

Abstract
MLL-rearranged leukemia represents approximately 5-10% of adult AML cases and nearly half of all infant/pediatric acute leukemia cases. These leukemias have a poor prognosis, and as of yet there are no approved targeted therapeutic options. The rearrangement in the MLL gene leads to aberrant expression of MLL-fusion proteins, which are transforming in murine bone marrow, and in particular on stem cells and myeloid progenitors derived from bone marrow or fetal liver. The commonality of the MLL-fusions is the in-frame fusion of 8-11 N-terminal exons of MLL1 (KMT2a) with C-terminus of a partner fusion gene. Currently over 80 different fusion partners have been described. This protocol will focus on bone marrow-derived models only, using one particular MLL fusion, MLL-AF9, which we have used in our studies previously. These models have proven effective for drug screening, as well as for predicting clinical response.
Keywords: Murine leukemia model, AML, bone marrow transplant, retroviral transduction techniques, cancer stem cells, myeloid progenitor cells, leukemia therapeutics
Unit introduction
For two decades MLL fusion proteins have been known to be transforming when ectopically expressed in mouse bone marrow (Lavau, Szilvassy, Slany, & Cleary, 1997). While the methodologies have evolved, the premise for generating MLL-driven acute myelogenous leukemia (AML) in mice has remained fairly constant. Hematopoietic stem and progenitor cells are isolated and transduced with a retrovirus driving expression of an MLL-fusion protein. The infected cells are reintroduced into recipients that will then become leukemic. Researchers use a wide range of hematopoietic populations for retroviral gene transduction. The most straightforward method is to utilize 5-fluorouracil- (5-FU) treated total bone marrow as the target population for viral infection. This is the method described here.
Flow-sorted hematopoietic stem cell fraction (Lineage-negative Sca1+ c-Kit+ CD34-) and Granulocyte-Macrophage progenitors (Lineage-negative Sca1- c-Kit+ CD34+ CD16/32+) are also transformable by the MLL fusions (Cozzio et al., 2003; Krivtsov et al., 2006). It is important to rationalize the specific cell fraction that will be used based on requirements of the experiments and downstream analyses. (For labs that do not have easy access to a FACS-based cell sorter, it is possible to use magnetic bead depletion of lineage-committed cells followed by Miltenyi bead selection of c-Kit (CD117)-positive cells as a surrogate for hematopoietic precursor cells) (Berson et al., 1996).
The earliest version of this experiment utilized an MLL-ENL expressing vector in the pMSCV backbone (Lavau et al., 1997). Our work has focused on MLL-AF9, which comprises 1395 amino acid (aa) residues from the N-terminus of MLL1 (KMT2A) fused in-frame to 91 aa from the C-terminus of AF9 (from Jay Hess, University of Michigan), originally cloned in pMSCVneo (Martin et al., 2003)) cloned into pMIG_1- (pMSCV containing vector tagged with IRES-GFP cassette). The size of this plasmid is approximately 10.5 kb, which barely meets the cut-off requirement for packaging into a retroviral envelope. Because of the size of the MLL-fusions it has proven difficult to use this technique for modeling leukemia mediated by MLL-AF4 since those plasmids are not efficiently packaged into retroviral envelopes. However, there have been several other successfully used models of MLL-fusion leukemia: MLL-ENL (Cozzio et al., 2003; Lavau et al., 1997), MLL-AF10 (DiMartino et al., 2002); MLL-ELL (DiMartino et al., 2000) MLL-AF6 (Deshpande et al., 2013) are among the more prevalent models.
When choosing an expression vector for an oncogene users need to know in what cell types this oncogene will be expressed and chose a vector with that cell type specific promoter. There are multiple virus based vectors available. Each of these has certain advantages and disadvantages such as genome integration, organism, cell lineage, and division specificity (Schott, Hoffmann, & Schambach, 2015). Given the need to express MLL-AF9 in murine hematopoietic cells, the preferred choice is a retroviral vector with MSCV backbone (pMIG1, AddGene # 9044). This type of retrovirus can only infect murine cells and therefore is safe for use by researchers in BL2 laboratory settings.
In order to monitor expression of the onco-protein, the use of a reporter is needed. There are several expression reporters available. These vectors can be defined into specific categories: 1. for in vitro/ex vivo work, antibiotic (e.g. Neomycin, Puromycin) resistance genes most often expressed from independent promoters; 2. fluorescent proteins (GFP, YFP, dsRed, Tomato) fused to the target gene of interest through an internal ribosomal entry site (IRES); 3. Reported surface antigens such as the extracellular part of human CD2 fused through an IRES for expression in murine cells; 4. for in vivo tracking, the use of luciferase expression vectors which are generally co-transfected along with the MLL-fusion. Each of these reporters has advantages and disadvantages. For example, when using GFP expressed as a single RNA with MLL-AF9 (due to the IRES in the pMIG vector), ex vivo FACS detection of oncoprotein-expressing cells can be performed using flow cytometry of peripheral blood during disease progression, or from bone marrow after the mouse is sacrificed. Antibiotic resistance genes cannot be tracked during disease progression, and can only be read out using ex vivo growth experiments such as methylcellulose colony counts. However, since the plasmids expressing antibiotic resistance genes do not generate a fluorescent marker, more colors can be used to analyze the resultant leukemia via flow cytometry.
As with all procedures involving live animals, experiments must fall under the guidelines of an institutional animal care and use committee (IACUC). The researcher must ensure that the experiments outlined here are performed per the IACUC protocols set up by the researcher's institute. A first time researcher would need to submit a protocol and/or proposal for the experiments planned, and the protocols would need IACUC approval before any animal work can commence.
Basic Protocol 1
Leukemic Transformation of Hematopoietic Stem and Progenitor Cells from Whole Bone Marrow
The production of the cells that will generate AML from mouse bone marrow requires a four step process:
Generation of the virus carrying the MLL-fusion oncogene
Isolation of the bone marrow cells that will transformed by the virally introduced oncogene
Infection of the isolated bone marrow cells
Assaying for infection success and for the number of infected cells that will be injected into recipient mice.
Steps one and two can be carried out concurrently, provided the virus is successfully produced in time to infect the newly isolated bone marrow.
Retrovirus production:
The first step toward transformation of healthy murine bone marrow is producing the virus that will carry the MLL-fusion oncogene. (Swift, Lorens, Achacoso, & Nolan, 2001)
Materials
-
-
Standard Tissue Culture (TC) environment: TC hood; incubator with 5% CO2 set to 37°C; TC centrifuge with bucket rotors; vortex mixer; Gilson pipets;
-
-
Maxiprep purified pMIG-MLL-AF9 plasmid 10-100 μg (AddGene #71443)
-
-
Maxiprep purified Ψ-Eco (Phoenix-ECO) plasmid (10-100 μg) containing gag, pol and packaging envelope, NOTE: please see the following web address for information on how to procure this cell line (created by Gary Nolan, Stanford University)https://web.stanford.edu/group/nolan/_OldWebsite/MTAs/mtas.html
-
-
HEK 293T cells that have not grown to confluence (ATCC CRL-3216)
-
-
IMDM media (Gibco / ThermoFisher Scientific)
-
-
Fetal Bovine Serum (FBS) (Gibco / ThermoFisher Scientific)
-
-
GlutaMAX (Gibco / ThermoFisher Scientific)
-
-
FuGENE 9 (Promega)
-
-
OptiMEM media (Gibco / ThermoFisher Scientific)
-
-
0.22 μm filter units with Luer lock (ThermoFisher Scientific)
-
-
1, 3, 5,10 ml syringes (BD Biosciences)
-
-
Needles (BD Biosciences)
-
-
10 cm tissue culture treated plates (Corning)
-
-
Flat bottom tissue culture treated 96 well plates (Corning - Costar)
-
-
6 well cell culture dishes (Corning)
-
-
Cryovials (Corning)
-
-
Ba/F3 cells (DSMZ #ACC 300)
-
-
Polybrene (Sigma/Millipore)
-
-
Murine IL-3, murine IL-6, murine SCF (Peprotech)Working vivarium (with protocols in place for your work)
-
-
C57BL/6 mice, female, 6 - 8 weeks old (Charles River, Jackson Labs, Taconic)
-
-
5-fluorouracil (Sigma)
-
-
surgical scissors and forceps
-
-
50 ml conical tubes (Falcon or Corning)
-
-
Phosphate Buffered Saline (PBS) (Gibco / ThermoFisher Scientific)
-
-
70% ethanol (ThermoFisher Scientific)
-
-
Red blood cell lysis buffer (Qiagen)
-
-
RPMI cell culture medium (Gibco / ThermoFisher Scientific)
-
-
Fetal Bovine Serum (FBS) (Gibco / ThermoFisher Scientific)
-
-
Cell strainers for 50 ml conical (Falcon - 352350)
-
-
Methylcellulose with cytokines (M3234 – Stem Cell Technologies; M3434 comes with cytokines pre-added)
-
-
Antibiotic – G418/neomycin, puromycin if applicable (Gibco)
-
-
Blunt-end needles (Stem Cell Technologies)
-
-
35 mm plates (Falcon)
Retrovirus production Procedure:
Plate 2.2×106 HEK293T cells per 10 cm tissue culture (TC) plate (4×104 per cm2) in IMDM supplemented with 10% FBS (no antibiotics). Incubate HEK293T cells overnight at 37°C (5% CO2) to achieve 30-50% confluency.
-
Day 0; Time 16:30” Prepare transfection media by combining the following in a 15 ml conical tube in this order:
- 1.5 ml of pre-warmed to 37°C OptiMEM media (the cells may dissociate from the plate if cold media is added);
- 90μl of FuGENE 6(9); mix gently;
- 10μg of each MLL-AF9 and Ψ-Eco plasmids
Mix gently. Bring the total volume to 2 ml with pre-warmed 37°C OptiMEM media. Leave for 15 min at room temperature to form micelles. Add 4 ml of pre-warmed IMDM media supplemented with 15% FBS (10% final concentration) to the 15 ml tube containing the FuGENE/plasmid mixture and mix gently.
Day 0; Time 16:45: Aspirate IMDM media from the plate with HEK293T cells and immediately add the transfection media from step 2. Place the plate into cell culture incubator (37°C/5% CO2) for 24 hours.
Day 1; Time 09:00: Aspirate and discard overnight media from HEK293T cells and replace with 6 ml of pre-warmed IMDM media supplemented with 10% FBS (no antibiotics) and incubate for >7 hours. At this point it worth checking if HEK293T cells are GFP+ under fluorescent microscope. If the cells are GFP-, then it is likely that the transfection was not successful and the transfection should be attempted again.
Day 1; Time 16:30 - Collect 6 ml of the conditioned media, containing retroviral supernatant into a 10 ml syringe, filter through 0.22μm filter and split into 500-1000μl aliquot in labeled cryo-vials, freeze on dry ice. Replace the media with 7 ml fresh, pre-warmed IMDM media supplemented with 10% FBS (no antibiotics).
Repeat the procedure two more times on Day 2; 09:00 and 16:30. Label the viral sup aliquots accordingly: “day1 collection”; “night1 collection”; “day2 collection”. Store the viral supernatants in -80°C freezer. Note: viruses are stable at -80°C for 3-6 months. Repetitive freeze/thaw cycles are not recommended and can cause large decreases in titer.
-
Before infecting bone marrow, it is important to know the concentration/titer of the viral particles expressed as “infectious units per ml” (IU/ml). This is generally done by infecting Ba/F3 cells (DSMZ) or any other non-adherent murine cell line.
In order for a retrovirus to penetrate a non-adherent cell, they need to gain access and remain in physical contact. There are two ways to increase the virus-cells contact time: 1) use retronectin-covered wells. This method is rather expensive and requires preparation of the plates ahead of time. 2) add polymer polybrene to the viral supernatant. The polybrene method is preferred since it is cheaper and, in our hands, the efficiency of both methods has been comparable.
To test titer of the viral supernatant, add 1×105 BA/F3 cells in 100 μl in each 4 wells of a 96-well plate. Ba/F3 cells require RPMI/10% FBS + 0.5 ng/ml of murine IL-3 for normal growth.
Add 80 μl of the viral supernatant to the first well; 10 μl of the viral sup to the second well and 2 μl of the viral supernatant to the third well and 0 μl of the viral sup to the fourth well. Add 2.8 μl of 0.5 mg/ml (1/25 of stock) polybrene to each well and bring the volumes of all wells to 200μl with IMDM media supplemented with 10% FBS (no antibiotics). Place the plate with the cells into cell culture incubator for at least 24 hours. Incubation longer than 48 hours is not generally necessary as the infection and viral integration should be complete, and growth kinetics of infected cells could skew the flow cytometry data if the infected cells possess a growth advantage over non-infected cells.
Harvest the infected cells by pipetting into a FACS tube., Wash by adding 4 ml PBS to the tube, spin at room temperature at 1700 rpm, then remove the PBS by gently pouring. Perform FACS by collecting fluorescence from FL1/GFP channel.
Calculate the IU/ml for three samples as (Number of Cells Plated × Fraction of Cells GFP+)/(ml virus used) = IU/ml. Therefore if 100,000 cells were plated, 10% were found to be GFP+, and 10 μl viral supernatant was used, the equation would give (100,000 cells × 0.1)/(0.01ml) = 1,000,000 IU/ml. Titer >500,000/ml is considered sufficient for infection of murine hematopoietic progenitors.
Depending on the experimental goal there might be a requirement to co-express in the murine cells MLL-AF9 (or MLL fusion of choice) with another oncoprotein such as FLT3-ITD, NRAS, KRAS, etc. In this case the retroviral supernatant needs to be produced and tested for the second oncogene. It is important to consider the second selection marker. For instance, if GFP is used for MLL-AF9, then Tomato or mCherry reporter proteins might be used for the second oncogene. The use of these two markers will allow selection of double-infected cells using fluorescence activated cell sorting (FACS).
Harvesting Bone Marrow
For experiments where whole bone marrow will be infected, it is critical to pretreat donor mice with 5-fluorouracil (5-FU) five days prior to bone marrow harvest. The 5-FU will act as a bone marrow ablative agent, thus causing the marrow to try to replenish itself (Szilvassy et al., 1996). The cells in the marrow will be actively cycling, which is the state necessary for successful retroviral integration.
NOTE: Female mice are generally more manageable than males, and are therefore used almost exclusively in these experiments.
NOTE: For each leukemic mouse that needs to be generated, normally one donor mouse is needed. After 5-FU treatment, each mouse will generally have roughly 107 bone marrow cells remaining in its femurs and tibias. After the infection and pre-incubation steps (below), the number of viable cells per donor will shrink to 1-2 × 106 cells. Normally, at least 1-2 × 106 infected cells are injected into recipient mice, so it is a good practice to maintain a 1:1 donor to recipient ratio.
Procedure:
5-FU is injected at a dose of 150 mg/kg ip from a solution of 8 mg/ml in PBS. This stock concentration used is based on the solubility of 5-FU.
-
12
Rodents are generally held head down with abdomen facing up for IP injections.
-
13
Hold the upper body of the mouse, wrap the tail around the lower forearm and stabilize the hind end of the animal to help restrain the animal and expose the abdomen.
-
14
If possible, inject into the left side of the abdomen to help avoid the cecum. Use a small needle: 27G is appropriate
-
15
Once the needle is inserted, draw back gently on the syringe plunger to confirm no blood, urine or intestinal material is aspirated. Depress plunger firmly to expel dose. Withdraw needle.
Five days after 5-FU administration, harvest bone marrow
Femurs generally give adequate numbers of cells, but tibias may also be harvested
-
16
Euthanize the mouse as per IACUC guidelines and spray the area for marrow harvest with 70% ethanol
-
17
Pull fur and skin away from hind legs
-
18
Cut tissue above and below to expose bones
-
19
Cut bone at ankle (if tibia is to be collected) and scrape away any remaining soft tissue, then cut below knee – place clean bone into 50 ml conical tube containing 25 ml RPMI media at room temperature.
-
20
Cut above knee and below hip for femur, again scraping away soft tissue and place bone in conical tube.
After all bones are collected, marrow can be flushed with RPMI/10% FBS/Penicillin-Streptomycin using a syringe (generally 10 ml or greater) with a 26-1/2G needle
-
21
Hold the bone with forceps over a clean 50 ml conical tube. Note: this is generally done at room temperature, and may be done on a benchtop or in a cell culture hood, depending on preference.
-
22
Insert the needle into an end of the bone: if needle does not enter easily, it may be necessary to trim the end of the bone with scissors to allow needle entry
-
23
Flush the bone until it appears clear (1-2ml/bone is generally sufficient)
-
24
After all bones are flushed, centrifuge at 1700 RPM (500× g) at room temperature for 5 minutes, remove supernatant
-
25
Add 10 ml red blood cell lysis buffer to each tube and place on ice for 10 minutes: solution should become clear red
-
26
Stop the lysis by adding 25 ml RPMI/10%FBS and gently mix by rotating the tube manually
-
27
Run cell solution through a 70 μm cell strainer (Falcon into a clean 50 ml conical tube and centrifuge at 1700 RPM (500× g) at RT for 5 minutes, then discard the supernatant
-
28
Resuspend the cells in PBS, spin at 1700 RPM for 5 minutes at room temperature and then aspirate the supernatant, then resuspend cells as follows:
-
29
For generation of primary leukemias, resuspend at 1 – 2 × 107 cells per ml in RPMI/10%FBS supplemented with 6 ng/ml mIL3, 10 ng/ml mIL6, and 100 ng/ml mSCF in 6 well plates at 106 cells per ml media for overnight incubation for infection the following day
-
30
For analysis of primary or secondary leukemias, resuspend PBS and determine cell concentration, then prepare cells for freezing, flow cytometry or other procedure.
Viral infection of bone marrow cells
(see above for materials)
Procedure:
-
31
Based on the above calculations for IU/mL, resuspend bone marrow cells or sorted progenitors so that each cell is being infected with 2 viral particles (multiplicity of infection = 2). From the IU/ml value generated in step 11, if the IU is 106/ml, 2 ml of viral supernatant (2 × 106 IU) would be added to 106 cells, giving 2 IU/cell. To the viral supernatant, add 6 ng/ml mIL3, 10 ng/ml mIL6, and 100 ng/ml mSCF, and polybrene to a final concentration of 5 μg/mL. Use an appropriate plate size for the number of cells being infected.
-
32
Spin the plate at 1,800-2000 rpm (define xg for 90-120 minutes at 37°C (termed spinoculation: spin + inoculation. Note: wrapping the plates in plastic wrap is a good idea to prevent viral contamination within the centrifuge, and keep the cells from being contaminated as well.
-
33
Incubate the cells for 4 hours at 37°C/5%CO2?.
-
34
Wash the cells in PBS (explain briefly)
-
35
Replate the cells in RPMI/10%FBS supplemented with 6 ng/ml mIL3, 10 ng/ml mIL6, and 100 ng/ml mSCF. Keep cells between 1-2 × 107cells/ml. Incubate overnight at 37°C/5%CO2.
-
36
Harvest the cells by pipetting into a clean 15 ml tube, and spin the tube at 1700 rpm, 5 minutes, room temperature. Aspirate off the media, and resuspend in PBS, and repeat the spin. Aspirate the PBS, add fresh PBS (keeping the cells at 5 × 107 gives 106 cells per 200 μl, which is a good volume for injection) and implantinto mice.
Viral efficiency testing using methylcellulose
For assessing infection efficiency, roughly 10,000 cells should be set aside and grown in semisolid media (methylcellulose) containing myeloid-specific growth factors: 100 ng/ml mSCF, 6 ng/ml mIL-3, and 10 ng/ml mIL-6 and selection antibiotic, if applicable. As every colony arises from a resistant (infected) cell, the number of colonies per cell number plated is the infection efficiency. If GFP is used, plate fewer harvested cells and perform flow cytometry for % GFP positive cells for a measure for efficiency
Procedure
-
37
Using a 2 mL Eppendorf tube, pipet in the cells, then add the cytokines and selection agent if necessary as indicated in the Unit Introduction.
-
38
Using a 3 ml syringe with a blunt end needle, bring up approximately 1.8 ml methylcellulose, avoiding bubbles
-
39
Resuspend the cell and cytokine mixture with the methylcellulose without introducing air bubbles into the mixture, and draw up into the syringe, and plate.
-
40
Keep plates in a small closed chamber with an open plate with 4-5 ml PBS to ensure the methylcellulose does not dry out. Incubate at 37° C/5%CO2.
-
41
After one week in culture, count colonies using an inverted light microscope with a 4× objective. Note: it is very useful to either mark the underside of the plate being counted or, place the plate in a tray with a grid to track your place on the plate while counting.
-
42
If monitoring GFP positivity, add 3 ml RPMI to each well/plate and incubate at 37°C for 15 minutes to dissolve the methylcellulose. Pipet up and down multiple times to fully break up the methylcellulose, then add to a 15 ml conical tube. Fill the conical tube with RPMI to further wash.
-
43
Rewash the cells with PBS, then prepare for flow cytometry by aliquoting into FACS tubes on ice.
Basic Protocol 2
Implantation of MLL-AF-9 Expressing Mouse Cells Into Recipient Mice
Recipient mice must be irradiated, and the proper amount of irradiation should be determined empirically, as each strain has a slightly different sensitivity to irradiation. An excellent primer for murine bone marrow transplant assays is Duran-Struuck and Dysko, (Duran-Struuck & Dysko, 2009).
Mouse irradiaton protocol
In general, either a sublethal (600 Rads) or lethal (1000 Rads) dose of irradiation for C57/B6 mice is given 4-24 hours prior to transplantation. Often, to avoid toxicity the lethal dose is split (4 hours) in two doses of 500 Rads each. Lethality is brought on by bone marrow failure, which is rescued with healthy, transplanted bone marrow.
Move mice from home caging to housing appropriate for the irradiator being used
Place housing with mice inside the irradiator, and irradiate the mice with either half of a lethal dose (500 rads) for primary leukemias, or a sublethal dose (600 rads) for secondary leukemias. The amount of time needed to generate these amounts of radiation will be dependent on the specific irradiator being used.
After irradiation, move the miceback to home caging for regular husbandry.
If mice require lethal radiation, the first of the two half-doses may be administered the day before inoculation, and the second should be administered 1 – 2 hours before inoculation.
Note: Mice receiving transplants from whole bone marrow should receive a lethal dose of irradiation, while mice receiving secondary leukemic transplants, or mice only receiving transduced progenitors should receive a sub-lethal dose, generally half the lethal dose. In this case, the sub-lethal (600 Rads) dose is toxic to a portion of the recipient bone marrow, thereby making space for the transplanted cells in the bone marrow niche.
Implantation protocol
Recipient mice should be at least 6 weeks old, (∼ 20 grams) for best results. These mice are generally most tolerant of therapeutic agents. Bone marrow/leukemic cells are introduced into recipient mice via intravenous injection into the lateral tail vein Injected volumes are kept to approximately 100 μl for ease of injection and reproducibility.
Materials list
-
-
Working vivarium (with protocols in place for your work)
-
-
C57BL/6 mice, female, 6 - 8 weeks old (Charles River, Jackson Labs, Taconic)
-
-
Irradiator capable of holding rodents
-
-
Irradiator safe housing – e.g. mouse pie cage (Braintree Scientific)
-
-
Ethanol wipes
-
-
Heat lamp (Braintree Scientific)
-
-
1 ml syringes (BD Biosciences)
-
-
Needles – 27G (BD Biosciences)
-
5
Warm the animal or tail (e.g., warm towel, warm water, heat lamp, or other heating devices) to make identification of the vein easier. Carefully observe the animal to avoid overheating or possible skin burns.
-
6
Select an appropriate syringe and needle (typically 26-27 gauge).
-
7
Draw 200 μl of the cells in PBS into the needle. While the cells remain on ice, allow the needles holding the cells to equilibrate to room temperature, so cold fluids are not injected into the mice.
-
8
Wipe the area with an antiseptic or ethanol wipe and start at the tip of the tail in case animal has to be injected again.
-
9
Insert the needle, bevel up, into the vein. If possible, pull back on the plunger and withdraw a small amount of blood into the hub to ensure correct position of the needle in the vein. If no blood enters the syringe, withdraw the needle and reposition.
-
10
Slowly depress the syringe plunger to expel the dose. If the needle is not properly in the vein, the tail will begin to swell (fluid is being expelled into the subcutaneous area)
-
11
Remove the needle and apply pressure to the puncture site until bleeding stops (5-10 seconds).
Basic Protocol 3
In Vivo Analysis of The MLL-Fusion Driven Mouse Leukemia Model for Disease Progression and Pharmacological Studies
Before using the MLL-AF9 or any other murine leukemia model for compound testing, the decision should be made as to whether testing on the de novo primary leukemia or an established secondary leukemia is more relevant for a particular project goal. While primary and secondary leukemias are phenotypically similar (Stubbs et al., 2008), there are advantages and disadvantages to consider. Testing compounds on a primary leukemia may provide more physiological relevance, but data may be more difficult to interpret due to the wide window of disease latencies, which could span 9 – 14 weeks post cell injection. Testing on a secondary leukemia may be a more rigorous test for potential therapeutics with more consistent latencies, but these more aggressive stem cell-nriched leukemias have a tighter experimental window due to a more rapid decline in health of the mice that will not be correctable with therapeutics.. Regardless of the methodology, the MLL mouse model has been shown to be a very relevant tool for preclinical analysis of new therapeutic options for AML (Zuber et al., 2009).
Tracking engraftment/disease progression
This step is critical for determining when dosing with a potential therapeutic agent should commence. Beginning dosing before leukemia is detectable systemically either in the peripheral blood or by bioluminescent imaging does not recapitulate the experience of a leukemic patient, and therefore may not accurately assess compound efficacy. If dosing commences when the leukemic burden is too high, the experimental window closes and the lethality of the disease will prevent any meaningful readout on compound efficacy or tolerability. Pilot studies could be useful to determine the times to leukemia detection and to moribundity as a guide for a given dosing regimen.
Materials
-
-
Working vivarium (with protocols in place for your work)
-
-
C57BL/6 mice, female, 6 - 8 weeks old (Charles River, Jackson Labs, Taconic)
Capillary tubes (ThermoFisher Scientific)
Vacutainer tubes for blood collection (BD Biosciences)
Gauze pads
Red blood cell lysis buffer (Qiagen)
15 ml conical tubes (Corning)
PBS (Gibco)
Flow cytometry tubes (Falcon)
Fluorescein tagged CD45.1 and CD45.2 antibodies (Affymetrix/eBiosciences/ ThermoFisher Scientific)
Flow cytometer with appropriate fluorescence excitation/detection capabilities
In vivo grade luciferin (Promega)
1 ml syringes (BD Biosciences)
Needles – 27G (BD Biosciences)
Procedure
As mentioned in the Unit Introduction, there are different ways to track disease engraftment and progression based on the tools at hand. The most popular are by tracking the leukemic population in the peripheral blood by drawing blood from the mice and analyzing the blood by flow cytometry (steps 1- 10 below), or by using in vivo luminescence (steps 11 – 15).
Orbital sinus/plexus (retro-orbital) bleeding
Note: The same eye must not be used successively as the bleed site on a given animal (i.e., alternate between eyes). There must be at least one week between bleeds on the same eye of a specific animal.
Anesthesia is required for retro-orbital blood collection. Anesthetize the animal using an approved method according to your IACUC.
Insert the end of a capillary tube at the median angle of the orbital conjunctiva (inner corner of eye).
Gently rotate the capillary tube and apply pressure until it nicks the underlying tissue.
Blood will freely flow from through the tube into a blood collection tube, usually containing heparin and/or EDTA.
-
When collection is complete, remove the tube, apply pressure with a gauze pad to the area to stop bleeding, and remove any excess blood.
Normally at least 50μl blood is necessary for accurate testing, with 200μl being the upper limit of what is safe to draw from the mouse.
Ensure that the animal recovers uneventfully from anesthesia, and apply lubricant to the eye if necessary. If the mouse does not fully recover from anesthesia (signs include labored breathing, lethargy, impaired gait) the mouse should be euthanized to avoid unnecessary suffering.
Detection of leukemic engraftment by flow cytometry
-
7
Add 1ml of Red Blood Cell (RBC) lysis buffer for every 100μl of blood in 5ml FACS tube, incubate on ice for 10 minutes.
Note: watch for the lysate solution to turn from opaque to clear. This is a good sign that lysis of the red blood cells is occurring appropriately.
-
8
Add 4 ml of PBS, spin cells at 500× g or 1800 rpm at RT? for 5 minutes. Aspirate PBS, then wash again with 5 ml PBS.
If cells express GFP,-resuspend in 300 μl PBS and test for GFP positivity using flow cytometry. Keep cells on ice.
If cells need to be tested for CD45.1 or CD45.2 positivity continue as follows:
-
9
Divide cells into four tubes and bring volumes up to 300 μl with PBS/10% FBS
-
10
Add fluorescein-tagged CD45.1, CD45.2, or both antibodies (Affymetrix/eBiosciences) at 1:100 in 300 μl PBS 30 minutes on ice in dark. Leave one tube as an unstained control. Fill FACS tube with PBS and spin at 500× g for 5 minutes at RT?. Gently decant PBS and resuspend cells in 300 μl PBS. Cells are ready for analyses by flow cytometry.
Note: For CD45.1/CD45.2 detection, choose fluorescent tags that will have little to no spectral overlap (FITC and APC for example).
Note: Only single-positive cells should be viewed as positively staining. Above 0.1% (10 events out of 10,000) positivity is threshold for engraftment, and that mouse is eligible to be placed on study (see Figure 1).
Figure 1. Representative CD45.1 /CD45.2 hematopoietic cell FACS plot.
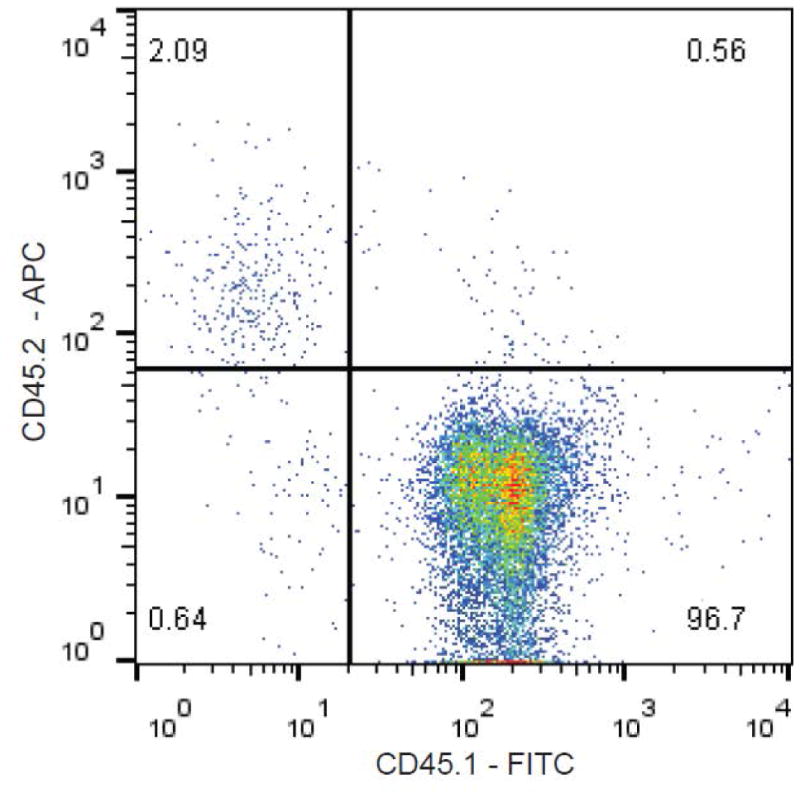
Shown are CD45.1 and CD45.2 levels found in a mouse that was chimeric for CD45.1+ and CD45.2+ hematopoietic cells. Typically, when the level of transplanted leukemia cells reaches 0.1% in the peripheral blood, a mouse is deemed to have an engrafted leukemia, and should be available for pharmacologic experiments.
Detection of leukemic engraftment by bioluminescence
Resuspend Luciferin in PBS for injection at 50 mg/kg
For imaging, weigh mice and anesthetize (per individual IACUC regulations).
Administer Luciferin by intraperitoneal injection at 50 mg/kg.
-
Image photonic emission using an in vivo imager.
Peak luminescence usually occurs 5-15 minutes after injection of luciferin.
Quantify total body bioluminescence (see Figure 2) by integrating the photonic flux (photons/sec) through a region of interest drawn around each mouse using appropriate software.
Figure 2. Monitoring bioluminescence for engraftment and drug efficacy.
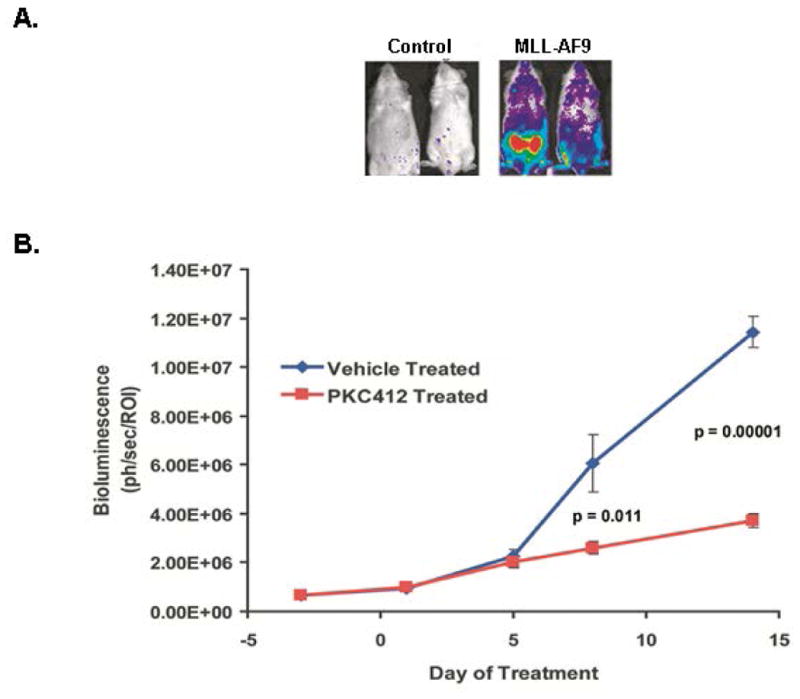
A. The MLL-AF9 mice shown here expressed the Luciferase gene. At roughly 10 weeks post injection of cells, mice were imaged for bioluminescence using the Xenogen (Perkin Elmer) IVIS system. Also shown are control mice that were injected with Luciferase-expressing cells that did not receive the MLL-AF9 expression vector. B. Primary leukemias injected into secondary recipients were intraperitoneally dosed once daily with the pan- kinase inhibitor PKC412 at 150 mg/kg when mice achieved a mean baseline of roughly 106 photons/second/ROI. Shown is the graph of luminescence over time during treatment of these mice with PKC412 (Stubbs et al., 2008).
Note: precise methods of luminescent monitoring will differ slightly due to differences in hardware and software.
Basic Protocol 4
In Vivo Analysis of Therapeutic Agents in Mice Bearing MLL-Rearranged AML
After mice are determined to be leukemic, the mice must be sorted into dosing cohorts that have similar average leukemic engraftment. Once this step is performed, dosing with a test article may commence until an appropriately chosen endpoint is met, based on the goals of the study.
Materials
-
-
Needles for intraperitoneal injection (27G is suitable) (BD Biosciences)
-
-
Needles for oral gavage (Braintree Scientific)
-
-
1 ml syringes (BD Biosciences)
-
-
50 ml conical tubes (Falcon)
Enrolling engrafted mice on a pharmacology study
To bin mice into dosing groups, we first rank the mice from highest to lowest amount of engraftment based on percentage fluorescence positivity in the peripheral blood or total body luminescence (see Detection of leukemic engraftment by flow cytometry and Detection of leukemic engraftment by bioluminescence).
Procedure
-
Then, based on the number of cohorts, sort mice as follows. For an experiment with four dosing groups:
% engraftment rank Cohort 1 1 2 2 3 3 4 4 5 4 6 3 7 2 8 1 Repeat until each cohort is filled. Ideally, each cohort should have at least seven mice. This is the minimum powering for an experiment that will ensure statistically significant results for any given changes due to pharmacology.
Final readout: survival
In general, there are two widely used criteria for determining the efficacy of a potential therapeutic agent in the MLL-AF9- (or other oncogene) driven murine leukemia model. The first is the most straightforward:survival benefit. Researchers can empirically determine the effects of a test compound on the ability of mice that receive it to survive longer than leukemic mice that receive a vehicle control.
-
3
Dosing begins upon confirmation of leukemia either by appearance in the peripheral blood (0.1% positivity) or a threshold value of luminescence being passed.
-
-
Dosing regimens are decided by the investigator; see Troubleshooting for potential concerns.
-
-
Dosing may last as long as tolerated, or until mice are moribund due to leukemic outgrowth.
-
-
-
4
Monitor mice for disease progression:
Note: to track leukemia progression, bioluminescence may be monitored throughout the duration of the experiment (as shown in Figure 2), with time points subject to allowable anesthesia limits as set by your IACUC. In general, monitoring more frequently than every 3-4 days may not be advisable due to health concerns for the mice involved. Likewise, if tracking progression by using peripheral blood/flow cytometry, bleeding of mice should occur infrequently (no more than once per week), and with the smallest volume of blood needed for successful analyses.
Note: Mice should be euthanized according to humane endpoints criteria set out by your IACUC. Generally, hunched posture, ruffled fur, labored breathing, weight loss, loss of mobility are early stage signs of moribundity. Later, mice will be non-responsive to stimuli and immobile. At the time of euthanasia, necropsy should be performed on each mouse to ensure that moribundity was brought on by leukemia: cardiac heart puncture for peripheral blood analysis (CBC), spleen weight, should be monitored as signs of leukemia.
-
-
Mice can be kept on study as long as they do not meet the criteria for humane endpoint euthanasia as stated above.
-
-
A dosing holiday may occur if it is determined that the mouse is not tolerating the test agent – this should be clearly noted.
-
-
-
5
Death of the mouse is NOT the endpoint. Mice should be humanely sacrificed when they are determined to be moribund, as noted previously.
-
-
As mice succumb to leukemia, note the number of days the mouse survived after transplant.
-
-
Construct Kaplan-Meier survival plots to show the survival times of each mouse in each dosing cohort.
-
-
-
6
Perform necropsy on each mouse to ensure that there were no irregularities, and that leukemia was the most likely cause of moribundity.
Basic Protocol 5
Final Readout: Leukemic Burden
The second criteria generally used to determine efficacy of a therapeutic agent is leukemic burden. Leukemic burden can be measured several ways, depending on the system being used. The most straightforward way of determining leukemic burden is by spleen weight. Isolating and weighing the spleens of mice sacrificed at the end of the study is a generally accepted surrogate for leukemic burden (see (Stubbs et al., 2008) Figure 6 for an example). Additional information is usually also necessary for a more critical analysis of the effects of a particular compound on leukemic growth. If a fluorescence-expressing viral vector is used (pMIG-MLL-AF9 for example), or if CD45.1/45.2 mice were used, then flow cytometry is performed on isolated bone marrow or spleen cells (below) from leukemic mice after the dosing interval is completed. The percentage of fluorescence-positive leukemia cells provides a measure of leukemic burden. It is also advantageous to be able to convert this percentage into an actual number of bone marrow cells, as therapy may add to bone marrow ablation, and therefore a straight percentage value would not be an accurate measure of drug efficacy or leukemic burden.
Materials
-
-
50 ml conical tubes (Falcon)
-
-
10 cm dish (Falcon)
-
-
Red blood cell lysis buffer (Qiagen)
-
-
10 ml syringes (BD Biosciences)
-
-
Needles – 18G (BD Biosciences)
-
-
RPMI (Gibco)
-
-
Fetal Calf Serum (Gibco)
-
-
Cell strainers (BD Biosciences)
-
-
Centrifuge (ThermoFisher Scientific)
-
-
Cytospin (ThermoFisher Scientific)
-
-
Glass slides (ThermoFisher Scientific)
Wright-Giemsa stain (Jorgensen Labs)
Generating single cell suspensions from mouse spleens
Note: this procedure may take place on a lab bench or in a tissue culture hood, depending on the amount of sterility trying to be maintained
Procedure
-
7
Place the spleen into a 10cm cell culture dish or petri dish on ice?
-
8
Add 5 mL red blood cell lysis buffer to the dish.
-
9
Using the back of a syringe, press firmly on the spleen until it begins to break down. Continue to press until the spleen is unable to be ground any further.
-
10
Allow the ground spleen to sit in RBC lysis buffer for 5-10 minutes on ice?
-
11
Add 10 mL RPMI/10%FBS to the plate.
-
12
12. Using a 10mL syringe with an 18G needle, aspirate the spleen cells up and down at least 5 times to make a single cell suspension.
-
13
Eject the cell suspension into strainer on top of a 50 mL conical to filter out debris.
-
14
Centrifuge the cells at 1700 rpm (500× g) for 5 minutes at RT or 4C?
-
15
Resuspend the cells in 25 mL PBS, then centrifuge again.
-
16
Resuspend the cells in PBS to what volume? and count.
-
17
Cells should then be ready for flow cytometry, cytospin, or other analyses.
Note: Slides made from use of a cytospin should be fixed and Wright-Giemsa stained according to manufacturer's protocol. Jorgensen Labs Jorvet Quick Dip (catalog number J-322) is very easy to use and gives a rapid readout.
Commentary
Background Information
Historically, potential therapeutics for acute myeloid leukemias (AMLs) have not fared well in the clinic. Clearly there is an unmet need in AML, with even the best clinical outcomes resulting in less than 50% 5-year survival rate. The subset of AML discussed here, leukemias driven by MLL fusion proteins, accounts for nearly 20% of pediatric AML, but has one of the poorest predicted outcomes of all types of pediatric AML (Balgobind et al., 2009). New targets are advancing into the clinic, with mixed success (Stein & Tallman, 2016), but clearly more work is needed. There is a need for models where the genetics are well defined in order to best identify on target effects of new therapies. While most AML cell lines have experienced genetic drift over the years and many have complex karyotypes and unclear driver mutations, MLL-rearranged mouse models have had their gene expression and epigenetic regulation thoroughly vetted (Krivtsov et al., 2013; Krivtsov et al., 2006; Somervaille & Cleary, 2006). Therefore, these models not only allow for the precise study of mechanisms of action for novel therapeutic agents, but also are very useful for study of several other aspects of leukemia biology pertinent to rational drug design, such as sensitivity of leukemia stem cells to a given agent, or myeloid differentiation as a therapeutic outcome.
Critical Parameters
For best viral production, 293T cells should be low passage, under 10 if possible, and not confluent at time of transfection.
Mice should be acclimated to the vivarium according to IACUC guidelines and be of sufficient age and weight (roughly 8 weeks old, near 20g) before injecting leukemic cells. The maximally tolerated in vivo dose and, if possible, a toxicity profile and pharmacokinetics of any compound being used in mice should be known before engaging in pharmacologic studies.
Tissue necrosis and cell death commences on sacrifice of the mouse so it is critical to perform necropsy and save cells/organs within 2 hours after final dose, and as close to immediately after sacrifice as possible for cleanest readouts.
Anticipated Results
The myeloid leukemia generated from the MLL-AF9 model, whether induced from hematopoietic stem/progenitor cells isolated from whole bone marrow, yields a very reproducible immunophenotype (Figure 3). Leukemic marrow is predominantly Mac1+/Gr1+, with an increase in c-kit positivity, and a greatly diminished B cell population as measured by B220 positivity when compared to normal bone marrow. This phenotype carries from primary to secondary leukemia as well (Stubbs et al., 2008). Latency of primary leukemia may vary depending on the oncogene used, the number of cells injected, and the efficiency of the viral transduction. In general, the earliest that primary recipients would become moribund is around 7 weeks post transplantation, but 10-15 weeks is still an anticipated timeframe for leukemic generation. Secondary leukemias are much more predictable in terms of latencies, and latency can be manipulated by altering the number of injected primary leukemic cells into the secondary recipient (see Figure 4). Results post dosing will vary with the therapeutic agent that is chosen, dosing schedule, therapeutic window, and toxicity.
Figure 3. Representative FACS plots of bone marrow from mice harboring MLL-AF9 driven AML.
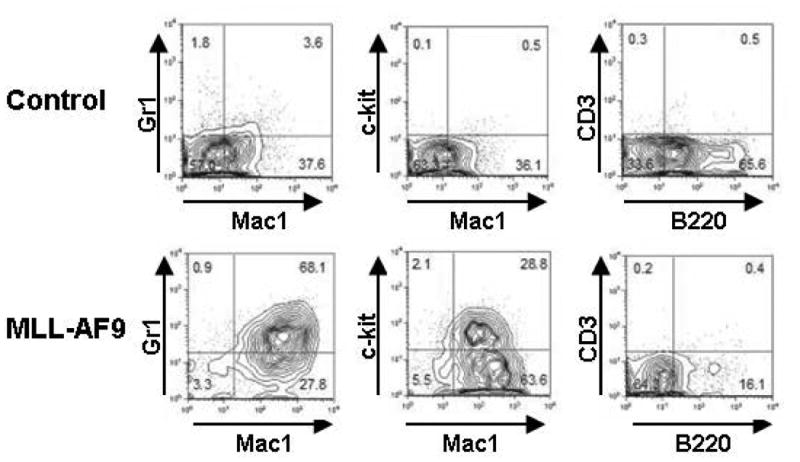
Bone marrow from leukemic mice has a distinct immunophenotype, differing from normal marrow. Shown is a representative FACS panel using the myeloid markers Mac1 (CD11b), Gr1 (Ly6G), the marker of less differentiated progenitors c-kit, and the B and T cell lineage markers B220 and CD3 respectively. Leukemic bone marrow has a large Mac1+/Gr1+ population, an expansion of c-kit+ cells, and a decrease in the B cell compartment (B220+ cells). This pattern can be seen in mice with either primary secondary leukemias. (Stubbs et al., 2008)
Figure 4. Typical latencies from transplant of primary leukemias into secondary recipients.
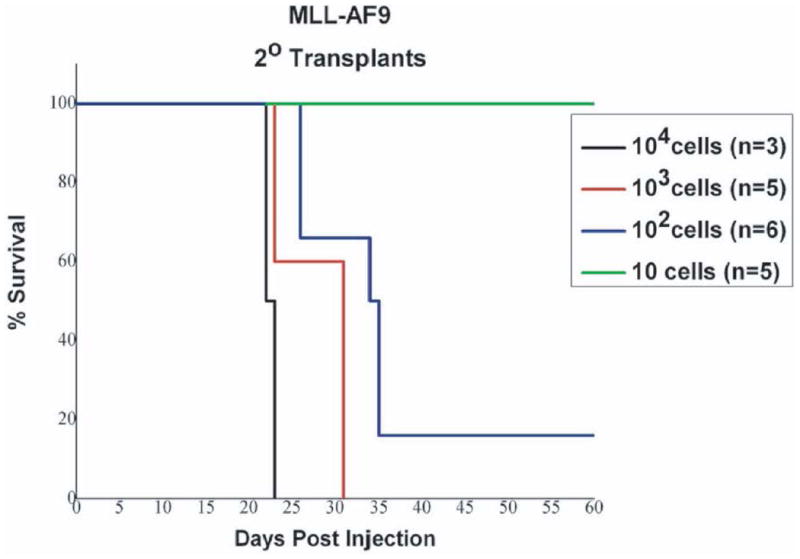
Kaplan Meier survival plot for mice injected with a primary MLL-AF9 derived leukemia (Stubbs et al., 2008). The latencies of disease shown here are directly related to the number of leukemic cells injected into the secondary recipient. These leukemias are more aggressive, with a much shorter latency than primary leukemias.
Troubleshooting
There are three major hurdles to clear for successfully using murine leukemias for determining efficacy of a potential therapeutic agent. If any of these critical areas of the protocol are not executed properly, the experiments will not yield meaningful (if any) results.
-
Primary recipient mice must become leukemic
There are several possibilities for why these protocols might not result in leukemic mice. First, if the virus produced is not high enough titer (infectious units/ml are too low), logically little to no transformation of the bone marrow will occur. To guard against this, viruses should be checked and the (IU/ml) should be determined using Ba/F3 cells prior to beginning mouse work. See “Retrovirus Production” in Basic Protocol 1. Further, after infection of donor bone marrow, if the number of colonies that grow out in methylcellulose is low or not GFP-positive, then the virus should be rechecked by re-infecting BaF3 cells and assaying for GFP positivity by flow cytometry as in “Retrovirus Production” Also, make sure the 5-FU has not expired and has been stored properly. If the 5-FU is not acting properly, the donor bone marrow will not be capable of infection.
-
Therapeutic window must be established
It is important to remember that leukemic cells are replacing healthy bone marrow, therefore compounds known to cause anemia or thrombocytopenia may be toxic at high doses. Therefore, it is critical that any therapeutic dose used selectively targets leukemia cells while largely leaving any healthy bone marrow intact. Likewise, off-target toxicity should be mitigated. Ensuring that a compound is pure (> 99.5%) and used at a concentration/dose consistent with its known mechanism will increase the likelihood of it engaging its specific target. Another way to minimize toxicity from the agent of choice is to maintain a minimum mouse size for experiments. Generally mice that are at least 8 weeks old and weigh over at least 18g are more able to contend with the effects of both the leukemia and the therapeutic agents being evaluated.
-
Therapeutic agent must change disease latency or leukemic burden
Depending on the compound under investigaton, either mouse survival or their leukemic burden should be affected as a read-out of compound efficacy against the myeloid leukemia. To assess the potential effectiveness of a compound in this model, it can be added to methylcellulose, along with primary (or secondary) leukemic cells to check its ability to reduce colony number, size, or type. If cell survival or differentiation status is observed in vitro/ex vivo at a concentration representing an achievable in vivo dose, there is a greater chance of seeing effects in vivo. This is not a direct correlation, but a trend.
Time Considerations
These protocols contain several parts that require lengthy incubations or numerous hours to accomplish, the first of which is virus production. To produce high-titer viral supernatants, plasmid preparations by maxiprep are preferred to obtain highest yield and purity. This will allow for the best transfection efficiency which should in turn lead to better viral production. To improve viral production, 293T cells require a few days to equilibrate after thawing and plating. The time from plasmid production, through transfection of 293T cells, to viral harvest will take approximately 5 days.
Another point to consider is that mice brought into the vivarium will not be cleared for use for several days, depending on local IACUC guidelines. So, when preparing for an experiment, make sure mice will be housed several days before initiating a study.
Bone marrow harvests are also lengthy endeavors. Taking femurs from 10 mice can take an hour, and the subsequent marrow harvest may take at least another hour depending on researcher proficiency. It is best to reserve an entire morning for harvesting mouse bone marrow.
One final scheduling consideration is that it is crucial to save relevant organs and cells quickly after the final compound dose. At the conclusion of a experiment, the sacrifice and necropsy (organ harvest, terminal blood draw, saving viable cells from marrow or spleens) of the mice on a study can require nearly an entire work day, depending on the number of people involved.
Acknowledgments
We would like to thank Dr. Scott Armstrong for his time, his instruction, and his patience as he taught us this model. Work from the Armstrong laboratory was supported by grants to Scott Armstrong from the NIH (CA92551), the Leukemia Lymphoma Society, the Charles H. Hood Foundation, and the Damon Runyon Cancer Research Foundation.
References
- Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, van den Heuvel-Eibrink MM. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489–2496. doi: 10.1182/blood-2009-04-215152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson AE, Knobel KM, Rood D, Chen K, Lamons D, McNally MA, Lebkowski JS. Selection of murine lymphoid and hematopoietic cells using polystyrene tissue culture devices containing covalently immobilized antibody. Biotechniques. 1996;20(6):1098–1103. doi: 10.2144/96206pf02. [DOI] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17(24):3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Armstrong SA. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121(13):2533–2541. doi: 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood. 2002;99(10):3780–3785. doi: 10.1182/blood.v99.10.3780. [DOI] [PubMed] [Google Scholar]
- DiMartino JF, Miller T, Ayton PM, Landewe T, Hess JL, Cleary ML, Shilatifard A. A carboxy-terminal domain of ELL is required and sufficient for immortalization of myeloid progenitors by MLL-ELL. Blood. 2000;96(12):3887–3893. [PubMed] [Google Scholar]
- Duran-Struuck R, Dysko RC. Principles of bone marrow transplantation (BMT): providing optimal veterinary and husbandry care to irradiated mice in BMT studies. J Am Assoc Lab Anim Sci. 2009;48(1):11–22. [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, Armstrong SA. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27(4):852–860. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Armstrong SA. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16(14):4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ME, Milne TA, Bloyer S, Galoian K, Shen W, Gibbs D, Hess JL. Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell. 2003;4(3):197–207. doi: 10.1016/s1535-6108(03)00214-9. [DOI] [PubMed] [Google Scholar]
- Schott JW, Hoffmann D, Schambach A. Retrovirus-based vectors for transient and permanent cell modification. Curr Opin Pharmacol. 2015;24:135–146. doi: 10.1016/j.coph.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10(4):257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood. 2016;127(1):71–78. doi: 10.1182/blood-2015-07-604538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, Armstrong SA. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia. 2008;22(1):66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift S, Lorens J, Achacoso P, Nolan GP. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol. 2001;Chapter 10(Unit 10):17C. doi: 10.1002/0471142735.im1017cs31. [DOI] [PubMed] [Google Scholar]
- Szilvassy SJ, Weller KP, Chen B, Juttner CA, Tsukamoto A, Hoffman R. Partially differentiated ex vivo expanded cells accelerate hematologic recovery in myeloablated mice transplanted with highly enriched long-term repopulating stem cells. Blood. 1996;88(9):3642–3653. [PubMed] [Google Scholar]
- Zuber J, Radtke I, Pardee TS, Zhao Z, Rappaport AR, Luo W, Lowe SW. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23(7):877–889. doi: 10.1101/gad.1771409. [DOI] [PMC free article] [PubMed] [Google Scholar]