

Abstract
Understanding the mechanisms of platinum compound resistance, including cisplatin resistance, has important implications for improving cancer treatments. Previous studies identified a potential role for mitogen-activated protein kinase phosphatase-1 (MKP-1) in cisplatin resistance. This work focuses on the regulation of poly(ADP-ribose) polymerase-1 (PARP-1) expression by MKP-1. We found that MKP-1 overexpression stimulates PARP-1 and poly(ADP-ribose) (PAR) protein expression and cisplatin resistance while its downregulation suppresses PARP-1 and PAR protein expression and cisplatin resistance. Silencing MKP-1 promoted PARP-1 ubiquitination, which decreased PARP-1 protein levels. We also found that silencing c-Jun N-terminal kinase 1/2 (JNK1/2) decreased PARP-1 ubiquitination while increasing total PARP-1 protein levels. Furthermore, we showed that acquired cisplatin resistant ovarian cancer cells expressed the high levels of MKP-1 and PARP-1 proteins, and that silencing MKP-1 or PARP-1 increased cisplatin sensitivity in resistant cells. Notably, the pharmacologic inhibition of PARP activity restored cisplatin sensitivity in MKP-1 overexpressing cells. Thus, this work indicates that suppression of JNK1/2 activity by MKP-1 maintains PARP-1 levels and suggests that MKP-1-mediated cisplatin resistance can be bypassed by PARP-1 inhibition.
Keywords: MKP-1, JNK, PARP, ubiquitination, cisplatin resistance, ovarian cancer
Introduction
Platinum compounds, including cisplatin/carboplatin, in combination with taxane are the current standard of care for treating ovarian cancer patients. Most patients with ovarian cancer initially respond well to platinum-based regimens but refractory tumors can emerge because of the development of chemoresistance 37. For example, up to 80% of ovarian cancer patients initially responded to platinum-based chemotherapy, but most of the responsive patients developed recurrent tumors within two years 42. Thus, understanding the molecular mechanism of platinum compound resistance, including cisplatin resistance, has important implications for improving the treatment of ovarian cancer patients. Currently, the mechanisms of cisplatin resistance are not fully understood, but a number of proteins/pathways that are implicated in cisplatin resistance have been identified 2, 8, 10, 11, 20, 22, 23, 27, 34. These pathways regulate a variety of cellular responses, ranging from DNA repair and the cell cycle to apoptosis 2, 8, 10, 11, 20, 22, 23, 27, 34. Among these pathways, the activation of the mitogen-activated protein kinase (MAPK) pathways modulates cisplatin sensitivity 7, 48.
MAPKs encompass a family of kinases that are subdivided into three subfamilies: c-Jun N-terminal kinase 1/2 (JNK1/2), p38, and extracellular-signal-regulated kinase (ERK) 12, 21. MAPKs orchestrate many of the short- and long-term cellular responses that occur in response to extracellular stimuli. MAPKs are activated and deactivated through the reversible phosphorylation/dephosphorylation of both threonine and tyrosine residues of the TXY motif. Once phosphorylated/activated, MAPKs can phosphorylate a variety of proteins/substrates. In doing so, the related signaling pathways are activated, and the subsequent cellular responses are achieved 12, 21. For example, the activation of JNK promotes programmed cell death 6. On the other hand, MAPKs are inactivated by dephosphorylation that is mediated by the MAPK phosphatase (MKP) family members.
MKP-1 (also known as DUSP1) is a member of the MKP family and is an endogenous regulator of MAPKs 25, 51. MKP-1 can remove phosphate groups from both phospho-threonine and phospho-tyrosine residues on JNK1/2, p38, and ERK, thereby inactivating MAPK signaling 25, 51. Previous studies have shown that MKP-1 is overexpressed in several types of cancer, including ovarian and lung cancers 35, 39, 43, 51. When MKP-1 expression was increased, it inhibited programmed cell death induced by several stimuli 7, 17, 24, 44, 48, but the underlying mechanism is not fully understood.
The post-translational modification of proteins by poly(ADP-ribosylation) is mediated by a family of proteins designated the poly(ADP-ribose) polymerases (PARPs). PARP-1 is a member of the PARP family that mediates post-translational modification of proteins by poly(ADP-ribosylation) 15, 29. In response to DNA damage, PARP-1 is recruited to single strand breaks to trigger the poly(ADP-ribosylation) of multiple substrates, which leads to the activation of a variety of cellular responses 15, 49. It has been shown that activation of PARP-1 plays a critical role in mediating DNA base excision repair 15, 49. PARP-1 is overexpressed in cancers, including cancer cells that have acquired cisplatin resistance and thereby it has been explored for its therapeutic potential in cancer treatment 1, 31. Studies with PARP inhibitors have shown encouraging results against some, but not all cancers 5, 14, 16. Therefore, understanding the mechanisms of the regulation of PARP protein levels and activity are critical for facilitating the development of PARP inhibitors for cancer treatment.
The present study focused on understanding the mechanisms of MKP-1-mediated cisplatin resistance in ovarian cancer cells, which lead to the identification of a previously unknown mechanism by which MKP-1 stabilizes PARP-1 protein via inhibition of JNK.
Results
MKP-1 stimulates PARP-1 and PAR expression and suppresses programmed cell death and cisplatin sensitivity
To study the mechanism by which MKP-1 promotes cisplatin resistance, we overexpressed MKP-1 in TOV112D cells, an ovarian cancer cell line that expresses a moderate level of MKP-1 among several human ovarian cancer cell lines, including OVCA432, CAOV3, OVCA420, RMG-1 and OV433 cells 44, and found that forced expression of exogenous MKP-1 suppressed cisplatin-induced PARP-1 cleavage (a hallmark of apoptosis) and cisplatin sensitivity, relative to the vector control cells (Fig. 1A and B). This overexpression of MKP-1 suppressed reproductive cell death and programmed cell death after cisplatin treatment, determined by colony formation assay and flow cytometry analysis, respectively (Fig. 1C and D). Interestingly, when MKP-1 was overexpressed, the protein levels of PARP-1 and its enzymatic product PAR were also elevated as compared to cells transfected with the control vector (Fig. 1A). To confirm the impact of MKP-1 on PARP-1 protein expression, we silenced MKP-1 with short hairpin RNA (shRNA) in CAOV3 cells, an ovarian cancer cell line that expresses a relatively high level of MKP-1. Fig. 1E shows that silencing MKP-1 reduced the levels of PARP-1 and PAR compared to cells transfected with control shRNA. Consistently, silencing MKP-1 promoted PARP-1 cleavage and increased apoptotic cell population (Fig. 1E and H), suggesting that downregulation of MKP-1 enhances cisplatin-induced programmed cell death. Furthermore, downregulation of MKP-1 by shRNA decreased cell viability and colony formation compared to control cells after cisplatin treatment (Fig. 1F and G). Since both MKP-1 and PARP-1 promote cisplatin resistance 33, 48, these results support the notion that increased PARP-1 expression may be one of the mechanisms by which MKP-1 promotes cisplatin resistance, which was previously unknown.
Figure 1. MKP-1 confers cisplatin resistance and MKP-1 levels correlate with PARP-1 and PAR levels in ovarian cancer cells.

(A) Immunoblot analyses of the amounts of the indicated proteins in TOV112D cells stably overexpressing pNTAP-MKP-1 (TAP-MKP-1) or pNTAP (vector) exposed to 2 μg/mL cisplatin for 24 hr. Exo-MKP-1, exogenous (transfected) MKP-1; Endo-MKP-1, endogenous MKP-1. (B) MTT analyses of cell viability of TOV112D cells treated with cisplatin for 4 days. Data represent mean ± S.D. (error bars) of three independent experiments. **, p < 0.001, statistically significant. (C) Representative results of colony formation assays of three independent experiments. Cells in (A) were treated with 0.5 μg/ml cisplatin. (D) Analyses of apoptosis. Cells in (A) were harvested after treatment with 2 μg/mL cisplatin for 16 hr and stained with Annexin V and Propidium Iodide (PI). Numbers in right two quadrants refer to the percent Annexin+/PI− and Annexin+/PI+ (apoptotic) cells in this representative experiment. (E) Immunoblot analyses of the amounts of the indicated proteins in MKP-1 silenced CAOV3 cells (sh-MKP-1) and control non-target CAOV3 cells (Non-target) after treatment with 1 μg/mL cisplatin for 24 hr. (F) MTT analyses of cell viability after 72-hr treatment. (G) Colony formation assay of cells in (E) after treatment with 1 μg/ml cisplatin. (H) Analyses of apoptosis. Cells in (E) were left untreated or exposed to cisplatin and analyzed as described in (D).
MKP-1 suppresses MAPK activities to maintain PARP-1 and PAR levels
It is established that MKP-1 exerts its biological functions by inhibiting MAPK activities 26, 51. Thus, we hypothesized that positive regulation of PARP-1 expression by MKP-1 is the consequence of MKP-1-mediated inhibition of MAPK activities. To test this hypothesis, we exposed mouse embryonic fibroblasts (MEFs) derived either from MKP-1-deficient mice or wild-type mice to cisplatin and then assessed the activation of the MAPK pathways. Fig. 2A shows that cisplatin treatment stimulates ERK, p38, JNK, CREB, and c-JUN phosphorylation in cisplatin-treated cells, suggesting the ERK, p38, and JNK pathways are activated 44. In agreement with Fig. 1E, we detected higher amounts of PARP-1 and PAR proteins in untreated-MKP-1+/+ MEFs but lower amounts of cisplatin-induced cleaved PARP-1 protein in cisplatin-treated MKP-1+/+ MEFs as compared to MKP-1−/− MEFs under the same treatment conditions (Fig. 2A). In addition, MKP-1−/− MEFs were more sensitive than MKP-1+/+ MEFs to cisplatin treatment, as evidenced by reduced cell viability (Fig. 2B).
Figure 2. A role for MKP-1 in the activation of the MAPK pathways, PARP-1 and PAR expression, and cisplatin resistance.
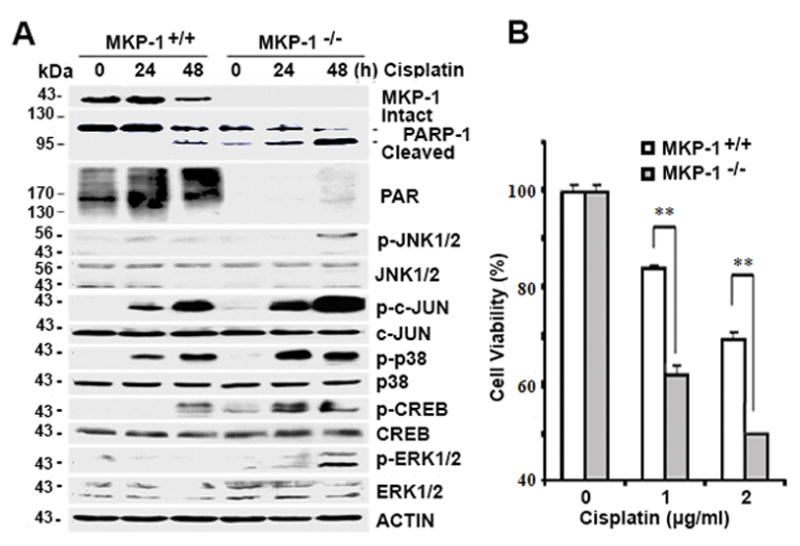
(A) Immunoblot analyses of the amounts of the indicated proteins in MKP-1+/+ and MKP-1−/− MEF cells after treatment with 2 μg/mL cisplatin for indicated time periods. (B) MTT analyses of the viability in MEFs after treatment with cisplatin at indicated concentrations for 72 hr.
Because PARP-1 is involved in cisplatin resistance 33, we asked which MAPK regulates PARP-1 expression to mediate cell survival. Fig. 3A shows that the suppression of the JNK pathway by SP600125 or the p38 pathway by SB203580, but not the ERK pathway by the MEK (MAPK/ERK kinase) inhibitor U0126, reduced cisplatin-induced growth inhibition compared to control cells. This result suggests that either JNK or p38 is involved in MKP-1-dependent PARP-1-mediated cell survival. To investigate the mechanism by which MAPKs regulate PARP-1 expression, we exposed MKP-1+/+ and MKP-1−/− MEFs to U0126, SP600125 or SB203580, and then used immunoblot assays to assess the amounts of PARP-1/PAR proteins. Using this approach, we found that the suppression of JNK but not p38 or ERK activity resulted in partial restoration of PARP-1 and PAR protein expression in MKP-1−/− MEFs (Fig. 3B). In concert with these findings, we found that the JNK downstream substrate c-JUN was phosphorylated and that the level of its phosphorylation was correlated with reduced amounts of PARP-1 protein (Fig. 3C). These results support our hypothesis that JNK is the protein that is responsible for suppressing PARP-1 expression.
Figure 3. Effects of MKP-1 deletion and MAPK inhibition on cisplatin sensitivity and PARP-1 and PAR expression.
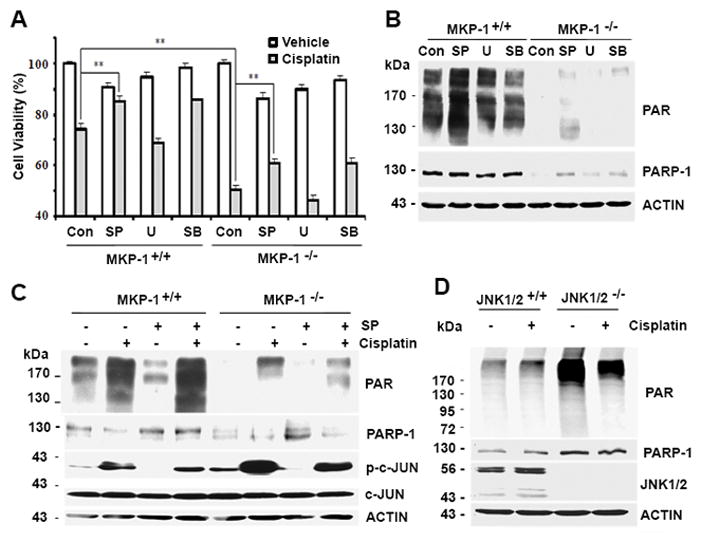
(A) MTT analyses of cell viability in MKP-1+/+ and MKP-1−/− MEF cells after treatment with 2 μg/mL cisplatin with or without 10 μM U012 6 (U), 10 μM SB203580 (SB) or 10 μM SP600125 (SP) for 48 hr. (B) Immunoblot analyses of PARP-1 and PAR levels in MEF cells treated as in (A) for 24 hr. (C) Immunoblot analyses of the amounts of the indicated proteins in MKP-1+/+ and MKP-1−/− MEF cells after treatment with 2 μg/mL cisplatin with or without 10 μM SP600125 (SP) for 24 hr. (D) Immunoblot analyses of PARP-1, PAR and JNK1/2 levels in JNK1/2+/+ MEFs and JNK1/2−/− MEFs exposed to 2 μg/mL cisplatin for 24 hr.
To define the direct role of JNK in the regulation of PARP-1 levels, we assessed the amounts of PARP-1 protein in JNK1/2−/− MEFs. As expected, JNK1/2 were not detected in JNK1/2−/− MEFs, as compared to JNK1/2+/+ MEFs. However, JNK1/2−/− MEFs expressed higher amounts of PARP-1 and PAR proteins than that in JNK1/2+/+ MEFs (Fig. 3D). Based on these data, we argue that JNK1/2 negatively regulates PARP-1 and PAR expression.
PARP-1 ubiquitination involves JNK
It is well established that the ubiquitination-mediated pathway can cause protein degradation. Decreased PARP-1 protein levels in cells with MKP-1 downregulation or deletion led us to ask if MKP-1 regulates PARP-1 ubiquitination/degradation to maintain the MKP-1 level. Accordingly, CAOV3 cells stably transfected with MKP-1 shRNA or control shRNA were used to assess the impact of MKP-1 silencing on PARP-1 ubiquitination. Fig. 4A (left panel) shows that increased amounts of PARP-1 ubiquitination were detected in MKP-1 silenced cells compared to cells transfected with control shRNA. Importantly, the level of ubiquitinated PARP-1 was inversely correlated with a lower level of total PARP-1 protein. Similarly, higher levels of total PARP-1 protein and lower levels of ubiquitinated PARP-1 protein were detected in MKP-1+/+ MEFs than in MKP-1−/− MEFs (Fig. 4A, right panel). Therefore, downregulating or deleting MKP-1 in both CAOV3 and MEF cells promotes PARP-1 ubiquitination.
Figure 4. MKP-1 and JNK regulate PARP-1 levels and ubiquitination.

(A) Immunoblot analyses of total and ubiquitinated PARP-1. Cell lysates were prepared from non-target shRNA CAOV3 cells (Non), shRNA MKP-1 CAOV3 cells (MKP-1) (left panel), MKP-1+/+ MEFs and MKP-1−/− MEFs (right panel). PARP-1 antibody was used to perform immunoprecipitation, and antibodies against PARP-1 and ubiquitin (Ub) were used to perform immunoblotting. (B) MKP-1+/+ and MKP-1−/− MEFs were left untreated or exposed to 10 μM SP600125 (SP) for 24 hr, and cell lysates were prepared and subjected to immunoprecipitation with antibody to PARP-1. Immunoblot analyses were performed using antibodies against Ub and PARP-1. (C) Non-target shRNA CAOV3 cells (Non-target) and shRNA MKP-1 CAOV3 cells (sh-MKP-1) were transfected with either non-target siRNA (si-Con) or JNK1/2 siRNA (si-JNK) for 72 hr. Immunoprecipitation and immunoblotting were performed as in (A). (D) Cell lysates were prepared from JNK1/2+/+ and JNK1/2−/− MEFs. Immunoprecipitation with PARP-1 antibody and immunoblot analyses using antibodies against Ub and PARP-1 were performed.
Since JNK1/2−/− MEFs exhibited higher levels of PARP-1 and PAR as compared to JNK1/2+/+ MEFs (Fig. 3D), we reasoned that JNK is the key player that regulates PARP-1 ubiquitination. In this regard, the consequence of JNK suppression by SP600125 on PARP-1 ubiquitination was assessed in MEFs. Fig. 4B shows that the pharmacological inhibition of JNK by SP600125 reduced PARP-1 ubiquitination as compared to untreated cells. Importantly, the inhibition of JNK activity by SP600125 restored PARP-1 levels in both MKP-1+/+ and MKP-1−/− MEFs (Fig. 4B). To further address the importance of JNK in PARP-1 ubiquitination, we downregulated JNK1/2 expression by siRNA and showed that JNK1/2 silencing reduced PARP-1 ubiquitination while increasing total amounts of PARP-1 protein as compared to cells transfected with control siRNAs (Fig. 4C). These results support a role for JNK in regulating PARP-1 ubiquitination and subsequent degradation.
To further confirm the importance of JNK in PARP-1 ubiquitination, we examined the amounts of total and ubiquitinated PARP-1 protein in JNK1/2 null MEFs. Fig. 4D shows that the amounts of ubiquitinated PARP-1 protein were higher in JNK1/2+/+ MEFs than in JNK1/2−/− MEFs. Conversely, JNK1/2−/− MEFs expressed higher amounts of PARP-1 protein as compared to JNK1/2+/+ MEFs. These data led us to conclude that MKP-1 maintains the PARP-1 level by inactivating JNK-dependent PARP-1 ubiquitination and subsequent degradation.
JNK promotes PARP-1 phosphorylation
To identify the mechanism by which JNK suppresses PARP-1 expression, we first asked whether JNK associates with PARP-1. We used PARP-1 antibody to perform immunoprecipitation to address if PARP-1 antibody can pull down JNK. Indeed, we showed that PARP-1 antibody pulled down similar amounts of total JNK proteins in both types of MEFs in the presence and absence of cisplatin treatment (Fig. 5A). We also confirmed that the amounts of PARP-1 protein were higher in MKP-1+/+ MEFs than that in MKP-1−/− MEFs. Notably, PARP-1 antibodies pulled down substantial amounts of phosphorylated serine in MKP-1−/− MEFs, but only negligible amounts of phosphorylated serine in MKP-1+/+ MEFs receiving cisplatin treatment (Fig. 5A). In agreement, we showed that PARP-1 and phosphorylated serine were co-immunoprecipitated with JNK in JNK1/2+/+ MEFs but not in JNK1/2−/− MEFs (Fig. 5B). More importantly, JNK1/2−/− MEFs expressed higher amounts of PARP-1 protein than JNK1/2+/+ MEFs. Thus, these studies identify an interaction between JNK and PARP-1, suggesting that such interaction may promote PARP-1 phosphorylation, ubiquitination, and subsequent degradation.
Figure 5. Interactions between JNK and PARP-1, and effects of JNK deletion on PARP-1 protein expression and serine phosphorylation.
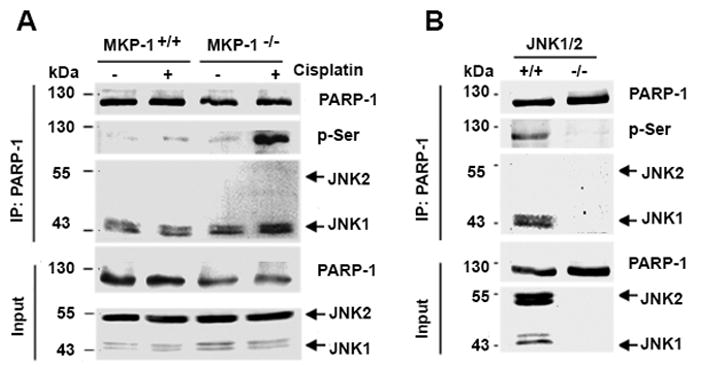
(A) Immunoprecipitation with PARP-1 antibody and immunoblotting with antibodies against PARP-1, phosphorylated serine (P-Ser) or JNK1/2 were performed using cell lysates prepared from MKP-1+/+ and MKP-1−/− MEFs treated with 2 μg/mL cisplatin for 24 hr. (B) Immunoprecipitation with PARP-1 antibody and immunoblotting with antibodies against PARP-1, phosphorylated serine, or JNK1/2 were performed using cell lysates prepared from JNK1/2+/+ and JNK1/2−/− MEFs.
Higher levels of MKP-1 and PARP-1 correlate with acquired cisplatin resistance in ovarian cancer cells
In addition to the role of the MKP-1-PARP-1 axis in intrinsic cisplatin resistance, we suspected that this regulatory mechanism also regulates acquired cisplatin resistance. To test this possibility, we employed the OV433-P/OV433-CR model to assess the relationship between the MKP-1 and PARP-1 levels and acquired resistance in ovarian cancer cells 45. Consistent with our previous work 45, cisplatin inhibited the growth of OV433-P cells but not that of OV433-CR cells (Fig. 6A). In this cell model, cisplatin sensitivity was correlated with cisplatin-induced apoptosis, as evidenced by the generation of cleaved PARP-1 in OV433-P cells but not in OV433-CR cells whose full-length PARP-1 remained intact (Fig. 6B). By examining the levels of PARP-1 and PAR proteins, we detected higher amounts of PARP-1 and PAR proteins in cisplatin resistant OV433-CR cells. By contrast, we detected lower amounts of PARP-1 and PAR proteins in chemosensitive OV433-P cells (Fig. 6B). Next, we compared the amounts of p-JNK (activated) protein in these two cell lines. As expected, cisplatin treatment led to JNK phosphorylation/activation, but the amounts of phosphorylated JNK protein was higher in OV433-P than OV344-CR cells (Fig. 6B) Importantly, we showed that both cell lines expressed similar amounts of total JNK proteins (Fig. 6B). By demonstrating the positive regulation of PARP-1 expression by MKP-1 in both intrinsic and acquired cisplatin resistance, our results strongly suggest that suppression of PARP-1 activity may be a viable strategy to bypass cisplatin resistance in ovarian cancer cells with MKP-1 overexpression.
Figure 6. The expression of MKP-1, PARP-1, PAR and JNK1/2 proteins and effects of MKP-1 or PARP-1 silencing on cisplatin sensitivity.
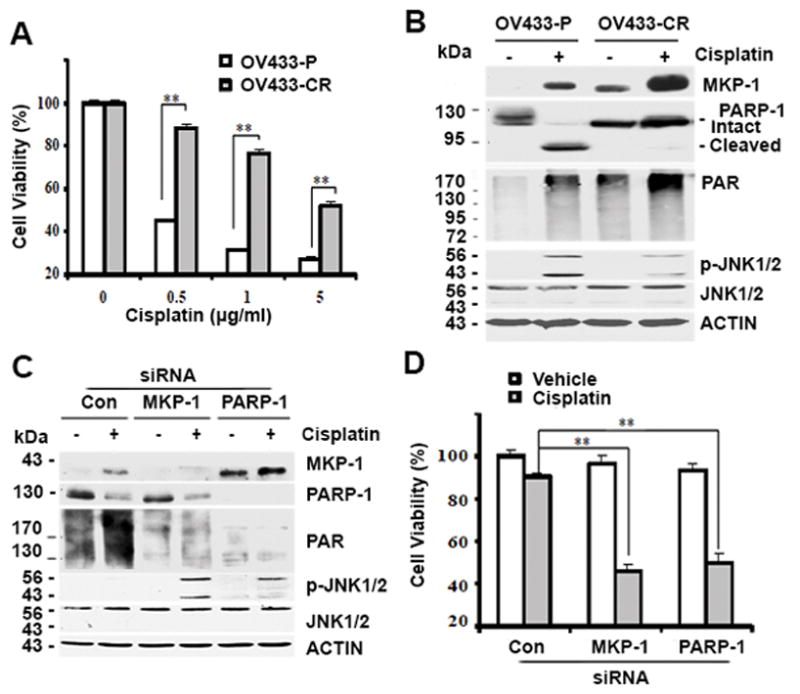
(A) Cell viability was determined by MTT assays in OV433-P and OV433-CR cells after treatment with cisplatin for 72 hr. (B) Immunoblot analyses of MKP-1, PARP-1, PAR, P-JNK1/2 and JNK1/2 levels in OV433-P and OV433-CR cells after treatment with 5 μg/mL cisplatin for 24 hr. (C) Immunoblot analyses of the amounts of the indicated proteins in OV433-CR cells. Cells transfected with control non-target siRNA (Con) or siRNA against MKP-1 or PARP-1 were exposed to 5 μg/mL cisplatin for 24 hr. (D) MTT analyses of cell viability in cells in (C) after treatment with 5 μg/mL cisplatin for 48 hr.
Suppression of PARP-1 activity enhances cisplatin’s anticancer activity in acquired cisplatin-resistant cells
To explore the possibility that targeting the MKP-1-PARP-1 axis overcomes acquired cisplatin resistance, we assessed the effects of knockdown of MKP-1 or PARP-1 on cisplatin-induced growth inhibition in resistant OV433-CR cells. Fig. 6C shows that silencing MKP-1 increased cisplatin-induced JNK1/2 phosphorylation/activation (p-JNK1/2), which was less in non-target transfected cells. Silencing MKP-1 decreased PARP-1 and PAR levels while silencing PARP-1 did not impact MKP-1 levels, indicating that PARP-1 is a downstream effector of MKP-1 (Fig. 6C). In addition, silencing MKP-1 or PARP-1 increased cisplatin-induced growth inhibition while such effects were not observed in cells transfected with control siRNA (Fig. 6D). These data identify a good correlation between MKP-1-dependent PARP-1 expression and acquired cisplatin resistance in ovarian cancer cells.
Since inhibitors for MKP-1 are not available, we asked if PARP-1 inhibition could bypass MKP-1-mediated cisplatin resistance. To address this question, we exposed TOV112D ovarian cancer cells transfected with either a control plasmid or MKP-1-expression plasmid (Fig. 1A) to cisplatin, the PARP inhibitor ABT888, or cisplatin plus ABT888. Using the MTT assay, we showed that cisplatin effectively suppressed the growth of vector control cells and ABT888 did not enhance cisplatin-induced cell killing (Fig. 7). By contrast, even though MKP-1 overexpressing cells were able to tolerate cisplatin treatment, ABT888 was able to enhance cisplatin-induced growth inhibition (Fig. 7). In fact, ABT888 alone did not have obvious effects on the growth of both cell lines. Therefore, these data indicate that MKP-1-mediated cisplatin resistance can be overcome by PARP-1 inhibition.
Figure 7. Inhibition of PARP-1 activity partially restores cisplatin sensitivity in MKP-1-overexpressing ovarian cancer cells.
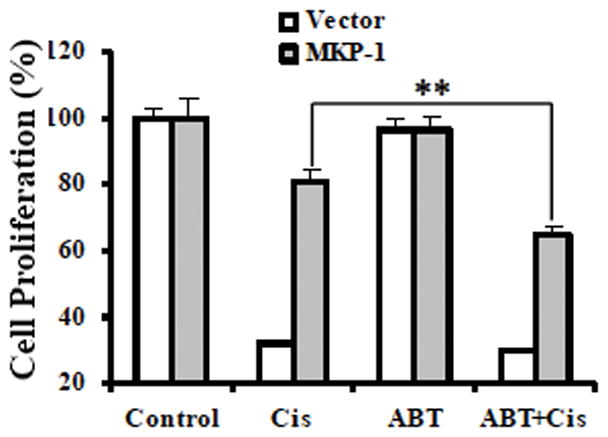
MKP-1-overexpressing or control vector TOV112D cells were left untreated or exposed to cisplatin (Cis) at 1 μg/mL, 20 μM PARP inhibitor ABT888 (ABT), or cisplatin plus ABT888 (ABT+Cis) for 3 days, and cell viability was determined by MTT assays.
Discussion
In this study, we have demonstrated for the first time that in ovarian cancer cells, MKP-1-mediated cisplatin resistance is PARP-1 dependent, and that this mechanism can be overcome by the inhibition of PARP-1 activity.
Previous studies showed that the activation of JNK plays an important role in cisplatin-induced apoptosis 4, 36. It was also shown that DNA damage induces apoptosis via the JNK pathway and MKP-1 19, suggesting an important role for the MKP-1-JNK1/2 pathway in regulating DNA damage-induced apoptosis. Further studies from ovarian cancer cells identified a good correlation between increased MKP-1 expression and cisplatin resistance 44, 45, 48, but the underlying mechanism remains to be defined. This study shows that overexpression of MKP-1 increases PARP-1 and PAR protein levels and confers cisplatin resistance. Conversely, silencing endogenous MKP-1 by shRNA reduces PARP-1 levels but enhances cisplatin-induced programmed cell death. These data suggest that PARP-1 is responsible for MKP-1-mediated cisplatin resistance.
PARP-1 is well known for its role in DNA repair 15. It has been shown that base excision repair is PARP-1-dependent 38. In normal cells, when base excision repair is inhibited, damaged DNA can be repaired through alternative pathways such as homologous recombination. However, in cancer cells homologous recombination is often defective as a result of additional mutations (e.g., mutations in BRCA1/2) or epigenetic events 32. Consequently, these cancer cells cannot repair their damaged DNA in the presence of PARP inhibitors, leading to cell death. Based on these studies, a number of chemically distinct PARP inhibitors have been synthesized and explored for their therapeutic potential in cancer treatment, either as single agents or as part of combination regimens 1, 31. PARP inhibitors have been shown to have effects on some, but not all ovarian cancers 5, 14, 16, and the underlying mechanism is not fully understood. In ovarian cancer cells, in vitro studies indicated that MKP-1 overexpression can cause cisplatin resistance 48, and our data showed that cells with increased MKP-1 expression also have high PARP-1 levels. These data suggest that PARP-1 is a downstream effector of MKP-1.
Although many patients with ovarian cancer initially respond well to platinum-based regimens, but refractory tumors emerge because of the development of resistance to the treatments 3. We showed that acquired cisplatin resistant OV433-CR cells, relative to OV433-P cells, express higher levels of both MKP-1 and PARP-1. Importantly, the inhibition of PARP-1 activity partially sensitized acquired resistant cells to cisplatin-induced growth inhibition. Therefore, we conclude that the inhibition of PARP-1 activity can render resistant cells sensitive to cisplatin.
It has been shown that cisplatin resistant cells express a higher level of PARP-1 33, but the mechanism of this higher expression is unclear. We showed that increased MKP-1 expression could suppress JNK1/2 activity. Because the activation of JNK1/2 is partially responsible for cisplatin-induced apoptosis, decreased JNK1/2 activity by MKP-1 overexpression can decrease cisplatin sensitivity in cancer cells. We also showed that JNK can interact with PARP-1 and that PARP-1 phosphorylation on serine residues is increased in MKP-1 −/− MEFs. This is consistent with previous studies showing that MAPKs, including JNK, phosphorylate PARP-1 to regulate PARP-1’s functions 18, 52. It would be interesting to determine whether JNK phosphorylates serine 257 and serine 782 to stimulate PARP-1 degradation because these two sites were predicted to be phosphorylated by JNK1/2 18. In addition, we showed that MKP-1 inactivated JNK1/2, which leads to decreased PARP-1 ubiquitination and thus maintains total PARP-1 levels. This result strongly suggests that JNK1/2 promote PARP-1 ubiquitination. Thus, we speculate that JNK1/2 can phosphorylate PARP-1, which in turn promotes PARP-1 ubiquitination and subsequent degradation. When the level of MKP-1 is higher, it inhibits JNK1/2-mediated PARP-1 phosphorylation and subsequent ubiquitination (Fig. 8), but this hypothesis requires further investigation. Since JNK can phosphorylate many proteins including members of the Bcl-2 family (e.g., Bcl-2 and Bax) to induce apoptosis 13, 30, we propose that JNK-mediated apoptosis is the consequence of the collective effects of many proteins that regulate cell death including PARP-1 in this study (Fig. 8).
Figure 8.

A proposed model for the mechanism by which MKP-1 inactivates JNK1/2-mediated PARP-1 phosphorylation, ubiquitination and degradation, which leads to cisplatin resistance.
In this study, we showed a role for constitutive PARP-1 in regulating cisplatin-induced apoptosis. It has been shown that some components of nucleotide excision repair (NER), including XFP and XPG, can be transcriptionally regulated by AP-1 9, 40, leading to an increase in NER activity. Because c-Jun, a component of AP-1, is a substrate of JNK, it is possible that the MKP-1-JNK1/2-AP-1 axis regulates NER, and that PARP-1 protein stabilization is a side effect of reduced toxicity and reduced PARP-1 cleavage.
In summary, this work identifies PARP-1 as a key component to mediate MKP-1-dependent cisplatin resistance. We also show that JNK1/2 are key components that regulate PARP-1 phosphorylation and subsequent degradation, which contributes to cisplatin sensitivity. Considering that MKP-1 or PARP-1 plays an important role in cisplatin resistance, this study not only provides mechanistic insights into how MKP-1 regulates cisplatin resistance, but also provides a therapeutic strategy to use PARP-1 inhibitors to bypass MKP-1-mediated cisplatin resistance. It has been shown that ovarian cancer cells that acquire ability to tolerate cisplatin insults express a high level of MKP-1 protein. MAPK phosphatases, including MKP-1, are currently regarded as “undruggable” targets, and developing small molecular inhibitors for MKP-1 has been challenging. PARP inhibitors are currently being used in clinic or clinical trials for the treatment of ovarian and breast cancer. Thus, targeting PARP-1 to overcome MKP-1-mediated cisplatin resistance is highly translational and clinically significant. Future work will determine how JNK1/2 regulate PARP-1 phosphorylation and how phosphorylation affects PARP-1 ubiquitination and subsequent degradation.
Materials and Methods
Reagents
SP600125 (CAS129-56-6) and anti-PAR antibody (AM80) were purchased from Calbiochem (San Diego, CA). U0126 (V1121) was provided by Promega (Madison, WI). ABT888 (ALX-270-444) was from Enzo Life Sciences (Farmingdale, NY). FITC Annexin V Apoptosis Kit (556547) was from BD Biosciences (Franklin Lakes, NJ). Anti-actin (AC-74) antibody, cisplatin (P4394) and SB203580 (S8307) were purchased from Sigma (St. Louis, MO). Antibodies for MAPKs and PARP-1 (9532) were provided by Cell Signaling Technology (Danvers, MA). The MKP-1 (SC-370) and ubiquitin (Ub) (SC-8017) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell lines
Three human ovarian cancer cell lines used in this study were described previously 28. TOV112D cells stably expressing MKP-1 and control vector, CAOV3 cells expressing MKP-1 shRNA and control shRNA, and parental- and cisplatin-resistant OV433 cells were described previously 47. MKP-1-deficient mouse embryonic fibroblasts (MEFs) (MKP-1−/− MEFs) and wild-type MEFs (MKP-1+/+ MEFs) were described elsewhere 48. JNK1/2-deficient MEFs (JNK1/2−/− MEFs) and wild-type MEFs (JNK1/2+/+ MEFs) were described previously 41.
Cell viability, colony formation and apoptosis assays
Cell viability was determined by the MTT (Sigma, M5655) assay, as described previously 46. Colony formation was performed, as described elsewhere 50. Apoptosis was evaluated with a FITC Annexin V Apoptosis Kit (556547) according to the manufacturer’s instruction (BD Biosciences).
Immunoprecipitation (IP) and immunoblot analysis
Preparation of whole cell lysates, immunoprecipitation (IP) of PARP-1 and immunoblot analysis were performed, as described previously 47.
Silencing JNK1/2, MKP-1 and PARP-1 by small interfering RNA (siRNA)
All siRNA reagents including siRNA specific to MKP-1 (L-003484-02), PARP-1 (M-006656-01) and control siRNA (D-001810-10) were obtained from Dharmacon Research (Lafayette, CO). JNK1/2 siRNA (16104) was purchased from Ambion (Austin, TX). Gene silencing effects on JNK1/2, MKP-1 and PARP-1 expression were evaluated by immunoblotting.
Data analysis
Data analysis was performed using two-sided unpaired Student’s t test, as described previously 47.
Acknowledgments
Financial support: This work was, in part, supported by National Institute of Health (R01CA174949)
This work was, in part, supported by the NIH (R01CA174949).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Annunziata CM, O’Shaughnessy J. Poly (ADP-ribose) polymerase as a novel therapeutic target in cancer. Clin Cancer Res. 2010;16:4517–4526. doi: 10.1158/1078-0432.CCR-10-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beale PJ, Rogers P, Boxall F, Sharp SY, Kelland LR. BCL-2 family protein expression and platinum drug resistance in ovarian carcinoma. Br J Cancer. 2000;82:436–440. doi: 10.1054/bjoc.1999.0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC, Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–679. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brozovic A, Fritz G, Christmann M, Zisowsky J, Jaehde U, Osmak M, et al. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int J Cancer. 2004;112:974–985. doi: 10.1002/ijc.20522. [DOI] [PubMed] [Google Scholar]
- 5.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 6.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 7.Chattopadhyay S, Machado-Pinilla R, Manguan-Garcia C, Belda-Iniesta C, Moratilla C, Cejas P, et al. MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene. 2006;25:3335–3345. doi: 10.1038/sj.onc.1209364. [DOI] [PubMed] [Google Scholar]
- 8.Cheng TC, Manorek G, Samimi G, Lin X, Berry CC, Howell SB. Identification of genes whose expression is associated with cisplatin resistance in human ovarian carcinoma cells. Cancer chemotherapy and pharmacology. 2006;58:384–395. doi: 10.1007/s00280-005-0171-8. [DOI] [PubMed] [Google Scholar]
- 9.Christmann M, Tomicic MT, Origer J, Aasland D, Kaina B. c-Fos is required for excision repair of UV-light induced DNA lesions by triggering the re-synthesis of XPF. Nucleic Acids Res. 2006;34:6530–6539. doi: 10.1093/nar/gkl895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cullen KJ, Newkirk KA, Schumaker LM, Aldosari N, Rone JD, Haddad BR. Glutathione S-transferase pi amplification is associated with cisplatin resistance in head and neck squamous cell carcinoma cell lines and primary tumors. Cancer Res. 2003;63:8097–8102. [PubMed] [Google Scholar]
- 11.Dabholkar M, Vionnet J, Bostick-Bruton F, Yu JJ, Reed E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J Clin Invest. 1994;94:703–708. doi: 10.1172/JCI117388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 13.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103:334–346. doi: 10.1093/jnci/djq509. [DOI] [PubMed] [Google Scholar]
- 15.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283:593–596. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- 16.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 17.Franklin CC, Srikanth S, Kraft AS. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci USA. 1998;95:3014–3019. doi: 10.1073/pnas.95.6.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gagne JP, Moreel X, Gagne P, Labelle Y, Droit A, Chevalier-Pare M, et al. Proteomic investigation of phosphorylation sites in poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase. J Proteome Res. 2009;8:1014–1029. doi: 10.1021/pr800810n. [DOI] [PubMed] [Google Scholar]
- 19.Hamdi M, Kool J, Cornelissen-Steijger P, Carlotti F, Popeijus HE, van der Burgt C, et al. DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene. 2005;24:7135–7144. doi: 10.1038/sj.onc.1208875. [DOI] [PubMed] [Google Scholar]
- 20.Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Molecular pharmacology. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 22.Johnson SW, Shen D, Pastan I, Gottesman MM, Hamilton TC. Cross-resistance, cisplatin accumulation, and platinum-DNA adduct formation and removal in cisplatin-sensitive and -resistant human hepatoma cell lines. Exp Cell Res. 1996;226:133–139. doi: 10.1006/excr.1996.0211. [DOI] [PubMed] [Google Scholar]
- 23.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 24.Keyse SM, Emslie EA. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature. 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- 25.Keyse SM. An emerging family of dual specificity MAP kinase phosphatases. Biochim Biophys Acta. 1995;1265:152–160. doi: 10.1016/0167-4889(94)00211-v. [DOI] [PubMed] [Google Scholar]
- 26.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–261. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 27.Kuo MT, Chen HH, Song IS, Savaraj N, Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26:71–83. doi: 10.1007/s10555-007-9045-3. [DOI] [PubMed] [Google Scholar]
- 28.Kwong J, Lee JY, Wong KK, Zhou X, Wong DT, Lo KW, et al. Candidate tumor-suppressor gene DLEC1 is frequently downregulated by promoter hypermethylation and histone hypoacetylation in human epithelial ovarian cancer. Neoplasia. 2006;8:268–278. doi: 10.1593/neo.05502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336:728–732. doi: 10.1126/science.1216338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18:80–86. doi: 10.1016/j.gde.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 32.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 33.Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73:2271–2280. doi: 10.1158/0008-5472.CAN-12-3000. [DOI] [PubMed] [Google Scholar]
- 34.Perego P, Giarola M, Righetti SC, Supino R, Caserini C, Delia D, et al. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996;56:556–562. [PubMed] [Google Scholar]
- 35.Rojo F, Gonzalez-Navarrete I, Bragado R, Dalmases A, Menendez S, Cortes-Sempere M, et al. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin Cancer Res. 2009;15:3530–3539. doi: 10.1158/1078-0432.CCR-08-2070. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Perez I, Murguia JR, Perona R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene. 1998;16:533–540. doi: 10.1038/sj.onc.1201578. [DOI] [PubMed] [Google Scholar]
- 37.Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39:3166–3175. doi: 10.1093/nar/gkq1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 40.Tomicic MT, Reischmann P, Rasenberger B, Meise R, Kaina B, Christmann M. Delayed c-Fos activation in human cells triggers XPF induction and an adaptive response to UVC-induced DNA damage and cytotoxicity. Cell Mol Life Sci. 2011;68:1785–1798. doi: 10.1007/s00018-010-0546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 42.Tummala MK, McGuire WP. Recurrent ovarian cancer. Clin Adv Hematol Oncol. 2005;3:723–736. [PubMed] [Google Scholar]
- 43.Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003;191:229–237. doi: 10.1016/s0304-3835(02)00612-2. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Zhou JY, Wu GS. ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 2007;67:11933–11941. doi: 10.1158/0008-5472.CAN-07-5185. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Zhou JY, Zhang L, Wu GS. Involvement of MKP-1 and Bcl-2 in acquired cisplatin resistance in ovarian cancer cells. Cell Cycle. 2009;8:3191–3198. doi: 10.4161/cc.8.19.9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289:17163–17173. doi: 10.1074/jbc.M114.558288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Zhou JY, Kho D, Reiners JJ, Jr, Wu GS. Role for DUSP1 (dual-specificity protein phosphatase 1) in the regulation of autophagy. Autophagy. 2016;12:1791–1803. doi: 10.1080/15548627.2016.1203483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Z, Xu J, Zhou JY, Liu Y, Wu GS. Mitogen-activated protein kinase phosphatase-1 is required for cisplatin resistance. Cancer Res. 2006;66:8870–8877. doi: 10.1158/0008-5472.CAN-06-1280. [DOI] [PubMed] [Google Scholar]
- 49.Whitacre CM, Hashimoto H, Tsai ML, Chatterjee S, Berger SJ, Berger NA. Involvement of NAD-poly(ADP-ribose) metabolism in p53 regulation and its consequences. Cancer Res. 1995;55:3697–3701. [PubMed] [Google Scholar]
- 50.Wu GS, Burns TF, McDonald ER, Jiang W, Meng R, Krantz ID, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–143. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 51.Wu GS. Role of mitogen-activated protein kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev. 2007;26:579–585. doi: 10.1007/s10555-007-9079-6. [DOI] [PubMed] [Google Scholar]
- 52.Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, Shen HM. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ. 2007;14:1001–1010. doi: 10.1038/sj.cdd.4402088. [DOI] [PubMed] [Google Scholar]