

Abstract
Chronic Hepatitis B Virus (HBV) infection is a major risk factor for hepatocellular carcinoma (HCC) and current treatments for CHB and HCC are perfectible. Herein, we identified cellular Serine/Threonine Polo-like-kinase 1 (PLK1) as a positive effector of HBV replication. The aim of this study was to demonstrate the proviral role of PLK1 in HBV biosynthesis and validate PLK1 inhibition a potential antiviral strategy.
To this end, we employed physiologically relevant HBV infection models of Primary Human Hepatocytes (PHH) and differentiated HepaRG cells, in conjunction with pharmacologic PLK1 inhibitors, siRNA-mediated knockdown, and overexpression of constitutively active PLK1 (PLK1CA). In addition, humanized liver FRG mouse model was used to determine antiviral effect of PLK1 inhibitor BI-2536 on HBV infection in vivo. Lastly, in vitro PLK1 kinase assays and site-directed mutagenesis were employed to demonstrate HBV core protein (HBc) is a PLK1 substrate.
We demonstrate HBV infection activated cellular PLK1 in PHH and dHepaRG cells. PLK1 inhibition by BI-2536 or siRNA-mediated knockdown suppressed, whereas overexpression of PLK1CA increased HBV DNA biosynthesis, supporting PLK1 effects on viral biosynthesis are specific, and PLK1 is a proviral cellular factor. Significantly, BI-2536 administration to HBV-infected humanized liver FRG mice strongly inhibited HBV infection, validating PLK1 as a novel antiviral target in vivo. The proviral action of PLK1 is associated with the biogenesis of the nucleocapsid, as BI-2536 leads to its decreased intracellular formation/accumulation. In this respect, our studies identified HBc as a PLK1 substrate in vitro, and mapped PLK1 phosphorylation sites on this protein.
PLK1 is a proviral host factor that could be envisaged as a target for combined antiviral and antitumoral strategies against HBV infection and HBV mediated carcinogenesis.
Keywords: Hepatitis B virus, polo-like-kinase 1, core protein, capsid-associated reverse transcription, BI-2536
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is an independent risk factor for development of hepatocellular carcinoma (HCC)(1, 2). Liver cancer is the 5th and 7th most common cancer worldwide according to gender, and the 3rd in terms of death. With approximately 250 million people chronically infected with HBV, according to the latest World Health Organization (WHO) estimation, many patients with chronic hepatitis B (CHB) are expected to progress to HCC, despite existing therapies(1). Although a prophylactic vaccine is available, new infections still happen because vaccination campaigns are not always well-implemented, and the vaccine it is not always 100% protective(2). Furthermore, children born of infected mothers still become chronically infected in endemic areas(2). Current treatments include use of Peg-IFNα and/or antiviral nucleoside analogs; the latter require lifelong administration, which can result in viral resistance when drugs with low genetic barrier are used(3). New and effective therapies are urgently needed(3). In particular, drugs that target both viral replication and prevent neoplastic transformation could represent a valuable addition to the therapeutic arsenal.
Replication of HBV within hepatocytes involves both nuclear and cytoplasmic events(4). Following viral entry, nucleocapsids are transported to the nucleus, releasing the viral DNA and core/capsid protein (HBc) into the nucleoplasm. There, the viral genome, initially a relaxed-circular partially-double-stranded DNA (rcDNA), is converted into covalently-closed-circular dsDNA (cccDNA), responsible for viral persistence. This nuclear form of the genome serves as template for viral RNA transcription, including of pre-genomic RNA (pgRNA)(4). In the cytoplasm, pgRNA interacts with HBV polymerase, is packaged into capsids where it is reverse-transcribed to generate newly synthesized rcDNA(4). HBc plays an essential and dynamic role in HBV biosynthesis, including selective binding to pgRNA in association with viral polymerase, encapsidation and reverse-transcription of pgRNA, transport of mature capsids to the site of viral envelopment for virion generation, and transport to the nucleus for recycling(5). Although, the mechanism by which HBc and capsid assembly regulate viral DNA synthesis is not fully understood, it is well established that phosphorylation of HBc has an important role in various aspects of pgRNA reverse-transcription(5, 6). Kinases that mediate HBc phosphorylations include CDK2 (Cyclin-Dependent Kinase 2), which phosphorylates serine/proline (SP) sites located at the C-terminus of HBc, including S155, S162 and S170(7, 8). Another study has identified additional serine/threonine (S/T) sites as having a role in regulating all aspects of HBV replication, and SRPK1 (Serine/Arginine Protein Kinase 1) as another kinase that phosphorylates HBc in vitro(9).
Our earlier studies, aiming at understanding the mechanism by which the HBV X protein (HBx) mediates oncogenic transformation of hepatocytes(10–12), led us to the study of Polo-like-kinase 1 (PLK1), a S/T kinase involved in cell cycle regulation(13, 14). In physiologic conditions, PLK1 is required for checkpoint recovery, mitotic entry and progression, whereas it is overexpressed in many human cancers, including HBV-mediated liver cancer(13, 14). We have shown that HBx activates PLK1 prematurely, allowing propagation of DNA damage to daughter cells by attenuating both DNA repair and p53 apoptosis(11, 12). In a cellular model of HBx-mediated transformation and in liver tumors of animals modeling HBx- and HBV-mediated hepatocarcinogenesis, expression of PLK1 is elevated(11, 12, 15, 16). However, whether PLK1 is activated during physiologic HBV infection of normal hepatocytes, and whether it has a role in establishment and maintenance of infection is unknown. To address this issue, in this study, we used HBV infection models of non-transformed primary human hepatocytes (PHH) and dHepaRG, a bipotential human de-differentiated hepatocyte cell line that upon differentiation supports HBV infection(17, 18). In this study we demonstrate the cellular PLK1 enzyme acts as a proviral host-factor for HBV replication in vitro, by favoring nucleocapsid formation and subsequent reverse transcription. In addition, we identified the viral core protein HBc as a PLK1 substrate in vitro, and mapped the sites of PLK1 phosphorylation. Lastly, we provide evidence that PLK1 inhibitors serve as a novel class of anti-HBV molecules in vitro, and validated the antiviral effect of PLK1 inhibitor BI-2536 in HBV infected liver humanized mice, in vivo.
METHODS
Chemicals, antibodies, and others reagents
All chemicals were purchased from Sigma Aldrich unless otherwise specified. PLK1 inhibitors (BI-2536, BI 6727, MLN 0905 and ON 01910) were from Selleckchem. Polyclonal HBc(19), and commercially available HBc antibodies were used for immunofluorescent staining and immunoblots. Rabbit polyclonal anti-HBc was from Dako (B0586) or Abcam (ab140243) and monoclonal anti-HBc from Abcam (ab8637; Clone C1). PLK1 antibodies used: rabbit polyclonal (ab21738) for detection of phospho-PLK1-S137 in WB and IF; rabbit monoclonal (ab115095) for detection of phospho-PLK1-T210 in WB and IF, and rabbit polyclonal (ab109777) for detection of PLK1. Small interfering RNA (SmartPool or sequence designed) were purchased from Dharmacon, transfected into dHepaRG or PHH using Darmafect-1 reagent, following manufacturer’s instructions. Tenofovir (TFV) and HBV capsid assembly inhibitors Bay41-4109(20) and AT130(21) were kindly provided by Gilead Sciences and Novira Pharmaceutics, respectively. IFN-α (i.e. Roferon) was from Hoffmann-La-Roche.
HepaRG cells and primary human hepatocytes & infection by HBV
Human liver progenitor HepaRG cells(18) were cultured for 2 weeks in complete William’s medium (Gibco) supplemented with 10% FCS Fetal Clone II serum (Thermo scientific), 50 U/mL penicillin/streptomycin (Gibco), 2 mM glutaMax (Gibco), 5 μg/mL human insulin (Sigma), and 5 × 10−5 M hydrocortisone hemisuccinate (UpJohn, SERB) at 37°C in humidified CO2 (5%) incubators and differentiated into hepatocyte-like cells by a treatment with 1.8% DMSO (Hybrid-max, Sigma) for two weeks as described(22). Engineered HepaRG cell lines were also used. HepaRG-TR-PLK1CA cell line enabling tetracycline-inducible expression of a constitutively active form of PLK1 (i.e. T210D mutant(23)) was constructed as described for HepaRG-TR-HBc cell line(24). Primary human hepatocytes (PHHs) were from surgical liver resections, after informed consent of patients (kindly provided by Pr. Rivoire (CLB, Lyon); agreement numbers DC-2008-99 and DC-2008-101). PHH were prepared as previously described(25) and cultured in complete William’s medium supplemented with 1.8 % of DMSO.
HBV infection and analysis of viral parameters during replication
Differentiated HepaRG cells (dHepaRG) and PHH were infected as previously described(26), with HBV inoculum prepared either from HepG2.2.15 or HepaAD38 cells. Intracellular accumulation of viral RNA and DNA, secretion of HBe and HBs antigens were monitored by qPCR, RTqPCR, southern blotting, and ELISA, as described(26). Briefly, HBeAg and HBsAg were quantified in culture medium by ELISA, using a chemiluminescence immunoassay kit (Autobio, China) following manufacturer’s instructions. Total DNA was purified from infected cells using MasterPure™ Complete DNA Purification Kit (Epicentre). Southern blot analysis of HBV DNA was performed using equal amounts of total DNA, electrophoresed on 1.1% agarose gels, hybridized to HBV DNA radiolabelled with [α-32P]CTP(22) and analyzed by autoradiography. Total RNA was extracted from infected cells using NucleoSpin® RNA (Macherey-Nagel) and transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, USA). Real-time PCR for total HBV DNA and cccDNA was performed using LightCycler 96 (Roche) as described(26, 27).
Transfections, plasmids and siRNAs
Transient transfections in HEK293T cells were carried out employing Lipofectamine 2000 (Invitrogen) as described by manufacturer. HBc-WT (EL43), HBc-3A (EL113) and HBc-3D (EL114) were kindly provided by Dr. D. Loeb, derived from EL43 plasmid(28). The CTD-GST fusion protein plasmids: GST-HCTD141 (CTD WT) and, the GST-HCTD141-AAAAAAA (CTD 7A)(7) were a kind gift from Dr. Hu. SiRNAs (Smart pool grade; Dharmacon) for PLK1, HBV and HCV (5–25nM) were transfected in HBV-infected HepaRG or PHH using Dharmafect I (Dharmacon) following manufacturer’s protocol.
Humanized FRG mouse model and HBV infection
All animal studies were reviewed and approved by a local ethical committee. The highly immunosuppressed FRG mice (Fah−/−/Rag2−/−/Il2rg−/−), deficient for T-, B- and NK-cells were used(29) and maintained in pathogen-free facility. High quality, cryopreserved human hepatocytes were purchased (BD, Biosciences) and injected via an intra-splenic route into 2 to 3 month old mice as described previously(30). Liver humanized Fah−/−/Rag2−/−/Il2rg−/− mice featuring serum production of human albumin, at least 5 mg/mL, were infected with 200 μl of HBV inoculums (1.108 veg to 1.109 veg in PBS) via intra-peritoneal route(30).
Mice were treated by intra-peritoneal injection of BI-2536 (10mg/kg/twice a week) for a month. Serum was harvested every week by retro-orbital bleeding and stored at −80°C in aliquots for further antigenemia and viremia analysis. Mice were sacrificed at week 8 post-infection and hepatic tissues were frozen and processed for virologic parameter analyses or fixed in formalin and embedded in paraffin for immune-staining.
Capsid migration assay
The intracellular formation/accumulation of HBV nucleocapsid in in vitro infected hepatocyte or in mouse derived liver resection was accessed from cell or liver lysate by native agarose gel electrophoresis followed by transfer onto ECL membrane and western blot analysis, as previously described(6, 31).
In vitro PLK1 kinase assays
Assays were performed as previously described(10) using recombinant PLK1 (BPS Bioscience, Protein One). Core protein was immuno-purified from HepaRG-TR-HBc cell line or purchased from Meridian Life Science, Inc. Site-directed mutagenesis of putative PLK1 phosphorylation sites in HBc-WT and HBc-3D was performed employing the Quick-change Lightning site-directed mutagenesis Kit (Agilent). Point mutations in the GST-CTD-WT and GST-CTD-7A plasmids were introduced following the same procedure. Mutations were confirmed by DNA sequencing. For protein staining, PageBlue™ Protein Staining Solution (ThermoFischer) was used following manufacturer’s protocol.
Statistical analysis
Statistical analysis was performed using two-way Anova, t tests, or nonparametric Mann-Whitney tests using the GraphPad Prism software. For all tests, p-value ≤0.05 (*), ≤0.01 (**), and ≤0.001 (***) were considered as significant.
RESULTS
PLK1 is activated by HBV infection in non-dividing/differentiated hepatocytes
Our earlier studies demonstrated that i) HBx activates the mitotic S/T kinase PLK1, in a conditional HBx-expressing cell line(11), ii) PLK1 activation initiates proteasomal degradation of chromatin modifying nuclear proteins SUZ12 and ZNF158(10), and iii) SUZ12 downregulation in HBV replicating hepatocytes results in expression of hepatic cancer stem cell markers and pluripotency genes(32). We have also shown activation of PLK1 in HBV-replicating HepAD38 cells(10), further suggesting a link between HBV infection and PLK1 activation. However, it remained to be determined whether PLK1 activation occurs in the context of physiologic infection of non-dividing, differentiated, and non-transformed hepatocytes. To this end, primary human hepatocytes (PHH) and differentiated HepaRG (dHepaRG) were infected with HBV, and expression and activation of PLK1 was quantified. Upon infection of dHepaRG cells, PLK1 mRNA increased by15-fold 24hr post-infection (p.i.), followed by a constant level of expression of 3-to 5- fold from 48h to 168h p.i. (Figure 1A). This resulted in a transient increase in PLK1 protein levels (Fig. 1B). More interestingly, an increase in PLK1 phosphorylation on S137 and/or T210, indicative of PLK1 activation, was detected as a function of HBV infection by immunoblots (Fig. 1B), and immunofluorescence microscopy (Fig. 1C) employing phospho-specific PLK1 antibodies. Remarkably, this activation of PLK1 by HBV infection was also detected by immunoblots of lysates from various preparations of PHH (representative blots are shown; Fig. 1D).
Figure 1. HBV infection activates PLK1.
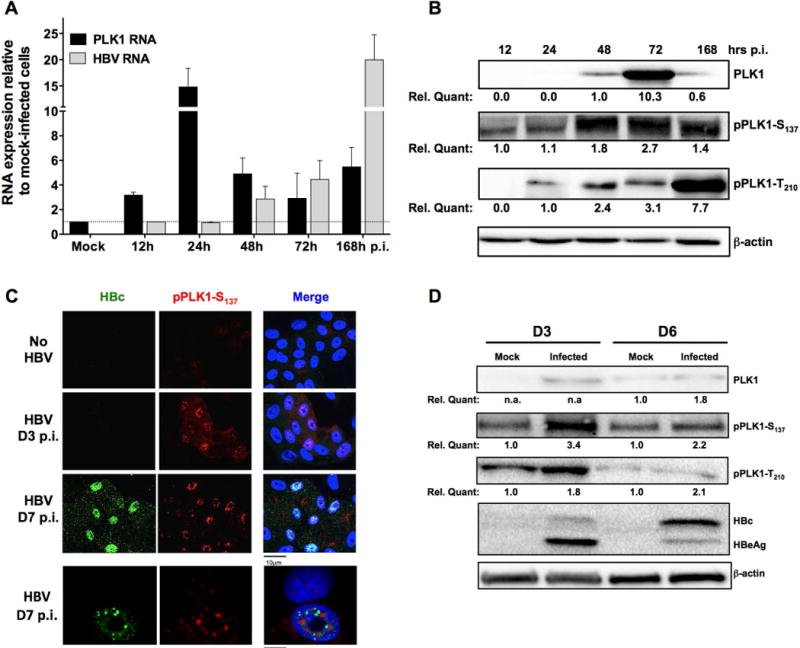
dHepaRG cells (A, B and C) or PHH (D) were infected with low dose (100 vge/cell) or high dose (1000 vge/cell) HBV. A) Cells were harvested at indicated time points, RNA extracted and subjected to RT-qPCR. Fold induction of mRNA expression level of PLK1 and HBV were normalized to housekeeping genes, compared to mock infection. B) Immunoblot of PLK1 and phosphorylated PLK1 (pPLK1-S137 and pPLK1-T210) using whole cell extracts (WCE) of mock- or HBV-infected dHepaRG cells isolated at indicated time points post-infection (p.i.). Quantification by chemiluminescence was done with a ChemiDoc XRS+ system (Biorad). C) Immunofluorescence microscopy of indicated proteins +/− HBV infection in dHepaRG cells at different time p.i. Cells were fixed by 2% PFA and stained with indicated antibodies. D) Immunoblots of PLK1 and phosphorylated PLK1 using WCE from mock- or HBV-infected PHH cells.
PLK1 inhibitors, including BI-2536, suppress HBV DNA accumulation in persistently HBV-infected hepatocytes
To test whether PLK1 activation has a proviral effect, dHepaRG cells were infected with HBV virions and on day-7 post-infection (7 d.p.i.), when infection had reached a “plateau” in terms of viral replication, cells were treated with various PLK1 inhibitors. Specifically, increasing concentration of ATP competitive (i.e. BI-2536 and BI 6727) or non-ATP competitive (i.e. MLN 0905 and ON 01910) PLK1 inhibitors were added for 3 days; accumulation of intracellular HBV DNA was measured by Southern blot and qPCR (Supplementary Figure 1A and 1B). Amongst all drugs tested, BI-2536 was the most active and selected for further studies, after monitoring batch-to-batch stability (Sup. Fig. 2). HBV infected dHepaRG treated with BI-2536 on day-7 p.i., exhibited no effect on levels of secreted HBe and HBs antigens, or intracellular viral RNA (Fig. 2A). Interestingly, BI-2536 significantly inhibited synthesis of total HBV DNA, with an effective concentration at 50% (EC50) of approximately 4 nM (Fig. 2A), in the absence of any toxicity (cytotoxic concentration at 50%, CC50) of ~10 μM Fig. 2B). Interestingly BI-2536 reduced the level of capsid (i.e. assembled HBc/core protein) detected by immunofluorescence microscopy (Fig. 2C) with an antibody recognizing HBc assembled into capsid, and not monomeric forms of HBc (Sup. Fig. 3). Finally, BI-2536 was shown to strongly (up to 95%) and dose-dependently reduce the formation/accumulation of HBV nucleocapsids in infected cells (Fig. 2D). Altogether these results demonstrate that PLK1 inhibition by BI-2536 affects the assembly of HBV capsids, as specific HBc assembly inhibitors (such as BAY41-4119 or AT130) do, thereby inhibiting subsequent rcDNA synthesis. Similar results were also obtained in HBV infected PHH (Fig. 3), with an EC50 at ~ 50 nM. Interestingly, in this model, a trend toward an increase in intracellular viral RNA was observed (Fig. 3A), thus inversely correlating with total DNA decrease. This corroborates the hypothesis that rcDNA synthesis is likely inhibited by an impaired assembly of capsids around pgRNA.
Figure 2. Effect of BI-2536 on HBV replication in non-dividing and non-transformed dHepaRG.
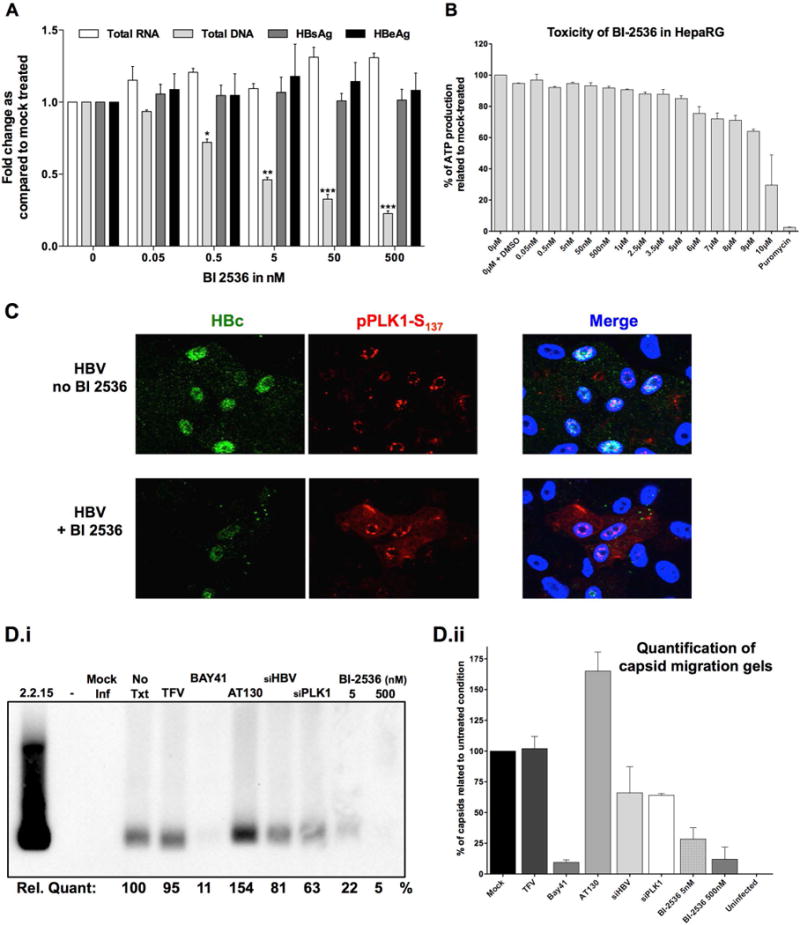
Differentiated HepaRG cell (A, B, C, and D) were infected with 100 vge/cell of HBV for 7 days followed by treatment with increasing concentration of PLK1 inhibitor BI-2536, as indicated, for 3 days. A) Secreted antigens HBsAg and HBeAg quantified by ELISA from supernatants of HBV infected dHepaRG cells, day-10 p.i. Total RNA and DNA extracted from infected cells, day-10 p.i., were analyzed by HBV-specific RT-qPCR or qPCR, normalized to housekeeping genes and compared to mock-treated cells. Results are presented as fold change in expression or secretion, compared to untreated controls and are mean ± SEM of at least 3 independent experiments. B) Cell viability of HepaRG cells measured using Cell Titer Glo One Solution Assay, with increasing concentration of BI-2536, as indicated. DMSO and puromycin were used as controls. C) Immunofluorescence microscopy of indicated proteins, +/− BI-2536 in infected dHepaRG cells. D) As in other panels, mock and infected dHepaRG were treated by biochemicals (No Txt= no treatment; TFV = tenofovir at 10μM; Bay41= Bay41-4109 at 10μM; AT130 at 10μM; BI-2536 at indicated concentration) or transfected by siRNA (25nM) targeting either HBV (siHBV) or PLK1 (siPLK1) for 3 days. Cell lysates were analyzed by native agarose gel electrophoresis, transferred onto ECL membrane, and immunoblotted with HBc antibody. (Di) immunoblot image, with it quantification underneath and (Dii) = quantification of 2 independent experiments by chemiluminescence with a ChemiDoc XRS+ system (Biorad).
Figure 3. Effect of BI-2536 on HBV replication in non-dividing and non-transformed PHH.
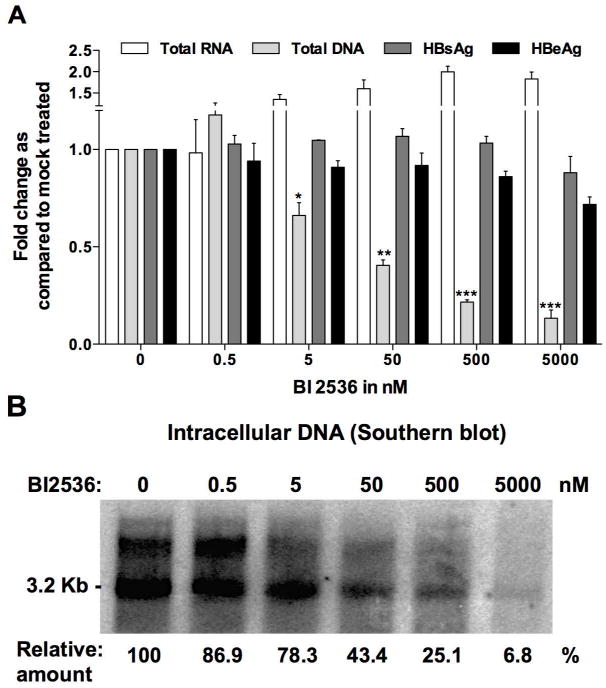
PHH (A, B, and C) were infected with 100 vge/cell of HBV for 4 days followed by treatment with increasing concentration of PLK1 inhibitor BI-2536, as indicated, for 3 days. A) Quantification of secreted HBsAg and HBeAg by ELISA using supernatants of HBV infected PHH on day 7 p.i. Total RNA and DNA extracted from HBV infected cells, day-7 p.i., were analyzed by HBV-specific RT-qPCR or qPCR, normalized to housekeeping genes, and compared to mock-treated cells. Fold change in expression or secretion quantified relative to untreated controls represent mean ± SEM of at least 3 independent experiments. B) A representative Southern blot is shown (n=3). Total DNA extracted from cells on day-7 p.i., was analyzed by agarose gel electrophoresis, transferred onto nylon membrane, and hybridized to radioactive HBV probe as previously described(22).
We also analyzed the effect of BI-2536 on the establishment of HBV infection and found that the antiviral effect was even greater with not only an effect on neosynthesis of HBV DNA but also effect on other measured viral parameters (Sup. Fig. 4).
Loss-of-function of PLK1 suppresses HBV DNA accumulation in infected hepatocytes
To conclusively establish that PLK1 is a proviral factor, we investigated effect of loss of function of PLK1 on HBV replication by siRNA-mediated knockdown of PLK1 (siPLK1). As controls, we employed siRNA targeting HBV transcripts (siHBV; positive control), and siRNA against HCV (siHCV) serving as negative control. The effectiveness of PLK1 knockdown by siRNA transfection was determined by quantification of PLK1 mRNA levels as a function of time after transfection (data not shown). Transfection of siPLK1 reduced by nearly 90% endogenous level of PLK1 mRNA at 72hr after transfection, while all non-targeting siRNAs had no effect on PLK1 mRNA levels (Fig. 4A). Transfection of increasing amounts of siPLK1, ranging from 5 to 25 nM in HBV-infected dHepaRG cells, resulted in a progressive reduction of 50 to nearly 70% in the accumulation of intracellular HBV DNA (Fig. 4A). By contrast transfection of 25nM siHCV had a minimal effect, while transfection of 25nM siHBV reduced HBV DNA by nearly 80% (Fig. 4A). With siPLK1, we again observed a decreased accumulation of assembled HBc within infected cells by IF, using an antibody recognizing assembled capsids (Fig. 4B), and by native capsid migration assay (Fig. 2D), thus confirming the potential relationship between proviral action of PLK1, formation of HBV capsids, and decrease in newly synthetized rcDNA. Similar results were also obtained in HBV-infected PHH cultures, transfected for 72 h with siRNAs targeting PLK1, HBV or an irrelevant target (Fig. 4C). Specifically, Southern blot analyses demonstrated a 50% reduction in HBV DNA by transfection of siPLK1, a 10% reduction with siHCV and 65% reduction with siHBV (Fig. 4D).
Figure 4. Effect of PLK1 knockdown on HBV replication in dHepaRG and PHH.
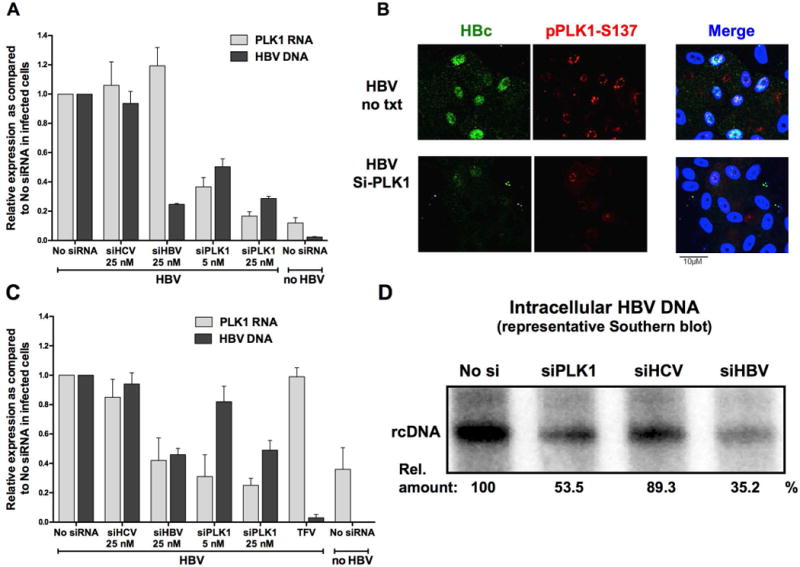
PHH or dHepaRG cells infected with 100 vge/cell of HBV for 7 days were transfected with siRNA (5 or 25 nM) targeting PLK1 (siPLK1) or HBV (siHBV) or HCV (siHCV), as indicated. A and C) Total RNA and DNA were extracted and subjected to RT-qPCR or qPCR with PLK1 or HBV primers. Quantification of PLK1 mRNA and HBV DNA is relative to absence of siRNA transfection (no siRNA). Results represent the mean ± SEM of at least 3 independent experiments. B) Immunofluorescence microscopy of indicated proteins, +/− siPLK1 transfection in HBV infected dHepaRG cells. Cells were fixed by 2% PFA at day 10 p.i., and stained with indicated antibodies. D) A representative Southern blot is shown. DNA was extracted from infected PHH transfected by indicated siRNA.
A modest gain-of-function of PLK1 is associated with increased HBV replication
To investigate the effect of gain-of-function of activated PLK1 on HBV replication, we generated a HepaRG cell line expressing a constitutively active form of PLK1 (PLK1CA) in a tetracycline inducible manner (i.e. HepaRG-TR-PLK1CA) (Sup Fig. 5A). HBV infected dHepaRG-TR-PLK1CA cells were induced to express PLK1CA on day-7 p.i. by addition of increasing amount of tetracycline (0, 16, 80, and 400 ng/ml) for 3 days. Quantification of viral DNA and secreted antigens was carried out day-10 p.i. Under these conditions of established infection, a modest expression of PLK1CA enhanced HBV replication by 1.5- to 2- fold, whereas no effect on HBeAg secretion was observed (Sup Fig. 5B). This result suggests that active PLK1 enhances formation of new viral genomes by reverse-transcription of pregenomic RNA (pgRNA), i.e., it exerts a positive effect on capsid-assisted reverse transcription of pgRNA.
Reduction of HBV viremia in liver-humanized infected FRG mice treated with BI-2536
To demonstrate the effect of BI-2536 on inhibition of HBV replication in vivo, we employed liver-humanized FRG mice(30) infected with HBV (5×108 vge/mouse). Once serum viremia reached 7 log10 vge/mL, antigenemia 5 log10 IU/mL for HBsAg and 2,500 NCU/mL for HBeAg at week-4 post infection, treatment was administered by intra-peritoneal injection of 10 mg/kg of BI-2536 (or same concentration of DMSO diluted in saline) bi-weekly for 4 weeks. Serum viremia and antigenemia were monitored at the end of weeks 1, 2, 4, 6, 7 and 8, while weeks 6, 7, and 8 were those under treatment. Treated mice exhibited at week-8 a mean reduction of 1.406 log10 ±0.26 in viremia (Fig. 5A), whereas minimal effect was observed on antigenemia (Fig. 5B and C). Intra-liver analyses, using samples collected at euthanasia, confirmed the reduction of total HBV DNA, while showing no change in HBV RNA and cccDNA, thus further confirming the antiviral phenotype of BI-2536 (Fig. 5D). If immunohistochemistry staining with a polyclonal anti-HBc antibody, which recognize all forms of HBc (Sup Fig. 3), revealed a slight decrease in accumulation of this protein at the end of the treatment period (representative IHC shown in Fig. 6A), an analysis of the accumulation of assembled capsid by native capsid migration assay confirmed the inhibitory effect of BI-2536 (representative IHC shown in Fig. 6B and C). This antiviral effect was obtained in the absence of toxicity as measured by monitoring mouse weight, secretion of human albumin in serum, and level of ALAT (Sup Fig. 6). It is worth noting that similar duration treatment with IFN-α (25 μg/kg/tw; 5 mice treated) or entecavir (ETV; 2.5μg/mouse/day in drinking water; 3 mice treated) in liver-humanized FRG mice led to a viremia reduction of 0.668 log10 ±0.18 or 1.919 log10 ±0.23 respectively (Sup Fig. 7). These results indicate that a mean reduction of 1.406 log10 ±0.26 in mono-therapy obtained with BI-2536 was relevant and in between performance of IFN-α and ETV in this model.
Figure 5. Effect of BI-2536 on HBV replication in liver humanized HBV-infected FRG mice.
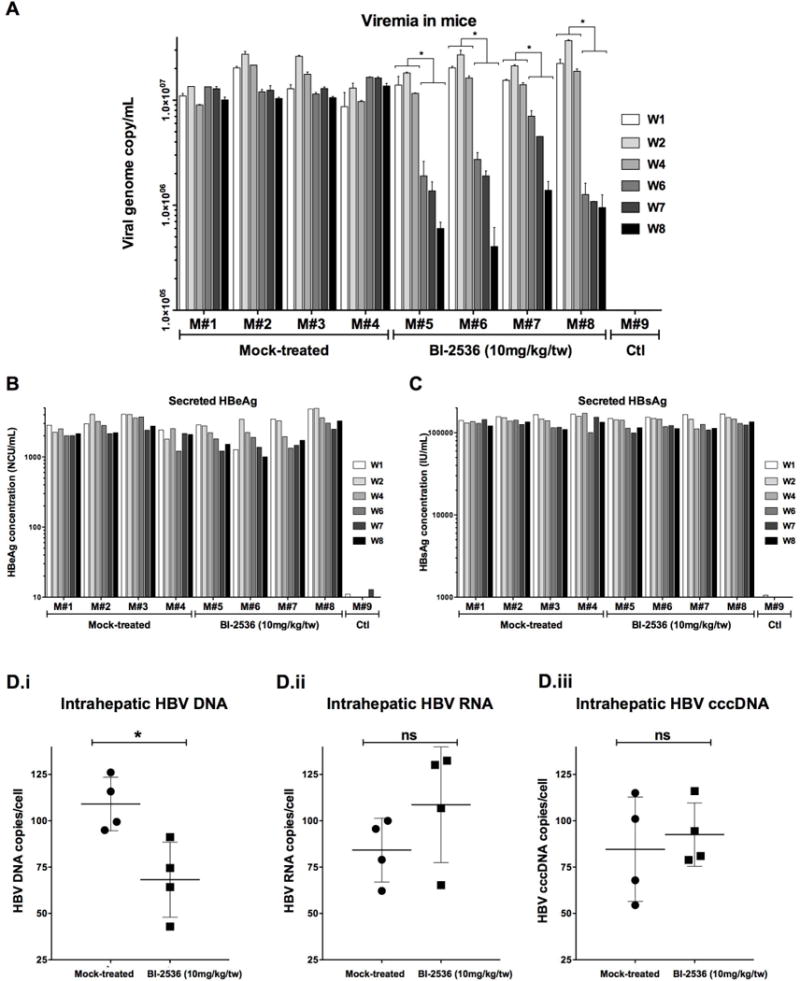
Eight mice, engrafted with human PHH for two months were infected with HBV (5.10e8 vge/mouse). On week-4 p.i., treatment was initiated as indicated. BI-2536 (10mg/kg) was injected twice per week intraperitoneally (IP) for 4 weeks. Blood was collected at the end of week-1, 2, 4, 6, 7, and 8 and viremia (A) and antigenemia (B and C) monitored by qPCR and ELISA. From liver pieces, intrahepatic HBV DNA (Di), RNA (Dii) and cccDNA (Diii) were quantified respectively by qPCR, RTqPCR, and cccDNA specific qPCR(27).
Figure 6. Effect of BI-2536 on capsid formation in liver-humanized HBV-infected FRG mice.
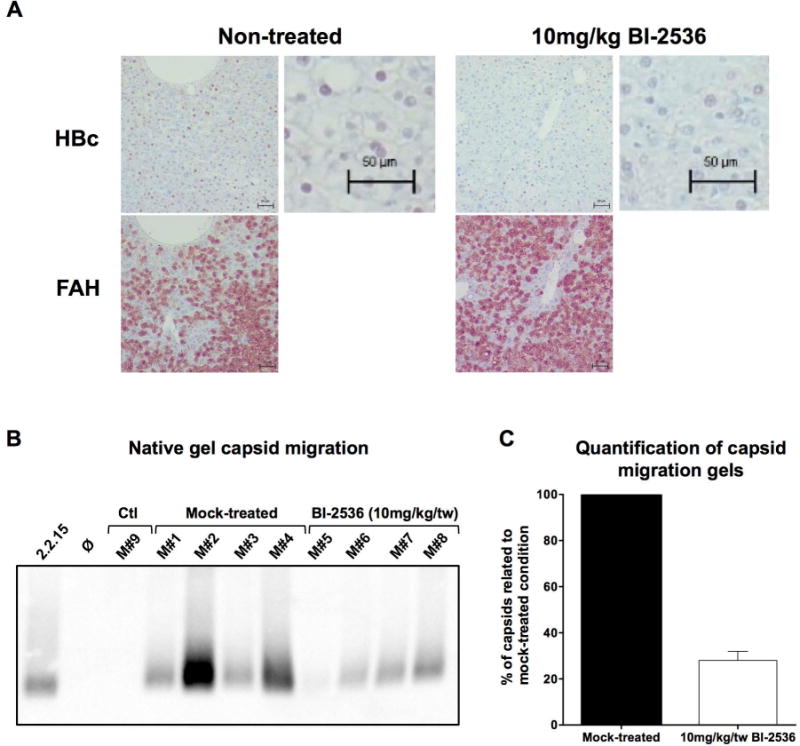
Immunohistochemistry staining (A) and native capsid migration assays (B) employing liver samples from the same mice as in Figure 5. A) Representative immunohistochemistry staining with anti-HBc Ab (Dako) and FAH Ab for non-treated and BI-2536 treated livers. B) Capsid migration assay employing extracts from mock-treated FRG mice (mouse numbers M#1- M#4) and BI-2536 treated animals (mouse # M#5-M38). C) Cumulative quantification of capsid migration signals from (B).
Effect of the combination of BI-2536 with approved or investigational anti-HBV drugs
Our in vitro and in vivo data demonstrated that PLK1 inhibitors could be used as efficient and safe anti-HBV drugs. PLK1 inhibitor, including BI-2536 seems to target the capsid-associated reverse-transcription of pgRNA into rcDNA. Other drugs also target this step of the viral life cycle, including nucleoside/nucleotide analogues (NA) and core inhibitors (also called core protein allosteric modulators; CpAM). But all these drugs do have different molecular targets, and therefore could be theoretically used in combination to enhance the inhibitory phenotype. In order to establish whether BI-2536 could be used in combination with NA or CpAM without leading to antagonistic effect, we combined these molecules to treat infected dHepaRG cells. A combination of BI-2536 and IFN-α was also included. As expected only BI-2536 + IFN-α treatment led to a reduction in HBeAg secretion (Fig. 7A), whereas all other combinations led to a sharp reduction of intracellular HBV DNA (Fig. 7B). No obvious antagonism was evidenced for all combinations tested, thus suggesting that these drugs could be further considered for combination studies.
Figure 7. Combination treatment of BI-2536 and tenofovir, IFN-α, or Bay41-4109.
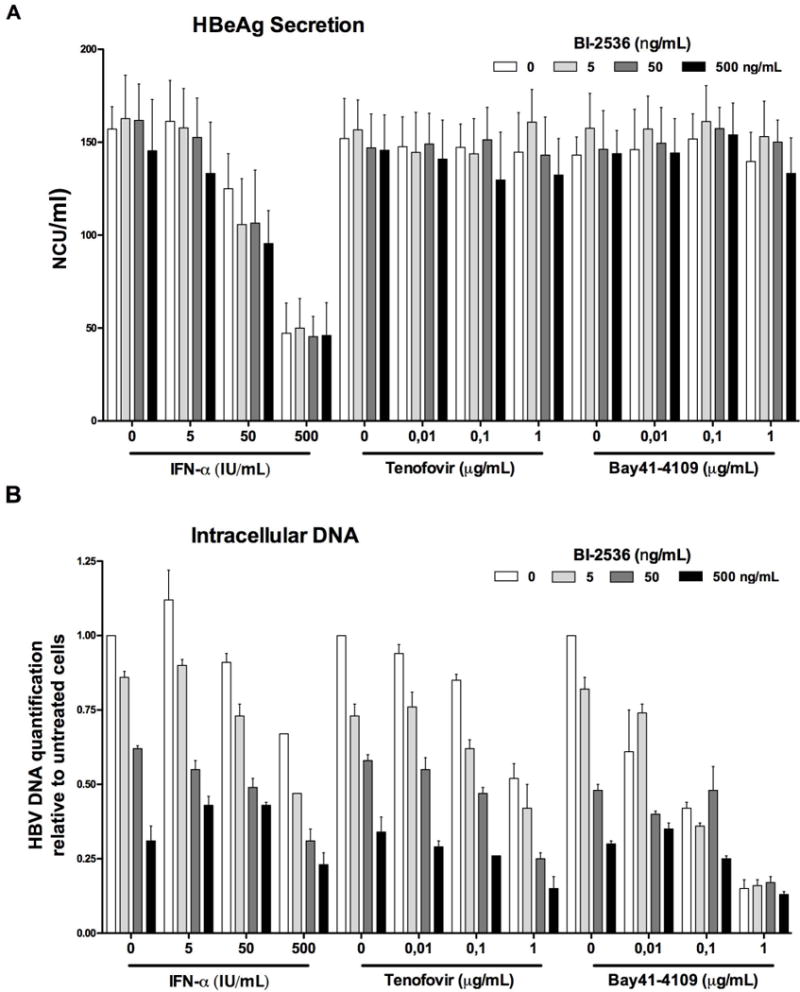
Differentiated HepaRG were infected with 100 vge/cell of HBV for 7 days, followed by treatment with indicated concentration of drugs for 3 days. A) HBeAg secretion in supernatant was monitored by ELISA. B) Intracellular HBV DNA was quantified by qPCR.
HBc is a substrate for PLK1 in vitro
During established infection, inhibition of PLK1 by treatment with BI-2536 suppressed only viral DNA synthesis (Fig. 2A, 3A, and 3B), suggesting that PLK1 may have a role in capsid-associated reverse-transcription of pgRNA into rcDNA. Conversely, inhibition of PLK1 could prevent these processes, based on capsid assembly. Since HBc has an essential and dynamic role in this process of capsid assembly, pgRNA encapsidation, and rcDNA biosynthesis, we investigated whether HBc is a PLK1 phosphorylation substrate.
To determine whether HBc is a direct target of PLK1, we performed in vitro kinase assays with purified recombinant HBc and PLK1 (Fig. 8). We observed in vitro PLK1 phosphorylation of HBc using either recombinant HBc or HBc isolated from HepaRG-TR-HBc cells (Fig. 8A); furthermore, addition of 500nM BI-2536 inhibited this phosphorylation (Fig. 8A, left panel). The C-terminal domain of HBc (CTD), spanning amino acids 143 to 183 (Fig. 8B), contains 8 S/T residues. We performed in vitro PLK1 kinase assays with GST-CTD fusion peptide isolated from transfected cells and featuring either wild type or mutated CTD sequences at all S/T sites. This approach was validated, by showing that fusion protein with 7 (not including T147) or all 8 S/T positions mutated exhibited absence of PLK1 phosphorylation (Fig. 8B). PLK1 requires a priming phosphorylation of its substrate, usually by CDK1 or CDK2 at SP sites. Interestingly, HBc is known to be phosphorylated by CDK2 at three SP phosphorylation sites (Fig. 8B). Accordingly, we tested whether HBc proteins containing S to A or S to D substitutions at all three SP positions (labeled as 3A or 3D) could serve as PLK1 substrates. Interestingly, only the phospho-mimetic substitutions, i.e., S155D, S162D, and S170D (referred to as 3D), exhibited phosphorylation by PLK1 (Fig. 8C). Next, we observed that S168A exhibited significantly reduced phosphorylation in the context of the phospho-mimetic substitutions at SP sites, whereas no difference in the phosphorylation by PLK1 was detected by T146A and T147A substitutions (Fig. 8D). Single point mutations at residues 168, 176 and 178, again in the context of the phospho-mimetic substitutions at SP sites, resulted in significant reduction of HBc phosphorylation by PLK1 (Fig. 8E). Therefore, we conclude that S168, S176 and S178 are PLK1 phosphorylation sites in vitro.
Figure 8. HBc is a phosphorylation substrate of PLK1 in vitro.
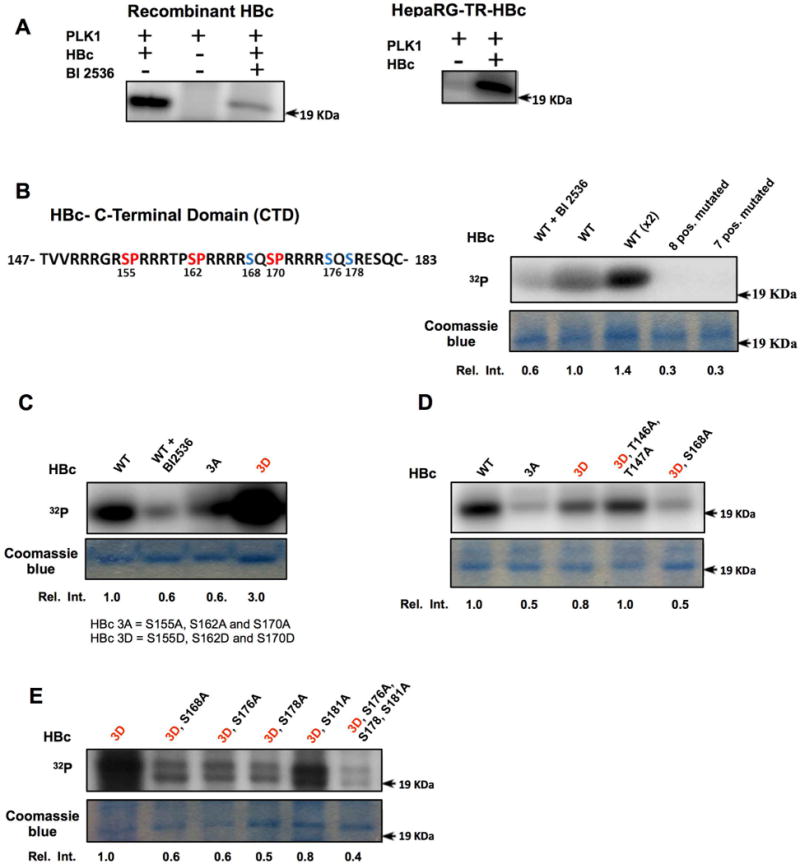
In vitro PLK1 kinase assays of wild type (WT) HBc and site-directed mutants. A) Recombinant HBc or immunoaffinity purified HBc isolated from HepaRG-TR-HBc cells, and PLK1 were used in in vitro PLK1 kinase assays. Reactions were performed with (+) or without (−) PLK1 inhibitor BI-2536 (500nM), as indicated, in the presence of γ32P-ATP, and analyzed by SDS-PAGE and autoradiography. B) Amino acid sequence of C-terminal domain (CTD) of HBc. Eight S/T residues are present in HBc CTD. CDK2 SP sites are indicated in red color, and putative PLK1 phosphorylation sites in blue. B, C, D and E) In vitro kinase assays of WT HBc and site-directed mutants with(+) or without (−) addition of BI-2536, as indicated. HBc 3A contains Ser to Ala substitutions of residues 155, 162 and 170. HBc 3D contains Ser to Asp substitutions of the same residues. Commassie blue staining of the same gel used for autoradiography is shown. Relative intensity quantified by ImageJ software is ratio of signal from in vitro kinase reaction signal versus the corresponding signal of commassie blue-stained band, expressed relative to WT (n=3).
DISCUSSION
The hepatitis B virus (HBV) is one of the most important human oncogenic virus(1, 2). Approximately 250 million individuals worldwide are chronically infected by HBV, having increased risk for developing hepatocellular carcinoma (HCC)(1, 2). If clinicians dispose of several anti-HBV drugs, the have limited chemotherapeutic options for HCC. It is thus crucial to identify novel molecules that target both virus biosynthesis and neoplastic transformation. In this study we provide evidence that the polo-like-kinase 1 (PLK1), a kinase already demonstrated to be involved in HCC, could represent potential target to tackle both HBV replication and liver oncogenesis.
Earlier studies demonstrated that HBx activates PLK1 in a hepatocyte model cell line(11), and that PLK1 was activated in the HepAD38 cellular model of HBV replication(10). However, this cellular model of viral replication does not model physiologic infection. In this study, using physiologically relevant HBV infection models comprised of primary human hepatocytes and the HepaRG cell line, we show that indeed, HBV infection leads to early activation of PLK1. Here, we show that PLK1 phosphorylation occurs upon HBV infection in non-dividing and differentiated cells; the mechanism of PLK1 activation during HBV infection is yet to be determined. HBx protein, which is expressed very early after HBV infection, remains the main candidate for this activation, likely mediating PLK1 activation via the p38MAKP(11, 33). Here we provide evidence for the functional advantage to the viral life cycle of this early activation of PLK1 by HBV infection.
Employing PLK1 pharmacological inhibitors and an RNA interference approach, we have shown that PLK1 enzymatic activity is crucial for maintenance of HBV replication in non-dividing hepatocytes. Indeed, BI-2536, an ATP-dependent PLK1 inhibitor, drastically inhibited HBV replication in vitro in both HepaRG cells and PHH, as well as in vivo, in liver humanized FRG mice, without toxicity Interestingly, the antiviral activity of BI-2536 was very potent with an EC50 ranging from 5 and 50 nM, depending on cell type used; such an antiviral efficacy is rarely obtained with host targeting agents (HTA) and corresponds to the efficacy of direct acting agents (DAA) including nucleoside analogues or assembly inhibitors. Accordingly, an in vivo antiviral activity was also obtained with BI-2536 in HBV-infected liver humanized FRG mice, and this antiviral activity was comparable to that obtained with entecavir or IFN-α in the same model. The RNAi approach confirmed the PLK1 specificity, as kinase inhibitors are often non-specific. The antiviral phenotype obtained by RNAi targeting PLK1 was also very strong in vitro. The use of such an approach in vivo using siRNA that could be specifically targeted to infected hepatocytes could represent an interesting alternative to pharmacological inhibitors, which may be quite toxic when administrated systemically. Recently, it has been shown that RNAi nanoparticles in complex with PLK1 siRNA, administered systemically, efficiently delivers PLK1 siRNA to the liver with no observable toxicity(34). Lastly, we have also shown that BI-2536 could be combined in vitro and presented no antagonism to currently approved (i.e. IFN and tenofovir) anti-HBV drugs and Bay41-4109(20), a representative core inhibitor; the latter being a novel class of inhibitors in current clinical development(5).
The molecular mechanism underlying the proviral role of PLK1 has not been completely determined in this study. Nevertheless, we have shown that a target of PLK1 is the capsid/core protein (HBc), responsible for capsid assembly around the pgRNA, and indirectly pgRNA reverse-transcription, as the latter occurs only within capsids. PLK1-mediated phosphorylation requires prior phosphorylated “primed” substrates. Here we presented evidence that HBc is a direct target of PLK1 in vitro, and importantly, efficient phosphorylation of HBc required prior phosphorylation at positions S155/S162/S170, sites shown to be phosphorylated by CDK2(7, 8). Our studies identified S168, S176 and S178 as PLK1 phosphorylation sites in vitro, dependent on prior phosphorylation of S155, S162 and S170. Thus, PLK1-mediated phosphorylation of HBc in a sequential phosphorylation process could directly mediate the dynamic events associated with capsid formation, pgRNA encapsidation and reverse transcription. Further mechanistic studies and molecular tools (phospho-specific antibodies) are required to fully delineate this mechanism of PLK1 involvement.
Many other viral proteins are directly phosphorylated by PLK1, including the non-structural 5A protein from HCV(35), the mump virus nucleoprotein(36), and the parainfluenza virus 5 (PIV5) protein(37). PLK1-mediated phosphorylation of the target can either lead to its degradation or change in function; therefore PLK1 can either act as a proviral or antiviral factor. For HCV, PLK1 seems to be a proviral factor, as it contributes to the hyper-phosphoryaltion of NS5A(35), which is a crucial event to switch from a replicative cycle (i.e. amplification of viral RNA) to a productive one (i.e. generation and secretion of novel virions). Nevertheless the use of anti-PLK1 drugs has only limited value to inhibit HCV replication, because the virus replicates better in vitro in dividing cells, thus precluding non-toxic PLK1 inhibition. But in the case of HBV, this framework for a potential use of PLK1 targeting drugs does exist. Indeed HBV only replicates in non-dividing and highly differentiated cells. Although we have shown here that an anti-HBV phenotype could be obtained in vivo, more experiments are warranted to further demonstrate the safety of such an HTA-based approach. Many PLK1 pharmacological inhibitors as well as strategies based on PLK1 silencing are in current development for treating several cancers(38); a repositioning of such drugs for the treatment of CHB patients could be foreseen. The fact that PLK1 inhibitors could reinforce the inhibition of the neosynthesis of encapsidated rcDNA, as nucleoside analogues already do, represent an excellent framework for the potential development of combination therapies.
Acknowledgments
The authors thank Lydie Lefrançois, Judith Fresquet and Maud Michelet for isolation of primary human hepatocytes, as well as the staff from Pr Michel Rivoire’s surgery room for providing us with liver resection. We also thank J.F. Henry, N. Aguilera and J.L. Thoumas from the animal facility (PBES, Plateau de Biologie Experimental de la Souris, ENS de Lyon), as well as G. Froment and B. Eckel for their technical help in handling mice. We thank the Vectorology platform (Inserm U1089, Nantes) for the production of the adeno-uPA vector and N. Gadot (Plateforme Anatomopathologie Recherche, Centre Léon Bérard, F-69373 Lyon, France) for the IHC analyses.
This work was supported by grants from ANRS (French national agency for research on AIDS and viral hepatitis; several grants from CSS4) and by INSERM core grants to FLC, FZ and DD. This work was also supported by the DEVweCAN LABEX (ANR-10-LABX-0061) of the “Université de Lyon”, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR) to FZ and DD, and NIH grant DK044533-19 to OA. The work in the laboratory of FL Cosset was further supported by the European Research Council (ERC-2008-AdG-233130-HEPCENT) and the LabEx Ecofect (ANR-11-LABX-0048). AD received a Chateaubriand Fellowship from the French Embassy in Washington DC.
List of abbreviations
- HBV
Hepatitis B Virus
- HCC
Hepatocellular Carcinoma
- PLK1
Polo-like-kinase1
- PHH
Primary Human Hepatocytes
- dHepaRG
differentiated HepaRG cells
- WHO
World Health Organization
- CHB
Chronic Hepatitis B
- HBc
HBV core antigen
- rcDNA
relaxed circular DNA
- cccDNA
covalently closed circular DNA
- pgRNA
pregenomic RNA
- CDK2
Cyclin Dependent Kinase 2
- SP
Serine/Proline
- SRPK1
Serine/Arginine Protein Kinase 1
- S/T
serine/threonine
- HBx
HBV X protein
- SUZ12
Suppressor of Zeste 12
- ZNF198
Zinc Finger, MYM-Type2
- p.i
post-infection
- d.p.i
days post infection
- HBs
HBV S antigen
- HBe
HBV E Antigen
- EC50
Effective Concentration 50%
- CC50
Cytotoxic concentration 50%
- siRNA
small interfering ribonucleic acid
- HCV
Hepatitis C virus
- HepaRG-TR-PLK1CA
HepaRG- tetracycline regulated-PLK1 constitutively active
- vge
virus genome equivalents
- NCU
- DMSO
Dimethylsufoxide
- qPCR
quantitative Polymerase Chain Reaction
- RTqPCR
Reverse transcription quantitative Polymerase Chain Reaction
- ETV
entecavir
- IFN-α
Interferon –α
- FRG
Fah−/−/Rag2−/−/Il2rg−/−
- CpAM
Core protein Allosteric Modulators
- CTD
C-terminal domain
- A
alanine
- D
aspartic acid
- WCE
Whole Cell Extract
- MAPK
Mitogen Activated Protein Kinase
- HTA
Host Targeting Agent
- DAA
Direct Acting Agent
- RNAi
Ribonucleic Acid interference
- PIV5
parainfluenza virus 5
- NS5A
Nonstructural 5A
Footnotes
Involvement of authors:
- study concept and design: AD, AF, OA, and DD
- acquisition of data: AD, AF, FF, PG, LNG, FA, NI, and DD
- analysis and interpretation of data: AD, AF, OA and DD
- drafting of the manuscript: AD, AF, OA and DD
- critical revision of the manuscript for important intellectual content: FLC, FZ, OA, and DD
- statistical analysis: AF, DD
Conflict of interest:
None for this study
References
- 1.Chan SL, Wong VWS, Qin S, Chan HLY. Infection and Cancer: The Case of Hepatitis B. Journal of Clinical Oncology. 2015 doi: 10.1200/JCO.2015.61.5724. [DOI] [PubMed] [Google Scholar]
- 2.Busch K, Thimme R. Natural history of chronic hepatitis B virus infection. Medical Microbiology and Immunology. 2015;204:5–10. doi: 10.1007/s00430-014-0369-7. [DOI] [PubMed] [Google Scholar]
- 3.Zoulim F, Durantel D. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb Perspect Med. 2015;5 doi: 10.1101/cshperspect.a021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley PM, editors. Field’s Virology. Vol. 2. Philadelphia: Lippincott Williams & Wilkins; 2015. p. 2185. [Google Scholar]
- 5.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Research. 2015;121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ning X, Basagoudanavar SH, Liu K, Luckenbaugh L, Wei D, Wang C, Wei B, et al. Capsid Phosphorylation State and Hepadnavirus Virion Secretion. Journal of Virology. 2017 doi: 10.1128/JVI.00092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu K, Ludgate L, Yuan Z, Hu J. Regulation of multiple stages of hepadnavirus replication by the carboxyl-terminal domain of viral core protein in trans. J Virol. 2015;89:2918–2930. doi: 10.1128/JVI.03116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol. 2012;86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J, Ullrich A, et al. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol. 2002;76:8124–8137. doi: 10.1128/JVI.76.16.8124-8137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Diab A, Fan H, Mani SK, Hullinger R, Merle P, Andrisani O. PLK1 and HOTAIR Accelerate Proteasomal Degradation of SUZ12 and ZNF198 during Hepatitis B Virus-Induced Liver Carcinogenesis. Cancer Res. 2015;75:2363–2374. doi: 10.1158/0008-5472.CAN-14-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Studach L, Wang WH, Weber G, Tang J, Hullinger RL, Malbrue R, Liu X, et al. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J Biol Chem. 2010;285:30282–30293. doi: 10.1074/jbc.M109.093963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Studach LL, Rakotomalala L, Wang WH, Hullinger RL, Cairo S, Buendia MA, Andrisani OM. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology. 2009;50:414–423. doi: 10.1002/hep.22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Archambault V, Lepine G, Kachaner D. Understanding the Polo Kinase machine. Oncogene. 2015;34:4799–4807. doi: 10.1038/onc.2014.451. [DOI] [PubMed] [Google Scholar]
- 14.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 15.Studach LL, Menne S, Cairo S, Buendia MA, Hullinger RL, Lefrancois L, Merle P, et al. Subset of Suz12/PRC2 target genes is activated during hepatitis B virus replication and liver carcinogenesis associated with HBV X protein. Hepatology. 2012;56:1240–1251. doi: 10.1002/hep.25781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang WH, Studach LL, Andrisani OM. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology. 2011;53:1137–1147. doi: 10.1002/hep.24163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marion MJ, Hantz O, Durantel D. The HepaRG cell line: biological properties and relevance as a tool for cell biology, drug metabolism, and virology studies. Methods Mol Biol. 2010;640:261–272. doi: 10.1007/978-1-60761-688-7_13. [DOI] [PubMed] [Google Scholar]
- 18.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petit MA, Pillot J. HBc and HBe antigenicity and DNA-binding activity of major core protein P22 in hepatitis B virus core particles isolated from the cytoplasm of human liver cells. J Virol. 1985;53:543–551. doi: 10.1128/jvi.53.2.543-551.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 21.Delaney WEt, Edwards R, Colledge D, Shaw T, Furman P, Painter G, Locarnini S. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob Agents Chemother. 2002;46:3057–3060. doi: 10.1128/AAC.46.9.3057-3060.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hantz O, Parent R, Durantel D, Gripon P, Guguen-Guillouzo C, Zoulim F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J Gen Virol. 2009;90:127–135. doi: 10.1099/vir.0.004861-0. [DOI] [PubMed] [Google Scholar]
- 23.Jang Y-J, Ma S, Terada Y, Erikson RL. Phosphorylation of Threonine 210 and the Role of Serine 137 in the Regulation of Mammalian Polo-like Kinase. Journal of Biological Chemistry. 2002;277:44115–44120. doi: 10.1074/jbc.M202172200. [DOI] [PubMed] [Google Scholar]
- 24.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lecluyse EL, Alexandre E. Isolation and culture of primary hepatocytes from resected human liver tissue. Methods Mol Biol. 2010;640:57–82. doi: 10.1007/978-1-60761-688-7_3. [DOI] [PubMed] [Google Scholar]
- 26.Luangsay S, Gruffaz M, Isorce N, Testoni B, Michelet M, Faure-Dupuy S, Maadadi S, et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol. 2015;63:1314–1322. doi: 10.1016/j.jhep.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 27.Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Lewellyn EB, Loeb DD. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol. 2011;85:1298–1309. doi: 10.1128/JVI.01957-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, et al. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bissig KD, Wieland SF, Tran P, Isogawa M, Le TT, Chisari FV, Verma IM. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest. 2010;120:924–930. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, et al. Secretion of Genome-Free Hepatitis B Virus – Single Strand Blocking Model for Virion Morphogenesis of Pararetrovirus. PLOS Pathogens. 2011;7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mani SK, Zhang H, Diab A, Pascuzzi PE, Lefrancois L, Fares N, Bancel B, et al. EpCAM-Regulated Intramembrane Proteolysis Induces a Cancer Stem Cell-like Gene Signature in Hepatitis B Virus-infected Hepatocytes. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarn C, Lee S, Hu Y, Ashendel C, Andrisani OM. Hepatitis B virus X protein differentially activates RAS-RAF-MAPK and JNK pathways in X-transforming versus non-transforming AML12 hepatocytes. J Biol Chem. 2001;276:34671–34680. doi: 10.1074/jbc.M104105200. [DOI] [PubMed] [Google Scholar]
- 34.McCarroll JA, Dwarte T, Baigude H, Dang J, Yang L, Erlich RB, Kimpton K, et al. Therapeutic targeting of polo-like kinase 1 using RNA-interfering nanoparticles (iNOPs) for the treatment of non-small cell lung cancer. Oncotarget. 2015;6:12020–12034. doi: 10.18632/oncotarget.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen YC, Su WC, Huang JY, Chao TC, Jeng KS, Machida K, Lai MM. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J Virol. 2010;84:7983–7993. doi: 10.1128/JVI.00068-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickar A, Zengel J, Xu P, Li Z, He B. Mumps Virus Nucleoprotein Enhances Phosphorylation of the Phosphoprotein by Polo-Like Kinase 1. J Virol. 2016;90:1588–1598. doi: 10.1128/JVI.02160-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun D, Luthra P, Li Z, He B. PLK1 down-regulates parainfluenza virus 5 gene expression. PLoS Pathog. 2009;5:e1000525. doi: 10.1371/journal.ppat.1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X. Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Translational Oncology. 2015;8:185–195. doi: 10.1016/j.tranon.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]