

Abstract
In the not too distant past, myofilament research attracted considerable attention after the discovery that mutations in myofilament genes cause cardiomyopathies. However, basic research has not been completely translated into therapies. This viewpoint discusses the need to develop innovative and integrative technologies and generate translatable models to uncover the complex biology of myofilament proteins to advance the field.
Keywords: myofilament protein, sarcomere physiology, contractile proteins, myocardial contractility, muscle contraction
The sarcomere, composed of myofilaments, is the fundamental contractile unit of striated skeletal and cardiac muscle.1 Myofilaments, occupying 70% of heart tissue are composed of thick and thin filament proteins. Post-translational modifications of myofilament proteins regulate the rate and force of contraction.2 Mutations in myofilament proteins cause contractile dysfunction leading to cardiomyopathies.3 Therefore, studying the structure and function of myofilament proteins is essential to understand basic muscle physiology. Since the late 1800s, scientists have studied myofilament biology, and interest in these proteins dramatically increased following the discovery of a point mutation in myosin that caused hypertrophic cardiomyopathy.3 From the late 1990s to the early 2000s, scientists, flush with research funding, made a number of significant discoveries. Unfortunately, continued reductions in funding and changing interests have significantly conspired to reduce the number of active muscle researchers and the enthusiasm to work in the muscle biology field. Yet, the need for muscle research remains high. The arrangement and interaction of thick and thin filament proteins in the myofilament are not completely defined.4 Increasing prevalence of heart failure demands new solutions. In response, the combination of advanced technology, improved scientific rigor, and interdisciplinary collaboration, including a renewed link to clinicians, is needed to reinvigorate myofilament research.
Advanced technologies are required to study the myofilament complex at the molecular level
The early pioneers of this field laid the fundamental groundwork for our understanding of myofilament properties by testing conditions that affect muscle behavior. These findings were almost always based on the development of new techniques to study muscle, but without the advantage of today’s accumulated knowledge and technology, such as fluorescence microscopy and basic molecular techniques.5 For example, the sliding filament and cross-bridge theories depended on the development and application of electron and dual interference contrast microscopies. Similarly, the length-tension relationship, which provided a molecular basis for the Frank-Starling Law, depended on developing tools for accurate and simultaneous measurement of muscle, or sarcomere, length and force generation.6 These were all revolutionary findings that have shaped our understanding of muscle function, clearly showing how advances in muscle research are critically linked to improvements in technology. Since these fundamental, but groundbreaking, discoveries, every decade has seen remarkable advancements in our understanding of muscle dynamics and function. While these advancements have been critical in furthering our knowledge of myofilament function in health and disease, our knowledge of the in vivo structure and function of myofilament proteins remains limited.7
Existing techniques and technological platforms used in the myofilament field have limitations.8 For example, at the single-cell level, we can measure the functional differences between control and sarcomeric mutants in pCa-force relationship studies. However, pCa-force relationships cannot entirely explain dysfunctions seen at the whole organ level. Thus, instead of detergent-skinned fiber force-pCa curves or measurement of cardiomyocyte contractility, we should be looking toward technologies that permit the resolution of intact myofilament structures in their native environment, e.g., intact trabeculae, papillary, or muscle strips, thereby allowing definitive analysis of their functional properties.9 For example, a number of researchers have elegantly used time-resolved X-ray diffraction along with measurement of contractile parameters to understand how myofilament proteins behave under specific conditions. Investigation of intact structures would allow more accurate assessments of myofilament proteins bearing pathogenic variants. However, these aforementioned techniques are incredibly specialized and expensive, and they require extensive skill and time to perform the experiments. As it stands, the study field suffers from a kind of myopia, addressing narrowly defined physiological questions instead of examining the same questions from a broader, more meaningful clinical context. Meanwhile, funding for research that only promises incremental discovery in a narrow area is diminishing. To spark renewed interest in myofilament studies, but from a reinvigorated translational/technological perspective, we should strive to establish effective crosstalk between bench scientists and clinicians, all from different disciplines.
At the level of bench science, we note the tremendous heterogeneity in cardiomyocyte sarcomere length and structure in different regions of the heart. Current in vivo studies investigate the physiology of myofilament (dys)function, but fail to address the mechanistic changes within the myofilament that lead to altered cardiac contractility. This calls for the development of new methods that can measure function at the organ level and provide resolution at the cellular and molecular levels, in turn allowing researchers to probe the myofilament complex in the context of the whole organ or organism. Solid-state nuclear magnetic resonance, electron spin resonance and fluorescence applications have broadened, resolution has improved, and bore sizes have been tailored to accommodate small animals. These technologies provide structural detail and insight into muscle diseases, but resolution of intact myofilament proteins in their native state has still not been solved. It is clear that many upstream regulators of myofilament function in vivo likely exist, but we cannot accurately recapitulate them using our current techniques.10 Similarly, current techniques make it difficult to understand how signaling downstream of myofilaments, e.g., feedback loops, regulate myofilament structure and function. To translate muscle function, as determined through established mouse models, to human disease, we must identify potential therapeutic targets in the context of normal in vivo myofilament function. Exploiting today’s technological advances to design discovery platforms able to determine such in vivo structural dynamics of myofilaments in high molecular detail is necessary to translate our structural and functional findings to real-world physiological consequences.
Translatable models and methods can verify and validate findings elucidated at the in vitro biochemical and isolated myofilament level
Current in vitro and myofilament studies contain many elements that can make them difficult to interpret biologically, thus restricting translation to human disease. Investigators in the myofilament field have utilized in vitro systems with protein constituents derived from a variety of species, which was rationalized because certain protein sources yielded better activity or were more readily available as research reagents. However, when performing alignments of protein sequences, it is quite evident that large differences can exist between protein isoforms throughout evolution. Recombinant full-length and truncated myofilament proteins generated in E. coli and insect cells are commonly used to study sarcomere machinery in in vitro reconstituted systems. However, the use of these proteins can be confounded owing to potential differences in protein folding and post-translational modifications between mammalian and bacterial cells. Even within a species, efforts should be taken to understand sources of data variability. For example, the use of different mouse strains can exhibit significant differences in cardiac disease presentation.11, 12 Additionally, animal age and sex, as well as varying transgenic copy numbers of myofilament proteins, are all variables that should be considered when drawing conclusions from these studies.13, 14
Importantly, human biopsy results should be interpreted with caution. Patient-to-patient variability, collection and storage methods, and transmural heterogeneity can contribute to inconsistencies. An exciting research area is the development of small molecules that activate/inhibit muscle contraction. Over the years, the specificity and effect of these molecules have been increasing. The rationale of targeting these molecules to the myofilament and, in particular, thick filament proteins rests on the idea that they should bypass potentially detrimental upstream signaling effects. For example, two recent and exciting discoveries were Omecamtiv mecarbil, a potent myosin activator, and MYK-461, an inhibitor of myosin ATPase. Omecamtiv mecarbil has been very successful in preclinical trials; however, its negative effects on the relaxation of the heart are concerning. MYK-461 was able to protect mouse models of hypertrophic cardiomyopathy from developing hypertrophy and reduce left ventricular outflow gradients in Maine Coon cats with hypertrophic cardiomyopathy. Whether MYK-461 translates into clinical studies is yet to be seen. Overall, these advances suggest that the development and study of these small-molecule therapies may result in a fruitful area of translatable research in the myofilament field.
In vitro motility assays are inherently intriguing. They allow observation of actin sliding over bound myosin. The kinetics measured may give some idea of contractile activity. Whether these data translate to in vivo measurements of actin-myosin dynamics is not yet clear.15 Skinned muscle assays consist of permeabilized sarcomeres that can measure contractile responsiveness to calcium and alterations in force production. However, the nature of the procedure strips away soluble factors and may affect post-translational modifications, possibly impacting the data being collected. Discrepancies in reproducibility exist between different laboratories. These might be explained by differences in protein purification and preparation, differences in animal species or strains, experimental setups, or calculation of calcium and magnesium concentrations in buffers. However, myofilament researchers appear to accept this lack of reproducibility as inevitable. As a community, developing guidelines to standardize components of assays can make the results more translatable. Inevitably, animal-based models cannot be an adequate substitute for all human diseases. However, the recent development of induced-pluripotent stem cells and patient-specific cardiomyocytes derived from these cells could provide another avenue of translatable models.
The future of the myofilament field depends on technological advancements, assay standardization, extending collaborations and using appropriate models
To advance and transform the field, myofilament researchers must extend their collaborations outside their areas of expertise so that larger, more biologically accurate studies can be performed (Figure 1). Technologies that determine the in vivo function and regulation of myofilaments should be pursued. Methods that can accurately reflect the myofilament milieu, such as the synergistic roles of regulatory proteins and modifiers should be considered. Additionally, regional and transmural cardiac differences should be contemplated—with mapping of myofilament differences being a goal. Only collaboration among myofilament researchers and clinicians from different disciplines can make these developments possible. Finally, the NIH should be encouraged to increase funding support for muscle physiology as a unique study area. Importantly, small laboratories focusing on developing techniques and important key questions should receive continuous funding to support their discoveries.
Figure 1. The future state-of-the-art myofilament field.
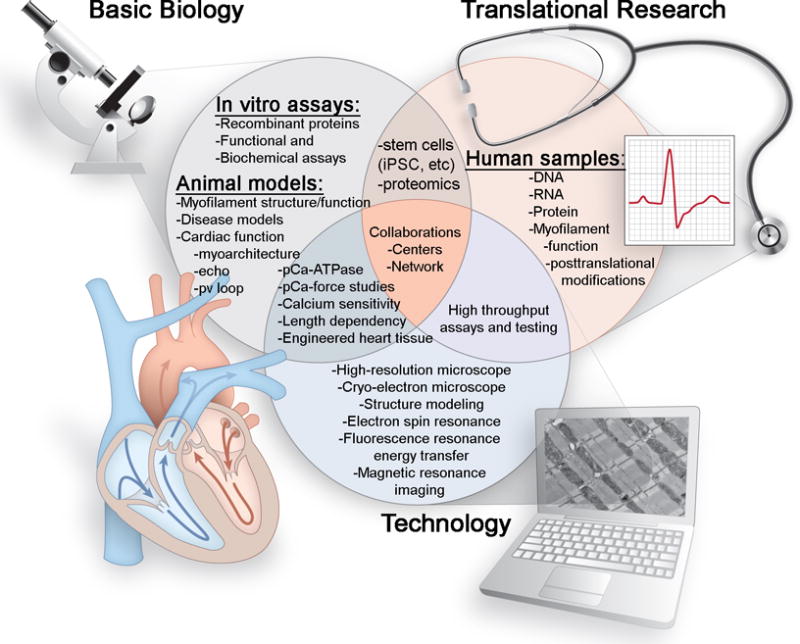
For the field to progress in an effective and rigorous manner, multimodal approaches to studying myofilament structure, function and regulation should be given increasing attention. Basic biology should be used along with advanced technologies to provide new pathways for translational research to understand and treat human cardiac diseases. The overall goal should be to generate consistent and reproducible unbiased observations that can build the foundation for these basic, clinical and translational studies in a collaborative manner.
Acknowledgments
Disclosures
Dr. Sadayappan was supported by National Institutes of Health grants (R01HL130356, R01HL105826, and K02HL114749), an American Heart Association catalyst award (17CCRG33671128), AstraZeneca, Inc., and Amgen.
Footnotes
The opinions expressed herein are not necessarily those of the editors or the American Heart Association.
References
- 1.Lin BL, Song T, Sadayappan S. Myofilaments: Movers and rulers of the sarcomere. Compr Physiol. 2017;7:675–692. doi: 10.1002/cphy.c160026. [DOI] [PubMed] [Google Scholar]
- 2.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-c in health and disease. J Mol Cell Cardiol. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: A β-cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 4.Huke S, Knollmann BC. Familial hypertrophic cardiomyopathy: Is the frank-starling law kaput? Circ Res. 2013;112:1409–1411. doi: 10.1161/CIRCRESAHA.113.301406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elting MW, Spudich JA. Future challenges in single-molecule fluorescence and laser trap approaches to studies of molecular motors. Dev Cell. 2012;23:1084–1091. doi: 10.1016/j.devcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Tombe PP, ter Keurs HE. Cardiac muscle mechanics: Sarcomere length matters. J Mol Cell Cardiol. 2016;91:148–150. doi: 10.1016/j.yjmcc.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duncker DJ, Bakkers J, Brundel BJ, Robbins J, Tardiff JC, Carrier L. Animal and in silico models for the study of sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:439–448. doi: 10.1093/cvr/cvv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marston SB. Why is there a limit to the changes in myofilament Ca2+-sensitivity associated with myopathy causing mutations? Front Physiol. 2016;7:415. doi: 10.3389/fphys.2016.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S, Woodhead JL, Zhao FQ, Sulbaran G, Craig R. An approach to improve the resolution of helical filaments with a large axial rise and flexible subunits. J Struct Biol. 2016;193:45–54. doi: 10.1016/j.jsb.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dewan S, McCabe KJ, Regnier M, McCulloch AD. Insights and challenges of multi-scale modeling of sarcomere mechanics in ctn and tm dcm mutants-genotype to cellular phenotype. Front Physiol. 2017;8:151. doi: 10.3389/fphys.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michele DE, Gomez CA, Hong KE, Westfall MV, Metzger JM. Cardiac dysfunction in hypertrophic cardiomyopathy mutant tropomyosin mice is transgene-dependent, hypertrophy-independent, and improved by beta-blockade. Circ Res. 2002;91:255–262. doi: 10.1161/01.res.0000027530.58419.82. [DOI] [PubMed] [Google Scholar]
- 12.Prabhakar R, Boivin GP, Grupp IL, Hoit B, Arteaga G, Solaro RJ, Wieczorek DF. A familial hypertrophic cardiomyopathy α-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33:1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 13.Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-c phosphorylation and cardiac function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase a phosphorylation of cardiac myosin binding protein-C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tardiff JC. Thin filament mutations: Developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]