10.1074/jbc.M110.100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu X, Vogel BE. A secreted protein promotes cleavage furrow maturation during cytokinesis. Curr Biol. 2011;21:114–119. doi: 10.1016/j.cub.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proctor SA, Minc N, Boudaoud A, Chang F. Contributions of turgor pressure, the contractile ring, and septum assembly to forces in cytokinesis in fission yeast. Curr Biol. 2012;22:1601–1608. doi: 10.1016/j.cub.2012.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arasada R, Pollard TD. Contractile ring stability in S. pombe depends on F-BAR protein Cdc15p and Bgs1p transport from the Golgi complex. Cell Rep. 2014;8:1533–1544. doi: 10.1016/j.celrep.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drakakaki G. Polysaccharide deposition during cytokinesis: Challenges and future perspectives. Plant Sci. 2015;236:177–184. doi: 10.1016/j.plantsci.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Xiao J, Goley ED. Redefining the roles of the FtsZ-ring in bacterial cytokinesis. Curr Opin Microbiol. 2016;34:90–96. doi: 10.1016/j.mib.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lutkenhaus J, Pichoff S, Du S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton (Hoboken) 2012;69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen CG, Wright AJ, Muller S. The role of the cytoskeleton and associated proteins in determination of the plant cell division plane. Plant J. 2013;75:258–269. doi: 10.1111/tpj.12177. [DOI] [PubMed] [Google Scholar]
- 17.Balasubramanian MK, Bi E, Glotzer M. Comparative analysis of cytokinesis in budding yeast, fission yeast and animal cells. Curr Biol. 2004;14:R806–818. doi: 10.1016/j.cub.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 18.Bhavsar-Jog YP, Bi E. Mechanics and regulation of cytokinesis in budding yeast. Semin Cell Dev Biol. 2017;66:107–118. doi: 10.1016/j.semcdb.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss EL. Mitotic exit and separation of mother and daughter cells. Genetics. 2012;192:1165–1202. doi: 10.1534/genetics.112.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida S, Bartolini S, Pellman D. Mechanisms for concentrating Rho1 during cytokinesis. Genes Dev. 2009;23:810–823. doi: 10.1101/gad.1785209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada A, Niwa H, Tsujita K, Suetsugu S, Nitta K, Hanawa-Suetsugu K, Akasaka R, Nishino Y, Toyama M, Chen L, et al. Curved EFC/F-BAR-domain dimers are joined end to end into a filament for membrane invagination in endocytosis. Cell. 2007;129:761–772. doi: 10.1016/j.cell.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 22.McDonald NA, Gould KL. Linking up at the BAR: Oligomerization and F-BAR protein function. Cell Cycle. 2016;15:1977–1985. doi: 10.1080/15384101.2016.1190893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittl PR, Schneider-Brachert W. Sel1-like repeat proteins in signal transduction. Cell Signal. 2007;19:20–31. doi: 10.1016/j.cellsig.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 24.DeMarini DJ, Adams AEM, Fares H, De Virgilio C, Valle G, Chuang JS, Pringle JR. A septin-based hierarchy of proteins required for localized deposition of chitin in the Saccharomyces cerevisiae cell wall. J Cell Biol. 1997;139:75–93. doi: 10.1083/jcb.139.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reyes A, Sanz M, Duran A, Roncero C. Chitin synthase III requires Chs4p-dependent translocation of Chs3p into the plasma membrane. J Cell Sci. 2007;120:1998–2009. doi: 10.1242/jcs.005124. [DOI] [PubMed] [Google Scholar]
- 26.Larson JR, Bharucha JP, Ceaser S, Salamon J, Richardson CJ, Rivera SM, Tatchell K. Protein phosphatase type 1 directs chitin synthesis at the bud neck in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:3040–3051. doi: 10.1091/mbc.E08-02-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozubowski L, Panek H, Rosenthal A, Bloecher A, DeMarini DJ, Tatchell K. A Bni4-Glc7 phosphatase complex that recruits chitin synthase to the site of bud emergence. Mol Biol Cell. 2003;14:26–39. doi: 10.1091/mbc.E02-06-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamei T, Tanaka K, Hihara T, Umikawa M, Imamura H, Kikyo M, Ozaki K, Takai Y. Interaction of Bnr1p with a novel Src Homology 3 domain-containing Hof1p. IMPLICATION IN CYTOKINESIS IN SACCHAROMYCES CEREVISIAE. J Biol Chem. 1998;273:28341–28345. doi: 10.1074/jbc.273.43.28341. [DOI] [PubMed] [Google Scholar]
- 29.Vallen EA, Caviston J, Bi E. Roles of Hof1p, Bni1p, Bnr1p, and Myo1p in cytokinesis in Saccharomyces cerevisiae. Mol Biol Cell. 2000;11:593–611. doi: 10.1091/mbc.11.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gladfelter AS, Pringle JR, Lew DJ. The septin cortex at the yeast mother-bud neck. Curr Opin Microbiol. 2001;4:681–689. doi: 10.1016/s1369-5274(01)00269-7. [DOI] [PubMed] [Google Scholar]
- 31.Oh Y, Schreiter J, Nishihama R, Wloka C, Bi E. Targeting and functional mechanisms of the cytokinesis-related F-BAR protein Hof1 during the cell cycle. Mol Biol Cell. 2013;24:1305–1320. doi: 10.1091/mbc.E12-11-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meitinger F, Palani S, Hub B, Pereira G. Dual function of the NDR-kinase Dbf2 in the regulation of the F-BAR protein Hof1 during cytokinesis. Mol Biol Cell. 2013;24:1290–1304. doi: 10.1091/mbc.E12-08-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moravcevic K, Alvarado D, Schmitz KR, Kenniston JA, Mendrola JM, Ferguson KM, Lemmon MA. Comparison of Saccharomyces cerevisiae F-BAR domain structures reveals a conserved inositol phosphate binding site. Structure. 2015;23:352–363. doi: 10.1016/j.str.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sung MK, Huh WK. Bimolecular fluorescence complementation analysis system for in vivo detection of protein-protein interaction in Saccharomyces cerevisiae. Yeast. 2007;24:767–775. doi: 10.1002/yea.1504. [DOI] [PubMed] [Google Scholar]
- 35.Markus SM, Omer S, Baranowski K, Lee WL. Improved Plasmids for Fluorescent Protein Tagging of Microtubules in Saccharomyces cerevisiae. Traffic. 2015;16:773–786. doi: 10.1111/tra.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo J, Vallen EA, Dravis C, Tcheperegine SE, Drees BL, Bi E. Identification and functional analysis of the essential and regulatory light chains of the only type II myosin Myo1p in Saccharomyces cerevisiae. J Cell Biol. 2004;165:843–855. doi: 10.1083/jcb.200401040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lesage G, Shapiro J, Specht CA, Sdicu AM, Menard P, Hussein S, Tong AH, Boone C, Bussey H. An interactional network of genes involved in chitin synthesis in Saccharomyces cerevisiae. BMC Genet. 2005;6:8–23. doi: 10.1186/1471-2156-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korinek WS, Bi E, Epp JA, Wang L, Ho J, Chant J. Cyk3, a novel SH3-domain protein, affects cytokinesis in yeast. Curr Biol. 2000;10:947–950. doi: 10.1016/s0960-9822(00)00626-6. [DOI] [PubMed] [Google Scholar]
- 39.Oh Y, Chang KJ, Orlean P, Wloka C, Deshaies R, Bi E. Mitotic exit kinase Dbf2 directly phosphorylates chitin synthase Chs2 to regulate cytokinesis in budding yeast. Mol Biol Cell. 2012;23:2445–2456. doi: 10.1091/mbc.E12-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ono N, Yabe T, Sudoh M, Nakajima T, Yamada-Okabe T, Arisawa M, Yamada-Okabe H. The yeast Chs4 protein stimulates the trypsin-sensitive activity of chitin synthase 3 through an apparent protein-protein interaction. Microbiology. 2000;146:385–391. doi: 10.1099/00221287-146-2-385. [DOI] [PubMed] [Google Scholar]
- 41.Cos T, Ford RA, Trilla JA, Duran A, Cabib E, Roncero C. Molecular analysis of Chs3p participation in chitin synthase III activity. Eur J Biochem. 1998;256:419–426. doi: 10.1046/j.1432-1327.1998.2560419.x. [DOI] [PubMed] [Google Scholar]
- 42.Donaldson AD, Kilmartin JV. Spc42p: a phosphorylated component of the S. cerevisiae spindle pole body (SPD) with an essential function during SPB duplication. J Cell Biol. 1996;132:887–901. doi: 10.1083/jcb.132.5.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meitinger F, Boehm ME, Hofmann A, Hub B, Zentgraf H, Lehmann WD, Pereira G. Phosphorylation-dependent regulation of the F-BAR protein Hof1 during cytokinesis. Genes Dev. 2011;25:875–888. doi: 10.1101/gad.622411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blondel M, Bach S, Bamps S, Dobbelaere J, Wiget P, Longaretti C, Barral Y, Meijer L, Peter M. Degradation of Hof1 by SCF(Grr1) is important for actomyosin contraction during cytokinesis in yeast. EMBO J. 2005;24:1440–1452. doi: 10.1038/sj.emboj.7600627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren G, Wang J, Brinkworth R, Winsor B, Kobe B, Munn AL. Verprolin cytokinesis function mediated by the Hof one trap domain. Traffic. 2005;6:575–593. doi: 10.1111/j.1600-0854.2005.00300.x. [DOI] [PubMed] [Google Scholar]
- 46.Roberts-Galbraith RH, Ohi MD, Ballif BA, Chen JS, McLeod I, McDonald WH, Gygi SP, Yates JR, 3rd, Gould KL. Dephosphorylation of F-BAR protein Cdc15 modulates its conformation and stimulates its scaffolding activity at the cell division site. Mol Cell. 2010;39:86–99. doi: 10.1016/j.molcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willet AH, McDonald NA, Bohnert KA, Baird MA, Allen JR, Davidson MW, Gould KL. The F-BAR Cdc15 promotes contractile ring formation through the direct recruitment of the formin Cdc12. J Cell Biol. 2015;208:391–399. doi: 10.1083/jcb.201411097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDermott MF. A common pathway in periodic fever syndromes. Trends Immunol. 2004;25:457–460. doi: 10.1016/j.it.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Bi E, Pringle JR. ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:5264–5275. doi: 10.1128/mcb.16.10.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular Biology. Methods Enzymol. 1991;194:933. [PubMed] [Google Scholar]
- 51.Lillie SH, Pringle JR. Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J Bacteriol. 1980;143:1384–1394. doi: 10.1128/jb.143.3.1384-1394.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 53.Longtine MS, McKenzie A, III, DeMarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 54.Lee S, Lim WA, Thorn KS. Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One. 2013;8:e67902. doi: 10.1371/journal.pone.0067902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang X, Luo J, Nishihama R, Wloka C, Dravis C, Travaglia M, Iwase M, Vallen EA, Bi E. Biphasic targeting and cleavage furrow ingression directed by the tail of a myosin-II. J Cell Biol. 2010;191:1333–1350. doi: 10.1083/jcb.201005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 57.Pringle JR. Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- 58.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wloka C, Vallen EA, Thé L, Fang X, Oh Y, Bi E. Immobile myosin-II plays a scaffolding role during cytokinesis in budding yeast. J Cell Biol. 2013;200:271–286. doi: 10.1083/jcb.201208030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okada S, Wloka C, Bi E. Analysis of protein dynamics during cytokinesis in budding yeast. Methods Cell Biol. 2017;137:25–45. doi: 10.1016/bs.mcb.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 61.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Localization of Hof1 in relation to Chs3 during the cell cycle. Related to Figure 1.
Movie S2. Interaction of Hof1 and Chs4 during the cell cycle as detected by BiFC. Related to Figure 1E.
Movie S3. Localization of Chs4 in relation to Spc42 during the cell cycle in WT and hof1Δ cells. Related to Figure 4A.
Movie S4. Localization of Chs4 in relation to Chs3 during the cell cycle in WT and hof1Δ cells. Related toFigure 4C.