

Abstract
We investigated the role of amygdala corticotropin-releasing factor (CRF) neurons in the perturbations of descending pain inhibition caused by neuropathic pain. Forced swim increased the tail-flick response latency in uninjured mice, a phenomenon known as stress-induced analgesia (SIA) but did not change the tail-flick response latency in mice with neuropathic pain caused by sciatic nerve constriction. Neuropathic pain also increased the expression of CRF in the central amygdala (CeAmy) and ΔFosB in the dorsal horn of the spinal cord. Next, we injected the CeAmy of CRF-cre mice with cre activated AAV-DREADD (Designer Receptors Exclusively Activated by Designer Drugs) vectors. Activation of CRF neurons by DREADD/Gq did not affect the impaired SIA but inhibition of CRF neurons by DREADD/Gi restored SIA and decreased allodynia in mice with neuropathic pain. The possible downstream circuitry involved in the regulation of SIA was investigated by combined injections of retrograde cre-virus (CAV2-cre) into the locus ceruleus (LC) and cre activated AAV-diphtheria toxin (AAV-FLEX-DTX) virus into the CeAmy. The viral injections were followed by a sciatic nerve constriction ipsilateral or contralateral to the injections. Ablation of amygdala projections to the LC on the side of injury but not on the opposite side, completely restored SIA, decreased allodynia and decreased ΔFosB expression in the spinal cord in mice with neuropathic pain. The possible lateralization of SIA impairment to the side of injury was confirmed by an experiment in which unilateral inhibition of the LC decreased SIA even in uninjured mice.
The current view in the field of pain research attributes the process of pain chronification to abnormal functioning of descending pain inhibition. Our results demonstrate that the continuous activity of CRF neurons brought about by persistent pain leads to impaired SIA, which is a symptom of dysregulation of descending pain inhibition. Therefore, an over-activation of amygdala CRF neurons is very likely an important contributing factor for pain chronification.
Keywords: stress-induced analgesia, central amygdala, chronic pain, neuropathic pain, corticotropin-releasing factor, norepinephrine, descending pain inhibition
Introduction
SIA can occur during or after a physical or psychological stressor and it decreases the conscious perception of pain in humans as well as the behavioral response to nociceptive stimuli in animals [1-3]. SIA depends on activation of inhibitory supraspinal projections to the dorsal horn of the spinal cord, which contains the first central nervous system synapses for nociceptive information. The inhibitory projections are collectively referred to as the descending pain inhibitory system or simply descending inhibition [4, 5]. Descending inhibition affects multiple nociceptive modalities including thermal, inflammatory and neuropathic pain. Descending inhibitory pathways are polysynaptic with significant contributions from the prefrontal and cingulate cortices, amygdala, ventrolateral periaqueductal gray (PAG), LC and rostral ventromedial medulla (RVM) [6]. Brainstem projections that contain norepinephrine or serotonin are a major part of the final inhibitory input to the dorsal horn. Norepinephrine and serotonin inhibit nociception via both pre- and postsynaptic mechanisms, which forms the basis for treating chronic pain with serotonin and norepinephrine reuptake inhibitors or receptor agonists [7]. The CeAmy plays an important role in the physiological response to multiple stressors including pain [8, 9]. The CeAmy receives processed sensory information through the basolateral amygdala (BLA) and a direct nociceptive input via the spino-parabrachial-amygdaloid pathway [10]. The CeAmy is a key element in descending inhibition of pain and is essential for robust SIA [11]. The projections of the CeAmy target the bed nucleus of stria terminalis (BNST) and several brainstem nuclei, including the LC [12, 13]. There is general agreement that CeAmy CRF neurons do not affect baseline sensory thresholds but their role in pain is not clear. Long lasting inflammatory and neuropathic pain increases CRF expression in the CeAmy [14-16]. CRF release in the CeAmy causes hypersensitivity via CRF1 receptor and analgesia via CRF2 receptor [17-19]. While low doses of endogenous CRF in CeAmy increase pain sensitivity [20], high doses of exogenous CRF are analgesic [21]. One hypothesis that summarizes the role of CRF in pain processing is that the CRF neurons in the amygdala may act as an on/off switch for chronic pain [22]. The amygdala CRF neurons are well situated for the role of pain switch because they not only receive and respond to nociceptive stimuli but also undergo plasticity in association with chronic nociceptive stimulation and are responsible for the central sensitization and hyperalgesia observed in chronic pain [19]. Furthermore, the CRF projections from the CeAmy to the LC provide a pathway by which information that reaches the amygdala can influence descending inhibition of pain [12, 23] and it has been well established that the LC and norepinephrine are essential for pain inhibition including SIA [24-26]. Still, the effects of chronic pain on norepinephrine signaling in the spinal cord are not clear with some reports demonstrating that chronic pain inhibits norepinephrine levels [27] and other showing that chronic pain enhances norepinephrine signaling in the spinal cord [28]. Recent studies show that augmented descending pain inhibition prevents pain chronification in neuropathic rats and that this prevention is, at least partially, norepinephrine dependent [29]. However, the role of CeAmy CRF neurons in regulation of norepinephrine transmission and descending pain inhibition during prolonged nociceptive input remains unexplored.
We tested the hypothesis that CRF neurons in the CeAmy contribute to pain chronification by affecting descending pain inhibition. First, we investigated the effects of long-lasting neuropathic pain on SIA, which depends on the activity of the descending pain inhibitory system. Second, we tested the effects of activation or inhibition of CeAmy CRF neurons on SIA in healthy mice and mice with neuropathic pain. Finally, we examined whether CeAmy projections to the LC are part of the circuitry that inhibits SIA in mice with neuropathic pain.
Materials and Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee at Rosalind Franklin University of Medicine and Science (North Chicago, IL) and adhered to the guidelines provided in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57BL/6J, B6(Cg)-Crhtm1(cre)Zjh/J and B6.Cg-Tg(Th-cre)1Tmd/J male mice, 9 to 10 weeks old, were purchased from a supplier (The Jackson Laboratory, Bar Harbor, ME) and were housed 3 to 5 mice per cage. All nociceptive tests were done in the morning after an hour of acclimatization to the testing room.
Neuropathic pain model
Surgeries were performed under isoflurane anesthesia and following aseptic technique. Peripheral nerve constriction surgery was performed by exposing the main branch of the sciatic nerve with forceps and carefully placing a 4 mm long piece of P90 nontoxic, sterile polyethylene tubing that was split lengthwise (inner diameter 0.86 mm and outer diameter 1.27 mm; Becton Dickinson Intramedic, Franklin Lakes, NJ) onto the nerve. After confirmation that the tubing remained loosely on the nerve, the nerve was returned to its position and the incision was closed with wound clips [30]. Analgesia (flunixin meglumine 2 mg/kg, subcutaneous) was given for the following three days. Control mice were anesthetized with isoflurane and the skin was shaved but the sciatic nerve was not exteriorized in order to prevent behavior alterations and stimulation of CeAmy activity, which is increased in sham animals that undergo surgical incisions of skin and muscle [31].
Application of pharmacogenetics
Stereotaxic surgeries were used to inject DREADD viral constructs into the CeAmy and/or LC. Animals were anesthetized with isoflurane and placed in a Stoelting stereotaxic apparatus. A longitudinal skin incision and removal of pericranial connective tissue exposed the bregma and lambda sutures of the skull. The coordinates for the CeAmy injections were - 1.3 mm, ± 2.5 mm and - 4.5 mm in respect to bregma while the LC injections were placed - 5.4 mm, ± 0.8 mm and - 4.5 mm to bregma. Small holes were drilled into the skull and the viral solution was injected via a 32 gauge needle connected to an infusion pump (Microsyringe pump, World Precision Instruments, Sarasota, FL) in volume of 0.2 μl over 5 minutes. All animals were given analgesic and fluids for 3 days after the surgery. B6(Cg)-Crhtm1(cre)Zjh/J mice, with Cre recombinase expression in CRF neurons, were injected bilaterally in the CeAmy with cre activated adeno-associated virus (serotype 5, pAAV-hSyn-DIO-hM3D(Gq)-mCherry and pAAV-hSyn-DIO-hM4D(Gi)-mCherry, titter ≥ 3×1012(UNC Vector core, Chapel Hill, NC), referred here as AAV-DREADD-Gq and Gi respectively). C57BL/6J mice were injected unilaterally with retrograde canine adeno-associated cre virus or CAV-2-cre, titter ≥ 2.5×1012 (Plateforme de Vectorologie de Montpellier, Montpellier, France) into the LC, and with cre activated AAV-diphtheria toxin (serotype 5 AAV-mCherry-FLEX-dtA, titter ≥ 3×1012 (UNC Vector core, Chapel Hill, NC) viral construct into the CeAmy. Further behavior and nociceptive tests were performed three to four weeks after the viral injections. During the course of these experiments an AAV variant (rAAV-CAG-eGFP-F2A-Cre, titer = 1×1012, NINDS Viral Production Core Facility, Bethesda, MD) with efficient retrograde transport became available [32]. Because of the potential benefits of working with AAV we repeated the experiment with retrograde labeling via LC injection and the SIA experiments using the retro-AAV. The results are very similar, providing further support for the initial observation and supporting the suggestion that retro-AAV may be an attractive alternative to CAV2-Cre for this type of experiments.
Nociceptive and Behavior testing
Mice were given at least one hour to acclimatize to the testing room before nociceptive or behavior testing. Tactile sensitivity was measured using von Frey filaments applied to the plantar surface of the hind paw through a mesh floor. Following the technique for assessment of mechanical allodynia described by Chaplan [33], six filaments with different stiffness were used for each measurement. The starting filament was always 1.0 g. A quick withdraw, shaking or licking of the paw were considered a positive reaction. The mechanical thresholds were calculated by Dixon's up and down method [34]. Mechanical thresholds were obtained before and in five-day increments following sciatic nerve constriction for up to 15 days.
Thermal pain thresholds were measured by a tail-flick test. Mice were gently restrained in a plastic laboratory tube covered in foil to block environmental stimuli and their tails freely projected out of the restrainer. Next, the mice were placed in the Tail-Flick Analgesia Meter Model 33 (IITC Life Science Instruments, Woodland Hills, CA) in which a high intensity light (Beam: 4; Sensitivity: 3.5) was directed at their tails. The timer on the meter shut off (automatic shut off at 25 seconds) immediately once the mice flicked their tails, and these times were recorded as response latencies.
Stress was induced by a forced swim. Mice were placed in a transparent cylinder, 30 cm in diameter, half filled with tap water (21-23°C) for 5 minutes. The mice were completely dried with paper towels before undergoing the tail-flick test that followed one minute after the forced swim. The amount of time the mice were actively swimming was also measured for each forced swim. The last 3 minutes of the 5-minute swimming session were analyzed and the idle time was calculated as a percentage.
Drugs
Injectable solution of flunixin meglumine (NSAID) was purchased from veterinary supplier Norbrook Inc. (Overland Park, KS) and was applied according to the recommendations for anesthesia and analgesia in laboratory animals by IACUC at RFUMS.
Clozapine N-Oxide (CNO) was purchased from Tocris (Ellisville, MO), dissolved in 200 μl DMSO, brought to volume with distillated water, aliquoted and stored at -20°C until use. CNO, an inert compound and the only ligand for the DREADD receptors, was injected intraperitoneally (1 mg/kg in 0.2 ml) or administered via drinking bottles wrapped in aluminum foil (1 mg/kg in 5 ml water) to activate (DREADD/Gq) or inhibit (DREADD/Gi) expressing neurons.
Immunohistochemistry
Mice were euthanized 24 hours after testing with an intraperitoneal injection of pentobarbital sodium (Vedco Inc., Saint Joseph, MO) and perfused transcardially with 1X phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA). The spinal cords were carefully dissected from the spinal column. The dissected spinal cords sections included the thoracic region, lumbar intumescence and cauda equina. The lumbar intumescence was clearly visible as the most widened area between the cauda equina and the thoracic region. The lumbar intumescence was then trimmed and the right side was marked with a small cut in the ventral part of the cord. The dissected brains were also marked on the right side. Next, the collected tissue, brains and spinal cords, were left overnight in 4% PFA at 4° C. After the overnight incubation in 4% PFA the brain tissue and spina cords were washed with PBS and sectioned into 40 μm thick sections using a Leica VT1000S vibrotome.
All brain and spinal cord sections were washed with 1X PBS, incubated in 3% H2O2 for 15 minutes, again washed with 1X PBS, and then incubated in a blocking solution (1X PBS, 0.05% Triton X-100, 3% normal donkey serum) for 2 hours at room temperature. Sections were incubated with primary antibodies diluted in the same blocking solution at 4°C for 48 hours. The dilutions of primary antibody were as follows: 1:2K rabbit antibody to CRF (anti-CRF; catalog # 5348, Sigma-Aldrich, MO), 1:2K rabbit antibody to ΔFosB (anti-ΔFosB; catalog # 14695; Cell Signaling Technology, Danvers, MA), 1:2K rabbit antibody to c-Fos (anti-c-Fos; catalog # 2250; Cell Signaling Technology, Danvers, MA), 1:5K mouse antibody against norepinephrine transporter (NET) (anti-NET; catalog # 1447, Phosphosulutions, Aurora, CO), 1:5K chicken antibody to mCherry, (anti-mCherry, catalog # CPCA-mCherry, EnCor Biotechnology Inc., Alachua, FL) and 1:2K chicken antibody to GFP (anti-GFP, catalog # A10262, Invitrogen, Carlsbad, CA). Following incubation with the primary antibody, sections for CRF, ΔFosB and c-Fos were incubated with biotinylated secondary antibodies (Jackson ImmunoResearch Inc. West Grove, PA), 1:2K dilution for 2 hours, followed by incubation in avidin-biotin complex (ABC kit, Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. Then, the sections were placed in tyramide conjugated Alexa Fluor 488 or Alexa Fluor 405 fluorescent dyes, 20 nmol concentrations for 12 minutes, followed by washes in 1X TRIS-buffered saline. Direct secondary antibodies conjugated to Alexa 595 or Alexa 488 and diluted 1:400 in 1X PBS/3% donkey serum were used to visualize mCherry, NET and GFP immunoreactivity.
Microscopy
Leica DM 5500B epifluorescence microscope was used to acquire 16-bit images of amygdala, while Zeiss 510 confocal microscope was used to obtain 16-bit images of the LC and dorsal horn of the spinal cord. CRF immunoreactivity (CRF-ir), ΔFosB immunoreactivity (ΔtFosB-ir) and c-Fos immunoreactivity (c-Fos-ir) were evaluated on six sections per animal. The atlas matched brain sections covered the CRF-ir expression in the CeAmy between bregma level -1.1 mm and -1.8 mm. ΔFosB-ir and c-Fos-ir were analyzed between bregma levels -5.3 and -5.5 mm, and included the expression of Fos markers in the LC and in surrounding LC-perinuclear zone [35].
Analysis of the immunohistochemistry results was done using the ImageJ software (ImageJ, NIH image, Research Service Branch, Bethesda, MD). CRF-ir was measured using the integrated density (ID) calculation in ImageJ. The region of the CeAmy was selected carefully using the draw tool. Next, the ID was measured from three different, but same sized areas in the background (BG). Our final ID per image was calculated using the formula: ID = (IDCeAmy) -((IDBG1+ID BG2+ID BG3)/3) in arbitrary units.
ΔFosB-ir and c-Fos-ir were measured as the number of immunofluorescent cells above background. First the microscopic images were converted to 8-bit gray scale images with white background. Next, the “Threshold” tool was set and laminae one and two of the dorsal horn or the LC perinuclear zone were selected with the draw tool as ROI. All particles above background in ROI were calculated using the “Analyze Particles” function.
Statistical analysis
Data are presented as mean ± SEM and analyzed with Graph Prism 7 software. Student's T-test was used for two-group comparisons, while the results of all other experiments were evaluated using Two-way ANOVA followed by Bonferroni post hoc test. The accepted level of significance was set at P < 0.05 in all tests.
Results
Neuropathic pain inhibits SIA, increases CRF-ir in the CeAmy and ΔFosB-ir in the dorsal horn of the spinal cord but decreases ΔFosB-ir in the LC
We evaluated mechanical paw withdrawal thresholds after two habituation sessions in which the mice were placed in a box with a mesh floor. Inserting a short piece of plastic tubing (cuff) around the left sciatic nerve lowered the mechanical withdrawal threshold from an average of 3.8 grams ± 0.72 to 0.09 grams ± 0.04, 0.24 grams ± 0.12 and 0.14 grams ± 0.05 on postoperative days 5, 10 and 15 respectively (Figure 1 A). The mechanical withdrawal thresholds of the nerve-cuffed mice during this period were significantly different from the withdrawal thresholds of the control mice (Repeated Measures Two-Way ANOVA, F1,9 = 41.2, P < 0.0001), as well as from their own baseline thresholds (Repeated Measures Two-Way ANOVA, significant for interaction time × cuff, F3,27 = 12.4, P < 0.0001) (Figure 1 A). The mechanical hypersensitivity that followed sciatic nerve cuffing was not associated with a change in the response latency in the thermal tail-flick test in otherwise naïve mice (Figure 1 B left).
Figure 1. Sciatic nerve constriction impaired SIA, increased CRF-ir in the central amygdala and ΔFosB-ir in the dorsal horn of the spinal cord.
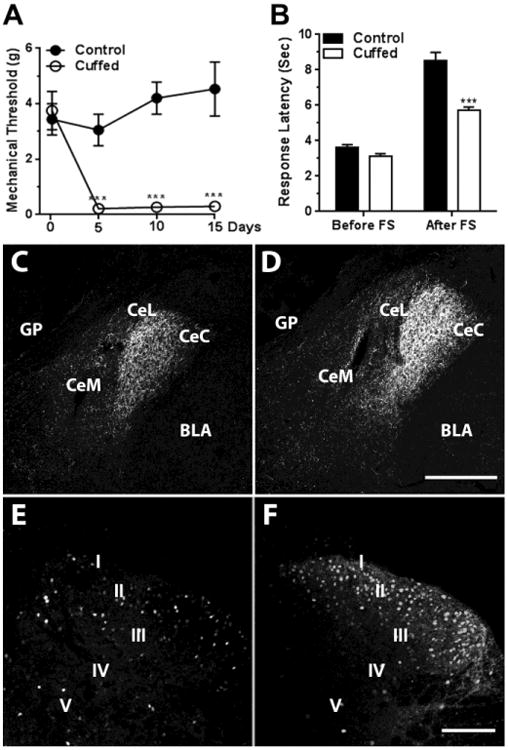
Panel A: Insertion of cuffs on the sciatic nerve decreased the mechanical paw withdrawal thresholds when compared to pre-surgical day 0 or when compared to the control group from day 5 to the end of the experiment at day 15. Two-Way ANOVA, *** - significant for interaction (time × cuff), F 3,27 = 12.4, P < 0.001 and for nerve-cuffed versus control at day 5, 10 and 15, Repeated Measures Two-Way ANOVA, F 1,9 = 41.2, P < 0.001. Panel B: Non-stressed, nerve-cuffed mice showed a normal response to thermal stimulus in a tail-flick test, but they were not able to generate SIA to the level of the control group after five minutes of forced swim. Two-Way ANOVA, *** - significant for interaction (pain × stress), F1,84 = 16.5, P < 0.001. Panels C and D: CRF-ir in the CeAmy was increased in nerve-cuffed mice (D) when compared to the controls (C); T-test, t16 = 7.2, P < 0.001. Panels E and F: Sciatic nerve constriction also increased ΔFosB-ir on the cuffed side of the dorsal horn of the spinal cord (F) when compared to the controls (E); T-test, t16 = 2.6, P < 0.05. Abbreviations: Roman numerals from I to V label the laminae of the dorsal horn. BLA -basolateral amygdala, CeC - centrocentral nucleus, CeM - cenrtromedial nucleus, CeL-centrolateral nucleus, FS – forced swim, GP - globus pallidus. Scale bar = 200 μm in D and 100 μm in F.
Having established a procedure that induces neuropathic hypersensitivity in which one measure of nociception, mechanical paw withdrawal sensitivity, is strongly affected and another measure, tail-flick response latency to a thermal stimulus, is unaffected, we went on to examine the effects of stress on these measures. We used a forced swim paradigm to induce stress. Both control and sciatic nerve-cuffed mice swam vigorously for the first two minutes after they were placed in water and floated for most of the remaining five-minute test session. The floating was interrupted by occasional bursts of vigorous swim. There were no detectable differences in the floating time between the two groups (control group mean: 76.6 % ± 6 versus the nerve-cuffed group mean: 77.9% ± 4; T-test, t42 = 0.2, P = 0.84). In contrast to the lack of difference in tail-flick response latency between control and sciatic nerve cuffed mice that were not subjected to the forced swim paradigm, the difference in tail-flick response latency was significantly different after the forced swim. For control mice, the tail-flick response latency increased from a baseline of 3.5 seconds ± 0.2 to 8.6 seconds ± 0.4, demonstrating the phenomena of SIA. For nerve-cuffed mice, the increase in tail-flick response latency was much less after a forced swim session. The response latency in nerve-cuffed mice increased from 3.2 seconds ± 0.2 to only 5.7 seconds ± 0.2 following a forced swim, Two-Way ANOVA significant for interaction, nerve cuff × swim stress, F1,84 = 16.5, P < 0.0001 (Figure 1 B).
Next, we looked for changes in neuronal protein synthesis that might help guide the investigation of the circuits and mechanisms that underlie the difference in SIA between mice with and without neuropathic pain. Published literature has shown that persistent pain causes increased CRF mRNA [15] and peptide content in the CeAmy [14], tyrosine hydroxylase (TH) immunoreactivity in the LC [36] and the markers of neuronal activity c-Fos and FosB labeling in the LC [37] and in the dorsal horn of the spinal cord [30, 38]. We evaluated the expression of CRF-ir in the CeAmy and ΔFosB-ir in the brain stem and dorsal horn of the spinal cord at the level of the lumbar intumescence. Neuropathic pain changed each of them. The CRF-ir integrated density in the CeAmy was significantly greater in the nerve-cuffed mice than in the control mice (control group mean: 0.93e6 arbitrary units ± 0.02e6 versus nerve-cuffed group mean: 1.3e6 arbitrary units ± 0.04e6, T-test, t16 = 7.2, P < 0.0001, Figure 1C and D). The left and right CeAmy nuclei showed similar change in the CRF-ir expression and were averaged for each mouse. However, the number of cells with ΔFosB-ir above the detectable threshold was significantly greater only in the left dorsal horn (ipsilateral to the cuffed nerve) in the mice with neuropathic pain when compared to uninjured mice (control group mean: 19.3 ± 4 ΔFosB-ir cells versus nerve-cuffed group mean: 55 ± 11 ΔFosB-ir cells, T-test, t16 = 3.3, P < 0.05, Figure 1E and F). ΔFosB-ir expression on the right side of the spinal cord (uninjured side) was not significantly different between the two groups (nerve-cuffed mean: 33 ± 8 ΔFosB-ir cells versus control group mean: 25 ± 4 ΔFosB-ir cells in control group, T-test, t16 = 0.6, P = 0.56).
The pattern of ΔFosB-ir expression in the LC was very different from that in the spinal cord. Despite one-sided injury, the expression of ΔFosB-ir was similarly affected on both sides of the brainstem and overall ΔFosB-ir was decreased in the LC of nerve-cuffed mice when compared to the control group (control group mean: 50 ± 9 positive cells versus nerve-cuffed group mean: 15 ± 3 positive cells, T-test, t16 = 3.3, P < 0.01, Figure 2).
Figure 2. Sciatic nerve constriction for fifteen days decreased expression of ΔFosB-ir in LC and perinuclear zone.
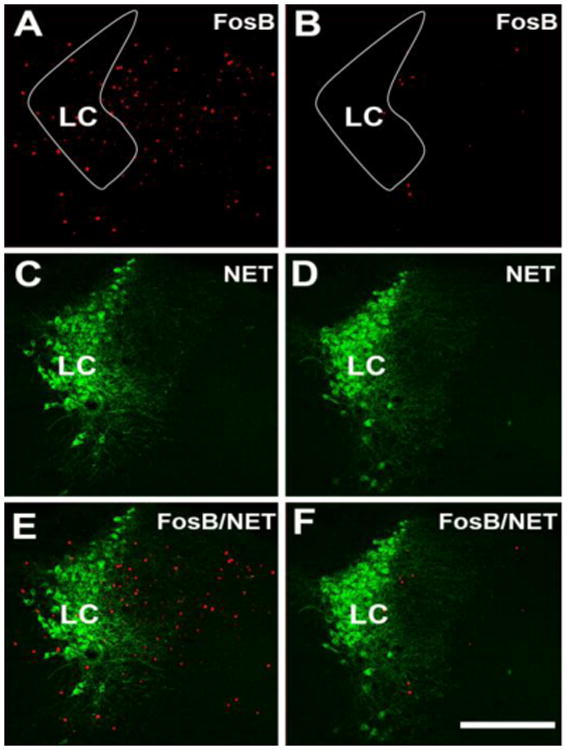
Panels A and B show ΔFosB-ir, C and D show NET-ir, and E and F show double ΔFosB/NET-ir of control (A, C and E) and nerve-cuffed mice (B, D and F) 15 days after insertion of the cuffs. The white contours in A and B outline LC proper. There were fewer ΔFosB labeled cells in the LC of the nerve-cuffed mice compared to the control mice; T-test, t16 = 3.3, P < 0.01. Abbreviation: LC – locus ceruleus. Scale bar = 200 μm.
We then addressed the question of whether relief from ongoing pain would affect the decreased SIA observed in the nerve-cuffed mice. The NSAID flunixin increased the mechanical paw withdrawal thresholds of nerve-cuffed mice from 0.3 grams ± 0.2 in the vehicle-only group to 2.8 grams ± 0.8 in a group that received flunixin (T-test, t16 = 3.5, P < 0.01, Figure 3 A) or the analgesic dose of flunixin increased mechanical paw withdrawal thresholds toward those seen in control mice. However, this dose did not alter baseline tail-flick response latency in the control or nerve-cuffed mice (control group mean: 4.5 seconds ± 0.2 versus flunixin group mean: 4.9 seconds ± 0.3; T-test, t18 = 1.01, P = 0.28). This lack of analgesic effect is congruent with the fact that while NSAIDs are drugs of choice for many types of chronic and inflammatory pain, they are relatively ineffective for treating acute pain, and would therefore not be expected to alter an acute nociceptive threshold. While the analgesic dose had no effect on the baseline tail-flick response latency, it did restore the phenomena of SIA in the nerve-cuffed mice. After a forced swim, the nerve-cuffed group treated with vehicle had a mean tail-flick response latency of 5.6 seconds ± 0.1 after forced swim, about half that of the control mice, and this increased with flunixin treatment to 9.6 seconds ± 0.2, nearly equal to that of the controls (Two-Way ANOVA, significant for interaction, pain × treatment, F1,44 = 4.7, P < 0.05, Figure 3 B).
Figure 3. Providing adequate analgesia restored SIA in mice with neuropathic pain.
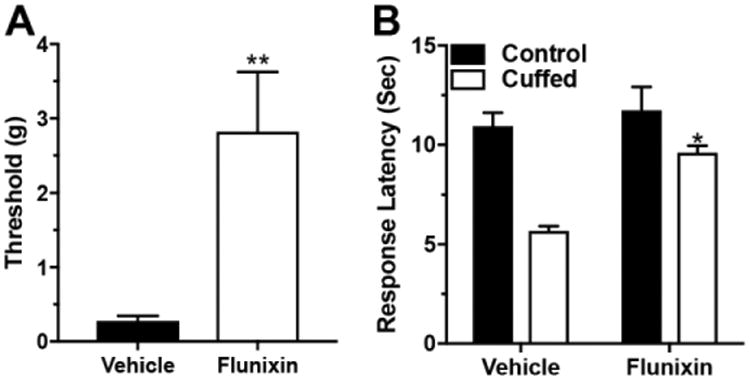
Panel A: Subcutaneous injection of the NSAID flunixin increased the mechanical paw withdrawal thresholds of nerve-cuffed mice toward a pre-surgical level, T-test, t16 = 3.5, P < 0.01. Panel B: Nerve-cuffed mice were able to generate SIA when pre-treated with analgesic. Two-Way ANOVA, * - significant for interaction (pain × drug), F1,44 = 4.7, P < 0.05.
Modulation of SIA by CRF neurons in pain-free and neuropathic states
It is well established that amygdala CRF neurons contribute to stress responses due to both physical and psychological stressors [39-41] and that the CeAmy is essential for SIA triggered by a variety of stressors [11, 42]. Based on the changes in amygdala CRF observed in the nerve cuff paradigm we investigated the possibility that CeAmy CRF neurons play a role in SIA in uninjured mice and/or in mice with neuropathic pain. First, we injected an AAV encoding a cre-activated excitatory DREADD (AAV-Cre-DREADD/Gq) bilaterally into the CeAmy of CRF-cre mice. A single injection of the DREADD ligand CNO did not affect the baseline tail-flick response latency of the CRF-cre mice. However, it did increase the tail-flick response latency after a forced swim. The mean tail-flick response latency after forced swim was 11 seconds ± 1.6 for the vehicle treated group and 17.7 seconds ± 1.3 for the CNO treated group (Two-Way ANOVA, significant for interaction, stress × treatment, F1, 30 = 5.3, P < 0.05, Figure 4 A). In contrast, the activation of amygdala CRF neurons in nerve-cuffed mice did not change the tail-flick response latency either before or after a forced swim. Before the forced swim, the mean tail-flick response latency was 2 seconds ± 0.6 for the vehicle treated group and 1.9 seconds ± 0.4 for the CNO treated group. After the forced swim the mean tail-flick response latency was 4.5 seconds ± 1.6 for the vehicle treated group and 6.4 seconds ± 5.1 for the CNO treated group, (Two-Way ANOVA, non-significant for interaction, stress × treatment, F1,32 = 2, P = 0.16).
Figure 4. Activation of CRF neurons in the CeAmy increased SIA in control mice, whereas the inhibition of amygdala CRF neurons for the full duration of the pain period relieved the mechanical allodynia and restored SIA in mice with neuropathic pain.
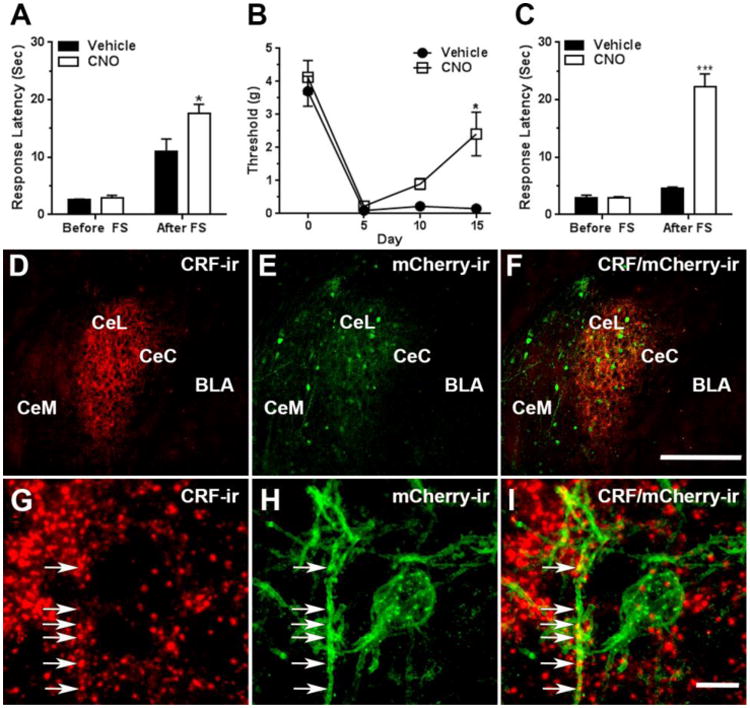
Panel A: Activation of CRF neurons expressing AAV-DREADD/Gq by a single injection of CNO did not change the tail-flick response latency of unstressed mice, but increased the response latency after a forced swim, Two-Way ANOVA, * - significant for interaction (stress × treatment), F1, 30 = 5.3, P < 0.05. Panel B: The mice expressing the inhibitory AAV-DREADD/Gi in CeAmy CRF neurons and treated with CNO in their drinking water developed mechanical allodynia after cuff placement, but their mechanical paw withdrawal threshold increased by day 15 of the CNO treatment, Two-Way Repeated Measures ANOVA, * - significant for interaction (treatment × subjects), F3,21 = 3.2, P < 0.05. Panel C: While the inhibition of CRF neurons for the entire pain period did not change tail-flick response latency of unstressed mice, it increased the tail-flick response latency after a forced swim, Two-Way ANOVA, *** - significant for interaction (stress × treatment), F1, 40 = 85.6, P < 0.001.
Panels D to F: Expression of AAV-DREADD/Gi viral constructs in the CeAmy and their colocalization with CRF-ir after fifteen days of CNO treatment. Panel D: A dense network of CRF-ir fibers that primarily occupy CeL, but are also visible in neighboring CeC and CeM subdivisions of CeAmy. Panel E: The expression of AAV-DREADD/Gi is confined to CeAmy, where the colocalization of DREADD/Gi with CRF-ir is indicated by a yellow tone in panel F.
Panels G to I: The arrows in these high magnification images show the colocalization of CRF-ir positive fibers with mCherry-ir, the fluorescent reporter of AAV-DREADD/Gi.
Abbreviations: BLA - basolateral amygdala, CeC - centrocentral nucleus, CeM -cenrtromedial nucleus, CeL- centrolateral nucleus, FS – forced swim. Scale bar = 200 μm in D to F and scale bar = 10 μm in G to I.
One possible explanation for the observations that DREADD-based activation of amygdala CRF neurons increases the tail-flick response latency following swim stress in control mice, but not in nerve-cuffed mice is that the amygdala CRF signaling pathway is already activated in injured mice. We attempted to address this possibility in the next series of experiments. In the first experiment, we injected an AAV carrying a cre-activated inhibitory DREADD (AAV-cre-DREADD/Gi) bilaterally into the CeAmy of CRF-cre mice and three weeks later administered a single dose of CNO. Inhibition of CRF neurons did not change the tail-flick response latency in the control mice before a forced swim (vehicle treated group mean: 2.2 seconds ± 0.2 versus CNO treated group mean: 2.6 seconds ± 0.5) or after a forced swim (vehicle treated group mean: 11.3 seconds ± 6.8 versus CNO treated group mean: 11.1 seconds ± 5.7; Two-Way ANOVA, non-significant for interaction, stress × treatment, F1,34 = 0.8, P = 0.86).
Next, we attempted to continuously inhibit CeAmy CRF neurons throughout the duration of the experiment in order to investigate if inhibiting the activity of amygdala CRF neurons for the entire time after placement of the nerve cuff would affect the process of pain chronification. For this experiment we inserted the nerve cuff three weeks after stereotaxic injection of the AAV-cre-DREADD/Gi and immediately added CNO (calculated to result in consumption of 1mg/kg/day) to the drinking water. All mice developed mechanical hypersensitivity as demonstrated by a large decrease in their mechanical paw withdrawal threshold on the fifth postsurgical day. The withdrawal thresholds of the CNO treated group subsequently increased. This effect was observed on the tenth postsurgical day and became statistically significant when compared to the controls by the fifteenth postsurgical day, (vehicle treated group mean: 0.2 grams ± 0.2 versus CNO treated group mean: 2.4 grams ± 1.8; Two-Way Repeated Measures ANOVA, significant for interaction, treatment × subjects, F3,21 = 3.2, P < 0.05, Figure 4 B). The continuous CNO treatment also considerably increased the tail-flick response latency in nerve-cuffed mice after a forced swim (vehicle treated group mean: 4.5 seconds ± 0.2 versus CNO treated group mean: 22.3 seconds ± 2.1; Two-Way ANOVA, significant for interaction, stress × treatment, F1, 40 = 85.3, P < 0.0001, Figure 4 C). After completion of the experiments the injection site and viral expression was examined in all mice. The viral expression was almost completely limited to the CeAmy with only a few scattered cells in the medial amygdala. This expression pattern matches the previously described pattern of CRF immunoreactivity in rats [43, 44] and mice [45]. We also performed antibody labeling. While CRF immunohistochemistry does not show cell bodies in the mouse we observed colocalization between CRF immunolabeled fibers and fibers containing a marker expressed by the AAV-cre-DREADD/Gi (Figure 4 D to I). Thus, continuous DREADD-mediated inhibition of CeAmy CRF neurons decreases the persistence of mechanical hypersensitivity following sciatic nerve cuff placement and restores development of SIA induced by forced swim.
Ablation of CeAmy projections to the LC ipsilateral to nerve injury reduces mechanical hypersensitivity and restores SIA in mice with neuropathic pain
The CeAmy is the major source of CRF projections to the LC [23]. However, in addition to their projections to the LC and peri-LC area, CeAmy CRF neurons also project to the oval nucleus of the BNST, lateral hypothalamic area, ventrolateral PAG, parabrachial region and the nucleus of the solitary tract [46]. To specifically assess the potential role of CRF projections to the LC in the development of neuropathic pain, we selectively ablated the CeAmy neurons that project to the LC, either ipsilateral or contralateral to a cuffed sciatic nerve. Ablation was performed by injecting CAV2-cre virus, which undergoes relatively efficient retrograde transport after entering nerve terminals in the region of injection [47], into the LC and injecting an AAV that encodes cre-dependent expression of diphtheria toxin (AAV-FLEX-DTX) into the CeAmy. In mice without a nerve cuff, the tail-flick response latency after a forced swim was not affected by ablation on either side (left side ablation group mean: 11.5 seconds ± 3.3 versus right side ablation group mean: 16.1 seconds ± 3.7; Two-Way ANOVA, non-significant for interaction (stress × ablation side), F1,16 = 0.2, P = 0.27, Figure 5 A).
Figure 5. Ablation of CeAmy projections to the LC did not affect SIA in uninjured mice, but ablation of CeAmy projections to the LC on the side of sciatic nerve constriction decreased the mechanical allodynia and recovered SIA.
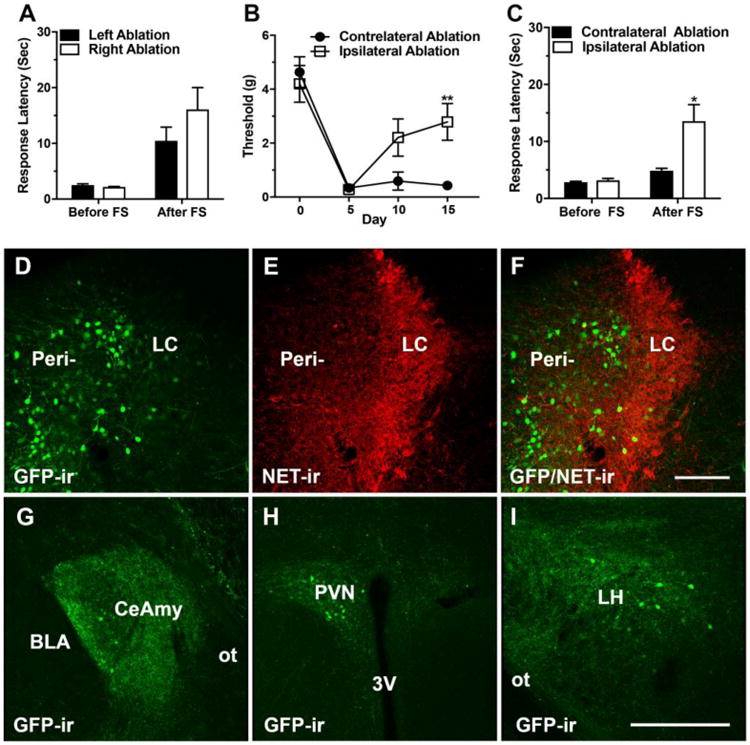
Panel A: SIA in uninjured mice, remains unaffected by ablation of the left or the right CeAmy projections to the LC. Two-Way ANOVA, non-significant for interaction (stress × ablation side), P > 0.05. Panel B: Mice with ablation of CeAmy projections to the LC ipsilateral to the nerve cuff developed mechanical allodynia, but their thresholds increased by postsurgical day 15, Two-Way Repeated Measures ANOVA, ** - significant for interaction (ablation side × subjects), F3,24 = 4.1, P < 0.01. Panel C: SIA was restored in nerve-cuffed mice after ablation of the CeAmy projections to the LC ipsilateral to the nerve cuff, Two-Way ANOVA, * - significant for interaction (stress × side), F1, 24 = 6.5, P < 0.05. Panels D to F show the injection of rAAV-CAG-eGFP-F2A-Cre (D) into the medial dendritic peri-LC zone (E and F). Panels G to I show the retrograde labeling of the CeAmy (G), PVN (H) and LH, (I) all of which are brain areas that unilaterally project to LC. Abbreviations: 3V – third ventricle, BLA - basolateral amygdala, CeAmy – central amygdala, FS – forced swim, GFP – green fluorescent protein, LH – lateral hypothalamus, LC – locus ceruleus, NET – norepinephrine extracellular transporter, ot – optic tract, PVN – paraventricular nucleus, Peri – peri-locus ceruleus zone. Scale bar = 100 μm in D to F and scale bar = 200 μm in G to I.
Ablation of CeAmy projections to the LC did affect the mechanical hypersensitivity of nerve-cuffed mice, but the effect was present only if the ablation was ipsilateral to the nerve injury. Mice with either ipsilateral or contralateral ablation of CeAmy projecting CRF neurons had profound mechanical hypersensitivity five days after nerve cuff placement. Mice with ipsilateral ablation showed an increase in mechanical paw withdrawal threshold at later time points, with a statistically significant difference between the ipsilateral and contralateral ablation groups on the fifteenth day after nerve cuff placement (contralateral ablation group mean: 0.4 grams ± 0.02 versus ipsilateral ablation group mean: 2.8 grams ± 0.9 (Two-Way Repeated Measures ANOVA, significant for interaction, ablation side × subjects, F3,24 = 4.1, P < 0.05, Figure 5 B). The tail-flick response latency in mice with nerve cuffs was not affected by ablation on either side before a forced swim. Nerve-cuffed mice with ablation of CeAmy projections to the LC that were ipsilateral to the nerve cuff showed robust SIA after a forced swim, as reflected by increased tail-flick response latency while the mice with contralateral ablation did not demonstrate significant SIA (contralateral ablation group mean: 4.9 seconds ± 0.2 versus ipsilateral ablation group mean: 13.7 seconds ± 1.2 (Two-Way ANOVA, significant for interaction, stress × side, F1, 30 = 5.9, P < 0.05, Figure 5 C).
The CAV2-cre virus that we used for ablation lacked fluorescent reporter and could not be visualized at the injection sites or in the CeAmy, thus it did not provide a complete depiction of the brain areas that were retrograde labeled after injection of the virus into LC. In order to verify the results of the ablation experiments we applied a second retrograde virus, rAAV-CAG-eGFP-F2A-Cre, with robust GFP expression. LC possesses a vast dendritic zone, referred as peri-locus ceruleus zone that receives a majority of the afferent projections to the nucleus including projections from CeAmy. Our injections of rAAV-CAG-eGFP-F2A-Cre were restricted to the medial peri-locus ceruleus area, where the virus labeled numerous neurons in the dendritic zone and LC proper (Figure 5 D to F). The spread of the retrograde virus into the forebrain matched the previous descriptions of known projections to LC and included the CeAmy, paraventricular nucleus of the hypothalamus (PVN), lateral hypothalamic area (LH) (Figure 5 G to I), BNST and sensory cortex [35]. Following the verification of the anatomical circuit, we combined the retro-AAV virus with AAV-FLEX-DTX for ablation and SIA experiments. The results of SIA matched very well with the results of the previous experiment (contralateral ablation group mean: 6 seconds ± 0.4 versus ipsilateral ablation group mean: 17.4 seconds ± 1.4, Two-Way ANOVA, significant for interaction, stress × side, F1, 28 = 11.2, P < 0.01).
The extent and the efficacy of CRF ablation was verified by immunostaining for CRF and comparing the optical density of CRF-ir in the ablated side with the non-ablated side (non-ablated side group mean: 6.39e6 arbitrary units ± 0.84e6 versus the ablated side group mean: 2.1e6 arbitrary units, ± 0.55e6; T-test, t14 = 4.3, P < 0.001 (Figure 6 A and B). The effects of perturbations in the CeAmy on mechanical paw withdrawal are probably mediated via descending input to the dorsal horn of the spinal cord, where the descending modulation interacts with incoming nociceptive information. To gain data relevant to this idea we compared the number of ΔFosB immunolabeled cells between treatment conditions. Mice with ablation ipsilateral to the nerve cuff showed a substantial reduction of ΔFosB-ir positive neurons in the nerve-cuffed side of the dorsal horn of the spinal cord. The dorsal horn of the contralateral CeAmy CRF neuron ablation group contained a mean of 134 ΔFosB-ir positive neurons ± 11, while the dorsal horn of the ipsilateral ablation group contained a mean of 78 ΔFosB-ir positive neurons ± 9; T-test, t27 = 3.9, P < 0.001 (Figure 6 C and D). Overall these results are similar to those from DREADD-based inhibition of all CeAmy CRF neurons, suggesting that in the sciatic nerve cuff paradigm, an intact ipsilateral CeAmy to LC CRF projection inhibits induction of SIA and supports development of mechanical hypersensitivity.
Figure 6. Injection of AAV-FLEX/DTX into the CeAmy and CAV2-cre into the LC decreased expression of CRF-ir in the CeAmy. The ablation of CRF neurons in CeAmy on the side of injury decreased the expression of ΔFosB-ir in the dorsal horn of the spinal cord in mice with sciatic nerve constriction.
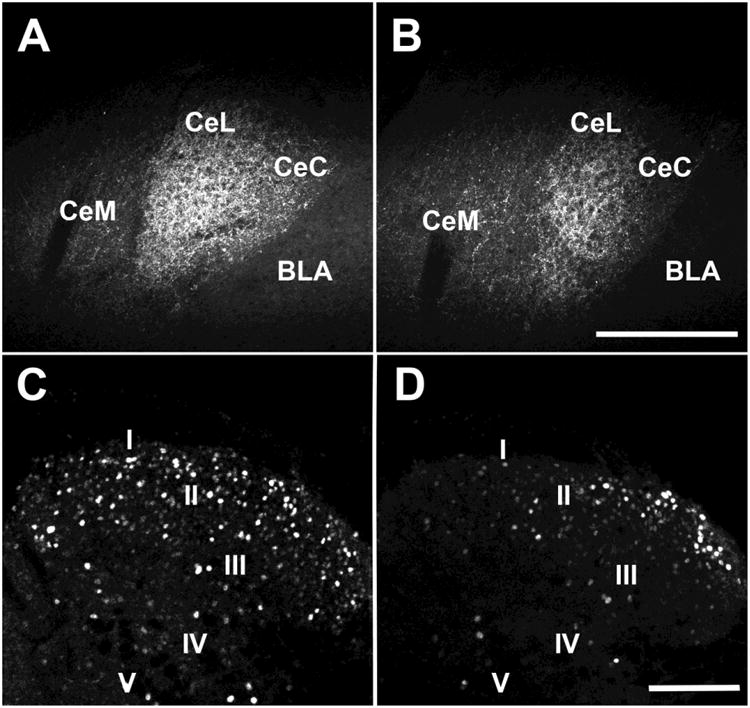
Panel A and B: CRF-ir expression in the contralateral CeAmy (A) and ipsilateral CeAmy (B) of nerve-cuffed mice; T-test, t14 = 4.3, P < 0.001. Panel C and D: ΔFosB-ir in the dorsal horn of the spinal cord after 15 days of neuropathic pain in mice with ablation contralateral to the nerve cuff (C) and ipsilateral to the nerve cuff (D), T-test, t27 = 3.9, P < 0.001. Abbreviations: BLA - basolateral amygdala, CeC - centrocentral nucleus, CeM -cenrtromedial nucleus and CeL- centrolateral nucleus. Roman numerals from I to V indicate spinal cord laminae. Scale bar = 200 μm in A and B and scale bar = 100 μm in C and D.
We confirmed that the ablation is restricted to the CeAmy projection neurons by evaluating the expression of ENK-ir in the CeAmy. ENK is expressed by a subpopulation of interneurons and the ENK-ir integrated density was not altered by the ablation (non-injected side group mean: 4.2e6 arbitrary units ± 0.4e6 versus injected side group mean: 4.6e6 arbitrary units ± 0.7e6; T-test, t14 = 0.48, P = 0.638, Figure 7).
Figure 7. While the ablation of neuronal projections from the CeAmy to the LC decreased CRF-ir expression, it did not affect the expression of ENK-ir.
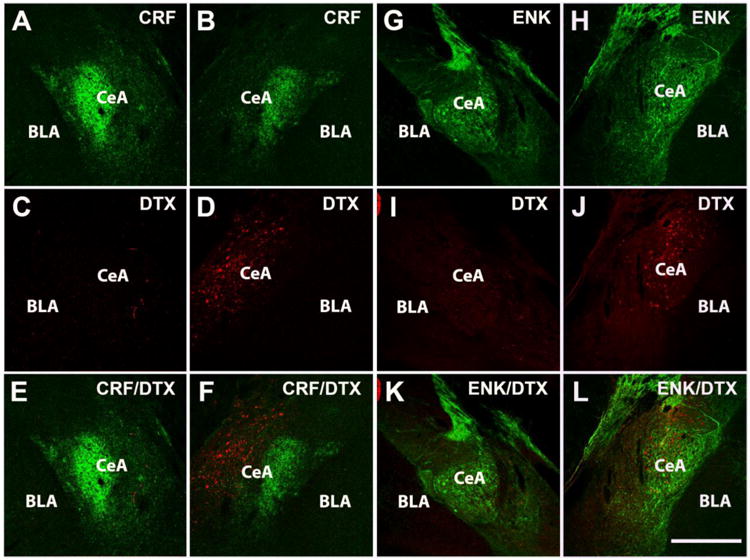
Panels A to F: Expression of CRF-ir and AAV-DTX-mCherry in CeAmy of non-injected side (A, C and E) and injected side (B, D and F). Panels G to L: Expression of ENK-ir and DTX-mCherry-ir in the CeAmy of non-injected side (G, I and K) and injected side (H, J and L). mCherry is expressed only by non-active neurons or neurons without cre recombinase, which was delivered retrogradely to CeAmy by CAV2 injection into the LC. Whereas CRF-ir decreased in the injected side when compared to the non-injected side (see figure 6 for statistical analysis), the expression of ENK-ir was similar in the CeAmy on both sides, T-test, t14 = 0.4, P >0.04. Abbreviations: BLA - basolateral amygdala, CeAmy – central amygdala, Scale bar = 200 μm.
Inhibition of a single LC is sufficient to impair SIA
Somatotropic organization of the descending pain modulatory system is described in human subjects [3] but is understudied in laboratory animals. Our ablation experiments demonstrated that restoration of SIA in mice with sciatic nerve cuffs requires a unilateral ablation of CeAmy projections to LC that must be ipsilateral to the nerve injury. This brings up the possibility that changes in function of the LC on one side can have profound effect on nociceptive function and SIA. To test this idea, we injected AAV-cre-DREADD/Gi into the left LC of TH-cre mice and three weeks later performed the thermal tail-flick assay before and after a forced swim. The baseline tail-flick response latency was similar in the vehicle treated and CNO treated group before a forced swim (vehicle treated group mean: 3.2 seconds ± 0.4 versus CNO treated group mean: 3.4 seconds ± 0.6) but the tail-flick response latency became substantially different after a forced swim (vehicle group mean: 14.1 seconds ± 2; CNO group mean: 7.5 seconds ± 2.1; Two-Way ANOVA, significant for interaction, stress × treatment, F1,28 = 5.1, P < 0.05, Figure 8). CNO treatment did not differentially affect the mechanical paw withdrawal thresholds between the vehicle and CNO treated groups or the thresholds of the right and left paw in the animals of the same treatment group (results not shown). We verified the inhibitory effect of cre-DREADD/Gi on LC activity by comparing c-Fos expression between vehicle treated and CNO treated mice. The expression of c-Fos-ir was significantly lower in the LC area of AAV-cre-DREADD/Gi injected mice treated with CNO than in the vehicle only group (vehicle treated group mean: 29 ± 6 c-Fos positive cells versus CNO treated group mean: 14 ± 3 c-Fos positive cells in LC area; T-test, t14 = 2.6, P < 0.05, Figure 9).
Figure 8. Inhibition of the left LC impairs SIA in uninjured mice.
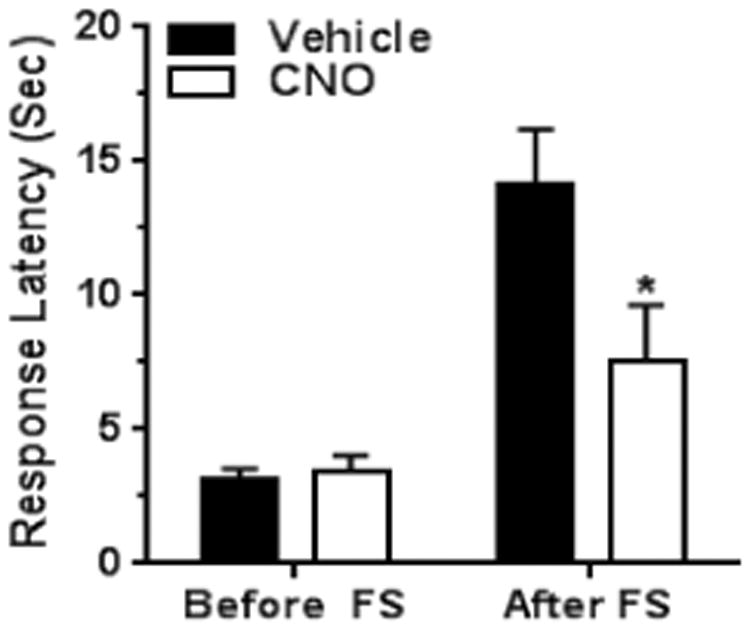
The baseline tail -flick response latency was not affected by the inhibition of LC neurons of mice without nerve cuffs and under non-stressful conditions. However, the inhibition of LC neurons significantly decreased the tail-flick response latency after a forced swim, Two-Way ANOVA, * - significant for interaction (stress × treatment), F1,28 = 5.1, P < 0.05. Abbreviation: FS – forced swim.
Figure 9. Activation of DREADD/Gi receptors in TH-cre mice injected into the LC area with AAV-DREADD/Gi decreased the expression of c-Fos-ir in the LC and perinuclear zone.

Panels A and B show c-Fos-ir, panels C and D show NET-ir, panels E and F show mCherry-ir and panels H and I show triple labeling of c-Fos (blue)/NET (green)/mCherry-ir (red) in vehicle-only treated (A, C, E and H) and CNO treated (B, D, F and I) mice. CNO treated group showed significantly lower c-Fos counts, T-test, t14 = 2.6, P < 0.05. The arrowheads point to cannula tracks left after the viral injections. Abbreviation: LC – locus ceruleus. Scale bar = 100 μm.
Discussion
We used a combination of neuropathic pain, induced by a cuff placed around the sciatic nerve, and stress, induced by forced swim, to investigate the role of amygdala CRF neurons in the descending modulation of pain. Descending modulation of pain consists of input from supraspinal regions to the dorsal horn of the spinal cord that decreases the effects of nociceptive signaling. SIA depends on this descending modulation and we therefore used an SIA model to assess the function of descending pain modulation during persistent neuropathic pain. SIA, assessed as an increase in thermal tail-flick response latency caused by a forced swim, was almost completely abolished in the presence of neuropathic pain. We attribute this loss to impaired descending pain inhibition. Providing adequate analgesia to the mice with nerve cuffs led to restoration of SIA, suggesting that ongoing nociceptive signaling inhibits SIA. We focused on the potential contribution of CeAmy CRF neurons to this disruption. Neither excitation nor inhibition of amygdala CRF neurons affected tail-flick response latency in non-stressed mice regardless of neuropathic pain. While activation of CeAmy CRF neurons increased SIA in uninjured mice, it did not affect the greatly decreased SIA in mice with sciatic nerve cuffs. Chronic pharmacogenetic inhibition of CeAmy CRF neurons led to recovery of SIA and somewhat decreased the mechanical hypersensitivity of nerve-cuffed mice. The effect on SIA due to CeAmy CRF neuronal inhibition was replicated by ablation of CeAmy neurons that project to the LC on the side of the nerve cuff. Ipsilateral ablation of CeAmy CRF neurons, confirmed by decreased expression of CRF-ir, restored SIA and decreased the mechanical hypersensitivity in mice with neuropathic pain. The ablation was associated with a parallel decrease in immunoreactivity for ΔFosB in the dorsal horn of the spinal cord on the side of the nerve cuff. In addition, SIA in normal mice was suppressed by unilateral pharmacogenetic inhibition of LC neurons. Taken together, these results support the hypothesis that persistent activation of CeAmy CRF neurons by long-lasting nociceptive input suppresses descending pain inhibition that would provide norepinephrine under other conditions to limit the development of hypersensitivity and contribute to the expression of robust SIA. This suggests the possibility that reduced activity of descending pain inhibition, potentially including the circuitries of CeAmy/CRF to the LC and LC/norepinephrine to the dorsal horn, contribute to pain chronification.
CeAmy CRF neurons and CeAmy projections to the BNST and brainstem are part of the circuitry that regulates endocrine and autonomic response to stress. Increases in CeAmy CRF signaling during chronic stress are thought to contribute to the development of anxiety [48, 49]. Different models of chronic pain show that pain increases CRF mRNA and peptide immunoreactivity in CeAmy [14, 15], and increases the CeAmy neurons response to peripheral mechanical stimulation via CRF1 receptor [19]. Here we confirmed that fifteen days of neuropathic pain increases CeAmy CRF-ir. However, our data do not support significant contributions of CeAmy CRF neurons to SIA in uninjured mice. The fact that the acute inhibition of the CRF neurons by DREADD/Gi or their ablation by AAV/DTX did not alter the tail-flick response latency after a forced swim suggests that these CRF neurons do not play a significant role in SIA under normal, pain-free conditions. The increased tail-flick response latency in stressed but uninjured mice that was observed after activation of CRF neurons by DREADD/Gq, was an addition to an already adequate SIA that was showed by the control group. This illustrates the power of the DREADD technique, rather than CRF participation in descending pain inhibition in uninjured mice. While activation of CRF neurons failed to affect SIA in mice with neuropathic pain, inhibition of CRF neurons restored SIA in these mice to levels comparable with those of uninjured mice.
The most parsimonious explanation for these seemingly disparate effects of CRF is based on the alterations in the CRF circuitry caused by persistent pain. As discussed above, studies have established that chronic pain changes not only the CRF content in the CeAmy but also the responsiveness of CeAmy neurons to mechanical stimulation, a phenomenon described as “central sensitization” [19]. Persistent pain causes sensitization of CeAmy neurons via CRF1 receptors, while the inhibitory role of CRF2 receptor is lost in the same pain conditions [19]. Rouwette et al. suggest that changes of CRF1 and CRF2 receptors expression in the CeAmy could underlie neuropathic pain [22]. A very recent paper demonstrated that chronic unpredictable stress alters the GABA neurotransmission between amygdala CRF neurons and inhibits CeAmy output to BNST [13]. It is plausible that persistent pain causes similar changes in CeAmy output that lead to inhibition of descending pain modulation and more specifically to inhibition of LC activity. The CeAmy is the main source of CRF projections to the LC and the observed CRF axons make both excitatory and inhibitory synaptic contacts with the LC neurons [12, 23]. In addition, many of the CeAmy projections form connections with a local population of GABA interneurons [35]. This variety of direct and indirect, excitatory and inhibitory synaptic connections provide an opportunity for pain associated plasticity and neurotransmitter changes in the CeAmy to cause a long lasting inhibition of the LC and decrease LC input to the dorsal horn of the spinal cord with an end result of insufficient inhibition of the ongoing nociception and impaired SIA. The idea that pain induces protracted inhibition of the LC by the CeAmy is supported by our ablation experiments. While the destruction of the projection neurons from CeAmy to the LC did not affect SIA in uninjured mice, the ablation of these projections restored SIA in mice with neuropathic pain. Removing inhibitory tone from the CeAmy very likely allowed inputs from other regions, such as the ventrolateral PAG and RVM, that normally contribute to descending pain modulation [50] to take effect. Therefore the lack of effect of CeAmy CRF neuronal inhibition or the ablation of CeAmy projections on SIA in uninjured mice may be explained by the engagement of PAG and RVM in SIA under pain-free circumstances.
32% of the CeAmy CRF-ir neurons were labeled following injection of the retrograde tracer fluorogold into a rat LC (unpublished data). Our observation that a great number of CRF neurons project to the LC is supported by other publications, which show that amygdala neurons provide a major CRF input to the LC [23, 51]. However, in this study, the ablation of CeAmy projections to the LC very likely was not restricted to CRF neurons and included some GABA projection neurons that may contain other neuropeptides and cotransmitters. Dynorphin is present in some amygdala CRF neurons and in some of the projections from the CeAmy to the LC [52]. We cannot exclude the possibility that our ablation experiment affected some of the dynorphin projections (with or without CRF) or any other of the great variety of neuropeptides that serve as cotransmitters in the CeAmy GABA neurons. However, the CeAmy contains two main non-overlapping neuronal populations, enkephalin interneurons and CRF neurons [35]. The fact that our ablation led to a significant decrease of CRF expression but did not affect the enkephalin expression in the CeAmy, makes a convincing argument that the ablation was largely restricted to a subpopulation of mostly CRF projection neurons.
Spinal nociceptive reflexes are under central control and LC projections to the spinal cord are part of this descending nociceptive control. We used the tail-flick test to assess a spinal reflex that is influenced by descending pain inhibition during SIA. The experiments with ablation of amygdala projections to the LC showed that these projection neurons provide inhibitory input to the LC and may unilaterally suppress norepinephrine release into the dorsal horn of the spinal cord. The experiment where we injected an inhibitory AAV/DREADD into the LC confirmed our expectation that inhibition of a single LC suffices to reduce SIA in pain free mice. This observation is consistent with the observations made by other researches that excitation of LC neurons ipsilateral to the injury relieves the symptoms of neuropathic pain in rats with chronic constriction injury [53]. The results of our experiments are also consistent with the anatomical description of ceruleospinal projections in mice. While the tracing studies done in rats and other species show a great variety of projection patterns that include LC projections to the dorsal and ventral horn and/or ipsilateral, contralateral and bilateral projection sites [37, 54], an injection with a retrograde tracer into the mouse spinal cord labeled only LC neurons on the ipsilateral side [55]. In any case, despite the differences observed among species, the LC is the main source for norepinephrine in the dorsal horn of the spinal cord and consequently is an important element in the descending pain inhibitory system, and our data emphasize its potential contributions to SIA and pain chronification.
The pathophysiology of chronic pain involves a vast array of neurotransmitters, brain circuits, glial factors, plasticity in receptor expression and neuronal excitability in the CNS and periphery. Different types of chronic pain may have quite different pathophysiological mechanisms. This study provides information about the contribution of CeAmy CRF neurons to neuropathic pain caused by constriction of a peripheral nerve. Our model of neuropathic pain induced changes in the expression of CRF by amygdala neurons, led to inhibition of LC function and subsequently to decreases in SIA and descending pain inhibition. These findings are consistent with the theory [40] that dysregulation of descending pain modulation is a major mechanism for chronification of neuropathic pain.
Highlights.
Chronic pain impairs stress-induced analgesia which depends on descending pain modulation
Continuous inhibition of amygdala CRF neurons recovers stress-induced analgesia and relieves pain
Over-activation of amygdala CRF neurons may contribute to the development of chronic pain
Acknowledgments
This work is supported by National Institute of Mental Health; award number R01MH105528 to E.D. Study concept, data acquisition and data analysis by M.A. under supervision by E.D. Technical support by T.M. Manuscript draft by M.A and E.D. We thank Dr. Ted Usdin (NIMH) for his excellent insights, rigorous discussion of the data and editing help. We also thank Alla Karpova for the generous contribution of the plasmid rAAV2-retro and Ted Usdin, Raymond Fields and the NINDS Viral Production Core Facility for production of the AAV-retro-construct.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lewis JW, Cannon JT, Liebeskind JC. Opioid and nonopioid mechanisms of stress analgesia. Science. 1980;208:623–625. doi: 10.1126/science.7367889. [DOI] [PubMed] [Google Scholar]
- 2.Long CC, Sadler KE, Kolber BJ. Hormonal and molecular effects of restraint stress on formalin-induced pain-like behavior in male and female mice. Physiol Behav. 2016;165:278–285. doi: 10.1016/j.physbeh.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benedetti F, Arduino C, Amanzio M. Somatotopic activation of opioid systems by target-directed expectations of analgesia. J Neurosci. 1999;19:3639–3648. doi: 10.1523/JNEUROSCI.19-09-03639.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer DJ, Price DD. Central nervous system mechanisms of analgesia. Pain. 1976;2:379–404. doi: 10.1016/0304-3959(76)90080-4. [DOI] [PubMed] [Google Scholar]
- 5.Liebeskind JC, Mayer DJ. Somatosensory evoked responses in the mesencephalic central gray matter of the rat. Brain Res. 1971;27:133–151. doi: 10.1016/0006-8993(71)90376-3. [DOI] [PubMed] [Google Scholar]
- 6.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 7.Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest. 2010;120:3779–3787. doi: 10.1172/JCI43766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliveira MA, Prado WA. Role of PAG in the antinociception evoked from the medial or central amygdala in rats. Brain Res Bull. 2001;54:55–63. doi: 10.1016/s0361-9230(00)00420-2. [DOI] [PubMed] [Google Scholar]
- 9.Carrasquillo Y, Gereau RWt. Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarhan M, Pawlowski SA, Barthas F, Yalcin I, Kaufling J, Dardente H, Zachariou V, Dileone RJ, Barrot M, Veinante P. BDNF parabrachio-amygdaloid pathway in morphine-induced analgesia. Int J Neuropsychopharmacol. 2013;16:1649–1660. doi: 10.1017/S146114571200168X. [DOI] [PubMed] [Google Scholar]
- 11.Werka T. Post-stress analgesia in rats with partial amygdala lesions. Acta Neurobiol Exp (Wars) 1994;54:127–132. [PubMed] [Google Scholar]
- 12.Van Bockstaele EJ, Colago EE, Valentino RJ. Corticotropin-releasing factor-containing axon terminals synapse onto catecholamine dendrites and may presynaptically modulate other afferents in the rostral pole of the nucleus locus coeruleus in the rat brain. J Comp Neurol. 1996;364:523–534. doi: 10.1002/(SICI)1096-9861(19960115)364:3<523::AID-CNE10>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.Partridge JG, Forcelli PA, Luo R, Cashdan JM, Schulkin J, Valentino RJ, Vicini S. Stress increases GABAergic neurotransmission in CRF neurons of the central amygdala and bed nucleus stria terminalis. Neuropharmacology. 2016;107:239–250. doi: 10.1016/j.neuropharm.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rouwette T, Vanelderen P, de Reus M, Loohuis NO, Giele J, van Egmond J, Scheenen W, Scheffer GJ, Roubos E, Vissers K, Kozicz T. Experimental neuropathy increases limbic forebrain CRF. Eur J Pain. 2012;16:61–71. doi: 10.1016/j.ejpain.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 15.Ulrich-Lai YM, Xie W, Meij JT, Dolgas CM, Yu L, Herman JP. Limbic and HPA axis function in an animal model of chronic neuropathic pain. Physiol Behav. 2006;88:67–76. doi: 10.1016/j.physbeh.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Rouwette T, Klemann K, Gaszner B, Scheffer GJ, Roubos EW, Scheenen WJ, Vissers K, Kozicz T. Differential responses of corticotropin-releasing factor and urocortin 1 to acute pain stress in the rat brain. Neuroscience. 2011;183:15–24. doi: 10.1016/j.neuroscience.2011.03.054. [DOI] [PubMed] [Google Scholar]
- 17.Itoga CA, Roltsch Hellard EA, Whitaker AM, Lu YL, Schreiber AL, Baynes BB, Baiamonte BA, Richardson HN, Gilpin NW. Traumatic Stress Promotes Hyperalgesia via Corticotropin-Releasing Factor-1 Receptor (CRFR1) Signaling in Central Amygdala. Neuropsychopharmacology. 2016;41:2463–2472. doi: 10.1038/npp.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji G, Neugebauer V. Pro- and anti-nociceptive effects of corticotropin-releasing factor (CRF) in central amygdala neurons are mediated through different receptors. J Neurophysiol. 2008;99:1201–1212. doi: 10.1152/jn.01148.2007. [DOI] [PubMed] [Google Scholar]
- 19.Ji G, Neugebauer V. Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol. 2007;97:3893–3904. doi: 10.1152/jn.00135.2007. [DOI] [PubMed] [Google Scholar]
- 20.Bourbia N, Ansah OB, Pertovaara A. Corticotropin-releasing factor in the rat amygdala differentially influences sensory-discriminative and emotional-like pain response in peripheral neuropathy. J Pain. 2010;11:1461–1471. doi: 10.1016/j.jpain.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Cui XY, Lundeberg T, Yu LC. Role of corticotropin-releasing factor and its receptor in nociceptive modulation in the central nucleus of amygdala in rats. Brain Res. 2004;995:23–28. doi: 10.1016/j.brainres.2003.09.050. [DOI] [PubMed] [Google Scholar]
- 22.Rouwette T, Vanelderen P, Roubos EW, Kozicz T, Vissers K. The amygdala, a relay station for switching on and off pain. Eur J Pain. 2012;16:782–792. doi: 10.1002/j.1532-2149.2011.00071.x. [DOI] [PubMed] [Google Scholar]
- 23.Tjoumakaris SI, Rudoy C, Peoples J, Valentino RJ, Van Bockstaele EJ. Cellular interactions between axon terminals containing endogenous opioid peptides or corticotropin-releasing factor in the rat locus coeruleus and surrounding dorsal pontine tegmentum. J Comp Neurol. 2003;466:445–456. doi: 10.1002/cne.10893. [DOI] [PubMed] [Google Scholar]
- 24.Tamano R, Ishida M, Asaki T, Hasegawa M, Shinohara S. Effect of spinal monoaminergic neuronal system dysfunction on pain threshold in rats, and the analgesic effect of serotonin and norepinephrine reuptake inhibitors. Neurosci Lett. 2016;615:78–82. doi: 10.1016/j.neulet.2016.01.025. [DOI] [PubMed] [Google Scholar]
- 25.Bohn LM, Xu F, Gainetdinov RR, Caron MG. Potentiated opioid analgesia in norepinephrine transporter knock-out mice. J Neurosci. 2000;20:9040–9045. doi: 10.1523/JNEUROSCI.20-24-09040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes SW, Hickey L, Hulse RP, Lumb BM, Pickering AE. Endogenous analgesic action of the pontospinal noradrenergic system spatially restricts and temporally delays the progression of neuropathic pain following tibial nerve injury. Pain. 2013;154:1680–1690. doi: 10.1016/j.pain.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuoka H, Suto T, Saito S, Obata H. Amitriptyline, but Not Pregabalin, Reverses the Attenuation of Noxious Stimulus-Induced Analgesia After Nerve Injury in Rats. Anesth Analg. 2016;123:504–510. doi: 10.1213/ANE.0000000000001301. [DOI] [PubMed] [Google Scholar]
- 28.Ma W, Eisenach JC. Chronic constriction injury of sciatic nerve induces the up-regulation of descending inhibitory noradrenergic innervation to the lumbar dorsal horn of mice. Brain Res. 2003;970:110–118. doi: 10.1016/s0006-8993(03)02293-5. [DOI] [PubMed] [Google Scholar]
- 29.De Felice M, Sanoja R, Wang R, Vera-Portocarrero L, Oyarzo J, King T, Ossipov MH, Vanderah TW, Lai J, Dussor GO, Fields HL, Price TJ, Porreca F. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain. 2011;152:2701–2709. doi: 10.1016/j.pain.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimitrov EL, Tsuda MC, Cameron HA, Usdin TB. Anxiety- and depression-like behavior and impaired neurogenesis evoked by peripheral neuropathy persist following resolution of prolonged tactile hypersensitivity. J Neurosci. 2014;34:12304–12312. doi: 10.1523/JNEUROSCI.0312-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morland RH, Novejarque A, Spicer C, Pheby T, Rice AS. Enhanced c-Fos expression in the central amygdala correlates with increased thigmotaxis in rats with peripheral nerve injury. Eur J Pain. 2016;20:1140–1154. doi: 10.1002/ejp.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, Huang CC, Gerfen CR, Schiller J, Dudman JT, Hantman AW, Looger LL, Schaffer DV, Karpova AY. A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron. 2016;92:372–382. doi: 10.1016/j.neuron.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 34.Dixon WJ. Staircase bioassay: the up-and-down method. Neurosci Biobehav Rev. 1991;15:47–50. doi: 10.1016/s0149-7634(05)80090-9. [DOI] [PubMed] [Google Scholar]
- 35.Dimitrov EL, Yanagawa Y, Usdin TB. Forebrain GABAergic projections to locus coeruleus in mouse. J Comp Neurol. 2013;521:2373–2397. doi: 10.1002/cne.23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bravo L, Torres-Sanchez S, Alba-Delgado C, Mico JA, Berrocoso E. Pain exacerbates chronic mild stress-induced changes in noradrenergic transmission in rats. Eur Neuropsychopharmacol. 2014;24:996–1003. doi: 10.1016/j.euroneuro.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Howorth PW, Teschemacher AG, Pickering AE. Retrograde adenoviral vector targeting of nociresponsive pontospinal noradrenergic neurons in the rat in vivo. J Comp Neurol. 2009;512:141–157. doi: 10.1002/cne.21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi SI, Levine JD, Basbaum AI. Effects of injury discharge on the persistent expression of spinal cord fos-like immunoreactivity produced by sciatic nerve transection in the rat. Brain Res. 1993;617:220–224. doi: 10.1016/0006-8993(93)91089-b. [DOI] [PubMed] [Google Scholar]
- 39.Akana SF, Chu A, Soriano L, Dallman MF. Corticosterone exerts site-specific and state-dependent effects in prefrontal cortex and amygdala on regulation of adrenocorticotropic hormone, insulin and fat depots. J Neuroendocrinol. 2001;13:625–637. doi: 10.1046/j.1365-2826.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- 40.Serrats J, Sawchenko PE. CNS activational responses to staphylococcal enterotoxin B: T-lymphocyte-dependent immune challenge effects on stress-related circuitry. J Comp Neurol. 2006;495:236–254. doi: 10.1002/cne.20872. [DOI] [PubMed] [Google Scholar]
- 41.Reyes BA, Zitnik G, Foster C, Van Bockstaele EJ, Valentino RJ. Social Stress Engages Neurochemically-Distinct Afferents to the Rat Locus Coeruleus Depending on Coping Strategy. eNeuro. 2015;2 doi: 10.1523/ENEURO.0042-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox RJ, Sorenson CA. Bilateral lesions of the amygdala attenuate analgesia induced by diverse environmental challenges. Brain Res. 1994;648:215–221. doi: 10.1016/0006-8993(94)91120-7. [DOI] [PubMed] [Google Scholar]
- 43.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 44.Merchenthaler I. Corticotropin releasing factor (CRF)-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides. 1984;5(1):53–69. doi: 10.1016/0196-9781(84)90265-1. [DOI] [PubMed] [Google Scholar]
- 45.De Francesco PN, Valdivia S, Cabral A, Reynaldo M, Raingo J, Sakata I, Osborne-Lawrence S, Zigman JM, Perello M. Neuroanatomical and functional characterization of CRF neurons of the amygdala using a novel transgenic mouse model. Neuroscience. 2015;289:153–165. doi: 10.1016/j.neuroscience.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray TS. Amygdaloid CRF pathways. Role in autonomic, neuroendocrine, and behavioral responses to stress. Ann N Y Acad Sci. 1993;697:53–60. doi: 10.1111/j.1749-6632.1993.tb49922.x. [DOI] [PubMed] [Google Scholar]
- 47.Soudais C, Laplace-Builhe C, Kissa K, Kremer EJ. Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J. 2001;15:2283–2285. doi: 10.1096/fj.01-0321fje. [DOI] [PubMed] [Google Scholar]
- 48.Albeck DS, McKittrick CR, Blanchard DC, Blanchard RJ, Nikulina J, McEwen BS, Sakai RR. Chronic social stress alters levels of corticotropin-releasing factor and arginine vasopressin mRNA in rat brain. J Neurosci. 1997;17:4895–4903. doi: 10.1523/JNEUROSCI.17-12-04895.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makino S, Shibasaki T, Yamauchi N, Nishioka T, Mimoto T, Wakabayashi I, Gold PW, Hashimoto K. Psychological stress increased corticotropin-releasing hormone mRNA and content in the central nucleus of the amygdala but not in the hypothalamic paraventricular nucleus in the rat. Brain Res. 1999;850:136–143. doi: 10.1016/s0006-8993(99)02114-9. [DOI] [PubMed] [Google Scholar]
- 50.Mason P. Central mechanisms of pain modulation. Curr Opin Neurobiol. 1999;9:436–441. doi: 10.1016/S0959-4388(99)80065-8. [DOI] [PubMed] [Google Scholar]
- 51.Van Bockstaele EJ, Colago EE, Valentino RJ. Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the co-ordination of emotional and cognitive limbs of the stress response. J Neuroendocrinol. 1998;10:743–757. doi: 10.1046/j.1365-2826.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- 52.Marchant NJ, Densmore VS, Osborne PB. Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: evidence of two distinct endogenous opioid systems in the lateral division. J Comp Neurol. 2007;504:702–715. doi: 10.1002/cne.21464. [DOI] [PubMed] [Google Scholar]
- 53.Muto ASA, Sakamoto Y, Suzuki H. Activation of NK1 receptors in the locus coeruleus induces analgesia through noradrenergic-mediated descending inhibition in a rat model of neuropathic pain. British Journal of Pharmacology. 2012;166:1047–1057. doi: 10.1111/j.1476-5381.2011.01820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruinstroop E, Cano G, Vanderhorst VG, Cavalcante JC, Wirth J, Sena-Esteves M, Saper CB. Spinal projections of the A5, A6 (locus coeruleus), and A7 noradrenergic cell groups in rats. J Comp Neurol. 2012;520:1985–2001. doi: 10.1002/cne.23024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang H, Paxinos G, Watson C. Projections from the brain to the spinal cord in the mouse. Brain Struct Funct. 2011;215:159–186. doi: 10.1007/s00429-010-0281-x. [DOI] [PubMed] [Google Scholar]