

Abstract
A mechanistic model of amyloid beta production, degradation, and distribution was constructed for mouse, monkey, and human, calibrated and externally verified across multiple datasets. Simulations of single‐dose avagacestat treatment demonstrate that the Aβ42 brain inhibition may exceed that in cerebrospinal fluid (CSF). The dose that achieves 50% CSF Aβ40 inhibition for humans (both healthy and with Alzheimer's disease (AD)) is about 1 mpk, one order of magnitude lower than for mouse (10 mpk), mainly because of differences in pharmacokinetics. The predicted maximal percent of brain Aβ42 inhibition after single‐dose avagacestat is higher for AD subjects (about 60%) than for healthy individuals (about 45%). The probability of achieving a normal physiological level for Aβ42 in brain (1 nM) during multiple avagacestat dosing can be increased by using a dosing regimen that achieves higher exposure. The proposed model allows prediction of brain pharmacodynamics for different species given differing dosing regimens.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Multiple AD treatments are developed targeting production of amyloid β. CSF and plasma Aβ are the main PD biomarkers in humans, so for understanding of brain PD, preclinical models are extensively used.
WHAT QUESTION DOES THIS STUDY ADDRESS?
☑ The questions this study address are 1) whether a mechanistic translational model can allow for prediction of short‐term GSI pharmacodynamics in humans, and 2) what inhibition levels can be achieved in human brain, given the information on the system and drug PK.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The mechanistic model allows comprehensive comparison of different species revealing the difference in Aβ transport and production. Different sensitivities of brain and BIF Aβ to drug AUC requires a specific schedule to normalize brain Aβ.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ Our model allows for more accurate translation of preclinical results to clinical data and optimization of therapeutic regimen. It provides a link between measured biomarkers and unobservable brain concentrations for estimation of the real drug effect on amyloid toxicity.
Cognitive decline in Alzheimer's disease (AD) is usually preceded by the accumulation of the pathologic amyloid beta (Aβ) protein in the brain. Both insoluble and soluble forms of Aβ may be neurotoxic. In familial forms of AD, genetic mutations may be responsible for modified (increased or decreased) Aβ turnover.1 Other hypothesized mechanisms (e.g., tau pathology, inflammatory response, vascular and metabolic dysfunction2, 3) for AD etiology are considered, but Aβ‐related toxicity participates in most of them. The Aβ hypothesis is being tested in multiple clinical trials evaluating drugs that can alter Aβ kinetics in humans. Passive immunotherapy against Aβ is tested in trials of bapineuzumab by Elan,4 Solaneuzumab by Eli Lilly, and crenezumab and gantenerumab by Roche/Genetech (Clinicaltrials.gov). Amyloid production inhibition efficacy is now tested for verubecestat by Merck, AZD‐3293 by Astra Zeneca, and JNU‐54861911 by Janssen. The influence on amyloid clearance pathways is tested for retinoid receptor agonists such as acitretin by Actavis/Allergan, and bexarotene by Ligand Pharmaceuticals. Gamma‐secretase inhibitors (GSI) avagacestat by BMS and semagacestat by Eli Lilly have also been tested and have shown no success.
Aβ is produced primarily in the endosome and plasma membrane of neurons5 and to a lesser extent in cells of other tissues.6, 7 Given the proximity of cerebrospinal fluid (CSF) to brain, for clinical trial purposes the change in the CSF Aβ level has been used as an indicator for brain Aβ modulation upon therapeutic intervention that targets brain Aβ production or clearance. Plasma Aβ has been monitored in early‐stage clinical trials as a quick endpoint for assessment of peripheral pharmacological activity. However, the relationship between brain, CSF, and plasma Aβ is not straightforward, and peripheral pharmacological activity does not necessarily translate into central pharmacological activity. Depending on the class of therapeutic, different patterns of Aβ kinetics, in plasma and CSF, have been reported. A pronounced rebound in plasma Aβ concentrations was observed for GSI, avagacestat, and semagacestat, in human8, 9 and in mouse,10 as well as in CSF for avagacestat doses 15–50 mg in humans11 and 30 mpk of semagacestat and GSI‐953 in wildtype mouse.10 No rebound in the brain Aβ concentrations was observed in the wildtype or transgenic mouse models10 for avagacestat, semagacestat, or GSI‐953 treatment.10 The quantitative understanding of the Aβ trafficking between brain, CSF, and plasma is of great importance for development of new anti‐AD therapy and AD diagnostics.
The majority of experimental data on Aβ distribution kinetics were obtained from in vivo mouse models12, 13 by monitoring radiolabeled Aβ concentrations in plasma, CSF, and brain. Additional preclinical models (mouse, monkey) have also been used to study Aβ responses in brain, CSF, and plasma following administration of Aβ‐modulating therapeutics in drug discovery.10, 14 Similar studies have also been conducted in healthy volunteers and AD patients to understand Aβ kinetics with or without pharmacological intervention.9, 15 Despite the availability of these data, there have been limited efforts in developing a quantitative understanding of Aβ kinetics across species.
Three types of mathematical models on Aβ kinetics exist in the literature, all with limited utility in providing a quantitative, holistic, cross‐species understanding. The first type is the semimechanistic pharmacokinetic/pharmacodynamic (PK/PD) modeling that focuses on characterizing the pharmacodynamics of Aβ‐modulating agents10, 16, 17 in preclinical and clinical experiments. They usually do not include such details as description of γ‐secretase as an enzyme catalyzed process and Aβ transport across the blood–brain barrier (BBB) and blood–CSF barrier (BCSFB). The second type of model (e.g. Ref. 18), describes the accumulation and distribution of Aβ in the brain, CSF, and plasma, throughout the course of AD treatment, but calibrated against a limited amount of data. The third type is focused on analyzing stable isotope labeling kinetic (SILK) data,19 limited by the scope of data without any extrapolation to the brain compartment.
To further the understanding of Aβ kinetics across species, and more important, to create a tool to enable drug discovery and development in AD, we present a translational mechanistic model of Aβ synthesis, degradation, and distribution.
METHODS
The model describes Aβ forms (Aβ40(42), labeled Aβ40(42)) in brain cells (BC), brain interstitial fluid (BIF), CSF, plasma (PL), and other tissues (OT), and C99 in BC, BIF, and OT (Figure 1 a). Concentration of species changes due to synthesis, distribution, and degradation. The description (model structure, rate equations) of the other amyloid forms (Aβ42, labeled Aβ40, labeled Aβ42) is identical to that for Aβ40 (Figure 1 a) in the full model (Figure 1 b). The final model consisted of 26 ODEs for Aβ and C99, ODEs and explicit function for PK of GSIs, 70 rate laws, and 70 parameters (Supplement A, Table S1). The description of different experimental conditions (labeled Aβ injection or labeled leucine infusion), specific initial values, or input of additional rates is detailed in Supplement B.
Figure 1.
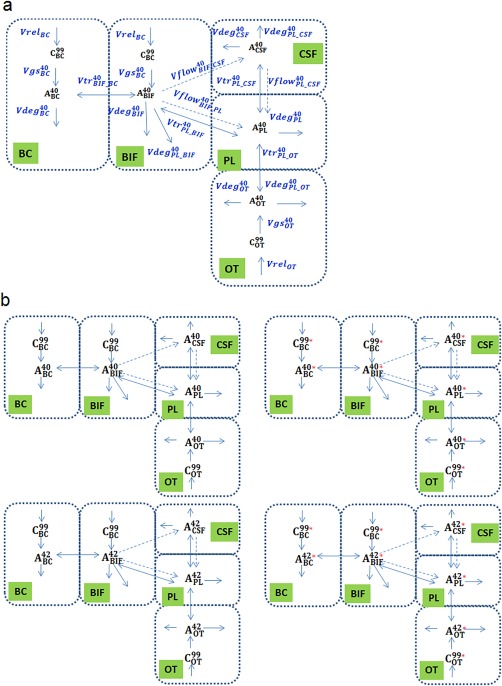
Schematic representation of processes considered in the model. (a) Processes related to endogenous Aβ40. Dashed arrows stand for transport with bulk flow. Solid arrows designate reactions, biosynthesis, degradations and transport mediated by proteins (uptake, efflux, and transcytosis). List of abbreviations: , , are C99 in brain cells (BC), brain interstitial fluid (BIF) and other tissues (OT), respectively, , , , , are Aβ40 in BC, BIF, CSF, PL, and OT, respectively. Processes designation: synthesis of amyloid β precursor protein C99 in BC, BIF, OT , , , respectively; transformation of C99 to Aβ (Aβ hereafter refers to both Aβ40 and Aβ42 unless specified) catalyzed by γ‐secretase in BC, BIF, and OT (processes , , ); Bulk phase (nonreceptor‐mediated) degradation of Aβ in BC, BIF, and OT (processes , , ); transport of Aβ between BC and BIF (processes ); transport of Aβ with bulk flow from BIF to cerebrospinal fluid (CSF), from BIF to plasma (PL) and from CSF to PL (processes , , ); protein‐mediated transport of Aβ via BBB (between PL and BIF), via BCSFB (between PL and CSF) and between PL and OT (processes , , ); degradation of Aβ during passage through BBB (between PL and BIF), BCSFB (between PL and CSF) and between PL and OT (processes , , ). (b) Complete scheme of the model for all species: left part, endogenous Aβ species; right, labeled Aβ. Processes are analogous to (a), but names are not given for simplification. Red asterisk indicates 13C‐label or 125I‐label. List of abbreviations: , , and , , are endogenous and 13C‐labeled C99 in BC, BIF, and OT, respectively; , , , , and , , , , are endogenous and 13C‐labeled (125I‐labeled) Aβ40 in BC, BIF, CSF, PL, and OT, respectively; , , , , and , , , , are endogenous and 13C‐labeled (125I‐labeled) Aβ42 in BC, BIF, CSF, PL, and OT, respectively.
The model has the same structure (variables, compartments, and rate equations) for all the species (mouse, monkey, and human).
Mechanisms of amyloid aggregation in BIF are not described, so this model is not applicable for longer‐duration simulations and the mechanistic description of disease progression. The pathological AD state is treated as steady state with altered values of Aβ production20, 21 and clearance in brain (see details in Supplement A).
Interspecies scaling
Interspecies translation of model parameters was performed using allometric scaling (a generic equation as below):
where P and P0 are reaction rate constants of species with body weight BW and BW0, respectively, and n is the scaling exponent. The allometric scaling alone may not allow for satisfactory translation from rodents to primates, so additional scaling coefficients for groups of processes were incorporated (Supplement A).
Model calibration and evaluation
The model calibration steps across different data types (Table 1) involved the Hooke‐Jeeves method22 implemented in the DBSolve Optimum package23 v. 36.
Table 1.
Description of stages of model construction and external verification
| Step # | Description of step | Type of data used in the step | Number of points | Parameter identification | |
|---|---|---|---|---|---|
| fitting | EV* | ||||
| 1 | Development of the PK sub‐model for Avagacestat and Semagacestat in mouse and human Supplementary B.1 | (i) mouse PK data for Avagacestat | 14 | 0 | PK description parameters were fitted |
| (ii) human PK data for Avagacestat (for two‐compartmental model) | 33 | 0 | |||
| (iii) human PK data for semagacestat | 18 | 0 | |||
| 2 |
Development of sub‐model describing Aβ distribution with bulk flows in mouse. Supplementary B.2.1 |
(ii) in vivo data on Inulin distribution after BIF administration | 12 | — | 3 parameters were fitted |
| 3 | Development, verification and validation of model describing mouse dynamics of production and distribution of endogenous and 125I‐labeled Aβ Supplementary B.2.2 | (i) 125I‐Aβ40/125I‐ Aβ42 distribution after BIF or PL administration | 42 | 20 |
bulk phase Aβb clearance was partially estimated from the literature data 25 parameters responsible for synthesis, degradation and distribution of amyloid β were fitted |
| (ii) 125I‐ Aβ40 clearance from brain after BIF administration | 4 | 1 | |||
| (iii) Steady state concentrations of Aβ40/Aβ42 in brain, CSF and PL | 5 | 0 | |||
| (iv) CSF, PL and brain Aβ40/Aβ42response after Avagacestat administration | 32 | 32 | |||
| 4 | Translation of mouse model to healthy human: verification and validation of the model describing human Aβ dynamics Supplementary B.3.1 | (i) physiological properties of human and mouse |
11 parameters of scaling from mouse to human were fitted Drug PK parameter taken from literature |
||
| (ii) SILK data, placebo and after Semagacestat administration | 25 | 103 | |||
| (iii) CSF and PL Aβ40/Aβ42 response after Semagacestat administration | 34 | 32 | |||
| (iv) Steady states of Aβ40/Aβ42 in BR(soluble), CSF and PL | 6 | — | |||
| (v) CSF and PL Aβ40/Aβ42 response after Avagacestat administration | — | 178 | |||
| 5 |
Description of AD state: verification of parameters different between healthy and AD Supplementary B.3.2 |
(i) steady state levels of Aβ40/Aβ42 in brain, CSF and plasma for AD individuals | 6 | — | 3 parameters describing release of Aβ in BC, and 2 parameters describing dummy efflux of Aβ in BIF to polymerization |
| 6 | Translation from human to monkey: validation of model describing monkey Aβ dynamics Supplementary B.3.3 | (i) physiological properties of human being and monkey | — | — | — |
| (ii) SILK data (12 hours, low leucine and 12 and 21 hours, high leucine), placebo | — | 44 | |||
| (iii) Steady states of Aβ40/Aβ42 in CSF and PL | — | 3 | |||
EV, external verification (comparison of data with predictions).
To evaluate its predictive ability, the model was employed to replicate datasets that were not used in model development (Table 1) with consideration of respective study conditions. For fitting the mouse model, we chose Aβ40 data in brain and CSF from an avagacestat dataset. The main goal was to describe human data as accurately as possible, so a human dataset (which is rich enough) was used for translation from rodents to primates: steady‐state concentrations, SILK data, and GSI PD were fitted by scaling factors for Aβ production and distribution (Table S2); then a monkey dataset (steady‐state values, SILK data) was used for external verification without scaling factor refitting. The semagacestat dataset24 was chosen for calibration among GSI data, as it was complemented by SILK kinetic data, while avagacestat human PD data were used for external verification.
Description of PK of avagacestat and semagacestat
The PK time courses that drove the systems model for avagacestat and semagacestat were implemented using compartmental PK modeling or explicit functions where appropriate (see Supplement B) for reproduction of observed PK data. Values of IC50 measured in vitro 10 were used in equations for γ‐secretase inhibition (Supplement A,B). Due to lack of the brain PK data, we assumed for simplicity brain PK profiles analogous to plasma profiles with correction for brain penetration coefficients.
A full description of experimental facts and model assumptions, ODE system, rate laws, values of model parameters, and experimental data used for model calibration and validation is given in the Supplementary Materials.
Simulation design
Details of design of simulations to explore model behaviors, analyze properties of the system, and optimize therapeutic regimen are provided in Supplement C.
RESULTS
Model calibration and verification
Aβ kinetics and steady state in different species
The model satisfactorily reproduces the mouse steady‐state Aβ concentrations in different compartments (Figure 2 a), mouse avagacestat PD, the phase shift between brain and CSF Aβ40, the overshoot in CSF and plasma Aβ concentrations before returning to baseline level (Figure 2 b), and correctly reproduces the amplitude of Aβ40 decrease.
Figure 2.
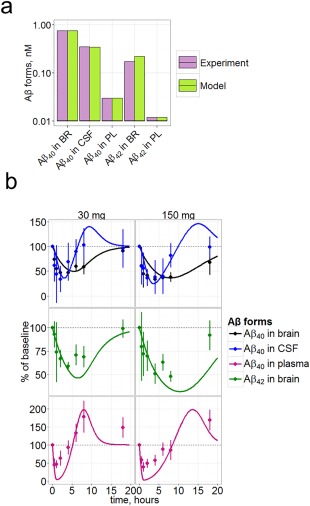
Verification of the model on the mouse data. (a) Steady state concentrations of Aβ40 and Aβ42 in mouse brain, CSF, and PL . Experimental data were taken from Ref. 45. Bars for experiments represent mean from across different animals (from 6 to 60 animals for different data items), bars for model represent average population model prediction. (b) Aβ40 (expressed in % of steady state baseline level) in brain, CSF, and plasma, and Aβ42 in brain in the mouse treated with a single dose of 30 or 150 mg/kg of avagacestat. Symbols represent data10 and curves model simulations. Plasma Aβ40 and brain Aβ42 were not used during the fitting.
The model adequately describes the difference of Aβ42 brain and CSF concentrations between healthy and AD individuals (Figure 3). Concentration of Aβ40 in healthy control human brain (Figure 3) are similar to those of mouse (Figure 2 a), but CSF Aβ40 is higher (more than 1 nM in healthy human vs. about 0.3 nM in mouse). Production and clearance of Aβ as measured by SILK data are also captured by the model (Supplement B).
Figure 3.
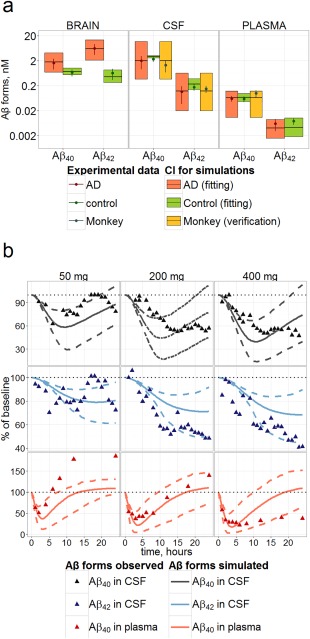
Verification of the model on the human and monkey data. (a) Steady‐state concentrations of Aβ40 and Aβ42 in brain, CSF, and PL for healthy (green) and AD (red) humans (used for fitting) and monkey (yellow, not used for fitting) predicted by the model (95% CI). Prediction for monkey was obtained by allometric scaling from human model. Experimental data (points with SE) were taken from Refs. 27, 28, 29, 30, 31,45–59. (b) Verification of the model against avagacestat data. Time dependence of Aβ40, Aβ42 in CSF and Aβ40 in plasma resulted from single administration of 50, 200 and 400 mg of avagacestat to healthy subjects. Aβ40 is expressed as % of steady state base level (placebo adjusted). Dots correspond to measured data taken from Refs. 8,11,17,50; lines denote confidence bands and median calculated by the model and Hessian for human‐fitted parameters.
Different variants of BW‐independent scaling parameters of synthesis, enzymatic, and transport reactions were tested. Satisfactory results were obtained by fitting 11 parameters for interspecies scaling (Table S2 of Supplement) and four parameters for scaling from healthy to AD state as specified in “Methods” (Table 2). Five parameters (Table 2) describe scaling from mouse to human and thus illustrate the magnitude of difference in Aβ synthesis between species.
Table 2.
Selected parameters describing differences between mouse and healthy and AD individuals
| Parameter | Description | Factor for healthy humans (95% CI) |
Factor for AD humans (95% CI) |
|
|---|---|---|---|---|
|
|
Scaling factor for gamma secretase Vmax in BC | 1.01 (0.34–2.86) | 32a | |
|
|
Scaling factor for gamma secretase Vmax in BIF | 8850 (4531–17278) | 8850 (4531–17278)c | |
|
|
Scaling factor for rate of Aβ precursor release in BC | 0.534 (0.28–1.01) | 5.18 (1.27–21.02) | |
|
|
Scaling factor for rate of Aβ precursor release in BIF | 4.55 (3.34–6.21) | 5.18 (1.27–21.02)b | |
|
|
Scaling factor for proportion of Aβ42/Aβ40 synthesis in BC | 13000 (2300–84269) | 15 (3.65–63.67) | |
|
|
Dummy polymerization rate constant of Aβ40 | 0 | 0.87 (0.01–56.08) | |
|
|
Dummy polymerization rate constant of Aβ42 | 0 | 1. (0.02–57.2) |
Example of equation for calculation of rate for production in BC in human is below: , where function switches the model from mouse ( to healthy ( ) and AD ( ) human. is analogous to but is calculated to scale from mouse to AD individuals. Complete list of functions is given in Supplement A, all scaling factors are given in the Table S2.
Parameter was not identifiable, thus it was fixed during hessian calculation.
Same as for BC (was not fitted separately).
Same as for healthy (not fitted).
After translation to primates, the model was externally verified on monkey data: body weight based allometric scaling alone was sufficient to describe the data (Figure 3 a, Supplemental Figure B11).
Description of human GSI data
GSI treatment prediction performance was verified comparing the clinical avagacestat single dose data for healthy individuals (Figure 3 b) with simulations of treatment at an IC50 value measured in vitro, and measured PBPK parameters (Table S2). Data for Aβ PD during GSI treatment often demonstrate strong fluctuations, which may be explained partially by diurnal Aβ oscillations, which were not accounted for in the model. However, the amplitude of inhibition (minimum of PD curve) falls within the 95% prediction band for each of measured quantities, except for Aβ42 inhibition, and exceeds the accuracy of prediction achieved in the work of Niva et al.17 We conclude that this model satisfactorily predicts inhibition.
Insights from simulations and implications for drug discovery and development
CSF and brain Aβ40 inhibition in human vs. mouse
We simulated the Aβ response to single‐dose avagacestat administration in the mouse and human (analogous to those presented in Figures 2 b, 3 b) to study the dependence of PD characteristics on dose and area under the curve (AUC). Model predictions were compared (Figure 4) to data used for fitting (mouse Aβ40 data) and external verification (human avagacestat PD data, mouse Aβ42 PD data).
Figure 4.
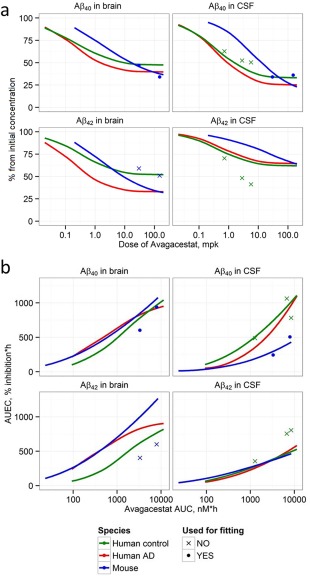
Comparison of avagacestat dose‐effect (a) and exposure‐response (b) relationships for mouse and humans (healthy and AD individuals). (a) Dose dependence of amplitude of Aβ40 and Aβ42 decrease resulting from single dose administration of avagacestat (expressed as % of steady state base level) in mouse (green line) healthy human (blue line), and AD human in brain (red line) and CSF; (b) dependence of Aβ40 and Aβ40 AUEC (area under effect curve) from avagacestat AUC (area under curve for concentration). Symbols correspond to measured data: circles correspond to data used for fitting,10 crosses correspond to data used for validation of human CSF predictions,8, 11, 17, 50 and mouse brain Aβ42. 10
The model adequately characterized the dose dependence for the “amplitude of Aβ40 inhibition” in CSF and brain (Figure 4 a), suggesting that 1) maximal inhibition (Imax) of avagacestat for brain Aβ40 is about 50% for healthy subjects and 60% for AD subjects; 2) avagacestat dosages, leading to the same magnitude of CSF Aβ40 decrease in mouse and human, are different; 3) although doses allowing to reach half‐maximal inhibition at the peak (ID50) for humans and mouse are substantially different, the model satisfactorily predicts the magnitude of human CSF Aβ40 inhibition in points not used for fitting; and 4) brain inhibition is lower than CSF for high dosages for all species.
Extrapolation from Aβ40 to Aβ42 pharmacodynamics
The Aβ42 PD data were not used for calibration of the model. The model simulations of the Aβ42 compartments tend to underestimate CSF Aβ42 inhibition (human data, Figure 4 a) and overestimate brain inhibition (mouse data). It appears that Aβ42 was not predicted satisfactorily based on Aβ40 data fitting even for the same species (mouse).
Exploration of exposure‐effect relationships
The resulting drug exposures corresponding to the observed single‐dose PD effects were similar for all species for brain Aβ40 inhibition (Figure 4 b). The differences in ID50 (Figure 4 a) between mouse and human are driven in part by species‐related PK properties. The large difference in CSF Aβ40 responses, as captured by area under the effect curves (AUEC) (Figure 4 b), may result from the different contributions of brain production to CSF concentration among species (Supplemental Figure C1). The brain Aβ pool is completely determined by brain production, while CSF and plasma Aβ may depend on production in other tissues (see details in Supplement C).
Efficacy of GSI on Aβ levels in the AD population compared with healthy individuals
To understand what potential therapeutic effects could be achieved on neuronal function and survival, we simulated inhibition for a range of dosages and compared the results with in vitro literature data describing the influence of Aβ on neuronal function. In vitro experiments25 suggest the enhancement of LTP (long‐term potentiation) in the presence of Aβ42 with a maximal effect around 200 pM and decrease of this effect for concentrations below 20 pM. These values correspond well with observed physiological levels of BIF Aβ42. We assumed that the Aβ42 concentration optimum for neuronal function in vivo would be similar as derived from in vitro experiments25, 26 and should not decrease below ∼10 pM. Significant cytotoxicity was observed at levels of intracellular soluble Aβ42 exceeding 1 nM.26 This conforms to the fact that the steady‐state brain concentrations of Aβ42 in healthy humans and mice are below 1 nM, while for AD subjects Aβ42 exceeds 1 nM (Figures 2 a, 3 a). We have specified in our simulations that 1 nM in brain cells would be the reference level for toxicity. Semagacestat and avagacestat demonstrate similar dose dependence for BIF Aβ inhibition in AD subjects during 3 days (Figure 5 a). A significant difference between drug efficacy (semagacestat vs. avagacestat) is observed for BC inhibition (Figure 5 a, bottom), with the central tendency of returning BC Aβ42 concentrations to physiological values. The 95% prediction confidence band for the brain Aβ42 inhibition by semagacestat reaches the region of safe concentration only at the highest doses simulated (Figure 5 a). For avagacestat, the median reaches 1 nM corresponding to ∼80% inhibition of Aβ42 concentration from about 8 nM at steady state level in brain for AD subjects (Figure 5 b).
Figure 5.
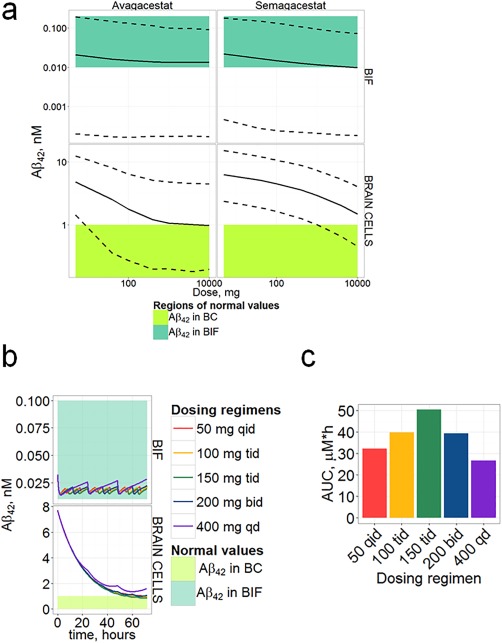
Simulations of Aβ42 (given in nM) maximal inhibition during multiple dosing (3 days) in AD subjects. (a) Comparison of predicted confidence bands (obtained by 4,200 replicates from log‐normal distribution of parameters using Hessian matrix) for BC and BIF Aβ42 minimal concentrations during 3 days of GSI administration once daily with levels supposed to be safe (or normal). Solid and dashed lines, confidence bands calculated by the model; colored regions, regions of physiologically safe values. Calculations were made for doses from 5 mg to 10,000 mg. (b) Simulation of Aβ42 inhibition dynamics for 3 days of different dosing regimens of avagacestat: comparison of Aβ42 in BC (upper panel) and BIF (lower panel) with normal values; q.d., once a day; b.i.d., twice a day; t.i.d., three times a day; q.i.d., four times a day. (c) AUC for different avagacestat dosing regimens.
Analysis of different GSI therapeutic regimens
Brain AUEC is more sensitive to plasma AUC than CSF AUEC at moderate doses (Figure 4) according to model predictions. This is explained through the model given the different dynamics of inhibition: Brain Aβ inhibition follows Aβ inhibition in CSF with some delay (Supplemental Figure C2), and both of them are delayed with respect to maximal drug concentration and maximal plasma inhibition. An optimized dosing regimen could lead to a potential therapeutic benefit due to higher AUC. We simulated 3 days of avagacestat treatment (analogous to previous section) with different dosing regimens. All simulated regimens with multiple daily dosing, even with a lower daily dose, provide better brain pharmacodynamics than the single dosing regimen (Figure 5 b). The forecasted brain cell Aβ concentrations fell below 1 nM on the third day of simulated therapy. Moreover, each regimen provided enough safety, as the minimal BIF concentration did not fall below the normal range of values. A q.d. regimen has the lowest AUC (Figure 5 c) even when compared with lower total daily dosing regimens. Higher AUC lead to higher maximal BC amyloid inhibition, but not maximal BIF inhibition.
DISCUSSION
The purpose of this study was 1) to apply the model to evaluate contributions of different sources of Aβ (synthesis in brain and other tissues) to its level in brain cells, brain interstitial fluid, CSF, and plasma; 2) to explore the translation of GSI mechanistic dynamics across mouse, monkey, and human species; and 3) to identify GSI administration regimens that would return Aβ to normal human (non‐AD) levels.
The developed model satisfactorily describes the kinetics of Aβ distribution and steady‐state levels in mouse, monkey, and human (healthy subjects and AD patients).
Model calibration efforts confirmed that conventional allometric scaling, of reaction rates, was not sufficient to translate the model from mouse to humans, reflecting that significant differences exist between these species that may not be explained solely by body weight. They relate not only to the production of Aβ, but also to its degradation and transport. Dissimilarity of these processes between species was found in three parameters other than for Aβ production. Bulk flow from BIF to CSF in humans differs by an order of magnitude from the value calculated by allometric scaling, reflecting possible involvement of other mechanisms in brain Aβ fluxes. Plasma‐CSF Aβ exchange is greater in humans according to the model, suggesting possible differences in BCSFB architecture across species. In contrast to mouse‐human translation, body weight‐based allometric scaling is sufficient for the translation between human and monkey, therefore the monkey may be a better preclinical in vivo model for biomarker translation.
The Aβ42/Aβ40 ratio is higher in human brain than in plasma and CSF (see Figure 3 with illustration of data from different references27, 28, 29, 30, 31). Moreover, the CSF Aβ40 level is much higher in humans than in mouse, while brain concentrations are similar. Plasma Aβ40 concentrations in human and mouse are similar (about 0.05 nM, Figures 2 a, 3 a), thus the higher Aβ40 level in human CSF reflects higher brain production. Higher Aβ42 level in human brain should reflect reduction of the portion of Aβ40 produced in brain cells. A balance between increase of Aβ42/Aβ40 in brain and high CSF Aβ40 level leads to a higher parameter value of (Table 2). The possibility that the higher level of CSF Aβ in humans is due to lower degradation seems unlikely, as the model correctly describes Aβ clearance after GSI administration. Many minor differences may exist between mouse and humans, but we have chosen only a few parameters identifiable given the dataset. According to the same reasoning, the AD state may be a result of slight changes in an extended set of processes, but here it was described by changing production and aggregation addition only (Table 2). This is in line with an increase of β‐secretase activity32 and contribution of aggregation into equilibrium between soluble and insoluble forms in BIF. As there is no dynamic data for GSI in AD, it is not clear whether an increased BC concentration in AD is due to increased production or failure of intracellular transport or degradation, so calibration only on baseline data allows for determining only some effective parameters. This is an important limitation, which should further be eliminated by extension of calibration on new PD data.33
GSI treatment maximal effect is reproduced by the model, but some dynamic properties were not accurately described. The overshoot for human Aβ plasma concentration is underestimated for semagacestat (Supplement Figure B9) and low doses of avagacestat (Figure 3 b) and overestimated for higher avagacestat doses (Figure 3 b). Model predictions for CSF Aβ show a similar pattern but do not completely follow the data.
Problems with the PD description are observed in the mouse also (Figure 2 b): both brain and CSF curves predicted by the model lag behind the measured points during the decline phase. It can be assumed that the description of distribution between BIF and brain cells is simplified: exchange between these compartments is carried out by different mechanisms, including endocytosis and exocytosis,34, 35 while there is one hypothetical carrier in our model responsible for transport. Another possible explanation is that drug IC50 values measured in vitro may not reflect the physiological situation.
Comparison of the inhibition amplitudes in different species (Figure 4) leads us to the conclusion that CSF Aβ40 has approximately the same biomarker capacity for humans and mouse, slightly overestimating brain inhibition (for AD subjects CSF 75% Aβ40 inhibition corresponds to ∼60% brain Aβ40 inhibition for avagacestat dose of 10 mpk). Healthy subject CSF data underestimate the potential CSF inhibition level for AD subjects. Avagacestat doses of ∼2 mpk would lead to about 55% inhibition of Aβ40 in CSF of healthy controls, while about 65% inhibition in CSF and 50% inhibition in brain are predicted for AD subjects. Clinically tested dosages (below 150 mg avagacestat for AD36) do not allow achieving a normal concentration (Figure 5 a), but may lead to a BIF Aβ concentration decrease below the physiological level.
Aβ42 pharmacodynamics for one GSI (e.g., avagacestat) cannot be predicted based on the PD data for Aβ40 or Aβ42 from another GSI (e.g., semagacestat) directly (Figure 3) even for the same species (mouse or human). In our model GSI acts on the total secretase rate, and so Aβ pharmacodynamics is determined by such system properties as proportions of Aβ40/42 synthesis in different compartments and clearance (distribution). To describe higher inhibition of Aβ42 we should suppose a specific mechanism leading to changes in the proportion of Aβ40/42 production: drug interaction with presenilin37 or detailed analysis of drug action in different intracellular compartments. Analysis of AUEC and AUC (Figure 4 b) have shown that even the exposure–response relationship difference between species will not be explained by PK properties only and, moreover, the difference in the brain Aβ42 inhibition between AD and healthy individuals should be expected.
A difference in the exposure–response relationship between brain and CSF (Figure 4 , Supplement Figure C2) is expected to be due to the different Aβ half‐lives in brain and plasma and different contributions of plasma amyloid to brain and CSF (Supplement Figure C1). The overshoots of CSF Aβ concentration, predicted for all species, may originate simply from γ‐secretase substrate accumulation in our model, as we do not consider more complex enzymology.38 It is similar in general to the mechanism proposed previously,39 assuming overwhelmed α‐secretase processing by C99 and increased APP pool. Slight overshoot in the brain is observed only for healthy subjects probably because of the different relationship between synthesis and degradation of Aβ (Table 2 , Table S2).
Soluble nonfibrillar Aβ has been demonstrated to be more toxic to neurons than the aggregated form.26, 40, 41 The model presented here allows for direct comparison of the concentrations of soluble amyloid species, forecast from the model, and the proposed toxicity thresholds as defined by in vitro studies in such inaccessible compartments as BIF and even brain cells. It could facilitate understanding the reasons for the failure of many GSI clinical trials. PD simulations for a long time require: 1) more accurate PK description, as differences between PK on days 1 and 7 have been shown11; 2) description of amyloid aggregation and accumulation processes; 3) disease progression description. As all of these considerations are out of the scope of this analysis, we simulated PD for only 3 days of treatment and assumed that it will give a rough estimate of the results of trials, which can later be compared with the results of an extended model. Achieving brain Aβ levels corresponding to normal concentrations (Figure 5 a) requires very high avagacestat dosages conjugated with a risk of decrease below an optimal BIF level of Aβ. This effect, if it exists in vivo, would be independent of the mechanism of production inhibition (BACE or GS inhibition), as it is related to Aβ level decrease, but not to other pathways, e.g., Notch signaling inhibition, observed for semagacestat,42 or substrate accumulation.43 Saturation of effect at avagacetat dosages higher than 5,000 mg may be due to a decrease in bioavailability for higher dosages and absorption saturation assumed in the model (Supplement B.1). Differences between brain efficacy of semagacestat vs. avagacestat are due to the distinct plasma–brain penetration coefficients (0.05 for semagacestat vs. 4.35 for avagacestat10) and different PK profiles: long decay (Figure 2 (b), Supplement) together with high brain penetration and low IC50 of avagacestat allows retaining substantial brain inhibition for a much longer period of time. Our simulations have shown the importance of higher AUC for brain inhibition, but not for BIF inhibition (Figures 4 , 5), suggesting that PK properties are significant for brain PD.
The reason for the different dynamics in BIF and BC may be the exchange between BC and BIF: in the model, the equilibrium constant between rates of uptake and release was fitted to 10, in line with observed extensive uptake,44 which leads to faster depletion of BIF Aβ. Both brain and BIF concentrations depend mainly on brain production, and thus, the only way to provide a positive difference in BC and BIF PD, besides the specific dosing regimens, would be the development of a compound with a different intra‐ and extracellular IC50 or a drug influencing intra‐ and extraneuronal amyloid transport. Compounds should have a lower clearance rate to be able to retain high drug plasma concentrations for a longer period of time.
CONCLUSION
The proposed model can be considered a framework for exploration of the amyloid system, translating between species, hypothesis generation, and understanding therapeutic options targeting this system. Key problems in construction of this model are the choice of datasets that are optimal for model fitting and external verification, and handling multiple datasets and analyzing accuracy of predictions.
In the accompanying article, we extend our Aβ distribution model in such way as to take into account Aβ40 and Aβ42 aggregation with longitudinal time effects to describe AD progression over the decades.
Supporting information
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Acknowledgments
Current affiliation for Y Lu: Novartis Oncology Pharmacometrics, USA. The authors thank the ISB team for technical and theoretical support, Kirill Zhudenkov, Eugenia Kazimirova for technical support of calculations, and Veronica Voronova for graphical support of the Supplement.
Conflict of Interest
T.K. and O.D. are external consultants who were paid to do this research; H.B., T.N., and S.D. are employees of Pfizer; Y.L. was an employee of Pfizer.
Author Contributions
T.K., O.D., H.B, S.D., Y.L., and T.N. wrote the article; T.K., O.D., H.B., S.D., Y.L., and T.N. designed the research; T.K. and O.D. performed the research; T.K. and O.D. analyzed the data.
References
- 1. Jonsson, T. et al A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature 488, 96–99 (2012). [DOI] [PubMed] [Google Scholar]
- 2. Ittner, L.M. & Götz, J. Amyloid‐β and tau—a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72 (2011). [DOI] [PubMed] [Google Scholar]
- 3. Murray, I.V.J. , Proza, J.F. , Sohrabji, F. & Lawler, J.M. Vascular and metabolic dysfunction in Alzheimer's disease: a review. Exp. Biol. Med. (Maywood). 236, 772–782 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Salloway, S. et al Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N. Engl. J. Med. 370, 322–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frykman, S. et al Synaptic and endosomal localization of active gamma‐secretase in rat brain. PLoS One 5, e8948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beer, J. , Masters, C.L. & Beyreuther, K. Cells from peripheral tissues that exhibit high APP expression are characterized by their high membrane fusion activity. Neurodegeneration 4, 51–59 (1995). [DOI] [PubMed] [Google Scholar]
- 7. Hébert, S.S. et al Coordinated and widespread expression of gamma‐secretase in vivo: evidence for size and molecular heterogeneity. Neurobiol. Dis. 17, 260–272 (2004). [DOI] [PubMed] [Google Scholar]
- 8. Tong, G. et al Multicenter, randomized, double‐blind, placebo‐controlled, single‐ascending dose study of the oral γ‐secretase inhibitor BMS‐708163 (Avagacestat): tolerability profile, pharmacokinetic parameters, and pharmacodynamic markers. Clin. Ther. 34, 654–667 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Bateman, R.J. et al A gamma‐secretase inhibitor decreases amyloid‐beta production in the central nervous system. Ann. Neurol. 66, 48–54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu, Y. et al Quantitative pharmacokinetic/pharmacodynamic analyses suggest that the 129/SVE mouse is a suitable preclinical pharmacology model for identifying small‐molecule γ‐secretase inhibitors. J. Pharmacol . Exp. Ther. 339, 922–934 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Dockens, R. et al A placebo‐controlled, multiple ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of avagacestat (BMS‐708163) in healthy young and elderly subjects. Clin. Pharmacokinet. 51, 681–693 (2012). [DOI] [PubMed] [Google Scholar]
- 12. Shibata, M. et al Clearance of Alzheimer's amyloid‐ss(1–40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J. Clin. Investig. 106, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghiso, J. Systemic Catabolism of Alzheimer's A 40 and A 42. J. Biol. Chem. 279, 45897–45908 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Cook, J.J. et al Acute β‐secretase inhibition of nonhuman primate CNS shifts amyloid precursor protein (APP) metabolism from amyloid—production to alternative APP fragments without amyloid‐rebound. J. Neurosci. 30, 6743–6750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bateman, R.J. et al Human amyloid‐beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu, Y. et al Cerebrospinal fluid β‐amyloid turnover in the mouse, dog, monkey and human evaluated by systematic quantitative analyses. Neurodegen. Dis. 12, 36–50 (2013). [DOI] [PubMed] [Google Scholar]
- 17. Niva, C. et al Has inhibition of Aβ production adequately been tested as therapeutic approach in mild AD? A model‐based meta‐analysis of γ‐secretase inhibitor data. Eur. J. Clin. Pharmacol. 69, 1247–1260 (2013). [DOI] [PubMed] [Google Scholar]
- 18. Craft, D.L. , Wein, L.M. & Selkoe, D.J. A mathematical model of the impact of novel treatments on the A beta burden in the Alzheimer's brain, CSF and plasma. Bull. Math. Biol. 64, 1011–1031 (2002). [DOI] [PubMed] [Google Scholar]
- 19. Elbert, D.L. , Patterson, B.W. & Bateman, R.J. Analysis of a compartmental model of amyloid beta production, irreversible loss and exchange in humans. Math. Biosci. 261, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li, R. et al Amyloid beta peptide load is correlated with increased beta‐secretase activity in sporadic Alzheimer's disease patients. Proc . Natl. Acad. Sci. U. S. A. 101, 3632–3637 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wahlster, L. et al Presenilin‐1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer's disease. Acta Neuropathol. 125, 187–199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hooke, R. & Jeeves, T.A. “Direct search” solution of numerical and statistical problems. J. ACM 8, 212–229 (1961). [Google Scholar]
- 23. Gizzatkulov, N.M. et al DBSolve Optimum: a software package for kinetic modeling which allows dynamic visualization of simulation results. BMC Syst. Biol. 4, 109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bateman, R.J. et al A γ‐secretase inhibitor decreases amyloid‐β production in the central nervous system. Ann. Neurol. 66, 48–54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Puzzo, D. et al Picomolar amyloid positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang, Y. , McLaughlin, R. , Goodyer, C. & LeBlanc, A. Selective cytotoxicity of intracellular amyloid beta peptide1–42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 156, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jensen, M. , Schro, J. , Blomberg, M. & Engvall, B. Cerebrospinal fluid A beta42 is increased early in sporadic Alzheimer's disease and declines with disease progression. Ann. Neurol. 45, 504–511 (1999). [DOI] [PubMed] [Google Scholar]
- 28. Sjögren, M. et al Tau and Aβ42 in cerebrospinal fluid from healthy adults 21–93 years of age: establishment of reference values. Clin. Chem. 47, 1776–1781 (2001). [PubMed] [Google Scholar]
- 29. Xia, W. et al A specific enzyme‐linked immunosorbent assay for measuring beta‐amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 66, 190–199 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Konno, T. et al Coordinated increase of γ‐secretase reaction products in the plasma of some female Japanese sporadic Alzheimer's disease patients: quantitative analysis of p3‐Alcα with a new ELISA system. Mol. Neurodegener. 6, 76 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rembach, A. et al Changes in plasma amyloid beta in a longitudinal study of aging and Alzheimer's disease. Alzheimers Dement. 10, 53–61 (2014). [DOI] [PubMed] [Google Scholar]
- 32. Holler, C.J. et al BACE2 expression increases in human neurodegenerative disease. Am. J. Pathol. 180, 337–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kennedy, M.E. et al The BACE1 inhibitor verubecestat (MK‐8931) reduces CNS β‐amyloid in animal models and in Alzheimer's disease patients. Sci. Transl. Med. 8, 363ra150 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Cirrito, J.R. et al Synaptic activity regulates interstitial fluid amyloid‐β levels in vivo. Neuron 48, 913–922 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Lai, A.Y. & McLaurin, J. Mechanisms of amyloid‐beta peptide uptake by neurons: the role of lipid rafts and lipid raft‐associated proteins. Int. J. Alzheimers Dis. 2011, 1–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coric, V. et al Safety and tolerability of the γ‐secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 69, 1430 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Crump, C.J. et al BMS‐708,163 targets presenilin and lacks notch‐sparing activity. Biochemistry 51, 7209–7211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Svedruzic, Z.M. et al Modulators of γ‐secretase activity can facilitate the toxic side‐effects and pathogenesis of Alzheimer's disease. PLoS One 8, e50759 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ortega, F. , Stott, J. , Visser, S.A.G. & Bendtsen, C. Interplay between α‐, β‐, and γ‐secretases determines biphasic amyloid‐β protein level in the presence of a γ‐secretase inhibitor. J. Biol. Chem. 288, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dahlgren, K.N. Oligomeric and fibrillar species of amyloid‐beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053 (2002). [DOI] [PubMed] [Google Scholar]
- 41. Lambert, M.P. et al Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc . Natl. Acad. Sci. U. S. A. 95, 6448–6453 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Henley, D.B. , May, P.C. , Dean, R.A. & Siemers, E.R. Development of semagacestat (LY450139), a functional gamma‐secretase inhibitor, for the treatment of Alzheimer's disease. Expert Opin. Pharmacother. 10, 1657–1664 (2009). [DOI] [PubMed] [Google Scholar]
- 43. Bittner, T. et al Gamma‐secretase inhibition reduces spine density in vivo via an amyloid precursor protein‐dependent pathway. J. Neurosci. 29, 10405–10409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burdick, D. , Kosmoski, J. , Knauer, M.F. & Glabe, C.G. Preferential adsorption, internalization and resistance to degradation of the major isoform of the Alzheimer's amyloid peptide, Aβ1–42, in differentiated PC12 cells. Brain Res. 746, 275–284 (1997). [DOI] [PubMed] [Google Scholar]
- 45. Karelina, T. Amyloid beta distribution: detailed kinetic model for mouse scaled to monkey and human. Clin. Pharmacol. Ther. 93, S4–S4 (2013). [Google Scholar]
- 46. Wang, J. , Dickson, D.W. , Trojanowski, J.Q. & Lee, V.M. The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 158, 328–337 (1999). [DOI] [PubMed] [Google Scholar]
- 47. Shoji, M. et al The levels of cerebrospinal fluid Aβ40 and Aβ42(43) are regulated age‐dependently. Neurobiol. Aging 22, 209–215 (2001). [DOI] [PubMed] [Google Scholar]
- 48. Fukumoto, H. et al Age but not diagnosis is the main predictor of plasma amyloid β‐protein levels. Arch. Neurol. 60, 958 (2003). [DOI] [PubMed] [Google Scholar]
- 49. Head, E. et al Plasma amyloid‐β as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. J. Alzheimers Dis. 23, 399–409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tong, G. et al Effects of single doses of avagacestat (BMS‐708163) on cerebrospinal fluid Aβ levels in healthy young men. Clin. Drug Investig. 32, 761–769 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.