

Abstract
The Dicer family of ribonucleases plays a key role in small RNA-based regulatory pathways by generating short dsRNA fragments that modulate expression of endogenous genes, or protect the host from invasive nucleic acids. Beginning with its initial discovery, biochemical characterization of Dicer has provided insight about its catalytic properties. However, a comprehensive understanding of how Dicer’s domains contribute to substrate-specific recognition and catalysis is lacking. One reason for this void is the lack of high-resolution structural information for a metazoan Dicer in the apo- or substrate-bound state. Both biochemical and structural studies are facilitated by large amounts of highly purified, active protein, and Dicer enzymes have historically been recalcitrant to overexpression and purification. Here we describe optimized procedures for the large-scale expression of Dicer in baculovirus-infected insect cells. We then outline a three-step protocol for the purification of large amounts (3–4 mg of Dicer per liter of insect cell culture) of highly purified and active Dicer protein, suitable for biochemical and structural studies. Our methods are general and are extended to enable overexpression, purification and biochemical characterization of accessory dsRNA binding proteins that interact with Dicer and modulate its catalytic activity.
Keywords: RNAi, Dicer, Loqs-PD, R2D2, Baculovirus, Protein purification
1. Introduction
The Dicer family of class 3 Ribonuclease III (RNase III) enzymes cleaves double-stranded RNA (dsRNA) substrates into short dsRNA fragments that regulate expression of endogenous genes, or protect cells from foreign nucleic acids [1]. Dicer enzymes contain an amino-terminal DExD/H-box helicase domain, followed by a domain of unknown function (DUF 283), the Platform and PAZ domains, two signature RNase III motifs, and a dsRNA-binding motif (dsRBM) at the carboxyl terminus [2–6]. The catalytic core of Dicer, comprising the tandem RNase III domains and the dsRBM, is derived from bacterial Class 1 RNase III enzymes [7].
Some organisms, including Homo sapiens and Caenorhabditis elegans, express a single isoform of Dicer that processes both long dsRNAs and precursor microRNA (miRNA) hairpins into mature small interfering RNAs (siRNAs) and miRNAs, respectively [1,8]. On the other hand, arthropods such as Drosophila melanogaster express two Dicer paralogs that exhibit functional specialization and generate different types of small regulatory RNAs [9,10]. The Dicer-1 enzyme (dmDcr-1) processes pre-miRNA hairpins by cleaving a portion of the terminal stem-loop structure to generate a small dsRNA consisting of the mature miRNA base-paired to miRNA*, which is subsequently discarded [9,11]. Dicer-2 (dmDcr-2) on the other hand, cleaves long dsRNAs of either cellular or viral origin into siRNAs that mediate an endogenous or antiviral RNA interference (RNAi) response [9,12,13]. Endogenous siRNAs (endo-siRNAs) are produced by dmDcr-2 from long dsRNAs of cellular origin that arise from transposon-derived sense-antisense pairs, convergent transcription of protein-coding genes, or long stem-loop structures [13]. This function of dmDcr-2 is modulated in vivo by accessory dsRNA binding proteins (dsRBPs) such as Loquacious-PD (Loqs-PD) [13–15]. Apart from the endo-siRNA pathway, dmDcr-2 also cleaves long dsRNAs from exogenous sources such as viral replication intermediates, or ectopically- introduced long dsRNAs. The resulting exogenous siRNAs (exo- siRNA) trigger an antiviral RNAi response that silences viral transcripts [12,16–18]. The products of dmDicer-1 cleavage, the mature miRNA/miRNA* duplexes, are sorted to the Drosophila melanogaster Argonaute-1 (dmAgo-1) RNA-induced silencing complex (RISC) where they mediate translational repression of proteincoding transcripts. By contrast, endo- and exo-siRNAs are preferentially sorted to the Argonaute-2 (dmAgo-2) RISC, where they mediate sequence-specific gene silencing by target cleavage and degradation [8,19,20].
Initial biochemical characterization of Dicer was performed in vitro using Drosophila cell and embryo-extracts - these early experiments demonstrated that Dicer activity in lysates processed radiolabeled dsRNAs into siRNAs, ~21–23 nucleotides (nts) in length, in an ATP-dependent manner [3,21]. The structural features of the product siRNAs - 5′ phosphates and 3′ hydroxyls, with two nt overhangs at the 3′ end, was the first hint that they were produced by a member of the RNase III family of enzymes [2]. Dicer activities were also reported in worms [4,5,22] and plants [23].
Biochemical characterization of Dicer using purified, recombinant protein was first demonstrated by Provost, Radmark and colleagues [24] and Filipowicz and colleagues [25] using Homo sapiens Dicer (hsDcr). Both groups purified recombinant hsDcr using the baculovirus expression system in insect cells. Several key observations were made by these groups, foremost among these, the recapitulation of processing of dsRNA into siRNA using purified recombinant Dicer. Filipowicz and colleagues [25] further demonstrated that limited proteolytic digestion of hsDcr enhanced its cleavage efficiency. Importantly, experiments with purified recombinant hsDcr showed that processing of dsRNA did not require ATP [24,25], contrary to studies performed with Drosophila cell lysates or purified dmDcr-2 [3,21,26]. Consistent with this observation, subsequent kinetic analyses of hsDcr revealed that the amino-terminal helicase domain attenuates cleavage of dsRNA or pre-miRNA substrates with 2-nt 3′overhanging termini [27]. Specifically, deletion of the helicase domain substantially enhances the catalytic efficiency of hsDcr [27]. Further biochemical analyses revealed that hsDcr processes pre-miRNA substrates more efficiently compared to longer dsRNA substrates [28]. Unlike hsDcr, studies performed with purified recombinant dmDcr-2 showed a profound effect of the helicase domain on substrate-specific kinetics [29–32]. For example, dsRNA with blunt termini, promoted an optimal reaction mode of dmDcr-2, whereby the helicase domain enabled ATP- dependent, processive cleavage. In contrast, dsRNA with 3′ overhanging termini, which mimic the termini of pre-miRNAs, promoted a suboptimal, distributive cleavage reaction [29,32].
A comprehensive understanding of how Dicer’s domains contribute to substrate-specific recognition and cleavage requires high-resolution structural information. However, structural information on Dicer is currently limited to Dicer enzymes from lower eukaryotes [33], low-resolution electron microscopic (EM) reconstructions of human Dicer [34–36], and crystal structures of isolated domains [37,38]. The first structural insight towards understanding the three-dimensional architecture of Dicer came from the crystal structure of Giardia intestinalis Dicer [33]. While Giardia Dicer lacks the amino-terminal helicase domain, the crystal structure of this ‘mini’ Dicer revealed that the unique spatial arrangement of the PAZ domain, which anchors one end of a dsRNA, and catalytic RNase III domains, enables Dicer to function as a molecular ruler and produce dsRNA fragments of a specific length [33]. Further structural insights derive from low resolution (≥20–30 Å) EM studies of hsDcr, which revealed that hsDcr has an “L-shaped” architecture with the amino terminal helicase domain occupying the base of the L-shape [34–36]. Using a combination of domain-specific antibodies, tagged-proteins and deletion constructs, these studies also revealed that the terminus-recognition modules comprising the Platform and PAZ domains occupy the cap or the “tip” of the L-shape, whereas the catalytic core comprising the tandem RNase III domains occupy the central core of the enzyme [34,35]. However, there are currently no high-resolution structures of a full-length metazoan Dicer, and no RNA-bound structures of Dicer are available for any organism.
A bottleneck to biochemical and structural characterization of metazoan Dicers has been the difficulty in overexpressing and purifying large amounts of the active enzyme [39,40]. We overcame this bottleneck by systematically optimizing overexpression protocols of dmDcr-2 in baculovirus-infected insect cells [32]. Next, we established and optimized an efficient three-step purification protocol for dmDcr-2 that enabled us to isolate multimilligram amounts of highly purified dmDcr-2 per liter of baculovirus-infected insect cells - yields that far exceeded those of prior protocols [32]. The details of our expression and purification protocols are described below. Our protocols are straightforward, utilize commercially available resources, and have been optimized based on diverse trials and attention to detail. These protocols have greatly facilitated biochemical and structure- based experiments with Dicer.
2. Methods
2.1. Expression of Dicer proteins using an optimized baculovirus expression system
The Bac-to-Bac Baculovirus Expression System (Thermo Fisher Scientific) was used to generate high-titer recombinant baculoviruses expressing various proteins involved in gene-silencing, with modifications as described elsewhere [41–43]. For simplicity, the protocol described below focuses on generating high-titer recombinant baculovirus for Drosophila melanogaster Dicer-2 (dmDcr-2), but the methods described here are general, and with minimal optimization may be extended to other proteins. For example, we have used the same protocols to express and purify mutant variants and truncations of dmDcr-2, hsDcr, and accessory dsRBPs (Loqs-PD and R2D2 [26]) and dmAgo-2.
The pFastBac™1 vector (Thermo Fisher Scientific) was modified to include an N-terminal One-STrEP-FLAG (OSF) tag and a PreScission protease (GE Healthcare Life Sciences) cleavage site in frame with a Sma I (New England BioLabs Inc.) restriction endonuclease site: (MASWSHPQFEKGGGARGGSGGGSWSHPQ- FEKGFDYKDDDDKGTRSPLEVLFQGP; Strep-tag® II sequences (IBA Lifesciences) underlined, FLAG-tag sequence (Sigma-Aldrich) in italics, PreScission protease cleavage site in bold). The modified pFastBac™1 vector was a kind gift from Dr. Wesley I. Sundquist, University of Utah.
The pFastBac™1 vector was linearized by SmaI digestion, and the coding sequence for dmDcr-2 was seamlessly inserted using Gibson Assembly® (New England BioLabs Inc.) following the manufacturer’s protocol. All plasmid constructs were verified by sequencing.
Recombinant bacmid DNA, created by site-specific transposition of the expression cassette from the pFastBac™1 vector into a baculovirus shuttle vector (bacmid), was generated, isolated, and verified as described in the Bac-to-Bac Expression System instruction manual (Thermo Fisher Scientific).
The pUC/M13 forward and reverse primers were used to verify the full-length dmDcr-2 cDNA in the recombinant bacmid DNA by PCR (Fig. 1A). This protocol is described in detail in the manufacturer’s instruction manual and will not be repeated here.
Fig. 1.
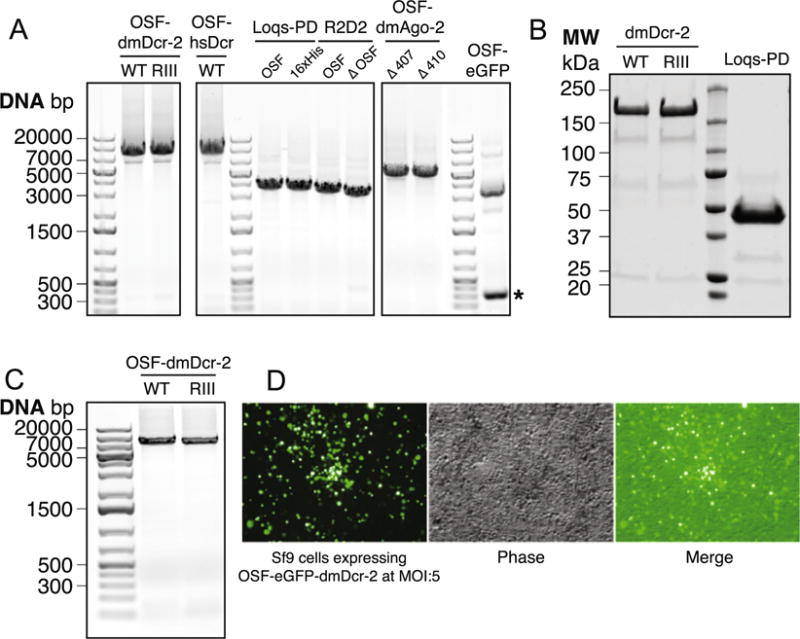
(A) PCR-based assay to analyze recombinant bacmid DNA by PCR: pUC/M13 Forward and Reverse primers were used to verify the presence of the expression cassette in the recombinant bacmid by PCR analyses. PCR products of the expected sizes were observed for OSF-dmDcr-2WT and OSF-dmDcr-2RIII (5331 + 2300 = 7631 bp), OSF- and 16xHis-Loqs-PD (1239 + 2300 = 3539 bp), OSF-R2D2 (1260 + 2300 = 3560 bp), ΔOSF-R2D2 (1098 + 2300 = 3398 bp), OSF-Δ407-dmAgo-2 (2792 + 2300 = 5092 bp) and OSF- Δ410-dmAgo-2 (2801 + 2300 = 5101 bp). The OSF-eGFP bacmid contained contaminating un-transposed bacmid, and PCR products for the recombinant bacmid (882 + 2300 = 3182 bp) and un-transposed bacmid (*, ~300 bp) were both observed - contaminated bacmids were specifically avoided for all downstream applications and are shown here specifically for representative purposes. PCR products were resolved on a 0.8% agarose gel and visualized by ethidium bromide staining. (B) OSF-dmDcr-2 and OSF-Loqs-PD expression from P1 virus: 25 ml cultures of Sf9 cells (2 × 106 cells/ml) were infected with recombinant P1 baculovirus at an MOI of 5 IU/cell and incubated for 48 h to allow recombinant protein expression. Cells were lysed and the proteins were purified on Strep-Tactin Sepharose affinity beads. Proteins were eluted in elution buffer (lysis buffer supplemented with 2.5 mM d-desthiobiotin and PreScission Protease as per manufacturer’s protocol). Eluted proteins were resolved by SDS-PAGE and visualized by Coomassie Brilliant Blue staining. (C) Baculoviral genomic DNA extraction and PCR analyses: Genomic DNA was extracted from recombinant P1 baculoviruses and pUC/M13 Forward and Reverse primers were used to verify the presence of the expression cassette by PCR analyses as described in Fig. 1(A). (D) Expression of OSF-eGFP-dmDcr-2 to determine optimal MOI: 25 ml cultures of Sf9 cells (2 × 106 cells/ml) were infected with recombinant P2 OSF-eGFP-dmDcr-2 baculovirus at an MOI of 5 IU/cell and incubated for 48 h to allow recombinant protein expression. Expression levels were monitored with wide-field fluorescence or phase-contrast microscopy.
Note: Recombinant bacmid DNA can be stored for up to 2 weeks at 4 °C in TE (10 mM TRIS pH 8.0, 1 mM EDTA) buffer. We do not recommend storing the purified bacmid DNA at −20 °C as the DNA is sheared during freeze/thaw cycles [44].
2.2. Generation of high-titer recombinant baculoviruses
2.2.1. General considerations for growing Spodoptera frugiperda (Sf9) cells
Suspension cultures of Sf9 cells (Cat# 94-001S), growing in ESF 921 media (Cat# 96-001), were purchased from Expression Systems. Cultures were passaged every two days and strictly maintained in log-phase growth for ~15 generations, at an optimal cell density of 1–5 × 106 cells/ml (27 °C, 135 rpm (~1×g); Eppendorf Innova Model 2300 large capacity platform shaker). Log-phase Sf9 cells are typically round in shape, uniform in size and do not form cell clumps. Over growth was carefully avoided. Morphological changes in Sf9 cells such as swelling (accumulation of tetraploid cells) and clumping (environmentally-stressed cells) are indications that a new culture should be started.
2.3. Protocol for generation of P0 and P1 virus
Initial (P0) virus stocks were generated by transfecting 1.2 μg recombinant bacmid DNA (Cellfectin II transfection reagent (Thermo Fischer Scientific; Cat# 10362100) and Transfection Medium (Expression Systems; Cat# 95-020) in 500 μl transfection mix) into Sf9 cells. Briefly, for each construct, 2.5 ml of Sf9 cells (1 × 106 cells/ml) supplemented with 10 ng/ml gentamicin were added to a well of a 24-well plate (Thomson Instrument Company, Cat# 931565-G-1X). “Solution A” was prepared by adding 1.2 mg recombinant bacmid DNA to 50 μl Transfection Medium. “Solution B” was prepared by adding 4 μl Cellfectin II to 50 μl Transfection Medium, incubated for 5 min (min), added to “Solution A”, and tapped gently to mix. The mixture was incubated at room temperature (RT) for 30 min. After incubation, 400 μl Transfection Medium was added to the tube and mixed gently. The transfection mixture was added to the cells dropwise, and the deep-well plate was covered with an air- porous plate seal (Thomson Instrument Company, Cat# 899410).
Transfected cells were incubated with shaking (300 rpm, 27 °C, 4 days) in a Vertiga-IM shaker (Thomson Instrument Company, Cat# 381150) to generate P0 viruses.
On day 4, 170 μl of cell culture from the P0 stock was added to 4 ml of Sf9 cells (1 × 106 cells/ml) in a fresh deep-well plate and incubated with shaking (300 rpm, 27 °C, 4 days) in the Vertiga- IM shaker to generate P1 viruses.
Cells were removed by centrifugation (700×g, 10 min). The supernatant (P1 virus) was transferred to a sterile 15 ml screw-capped tube and stored at 4 °C in a styrofoam box. P1 viruses are stable at 4 °C for ~6 months. A small volume (~250–500 μl) of P1 virus was archived at −80 °C for long-term storage. Note that there is an ~1 log drop in viral titer upon thaw [45].
Titers of the newly generated P1 viruses were determined by a flow cytometric approach by monitoring the cell surface expression of baculoviral gp64 by staining with anti-gp64 antibody [42,43,46]. Titering of the recombinant baculovirus was a critical step required to optimize dmDcr-2 protein production and is highly recommended. Titering allows reproducible viral amplification and optimal expression, and protocols are described elsewhere [46] and will not be repeated here. We observed P1 viral titers between 5 × 108 and 1 × 109 infectious units (IU)/ml for dmDcr-2 constructs.
2.4. Protocol to check dmDcr-2 expression with P1 virus
Before amplifying the P1 stock to generate P2 viruses, and optimizing conditions for large-scale dmDcr-2 expression, we checked for protein expression in small scale (25 ml) cultures using titered recombinant P1 viruses.
- 25 ml of Sf9 cells (1 × 106 cells/ml) were passaged in a Corning disposable polycarbonate Erlenmeyer flask with standard screw cap (Fisher Scientific; Cat# 10-041-18) and incubated with shaking (135 rpm, 27 °C) for ~1 day to allow cells to reach a density of 2 × 106 cells/ml. During incubation, the cap is slightly unscrewed to allow sufficient aeration. Cells at 2 × 106 cells/ml were infected with P1 virus at an MOI (multiplicity of infection; ratio of infective baculoviral particles to Sf9 cells) of 5 IU/cell using the general formula shown below.
In other words, 500 μl of P1 virus (viral titer ~5 × 108 IU/ml) was added to 25 ml of Sf9 cells at 2 × 106 cells/ml (5 × 107 total cells) and incubated with shaking (135 rpm, 27 °C) for 2 days. Cells were harvested at 48 h (700 × g, 10 min, 4 °C) and checked for dmDcr-2 expression and the ability to bind to StrepTactin® Sepharose resin (IBA Lifesciences; Cat# 2-1201-010). The cell pellet was lysed in 10 ml of lysis buffer (100 mM TRIS pH 8.0, 150 mM NaCl, 5mM MgCl2·6H2O, 5 mM KCl, 5% glycerol, 1mM TCEP (tris(2-carboxyethyl)phosphine (Gold Biotechnology, Cat# TCEP1; 1 M TCEP stock solutions were prepared and adjusted to pH 8.0 as per manufacturer’s protocol) supplemented with 0.5% Triton X-100, 20 nM Avidin (VWR International, Cat #189725), 10 μg/ml DNase I Grade II (Sigma Aldrich, Cat # 10104159001), complete EDTA-free protease inhibitor (Sigma Aldrich, Cat # 05056489001; 1 tablet per 25 ml lysis buffer), Protease Inhibitor Cocktail (Sigma Aldrich, Cat # p8340), and 1mM PMSF (phenylmethanesulfonyl fluoride), by douncing on ice. The lysate was centrifuged (5000 × g, 4°C, 45 min) and the supernatant was filtered through a low protein-binding filter (Corning 28mm diameter syringe filter, 0.45 μM pore, SFCA membrane; VWR International, Cat# 28200-026).
The filtered supernatant was bound to 50 μl of packed StrepTactin Sepharose resin (4 °C, 1 h), and washed with 10 ml of high salt buffer (100 mM TRIS pH 8.0, 1 M NaCl, 5 mM MgCl2·6H2O, 5 mM KCl, 5% glycerol, 1 mM TCEP) (4 °C, 10 min) followed by 10 ml of lysis buffer (4 °C, 10 min).
The protein was eluted in 30 μl elution buffer (lysis buffer supplemented with 2.5 mM d-desthiobiotin and PreScission Protease (GE Healthcare, Cat # 27-0843-01) as per manufacturer’s protocol) (4 °C, 2 h). A strong band of dmDcr-2 was observed by Coomassie Brilliant Blue staining following SDS-PAGE (Fig. 1B).
2.4.1. Troubleshooting tips
A recombinant baculovirus expressing OSF-tagged eGFP was used as an experimental control during generation of P0 and P1 viruses. If all previous steps have been successful, a fluorescent, green cell pellet was observed from P1 virus-infected Sf9 cells expressing eGFP during 25 ml small-scale expression trials.
If protein expression was not observed from 25 ml Sf9 cultures infected with the P1 virus, baculoviral genomic DNA was extracted [47] to verify the presence of the expression cassette using PCR. Briefly, 100 μl of the P1 virus was lysed in 100 μl viral lysis buffer (10 mM TRIS-HCl pH 7.6, 10 mM EDTA, 0.25% SDS) by gentle pipetting. 200 μl chloroform was added to the lysate and rotated gently for 3–4 min. The sample was centrifuged briefly (>14,000×g, 5 min). 150 μl supernatant from the aqueous phase was removed, and 15 μl of 3 M sodium acetate (pH 5.2) was added. The DNA was precipitated by adding 450 μl of 100% ethanol. The sample was centrifuged (≥14,000×g, 15 min), the supernatant was discarded, and the pellet dried (60 °C, 5 min). The genomic DNA was resuspended in 20 μl of water. 1 ml of the genomic DNA was used as template with the pUC/M13 forward and reverse primers to verify the expression cassette in the recombinant baculoviral DNA using PCR as described earlier (Fig. 1C).
2.5. Protocol to generate P2 virus
To generate the P2 virus stock, 50 ml of Sf9 cells at 1 × 106 cells/ml were passaged in a Corning disposable polycarbonate Erlenmeyer flask and incubated with shaking (135 rpm; 27 °C) for ~1 day to allow cells to reach a density of 2 × 106 cells/ml.
Cells at 2 × 106 cells/ml density were infected with P1 virus at an MOI of 0.1 IU/cell (20 μl of P1 virus with viral titer ~5 × 108 IU/ml to 50 ml Sf9 cells at 2 × 106 cells/ml density), incubated with shaking (135 rpm, 27 °C), and harvested after 3 days. Cells were removed by centrifugation (700 × g, 10 min, 4°C) and the supernatant (P2 virus) was filtered through a 0.45 μm, low protein-binding filter (Corning 28mm diameter syringe filter, 0.45 μM pore, SFCA membrane, Cat# 28200-026, VWR International). This step is critical to remove residual cell debris from the supernatant and maintain viral titer stability during long-term storage. P2 viruses were titered as described previously and stored at 4 °C for ~2 months. We observed P2 viral titers between 1 and 2 × 109 IU/ml for newly generated recombinant viruses.
2.6. Protocol for large-scale dmDcr-2 expression
For large-scale protein expression, 1000 ml of Sf9 cells at 1 × 106 cells/ml were passaged in a PYREX® 2800 ml Fernbach-Style Culture flask (Corning, Cat#4420-2XL) and incubated with shaking (135 rpm, 27 °C) for ~1 day to allow cells to reach a density of 2 × 106 cells/ml. To ensure adequate aeration, we do not recommend growing more than 1L cultures in 2.8L flasks.
Cells at 2 × 106 cells/ml density were infected with recombinant P2 viruses (viral titer ~1 × 109 IU/ml) at an MOI of 5 IU/- cell (10 ml of P2 virus per 1000 ml of Sf9 cells at 2 × 106 cells/ml density) and incubated with shaking (135 rpm, 27 °C) for 2 days. Cells expressing dmDcr-2 were harvested (700×g, 4 °C, 10 min) and pellets were stored at −80 °C in 50 ml conical screw-capped tubes; in our hands pellets can be successfully stored for years.
2.6.1. Troubleshooting hints
A systematic screen with P2 virus, using a wide range of MOI values (2 ≤ MOI (IU/cell) ≤10), should be carried out using 25 ml Sf9 cultures to determine the optimal MOI for protein expression in Sf9 cells. Additionally, for dmDcr-2, a N- terminal eGFP-tagged construct (OSF-eGFP-dmDcr-2) was initially used to monitor expression levels in Sf9 cells (Fig. 1D). An MOI of 5 IU/cell was found to be optimal for dmDcr-2 expression under the conditions tested. While not used for dmDcr-2 during optimization, partially purified eGFP-fused proteins may also be analyzed by fluorescence-detection size exclusion chromatography (FSEC) to estimate the approximate molecular mass and degree of monodispersity of the expressed protein [48].
If the expressed protein is insoluble, a solubility screen should be carried out using 25 ml cultures to identify conditions that may improve protein solubility. Typically, we systematically test a wide range of pH (6 ≤ pH ≤ 10), salt (100 mM ≤ NaCl ≤ 1000 mM), detergents, and additives, during lysis, for conditions that improve solubility. Alternatively, solubility enhancing proteins such as MBP and SUMOstar may be fused to the protein of interest to enhance expression levels and the soluble fraction of the expressed protein [49].
3. Large scale purification of dmDcr-2
General considerations
dmDcr-2 is a 198 kDa protein with multiple domains, each of which are predicted to carry out specialized functions (Fig. 2A). The three-step protein purification scheme for dmDcr-2 is outlined in Fig. 2B. OSF-dmDcr-2 was initially purified using Strep-Tactin affinity chromatography, the affinity tag was removed by PreScission Protease treatment, and the protein was purified to homogeneity by tandem-ion exchange and gel filtration chromatography. Typical yields were between ~3 and 4 mg/L of insect cell cultures. The purification procedure described below works equally well for mutant variants and truncations of dmDcr-2, hsDcr, and accessory dsRNA binding proteins of dmDcr-2 (Loqs-PD and R2D2). All purification steps were carried out in the cold room at ≤4 °C.
Fig. 2.
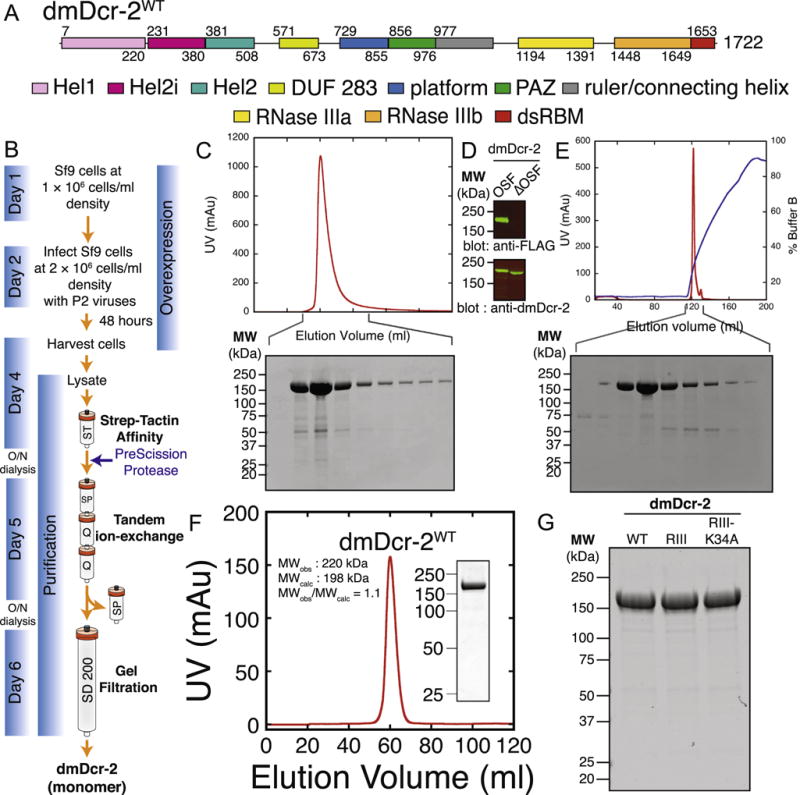
(A) Domain architecture of dmDcr-2: Colored rectangles depict conserved domains. Information from Expresso structure-based alignments [60], NCBI conserved domains [61], and crystallographic analyses [37,62] was used in defining domain boundaries. (B) Outline of OSF-dmDcr-2 protein overexpression and purification scheme: Sf9 cells at1 × 106 cells/ml were passaged in a flask (Day1)and incubated with shaking (135 rpm; 27 °C) for ~1 day to allow cells to reach a density of2 × 106 cells/ml. Cells at 2 × 106 cells/ml density were infected with recombinant P2 viruses (Day 2) and incubated with shaking (135 rpm, 27 °C) for 2 days. On day 4, cells expressing OSF-dmDcr-2 were harvested, lysed, purified by StrepTrap affinity chromatography, and dialyzed overnight in the presence of PreScission Protease to remove the N-terminal OSF-tag. dmDcr-2 was further purified by tandem ion-exchange chromatography, eluted from the anion-exchange column with a linear salt gradient, dialyzed overnight in protease cleavage buffer, and subjected to gel-filtration chromatography. Pellets harvested on day 4 may alternatively be stored at −80 °C in 50 ml conical screw-capped tubes for long-term use. O/N; overnight. (C) StrepTrap chromatogram and SDS-PAGE analysis of peak fractions: OSF-dmDcr-2 was eluted from a StrepTrap HP affinity column in lysis buffer supplemented with 2.5 mM d-Desthiobiotin, and peak fractions from the eluate were analyzed by SDS-PAGE and Coomassie Brilliant Blue staining. UV absorbance at 280 nm (arbitrary units, red trace) is plotted against elution volume (ml). (D) Western blot analyses of OSF-dmDcr-2 and dmDcr-2: Peak fractions from the StrepTrap HP affinity column were pooled and dialyzed overnight in the presence of PreScission Protease. Pre- (OSF-dmDcr-2) and post-cleavage (ΔOSF-dmDcr-2; referred to as dmDcr-2 throughout this article, after removal of the OSF tag) samples were analyzed by western blot analyses either using mouse anti-FLAG antibody (Cat # F3165, Sigma-Aldrich) (top-panel), or rabbit anti-dmDcr-2 antibody (Cat # ab4732, Abcam) (bottom panel). (E) Anion-exchange (Q) chromatogram and SDS-PAGE analyses of peak fractions: After removal of the OSF tag, dmDcr-2 was eluted from a Q HP column with a linear salt gradient (0.1-1 M KCl), and peak fractions from the eluate were analyzed by SDS-PAGE and Coomassie Brilliant Blue staining. UV absorbance at 280 nm (arbitrary units, red trace) is plotted against elution volume (ml). Blue trace, % Buffer B, linear salt gradient. (F) Gel filtration chromatogram and SDS-PAGE analyses of peak fraction: dmDcr-2 was subjected to gel filtration chromatography on a HiLoad 16/600 Superdex 200 prep grade gel filtration column pre-equilibrated in protease cleavage buffer. dmDcr-2 was eluted in1 CV (120 ml) of protease cleavage buffer. dmDcr-2 eluted as a monomer (MWobserved, 220 kDa; MWcalculated, 198 kDa; MWobserved/MWcalculated: 1.1). Inset: Coomassie-stained SDS-PAGE of the peak fraction from the Superdex 200 column. (G) Coomassie-stained SDS-PAGE of10 μg of purified, recombinant dmDcr-2WT, dmDcr-2RIII (D1217A, D1476A), and dmDcr-2RIII-K34A (K34A, D1217A, D1476A) (point mutations in parenthesis). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.1. Protocol for dmDcr-2 purification
Cell pellets from 3L of insect cell culture were removed from a −80 °C freezer and thawed in a 30 °C water bath for ~5- 7 min. The pellets were gently swirled by hand during thawing.
Cell pellets were resuspended in 5.5 times the pellet volume (55 ml for a 10 ml pellet from 1L insect cell culture) of ice- cold lysis buffer (100 mM TRIS pH 8.0, 150 mM NaCl, 5% glycerol, 5 mM MgCl2·6H2O, 5 mM KCl, 1 mM TCEP) supplemented with 0.5% Triton X-100, 20 nM Avidin (VWR International, Cat# 189725), 250 μg/ml DNase I Grade II (Sigma Aldrich, Cat# 10104159001), cOmplete EDTA-free protease inhibitor (Sigma Aldrich, Cat # 05056489001; 1 tablet per 25 ml lysis buffer), 0.7% (vol/vol) Protease Inhibitor Cocktail (Sigma Aldrich, Cat # p8340), and 1 mM phenylmethanesulfonyl fluoride (PMSF).
The cell suspension was poured into a 100 ml glass dounce (KONTES® Dounce Tissue Grinder, Kimble Chase; VWR International, Catalog # KT885300-0100), and cells were lysed by 50 strokes of douncing on ice.
Cell lysates were clarified by ultracentrifugation at 184,000×g (Beckman Ti 50.2 rotor, 4°C, 1 h).
The supernatant was filtered through a low protein-binding filter (Corning 28 mM diameter syringe filter, 0.45 μM pore, SFCA membrane; VWR International, Cat# 28200-026).
The filtered supernatant was bound to a 5 ml StrepTrap HP column (GE Healthcare, Cat# 28-9075-47) pre-equilibrated in lysis buffer (20 column volumes (CV)). The column was washed with lysis buffer (6 CV), high salt buffer (100 mM TRIS pH 8.0, 1 M NaCl, 5% glycerol, 5 mM MgCl2·6H2O, 1 mM TCEP, supplemented with 5 mM ATP to remove chaperones bound to dmDcr-2, (6 CV)), and a final wash with lysis buffer (6 CV).
OSF-dmDcr-2 was eluted in lysis buffer supplemented with 2.5 mM d-Desthiobiotin (Sigma Aldrich, Cat# D1411) (6 CV) (Fig. 2C). Fractions from the eluate were analyzed by SDS-PAGE (Fig. 2C). The StrepTrap HP column was cleaned (3 CV of 0.5 N NaOH followed by 3 CV of filter-sterilized water), regenerated in regeneration buffer (50 mM TRIS pH 8.0, 150 mM NaCl, 1 mM 2-(4-Hydroxyphenylazo) benzoic acid (Sigma Aldrich, Cat# H5126)) (5 CV), washed (10 CV of filter-sterilized water) and stored in 20% ethanol.
The eluate fractions containing OSF-tagged dmDcr-2 were pooled and dialyzed overnight (4°C) against 2L protease cleavage buffer (25 mM TRIS pH 8.0, 100 mM KCl, 5 mM MgCl2·6H2O, 5% glycerol, 1 mM TCEP) in a 6–8 kDa MWCO dialysis tubing (Fisher Scientific, Cat# 21-152-5). PreScission Protease was added to the pooled fractions of OSF-tagged dmDcr-2 (~ 1:100 by mass, enzyme: substrate) to remove the OSF-tag (dmDcr-2; throughout this article, after removal of the OSF tag, the protein is referred to with the name of the native protein). Pre- and post-cleavage aliquots are analyzed by western blot using a mouse anti-FLAG antibody (Sigma Aldrich, Cat# F3165) or a rabbit anti-dmDcr-2 antibody (Abcam, Cat# ab4732) (Fig. 2D). We find that the OSF-tag does not interfere with biochemical assays of dmDcr-2, but it remains possible that it could interfere with other analyses, such as structural and single-molecule studies. Hence, we recommend removing the OSF-tag, unless it is required for specific applications.
dmDcr-2 was further purified by tandem ion-exchange chromatography. The protein was applied to one 5 ml HiTrap SP HP column (GE Healthcare, Cat# 17115101) in line with two 5 ml HiTrap QHP columns (GE Healthcare, Cat# 17115301), pre-equilibrated with protease cleavage buffer. At pH 8.0, dmDcr-2 (pI: 6.42) flowed through the HiTrap SP HP (cation exchange) column and bound to the HiTrap Q HP (anion exchange) columns. The HiTrap SP HP column was intentionally placed above the HiTrap Q HP columns to capture residual contaminants purified with dmDcr-2 during affinity chromatography. After loading the sample, the tandem ionexchange columns were washed with 30 ml (2 CV) of protease cleavage buffer.
The HiTrap SP HP column (containing contaminants) was removed from the line and dmDcr-2 was eluted from the HiTrap Q HP column (10 CV, 100 ml) with a linear salt gradient (0.1 M-1 M KCl) in protease cleavage buffer (Fig. 2E). Fractions from the eluate were analyzed by SDS-PAGE (Fig. 2E).
Fractions containing dmDcr-2 were pooled and dialyzed overnight against 2L protease cleavage buffer in a 6–8 KDa MWCO dialysis tubing.
After overnight dialysis, dmDcr-2 was concentrated to ~4–6 mg/ml in a Vivaspin 20 ml 30K MWCO PES centricon (Sartorius Stedim Biotech, Intermountain Scientific (ISC)), Cat# F-2734-30C) and loaded onto a HiLoad 16/600 Superdex 200 prep grade gel filtration column (GE Healthcare, Cat# 28989335), pre-equilibrated with protease cleavage buffer. dmDcr-2 was eluted in 1 CV (120 ml) of protease cleavage buffer as a monomer (MWobserved, 220 kDa; MWcalculated, 198 kDa; MWobserved/MWcalculated: 1.1) (Fig. 2F). Pooled fractions were concentrated to ~4–6 mg/ml in a Vivaspin 20 ml 30K MWCO PES centricon, supplemented with 20% glycerol and flash-frozen in liquid nitrogen for long-term storage at −80 °C. Typical yields were between ~3 and 4 mg/L of culture. Protein purity, identity, and homogeneity were established using SDS-PAGE, mass spectrometry (LC/MS/MS), Edman sequencing and negative-stain electron microscopy (data not shown). Purified dmDcr-2 variants are shown (Fig. 2G).
4. Expression and purification of accessory dsRNA RNA binding proteins
In vivo, Loqs-PD interacts with dmDcr-2′s helicase domain [50,51] and facilitates cleavage of a subset of endogenous dsRNA precursors [13–15,52]. However, Loqs-PD is not required for producing exogenous siRNAs from certain viruses [18]. In previous biochemical studies, we showed that Loqs-PD modulates dmDcr- 2′s termini-dependence and enables dmDcr-2 to cleave substrates normally refractory to cleavage [32]. Ongoing studies in our laboratory are aimed at providing a mechanistic understanding of how Loqs-PD modulates dmDcr-2′s cleavage activity (Trettin et al., 2017; manuscript submitted).
4.1. Expression and purification of Loqs-PD from Sf9 cells
OSF-tagged Loqs-PD was expressed and purified from Sf9 cells using the same protocol as described for dmDcr-2 (Fig. 3A).
However, at pH 8.0, after removing the OSF tag, Loqs-PD (pl: 7.87) flowed through an anion-exchange column and bound to a cation-exchange column. Hence, tandem-ion exchange chromatography was performed by placing one 5 ml HiTrap Q HP column in line with two 5 ml HiTrap SP HP columns. The HiTrap Q HP column (containing contaminants) was removed from the line, and Loqs-PD was eluted from the HiTrap SP HP columns (10 CV, 100 ml) with a linear salt gradient (0.1 M- 1 M KCl) in protease cleavage buffer (Fig. 3A).
Loqs-PD was purified to homogeneity on a HiLoad 16/600 Superdex 200 prep grade gel filtration column (Fig. 3B). Loqs- PD eluted from the Superdex 200 column with an observed MW of 101.5 kDa (MWobserved/MWcalculated: 2.6). Ongoing studies in our laboratory indicate that Loqs-PD is a monomer (Tret- tin et al., 2017; manuscript submitted) but migrates aberrantly by gel-filtration. The purified protein was verified by intact- mass ESI/MS analyses. Based on accurate mass measurements, intact-mass ESI/MS analyses suggested that Loqs-PD, expressed in Sf9 cells, was mono-phosphorylated (Fig. 3C; mass of Phospho-Loqs-PD: 38688.7680 Da, mass of dephosphorylated Loqs-PD: 38608.8017 Da (error ±0.4 ppm); the difference in mass between the two species, 79.9663 Da, could be accurately attributed to a phosphate moiety. To identify the phosphorylation site on Loqs-PD, we performed tryptic digestion and analyzed the sample by LC/MS/MS. Based on a Mascot database search (Matrix Science) of the LC/MS/MS spectra, the putative phosphorylation site on Loqs-PD was assigned to the motif 213- SPSSS217, which occurs in the unstructured linker between the two dsRBMs. While the function of Loqs-PD phosphorylation is yet to be determined, interestingly, human TAR-RNA binding protein (TRBP) is also phosphorylated on serine residues (S142, S152, S283, S286) positioned in linkers between dsRBMs [53]. Phosphorylation of TRBP, mediated by the mitogen-activated protein kinase (MAPK) Erk, enhances Dicer·TRBP-mediated production of miRNAs by stabilizing the miRNA-generating complex [53]. Coincidently, the putative phosphorylation motif on Loqs-PD (213SP) is a consensus site for Erk-mediated phosphorylation.
Purified, recombinant, Loqs-PD formed a stable complex with dmDcr-2 as verified by analytical gel filtration chromatography (Fig. 3D). Briefly, 2-fold molar excess Loqs-PD was incubated with dmDcr-2 in binding buffer (50 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2·6H2O, 1 mM TCEP) overnight at 4°C. The complex was resolved on an analytical Superose-6 Increase 10/300 GL gel-filtration column (GE Healthcare Life Sciences) pre-equilibrated with binding buffer. 0.5 ml fractions were collected and analyzed by SDS-PAGE. dmDcr-2 eluted in 1 CV (24 ml) of binding buffer as a monomer (MWobserved, 231 kDa; MWcalculated, 198 kDa; MWobserved/MWcalculated: 1.16) (Fig. 3D, chromatogram, red trace; bottom, SDS-PAGE). However, in the presence of Loqs-PD, the dmDcr-2 Loqs-PD complex eluted from the gel filtration column with an apparent molecular weight (MWobserved) of 341 kDa (Fig. 3D; chromatogram, grey trace; top SDS-PAGE). As observed in the sizing chromatogram (elution volume, 13.67 ml for dmDcr-2 Loqs-PD compared to 14.11 ml for dmDcr-2 alone) and by SDS-PAGE, the peak fraction of dmDcr-2 shifted towards a higher molecular weight in the presence of Loqs-PD, indicating formation of a stable complex. Aliquots (100 μl) of the peak fraction of the dmDcr-2 Loqs-PD complex from the gel filtration column were treated with increasing concentrations of glutaraldehyde (stock concentration: 0.8 mM, 8% (w/v); final concentration: see Fig. 3D, bottom right), incubated on ice (30 min, 4°C) and quenched with 100 mM TRIS pH 8.0. The reaction was spun at maximum speed in a microcentrifuge (16,100 ×g, 7 min, 4 °C) to remove any aggregates, and the crosslinked proteins were resolved by SDS-PAGE and detected by staining with Coomassie Brilliant Blue. At ~0.025% (w/v; 2.5 μM) glutaraldehyde, the covalently cross-linked dmDcr-2 Loqs-PD complex migrated at ~250 kDa, indicative of the formation of a 1:1 stoichiometric complex (Fig. 3D, bottom right).
Fig. 3.
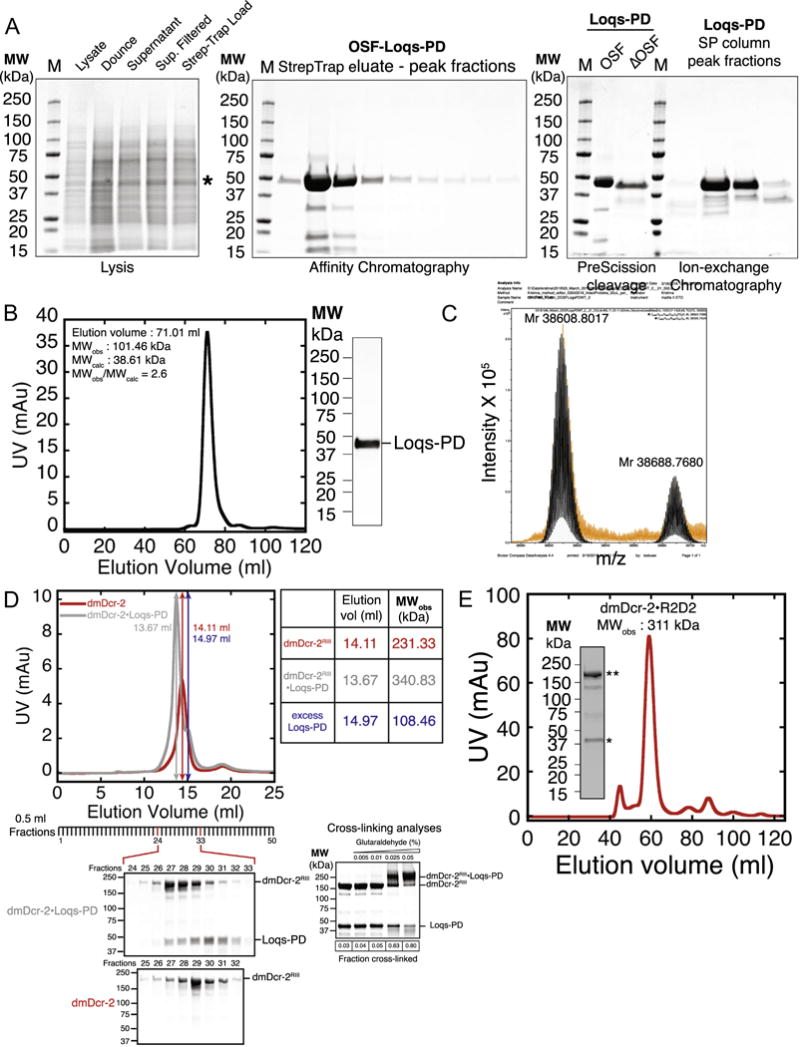
(A) Lysis, affinity, protease-cleavage, and ion-exchange-chromatography of Loqs-PD: (Left) Coomassie-stained SDS-PAGE analyses of lysate, dounced lysate, supernatant following clarification of the lysate by ultracentrifugation, supernatant filtered through a low protein-binding filter to remove residual cellular debris, and the clarified lysate loaded onto a StrepTrap HP affinity column; *, OSF-Loqs-PD. (Middle) Coomassie-stained SDS-PAGE analyses of peak fractions eluted from a StrepTrap affinity column. (Right) Coomassie-stained SDS-PAGE analyses of Loqs-PD before and after cleavage of the N-terminal OSF-tag by PreScission Protease (ΔOSF-Loqs-PD; referred to as Loqs-PD throughout this article, after removal of the OSF tag); peak fractions of Loqs-PD eluted from a cation-exchange column (SP-HP) following tandem ion-exchange chromatography. (B) Gel filtration chromatogram and SDS-PAGE analyses of peak fraction: Loqs-PD was subjected to gel filtration chromatography on a HiLoad 16/600 Superdex 200 prep grade gel filtration column. Loqs-PD was eluted in 1 CV (120 ml) of protease cleavage buffer and eluted with an observed molecular weight (MWobs) of 102 kDa; Right, Coomassie-stained SDS-PAGE of the peak fraction from the Superdex 200 column. (C) Intact-mass ESI/MS analyses of Loqs-PD: Loqs-PD was subjected to intact-mass ESI/MS analyses. Accurate mass measurements showed two peaks at 38688.7680 Da and 38608.8017Da (error ±0.4 ppm) (see text for details). (D) Gel filtration profiling of the dmDcr-2-Loqs-PD complex: dmDcr-2 alone or dmDcr-2 in complex with Loqs-PD (dmDcr-2-Loqs-PD), was subjected to gel filtration chromatography on a Superose-6 Increase 10/300GL gel filtration column. dmDcr-2 eluted with an MWobs of 231 kDa (elution volume, 14.11 ml, red trace); dmDcr-2·Loqs-PD eluted with an MWobs of 341 kDa (elution volume, 13.67 ml, red trace). Excess Loqs-PD eluted with an MWobs of 109 kDa (elution volume, 14.97 ml; vertical blue arrow indicates peak fraction of excess Loqs-PD elution); Fractions were subjected to SDS-PAGE analyses (below chromatogram) or SDS-PAGE analyses after cross-linking with glutaraldehyde. n = 2. (E) Gel filtration chromatogram and SDS-PAGE analyses of dmDcr-2 R2D2: dmDcr-2 R2D2 eluted from a HiLoad 16/600 Superdex 200 prep grade gel filtration column with a MWobs of 311 kDa. Inset, SDS-PAGE analyses of the peak fraction from the gel filtration column. **, dmDcr-2, *, R2D2. n = 2. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4.2. Co-expression and purification of dmDcr-2·R2D2 from Sf9 cells
In Drosophila melanogaster, an accessory dsRBP called R2D2 interacts with dmDcr-2′s helicase domain and is required for the passage of siRNAs from dmDcr-2 to dmAgo-2, although the mechanism is not well characterized [26,51,54–57]. In vivo, R2D2 forms a stable complex with dmDcr-2, such that depletion of dmDcr-2 from S2 cells co-depletes R2D2, without affecting R2D2 mRNA levels. These results suggest that R2D2 is unstable in the absence of dmDcr-2 [26]. R2D2 co-localizes with dmDcr-2 to cytoplasmic ribonucleoprotein granules called D2 bodies, which are hubs for loading endo-siRNAs onto dmAgo-2 [56]. dmDcr-2 and R2D2 form hetero-tetramers in the presence of siRNA, as observed by nativePAGE analyses [57].
We initially tried to purify recombinant OSF-tagged R2D2 from baculovirus-infected Sf9 cells. Unfortunately, without the OSF tag, gel-filtration chromatography and negative-stain EM experiments indicated that R2D2 was a soluble aggregate (data not shown).
Given the evidence that R2D2 is unstable in the absence of dmDcr-2, we attempted to co-express dmDcr-2 and R2D2. We co-infected Sf9 cells (2 × 106 cells/ml) with recombinant OSF- dmDcr-2 and untagged-R2D2 P2 viruses, each at an MOI of 3 IU/cell. We purified OSF-dmDcr-2 R2D2 by StrepTrap affinity chromatography, removed the OSF-tag on dmDcr-2 by overnight dialysis in the presence of PreScission Protease, and purified the dmDcr-2 R2D2 complex by gel filtration chromatography. dmDcr-2 R2D2 eluted with an observed molecular weight of 311 kDa (Fig. 3E) and was determined to be ~95% pure by mass spectrometry analyses.
5. Preparation of dsRNA for biochemical assays
dmDcr-2 recognizes dsRNA termini and initiates cleavage from the “ends” of long dsRNAs, using distinct catalytic modes that are dictated by the nature of the dsRNA terminus. For example, dmDcr-2 acts processively on dsRNA with blunt (BLT) termini, whereas it acts distributively on dsRNA with two-nucleotide 3′- overhanging (3′ovr) termini [29,32]. The termini preferences of dmDcr-2 dictate that dsRNA substrates used for in vitro assays must be carefully prepared to maintain precise termini. Below we describe protocols for preparing dsRNAs using sense and antisense strands of 106 nts; these protocols are applicable to the preparation of dsRNAs of other lengths.
5.1. dsRNA preparation
Construct design: To ensure precise and homogeneous termini for biochemical assays, 106-nt sense, and BLT and 3′ovr antisense strands were cloned into a pss419 plasmid (kindly provided by Dr. Sandra Searles, MRC Laboratory of Molecular Biology, Cambridge, UK), to position a hammerhead ribozyme sequence (68 nts) at the 5′ end and a Hepatitis Delta Virus (HDV) ribozyme sequence (65 nts) at the 3′ end of the 106 nt sequence [32]. A T7 RNA polymerase promoter was positioned at the 5′ end of the hammerhead ribozyme sequence.
RNA was transcribed in vitro using T7 RNA polymerase (37°C) using standard protocols [58]. Following transcription, the reaction was treated with Turbo DNase (Thermo Scientific, Cat # AM2238) to remove the DNA template, followed by Proteinase K (Thermo Scientific, Cat # AM2548) treatment, phenolchloroform extraction and ethanol precipitation.
The transcribed RNA was resuspended in ribozyme cleavage buffer (20 mM TRIS pH 8.0, 10 mM MgCl2·6H2O, 1mM TCEP) supplemented with T4 polynucleotide kinase (T4 PNK, New England Biolabs Inc. Cat # M0201S) and incubated (37°C, 2 h) to allow ribozyme cleavage. Cleavage by the hammerhead ribozyme leaves a hydroxyl group at the 5′ end of the 106-nt RNA, and cleavage by the HDV ribozyme leaves a 2′,3′-cyclic phosphate at the 3′ end of the 106-nt RNA. T4 PNK has a lesser known 2′,3′-cyclic phosphatase activity [59] and is used to remove the cyclic phosphate. After kinase treatment, the sample is treated with Proteinase K, phenol-chloroform extracted, and ethanol precipitated.
The 106-nt RNA is separated from the hammerhead and HDV ribozymes by denaturing PAGE (8% polyacrylamide, 8M urea, 1X TBE gel; 30W, 2 h), gel extracted and ethanol precipitated.
106-nt RNA was 32P-end labeled using γ-32P-ATP (Perkin Elmer) and T4 PNK using standard protocols, followed by gel purification after 8% denaturing PAGE. In our studies, 32P-end labeled RNAs were used for gel-shift analyses and single-turnover cleavage assays to specifically monitor siRNAs generated by the first cleavage event from the end [32]. However, other experiments, such as multiple-turnover cleavage assays require a uniformly labeled dsRNA. For such purposes, we added α-32P- ATP (Perkin Elmer) to in vitro transcription reactions to yield internally-labeled RNAs [32].
Equimolar amounts of labeled-sense and unlabeled-antisense RNAs were annealed in annealing buffer (50 mM TRIS, pH 8.0, 20 mM KCl) by placing the reaction on a heat block (95°C) and slow cooling ≥3 h. Double-stranded RNA (dsRNA) was gel purified after native-PAGE (8% polyacrylamide), gel extracted, ethanol precipitated and resuspended in annealing buffer for use.
5.2. Sequences of dsRNA substrates
106 dsRNAs:
106-nt sense RNA:
5′ggcaaugaaagacggugagcuggugauaugggauaguguucacccuuguuacaccguuuuccaugagcaaacugaaacguuuucaucgcucuggagugaauaccaa3′
106-nt BLT antisense RNA
5′uugguauucacuccagagcgaugaaaacguuucaguuugcucauggaaaacgguguaacaagggugaacacuaucccauaucaccagcucaccgucuuucauugcc3′
106-nt 3′ovr antisense RNA
5′gguauucacuccagagcgaugaaaacguuucaguuugcucauggaaaacgguguaacaagggugaacacuaucccauaucaccagcucaccgucuuucauugccaa3′
106-nt sense RNA annealed to 106-nt BLT antisense RNA =106 BLT-BLT dsRNA
106-nt sense RNA annealed to 106-nt 3′ovr antisense RNA =106 3′ovr-3′ovr dsRNA
6. Discussion
The described expression and purification protocols result from careful optimization at every step, and our analyses indicate they are applicable to a variety of dsRBPs. The three-step purification protocol described for dmDcr-2 enabled us to achieve ~90% purity after the affinity chromatography step, ~95% purity after the tandem ion-exchange chromatography step, and homogenously- purified dmDcr-2 after gel-filtration chromatography. The high- salt (1M NaCl) wash step during affinity chromatography and anion-exchange chromatography both removed bound nucleic acid from dmDcr-2 (the ratio of absorbance at 260 nm versus 280 nm is typically between 0.57 and 0.60 for dmDcr-2). The presence of 5 mM ATP during the high-salt (1M NaCl) wash step during affinity chromatography removed co-purifying chaperone proteins bound to dmDcr-2. In our hands the described protocols consistently produce high yields of active, well-behaved, highly-purified protein, and for dmDcr-2, this feature has greatly facilitated biochemical studies [32].
Our biochemical studies of dmDcr-2 provide insight into how dmDcr-2 discriminates between the “ends” of long dsRNA (dsRNA termini) to modulate alternate reaction states [29,32]. We find that dsRNA with BLT termini, which mimic the termini of certain viral dsRNAs, promote an optimal reaction whereby the helicase domain enables ATP-dependent, processive cleavage. In contrast, dsRNA with 3′ovr termini, which mimic the termini of some cellular dsRNAs, promote a suboptimal, distributive cleavage reaction. These conclusions derived from a variety of biochemical assays that are detailed elsewhere [32]. Biochemical assays included gel-shift assays to define dmDcr-2 binding to dsRNA termini, and single- and multiple-turnover cleavage assays to determine kinetic parameters that allow discrimination between distributive and processive reactions. Because our protocols resulted in higher yields than those used previously, we were also able to perform biochemical assays that require large amounts of protein, such as limited proteolysis; the latter postulated a helicase-dependent conformational change that accompanies recognition of BLT termini and the concomitant processivity. Direct assays of the helicase domain such as ATP hydrolysis are also described in our previous studies [32].
Acknowledgments
We thank the Bass lab for invaluable feedback; Viswanathan Chandrasekaran, Steven Alam and Costa Georgopoulos for advice on insect cell expression and protein purification. RNA and DNA oligonucleotides were synthesized by the DNA/Peptide Facility, part of the Health Sciences Center Cores at the University of Utah (CCSG P30CA042014). Mass spectrometry was performed at the Mass Spectrometry and Proteomics Core Facility at the University of Utah. Mass spectrometry equipment was obtained through NCRR Shared Instrumentation Grant # 1 S10 RR020883-01 and 1 S10 RR025532-01A1. This work was supported by funding to B.L. B from the National Institute of General Medical Sciences of the National Institutes of Health (R01GM121706) and the H.A. and Edna Benning Presidential Endowed Chair.
Appendix A. Equipment and supply lists
A.1. Chemicals and reagents
All chemicals and reagents were purchased from Sigma-Aldrich unless indicated.
A.2. Baculovirus expression system
A.2.1. Reagents
| Name | Source | Catalog Number |
|---|---|---|
| pFastBac™1 vector (Bac-to-Bac® Vector Kit) | Thermo Fisher Scientific | 10360-014 |
| MAX Efficiency® DH10Bac™ Competent Cells | Thermo Fisher Scientific | 10361012 |
| Sma I | New England BioLabs Inc. | R0141S |
| Gibson Assembly® Cloning Kit | New England BioLabs Inc. | E5510S |
| Sf9 cells | Expression Systems | 94-001S |
| ESF-921 Insect Cell Culture Medium | Expression Systems | 96-001 |
| Cellfectin II transfection reagent | Thermo Fischer Scientific | 10362100 |
| Transfection Medium | Expression Systems | 95-020 |
| Gentamicin | Sigma Aldrich | G1914 |
| Baculovirus Titering Kit | Expression Systems | 97-101 |
| gp64-PE Antibody | Expression Systems | 97-201 |
A.2.2. Equipment
| Name | Source | Catalog Number |
|---|---|---|
| Eppendorf Innova Model 2300 large capacity platform shaker | Fisher Scientific | 14-278-164 |
| 24-well plate | Thomson Instrument Company | 931565-G-1X |
| Air-porous Plate Seal | Thomson Instrument Company | 899410 |
| Vertiga-IM shaker | Thomson Instrument Company | 381150 |
| Corning® Costar® Ultra-Low attachment multiwall plates | Sigma Aldrich | CLS3474-24EA |
| Corning® disposable polycarbonate Erlenmeyer flasks with standard screw cap | Fisher Scientific | 10-041-18 |
| PYREX® 2800 ml Fernbach-Style Culture flask | Corning | 4420-2XL |
A.3. Large scale purification of dmDcr-2
A3.1. Reagents
| Name | Source | Catalog Number |
|---|---|---|
| StrepTactin® Sepharose resin | IBA Lifesciences | 2-1201-010 |
| Avidin egg-white | VWR International | 189725 |
| DNase I Grade II | Sigma Aldrich | 10104159001 |
| cOmplete EDTA-free protease inhibitor | Sigma Aldrich | 05056489001 |
| Protease Inhibitor Cocktail | Sigma Aldrich | p8340 |
| Corning 28 mm diameter syringe filter, 0.45 μM pore, SFCA membrane | VWR International | 28200-026 |
| d-Desthiobiotin | Sigma Aldrich | D1411 |
| 2-(4-Hydroxyphenylazo) benzoic acid | Sigma Aldrich | H5126 |
| Tris (2-carboxyethyl) phosphine | Gold Biotechnology | TCEP1 |
| PreScission Protease | GE Healthcare | 27-0843-01 |
| Monoclonal aNTI-FLAG M2 antibody | Sigma-Aldrich | F3165 |
| anti-Dcr-2 antibody | Abcam | ab4732 |
Note: Tris (2-carboxyethyl) phosphine (TCEP) stock solution was adjusted to pH 8.0 as per manufacturer’s protocol before use. The protocol is described in the manufacturer’s webpage: https://www.goldbio.com/documents/1085/TCEP+Stock+Solution.pdf
A.3.2. Equipment
| Name | Source | Catalog Number |
|---|---|---|
| KONTES® Dounce TissueGrinder, Kimble Chase | VWR International | KT885300-0100 |
| Beckman Centrifuge Tubes, Thickwall, polycarbonate 32 ml, 25 tubes | Beckman Coulter | 355631 |
| StrepTrap HP column, 5 ml | GE Healthcare | 28-9075-47 |
| Fisherbrand™ Regenerated Cellulose Dialysis Tubing | Fisher Scientific | 21-152-5 |
| Monoclonal aNTI-FLAG® M2 antibody produced in mouse | Sigma Aldrich | F3165 |
| HiTrap SP HP column, 5 ml | GE Healthcare | 17115101 |
| HiTrap Q HP column, 5 ml | GE Healthcare | 17115301 |
| Vivaspin 20 ml 30K MWCO PES centricon, Sartorius Stedim Biotech | Intermountain Scientific (ISC) | F-2734-30C |
| HiLoad 16/600 Superdex 200 pg | GE Healthcare | 28989335 |
| Superose-6 Increase 10/300 GL | GE Healthcare | 29-0915-96 |
A.4. RNA preparation and biochemical assays
γ-32P-ATP and α-32P-ATP were purchased from Perkin-Elmer
| Name | Source | Catalog Number |
|---|---|---|
| Turbo DNase | Thermo Scientific | AM2238 |
| Proteinase K | Thermo Scientific | AM2548 |
| T4 polynucleotide kinase | New England Biolabs Inc. | M0201S |
References
- 1.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. http://dx.doi.org/10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass BL. Double-stranded RNA as a template for gene silencing. Cell. 2000;101:235–238. doi: 10.1016/s0092-8674(02)71133-1. http://dx.doi.org/10.1016/s0092-8674(02)71133-1. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. http://dx.doi.org/10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 4.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. http://dx.doi.org/10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knight SW. A Role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. 2001;293:2269–2271. doi: 10.1126/science.1062039. http://dx.doi.org/10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu Rev Biophys. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. http://dx.doi.org/10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shabalina SA, Koonin EV. Origins and evolution of eukaryotic RNA interference. Trends Ecol Evol (Amst) 2008;23:578–587. doi: 10.1016/j.tree.2008.06.005. http://dx.doi.org/10.1016/j.tree.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomari Y, Zamore PD. Perspective: machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. http://dx.doi.org/10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- 9.Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. http://dx.doi.org/10.1038/nrm1387. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee K, Campos H, Kolaczkowski B. Evolution of animal and plant dicers: early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Mol Biol Evol. 2013;30:627–641. doi: 10.1093/molbev/mss263. http://dx.doi.org/10.1093/molbev/mss263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 12.Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, Imler JL. Essential function in vivo for Dicer-2 in host defense against RNA viruses in drosophila. Nat Immunol. 2006;7:590–597. doi: 10.1038/ni1335. http://dx.doi.org/10.1038/ni1335. [DOI] [PubMed] [Google Scholar]
- 13.Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. http://dx.doi.org/10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou R, Czech B, Brennecke J, Sachidanandam R, Wohlschlegel JA, Perrimon N, et al. Processing of Drosophila endo-siRNAs depends on a specific Loquacious isoform. RNA. 2009;15:1886–1895. doi: 10.1261/rna.1611309. http://dx.doi.org/10.1261/rna.1611309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartig JV, Esslinger S, Böttcher R, Saito K, Förstemann K. Endo-siRNAs depend on a new isoform of loquacious and target artificially introduced, high- copy sequences. EMBO J. 2009;28:2932–2944. doi: 10.1038/emboj.2009.220. http://dx.doi.org/10.1038/emboj.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marques JT, Carthew RW. A call to arms: coevolution of animal viruses and host innate immune responses. Trends Genet. 2007;23:359–364. doi: 10.1016/j.tig.2007.04.004. http://dx.doi.org/10.1016/j.tig.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Deddouche S, Matt N, Budd A, Mueller S, Kemp C, Galiana-Arnoux D, et al. The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in drosophila. Nat Immunol. 2008;9:1425–1432. doi: 10.1038/ni.1664. http://dx.doi.org/10.1038/ni.1664. [DOI] [PubMed] [Google Scholar]
- 18.Marques JT, Wang JP, Wang X, de Oliveira KPV, Gao C, Aguiar ERGR, et al. Functional specialization of the small interfering RNA pathway in response to virus infection. PLoS Pathog. 2013;9:e1003579. doi: 10.1371/journal.ppat.1003579. http://dx.doi.org/10.1371/journal.ppat.1003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomari Y, Du T, Zamore PD. Sorting of drosophila small silencing RNAs. Cell. 2007;130:299–308. doi: 10.1016/j.cell.2007.05.057. http://dx.doi.org/10.1016/j.cell.2007.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Förstemann K, Horwich MD, Wee L, Tomari Y, Zamore PD. Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell. 2007;130:287–297. doi: 10.1016/j.cell.2007.05.056. http://dx.doi.org/10.1016/j.cell.2007.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. http://dx.doi.org/10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 22.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 24.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Rådmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. http://dx.doi.org/10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J. 2002;21:5875–5885. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Q, Rand TA, Kalidas S, Du F, Kim HE, Smith DP, et al. R2D2, a bridge between the initiation and effector steps of the Drosophila RNAi pathway. Science. 2003;301:1921–1925. doi: 10.1126/science.1088710. http://dx.doi.org/10.1126/science.1088710. [DOI] [PubMed] [Google Scholar]
- 27.Ma E, MacRae IJ, Kirsch JF, Doudna JA. Autoinhibition of human dicer by its internal helicase domain. J Mol Biol. 2008;380:237–243. doi: 10.1016/j.jmb.2008.05.005. http://dx.doi.org/10.1016/j.jmb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakravarthy S, Sternberg SH, Kellenberger CA, Doudna JA. Substrate- specific kinetics of dicer-catalyzed RNA processing. J Mol Biol. 2010;404:392–402. doi: 10.1016/j.jmb.2010.09.030. http://dx.doi.org/10.1016/j.jmb.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welker NC, Maity TS, Ye X, Aruscavage PJ, Krauchuk AA, Liu Q, et al. Dicer’s helicase domain discriminates dsRNA termini to promote an altered reaction mode. Mol Cell. 2011;41:589–599. doi: 10.1016/j.molcel.2011.02.005. http://dx.doi.org/10.1016/j.molcel.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cenik ES, Fukunaga R, Lu G, Dutcher R, Wang Y, Tanaka Hall TM, et al. Phosphate and R2D2 restrict the substrate specificity of Dicer-2, an ATP-driven ribonuclease. Mol Cell. 2011;42:172–184. doi: 10.1016/j.molcel.2011.03.002. http://dx.doi.org/10.1016/j.molcel.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukunaga R, Colpan C, Han BW, Zamore PD. Inorganic phosphate blocks binding of pre-miRNA to Dicer-2 via its PAZ domain. EMBO J. 2014;33:371–384. doi: 10.1002/embj.201387176. http://dx.doi.org/10.1002/embj.201387176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha NK, Trettin KD, Aruscavage PJ, Bass BL. Drosophila dicer-2 cleavage is mediated by helicase- and dsRNA termini-dependent states that are modulated by Loquacious-PD. Mol Cell. 58(2015):406–417. doi: 10.1016/j.molcel.2015.03.012. http://dx.doi.org/10.1016/j.molcel.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacRae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, et al. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. http://dx.doi.org/10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 34.Lau PW, Guiley KZ, De N, Potter CS, Carragher B, MacRae IJ. The molecular architecture of human Dicer. Nat Publish Group. 2012;19:436–440. doi: 10.1038/nsmb.2268. http://dx.doi.org/10.1038/nsmb.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor DW, Ma E, Shigematsu H, Cianfrocco MA, Noland CL, Nagayama K, et al. Substrate-specific structural rearrangements of human Dicer. Nat Publish Group. 2013;20:662–670. doi: 10.1038/nsmb.2564. http://dx.doi.org/10.1038/nsmb.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang HW, Noland C, Siridechadilok B, Taylor DW, Ma E, Felderer K, et al. Structural insights into RNA processing by the human RISC-loading complex. Nat Publish Group. 2009;16:1148–1153. doi: 10.1038/nsmb.1673. http://dx.doi.org/10.1038/nsmb.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian Y, Simanshu DK, Ma JB, Park JE, Heo I, Kim VN, et al. A phosphate- binding pocket within the platform-PAZ-connector helix cassette of human Dicer. Mol Cell. 2014;53:606–616. doi: 10.1016/j.molcel.2014.01.003. http://dx.doi.org/10.1016/j.molcel.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du Z, Lee JK, Tjhen R, Stroud RM, James TL. Structural and biochemical insights into the dicing mechanism of mouse Dicer: a conserved lysine is critical for dsRNA cleavage. Proc Natl Acad Sci USA. 2008;105:2391–2396. doi: 10.1073/pnas.0711506105. http://dx.doi.org/10.1073/pnas.0711506105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma E, Zhou K, Kidwell MA, Doudna JA. Coordinated activities of human dicer domains in regulatory RNA processing. J Mol Biol. 2012;422:466–476. doi: 10.1016/j.jmb.2012.06.009. http://dx.doi.org/10.1016/j.jmb.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kidwell MA, Chan JM, Doudna JA. Evolutionarily conserved roles of the dicer helicase domain in regulating RNA interference processing. J Biol Chem. 2014;289:28352–28362. doi: 10.1074/jbc.M114.589051. http://dx.doi.org/10.1074/jbc.M114.589051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luckow VA, Lee SC, Barry GF, Olins PO. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J Virol. 1993;67:4566–4579. doi: 10.1128/jvi.67.8.4566-4579.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. http://dx.doi.org/10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanson MA, Brooun A, Baker KA, Jaakola VP, Roth C, Chien EYT, et al. Profiling of membrane protein variants in a baculovirus system by coupling cell-surface detection with small-scale parallel expression. Protein Expr Purif. 2007;56:85–92. doi: 10.1016/j.pep.2007.06.003. http://dx.doi.org/10.1016/j.pep.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myers JW, Ferrell JE. Silencing gene expression with Dicer-generated siRNA pools. Methods Mol Biol. 2005;309:93–196. doi: 10.1385/1-59259-935-4:093. http://dx.doi.org/10.1385/1-59259-935-4:093. [DOI] [PubMed] [Google Scholar]
- 45.Jorio H, Tran R, Kamen A. Stability of serum-free and purified baculovirus stocks under various storage conditions. Biotechnol Prog. 2006;22:319–325. doi: 10.1021/bp050218v. http://dx.doi.org/10.1021/bp050218v. [DOI] [PubMed] [Google Scholar]
- 46.Mulvania T, Hayes B, Hedin D. A flow cytometric assay for rapid accurate determination of baculovirus titers. BioProcess J. 2004;3:47–53. http://dx.doi.org/10.12665/j33.mulvania. [Google Scholar]
- 47.McCarthy CB, Romanowski V. A simplified method for the extraction of baculoviral DNA for PCR analysis: a practical application. J Virol Methods. 2008;148:286–290. doi: 10.1016/j.jviromet.2007.11.004. http://dx.doi.org/10.1016/j.jviromet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Struct/Fold Des. 2006;14:673–681. doi: 10.1016/j.str.2006.01.013. http://dx.doi.org/10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 49.Liu L, Spurrier J, Butt TR, Strickler JE. Enhanced protein expression in the baculovirus/insect cell system using engineered SuMo fusions. Protein Expr Purif. 2008;62:21–28. doi: 10.1016/j.pep.2008.07.010. http://dx.doi.org/10.1016/j.pep.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyoshi K, Miyoshi T, Hartig JV, Siomi H, Siomi MC. Molecular mechanisms that funnel RNA precursors into endogenous small-interfering RNA and microRNA biogenesis pathways in Drosophila. RNA. 2010;16:506–515. doi: 10.1261/rna.1952110. http://dx.doi.org/10.1261/rna.1952110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartig JV, Förstemann K. Loqs-PD and R2D2 define independent pathways for RISC generation in Drosophila. Nucleic Acids Res. 2011;39:3836–3851. doi: 10.1093/nar/gkq1324. http://dx.doi.org/10.1093/nar/gkq1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mirkovic-Hösle M, Förstemann K. Transposon defense by endo-siRNAs, piRNAs and somatic pilRNAs in Drosophila: contributions of Loqs-PD and R2D2. PLoS One. 2014;9:e84994. doi: 10.1371/journal.pone.0084994. http://dx.doi.org/10.1371/journal.pone.0084994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA- generating complex mediates MAPK/Erk signaling. Cell. 2009;139:112–122. doi: 10.1016/j.cell.2009.06.044. http://dx.doi.org/10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomari Y. A protein sensor for siRNA asymmetry. Science. 2004;306:1377–1380. doi: 10.1126/science.1102755. http://dx.doi.org/10.1126/science.1102755. [DOI] [PubMed] [Google Scholar]
- 55.Pham JW, Pellino JL, Lee YS, Carthew RW, Sontheimer EJ. A Dicer-2- dependent 80s complex cleaves targeted mRNAs during RNAi in Drosophila. Cell. 2004;117:83–94. doi: 10.1016/s0092-8674(04)00258-2. http://dx.doi.org/10.1038/nrm1387. [DOI] [PubMed] [Google Scholar]
- 56.Nishida KM, Miyoshi K, Ogino A, Miyoshi T, Siomi H, Siomi MC. Roles of R2D2, a cytoplasmic D2 body component, in the endogenous siRNA pathway in Drosophila. Mol Cell. 2013;49:680–691. doi: 10.1016/j.molcel.2012.12.024. http://dx.doi.org/10.1016/j.molcel.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 57.Liang C, Wang Y, Murota Y, Liu X, Smith D, Siomi MC, et al. TAF11 assembles the RISC loading complex to enhance RNAi efficiency. Mol Cell. 2015;59:807–818. doi: 10.1016/j.molcel.2015.07.006. http://dx.doi.org/10.1016/j.molcel.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brunelle JL, Green R. In vitro transcription from plasmid or PCR-amplified DNA. Meth Enzymol. 2013;530:101–114. doi: 10.1016/B978-0-12-420037-1.00005-1. http://dx.doi.org/10.1016/B978-0-12-420037-1.00005-1. [DOI] [PubMed] [Google Scholar]
- 59.Morse DP. [5] Identification of messenger RNAs that contain inosine. Elsevier; 2000. http://dx.doi.org/10.1016/s0076-6879008044-9. [DOI] [PubMed] [Google Scholar]
- 60.Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, et al. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-coffee. Nucleic Acids Res. 2006;34:W604–8. doi: 10.1093/nar/gkl092. http://dx.doi.org/10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–9. doi: 10.1093/nar/gkq1189. http://dx.doi.org/10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. http://dx.doi.org/10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]