

Summary
Acinetobacter oleivorans DR1 can utilize C12–C30 alkanes as a sole carbon source but not short‐chain alkanes (C6, C10). Two copies of each alkB‐, almA‐ and ladA‐type alkane hydroxylase (AH) are present in the genome of DR1 cells. Expression and mutational analyses of AHs showed that alkB1 and alkB2 are the major AH‐encoding genes under C12–C30, and the roles of other almA‐ and ladA genes are negligible. Our data suggested that AlkB1 is responsible for long‐chain alkane utilization (C24–C26), and AlkB2 is important for medium‐chain alkane (C12–C16) metabolism. Phylogenetic analyses revealed large incongruities between phylogenies of 16S rRNA and each AH gene, which implies that A. oleivorans DR1 has acquired multiple alkane hydroxylases through horizontal gene transfer. Transcriptomic and qRT‐PCR analyses suggested that genes participating in the synthesis of siderophore, trehalose and poly 3‐hydroxybutyrate (PHB) were expressed at much higher levels when cells used C30 than when used succinate as a carbon source. The following biochemical assays supported our gene expression analyses: (i) quantification of siderophore, (ii) measurement of trehalose and (iii) observation of PHB storage. Interestingly, highly induced both ackA gene encoding an acetate kinase A and pta gene encoding a phosphotransacetylase suggested unusual ATP synthesis during C30 alkane degradation, which was demonstrated by ATP measurement using the ΔackA mutant. Impaired growth of the ΔaceA mutant indicated that the glyoxylate shunt pathway is important when C30 alkane is utilized. Our data provide insight into long‐chain alkane degradation in soil microorganisms.
Introduction
A large number of hydrocarbonoclastic‐ and alkane‐degrading bacteria are widely distributed in nature (Liu et al., 2015). Their degrading mechanisms have been investigated owing to their ecological importance and the versatile applications of alkane‐degrading enzymes with economic benefits (Rojo, 2010). Four different pathways for aerobic alkane oxidation have been reported: terminal oxidation, subterminal oxidation, biterminal oxidation and the Finnerty pathway (Ji et al., 2013). The best‐characterized strategy for aerobic alkane degradation is terminal oxidation wherein through a series of oxidation steps, the alkane is transformed into alcohol, aldehyde and, finally, into fatty acid (Van Beilen et al., 2003). The first reaction mediated by alkane hydroxylase is known to be the rate‐limiting step, which converts alkane to fatty alcohol. Diverse alkane hydroxylases, from many different bacteria, have been characterized, revealing that they have different ranges of carbon‐chain length preferences with respect to alkane substrates (Van Beilen and Funhoff, 2007). Short‐chain alkanes (C1–C4) can be oxidized by soluble methane monooxygenase, particulate methane monooxygenase, propane monooxygenase and butane monooxygenase while integral‐membrane non‐haem di‐iron monooxygenase (AlkB or AlkM) and alkane hydroxylating cytochrome P450 appear to mediate the oxidation of short‐ and medium‐chain alkanes (C5–C16). In addition, the recently discovered novel monooxygenases, such as AlmA in Acinetobacter sp. DSM 17978 and LadA in Geobacillus thermodenitrificans NG80‐2, have gained attention because they can oxidize very long‐chain alkanes (> C20), indicating that these enzymes could potentially be used for bioremediation because of their unusual substrate ranges (Feng et al., 2007; Throne‐Holst et al., 2007).
Examination of novel alkane dioxygenase in Acinetobacter sp. M‐1 has revealed activity on the C10–C30 alkanes through postulated Finnerty pathway (Maeng et al., 1996). Genomic data do not reveal the substrate ranges of each alkane hydroxylase, and the regulation of gene expression involved in alkane degradation is not well understood in many known alkane degraders. Expression analysis of the alkane hydroxylase genes provides insight into how bacteria can utilize alkanes and which enzymes should be investigated for biotechnological applications. Expression of the alkM gene in Acinetobacter sp. ADP1 is variable depending on its carbon source, growth phase and inducer molecules, although rubredoxin and rubredoxin reductase, which are the components of the full alkane hydroxylase complex, are constitutively expressed (Ratajczak et al., 1998). In addition, Alcanivorax dieselolei B‐5, which contains at least four alkane hydroxylases, degrades a wide range of alkanes (C5–C36); these multiple alkane hydroxylases are coexpressed and were shown to degrade a broad range of alkanes with different chain length alkanes (Liu et al., 2011). Acinetobacter species have been extensively studied because of their versatile ability to degrade diverse hydrocarbons and their potential biotechnological applications (Jung and Park, 2015). It has been determined that many Acinetobacter strains have the ability to utilize long‐chain alkanes (> C20) as a carbon source (Wentzel et al., 2007). In addition, Acinetobacter calcoaceticus uses hydrophobic fimbriae, which assists in attachment to the hydrophobic surface of substrates (Rosenberg et al., 1982), and Acinetobacter venetianus RAG‐1 produces a kind of polysaccharide biosurfactants, known as an emulsan, which facilitates incorporation of alkanes into bacterial cells (Bach et al., 2003). However, the mechanisms of alkane transport and the enzymes involved in alkane degradation in other Acinetobacter species, including Acinetobacter oleivorans DR1, are not well understood (Kang and Park, 2009).
In this study, we determined the range of alkanes that strain DR1 can use as a carbon source. Expression patterns of putative alkane monooxygenases on a medium‐chain alkane (C12) to a long‐chain alkane (C30) were also monitored using quantitative reverse transcription polymerase chain reaction (qRT‐PCR) and Northern blot. In addition, the role of each gene was confirmed by gene deletion analysis. Phylogenetic analysis demonstrated that strain DR1 possesses six putative alkane monooxygenases. Finally, RNA‐seq analysis of strain DR1 in the presence of triacontane (C30) was conducted. Our analyses also provide considerable insight into metabolic and stress responses during bacterial long‐chain alkane metabolism.
Results
Substrate ranges of alkane degradation in A. oleivorans DR1
The DR1 cells are known to utilize diesel and hexadecane as carbon sources (Jung et al., 2011a; Kang et al., 2011); however, the range of degradable aliphatic alkanes is not known and the corresponding alkane monooxygenases have not been characterized. Growth tests on various substrates indicated that DR1 cells could utilize both medium‐ and long‐chain alkanes (C12–C30) but not short‐chain alkanes (C6–C10) (Fig. 1A, Figs S1A and S1B). Because of the extremely low water solubility of tested alkanes (less than 0.01 mg L−1 at ambient temperature), the maximum growth rate could be monitored through simple growth measurements using each alkane at 0.1% concentration. The highest growth rate was observed with hexadecane (C16, μmax = 1.38 h−1) followed by hexacosane (C24, μmax = 1.37 h−1), tetracosane (C26, μmax = 1.37 h−1) and tetradecane (C14, μmax = 1.33 h−1). The lowest growth rates were determined with dodecane (C12, μmax = 1.23 h−1) and triacontane (C30, μmax = 1.25 h−1), which had the shortest and longest carbon‐chain lengths among tested alkanes respectively (Fig. 1B). Although no growth was detected in case of hexane, its corresponding oxidized products (hexanol and hexanoic acid) could be utilized as carbon sources (Fig. S1A). In contrast, growth occurred only with decanoic acid as carbon source but not with decanol (Fig. S1B). In addition, further examination of decanol by survival and agar diffusion tests showed its high toxicity to DR1 strains. Only 0.1% of total cells survived 15‐min postdecanol treatment (Fig. S1D). Agar diffusion tests with decane and decanol confirmed the results of the survival test. A clear zone was not formed even when 100% decane was used; however, inhibition occurred following treatment with 50% and 100% decanol, showing 2.3‐ and 2.5‐cm clear zones respectively (Fig. S1E). A high concentration of decanol (> 50%) was required to suppress the growth of DR1 in the agar diffusion test, although only 0.1% decanol was sufficient to inhibit growth in the liquid survival test. This difference may be due to the limited solubility of decanol, which hindered its diffusion on the agar disc. Our data indicated that alkane monooxygenases in DR1 cells are specific for medium‐ and long‐chain alkane degradation.
Figure 1.
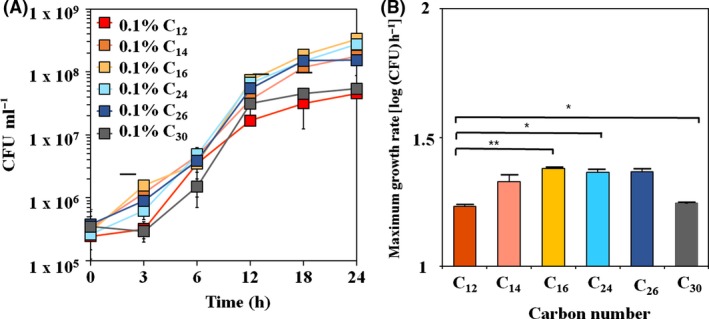
Determination of substrate range of the DR1 strain was carried out using growth curves on (A) medium‐chain length alkanes (C12–C16) to long‐chain length alkanes (C24–C30). (B) Comparison of maximum growth rate based on growth curves. Duplicate experiments were performed, and each dot indicates the average of the experiments. Error bars indicate standard deviation (SD). A t‐test was performed, and a P‐value less than 0.01 or 0.05 is marked by a double or single asterisk (**P < 0.01, *P < 0.05).
Expression analysis and knockout mutant studies of AlkB‐type alkane monooxygenases in the presence of triacontane
Sequence analysis has revealed the presence of homologs of alkane monooxygenases (alkB‐, almA‐ and ladA types) in strain DR1 (Jung et al., 2010). Based on amino acid alignments, two copies of three different types of alkane monooxygenases were identified (Table 1). Both alkB1 and alkB2 homologs possessed relatively high amino acid identities with alkMa (88%) and alkMb (93%) of Acinetobacter sp. M‐1, respectively. AlmA and LadA homologs have high identities with those of Acinetobacter sp. DSM 17874 and of Geobacillus denitrificans NG80‐2, respectively (Table 1). However, the DR1 genome does not seem to harbour any cytochrome P450‐type monooxygenase gene.
Table 1.
The result of alignment for alkane monooxygenase homologues between Acinetobacter oleivorans DR1 and other bacteria using blastp
| Gene seq identifier | Gene name | Protein/organism | Amino acid identity (%) | Length of amino acids | E‐value |
|---|---|---|---|---|---|
| AOLE_RS10590 | alkB1 | Alkane hydroxylase A (AlkMa)/Acinetobacter sp. M‐1 | 359/409 (88%) | 407 | 0 |
| AOLE_RS13400 | alkB2 | Alkane hydroxylase B (AlkMb)/Acinetobacter sp. M‐1 | 368/395 (93%) | 397 | 0 |
| AOLE_RS02255 | almA1 | Putative monooxygenase (AlmA)/Acinetobacter sp. DSM 17874 | 406/497 (82%) | 496 | 0 |
| AOLE_RS09555 | almA2 | 228/481 (47%) | 510 | 2e−159 | |
| AOLE_RS11290 | ladA1 | Monooxygenase (LadA)/Geobacillus thermodenitrificans NG80‐2 | 211/447 (47%) | 470 | 2e−154 |
| AOLE_RS11525 | ladA2 | 231/455 (51%) | 463 | 7e−168 |
Transcriptomic analysis of the strain DR1 cells grown on triacontane (hereinafter TRI) was conducted, and cells grown on 10 mm succinate (hereinafter SUC) were used as controls. The information obtained from the raw RNA‐seq profile is summarized in Table S1. All genes associated with the predicted alkane degradation pathway were upregulated. However, six alkane monooxygenase‐encoding genes were differentially expressed (Table S2). alkB1 and alkB2 were highly expressed (11.6‐fold and 7.2‐fold, respectively) with high RPKM values, indicating that AlkB‐type monooxygenases are important for TRI degradation in DR1 cells, although other AlmA‐ and LadA‐type enzymes are known to be involved in long‐chain alkane degradation in other bacterial species (Feng et al., 2007; Throne‐Holst et al., 2007). Induction of AlkB‐type enzymes for TRI degradation was also confirmed by qRT‐PCR and Northern blot analysis (Fig. 2). The alkB1 gene is more inducible in presence of C24–C26 alkanes, while alkB2 is highly expressed with C12–C16 (Fig. 2A and C). In addition, a growth defect of the alkB2 mutant grown on C12 and C16 alkanes (Fig. 3A and B) and that of the alkB1 mutant on the C24 alkane (Fig. 3C) indicated their significant roles in degrading medium‐ and long‐chain alkanes. Consistent with our RNA‐seq data, our qRT‐PCR and Northern blot data revealed that both alkB genes are induced with the C30 alkane (Fig. 2A and C). In addition, no growth was observed in the ΔalkB1 ΔalkB2 double‐knockout strain (Fig. 3D). Unexpected observation of this expression study implies that more complex mechanisms occur during C30 alkane degradation, unlike C24 and C26 alkane metabolism. Our mutational study also supports our observation showing that deletion of a single alkB gene does not affect the growth on the C30 alkane (Fig. 3D). This observation may be attributed to the compensation of each alkB gene deletion by the remaining alkB genes, which could be possible because of intrinsic slow growth and expression of both the alkB genes in the presence of TRI. Northern blot analyses were performed to reveal the level of alkB2 expression in the alkB1 mutant background under C30‐amended condition. Interestingly, the level of alkB2 gene expression in the ΔalkB1 mutant was greater than that in wild‐type cells (Fig. S2). However, our data also suggested that weak alkB1 transcription in the ΔalkB2 mutant might be sufficient for full growth of the mutant on TRI even in the absence of AlkB2 activity (Fig. 3D). Collectively, our data indicated that both alkB gene products play significant roles in the WT strain on TRI metabolism. Although our qRT‐PCR showed the induction of the almA genes during C24, C26 and C30 alkane metabolism, Northern blot analysis indicated a very low level of expression of both the almA genes (Fig. 2B and D). In addition, ladA genes appear to be not important for degradation of TRI because of their low level of expression (Table S2). Taken together, we observed that alkB‐type alkane hydroxylases are important for degradation of TRI, and the contribution of both almA and ladA types to C30 alkane degradation is negligible.
Figure 2.
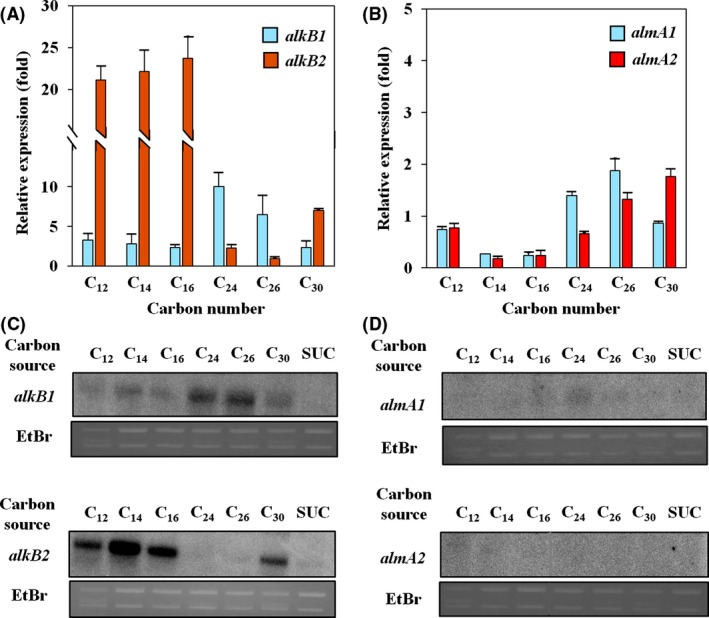
Relative expression levels of (A) alkB1 (cyan bar), alkB2 (red bar), (B) almA1 (cyan bar) and almA2 (red bar) along medium‐chain alkanes to long‐chain alkanes (C12, C14, C16, C24 and C30) were measured by qRT‐PCR. The fold changes are defined as the expressions of each gene with alkanes comparing to 10 mm succinate. The expression levels of each gene were normalized by 16S rDNA. Northern blotting was also performed to visualize the expression of (C) alkB and (D) almA. A consistent amount of total RNA was loaded, which is shown by the ethidium bromide‐stained gel (EtBr).
Figure 3.
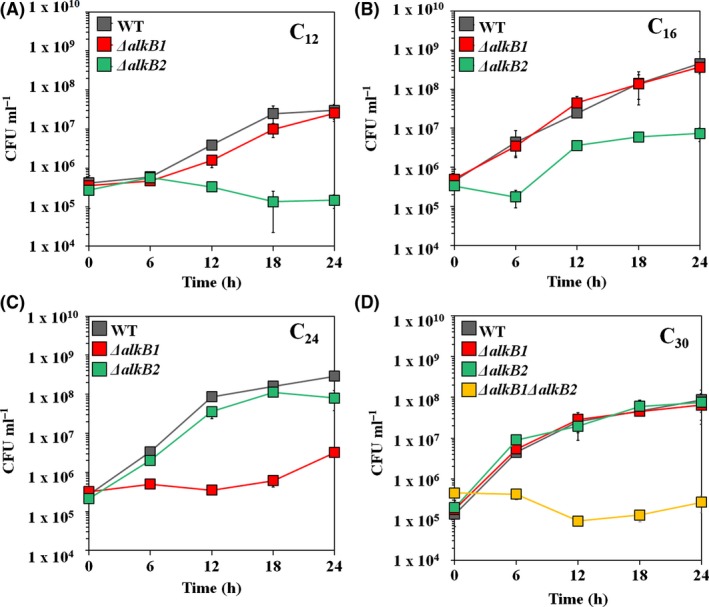
Comparative growth assay of wild type, alkB single‐ (ΔalkB1 and ΔalkB2) and double‐knockout mutants (ΔalkB1ΔalkB2) on (A) C12, (B) C16, (C) C24 and (D) C30 alkanes supplied with MSB media.
Phylogenetic analysis of alkane monooxygenases
Multiple copies of alkane monooxygenases are present in the DR1 strain possibly because of horizontal gene transfer and gene duplication events. Phylogenetic analysis showed that 13 of 19 γ‐proteobacteria species shared alkB‐type alkane monooxygenases (Fig. 4). In contrast, AlmA‐type and LadA‐type monooxygenases were present mostly in Actinobacteria (21 of 25 species) and Firmicutes (11 of 12 species). Although Geobacillus sp. MH‐1 belonging to Firmicute harbours only AlkB2 type alkane monooxygenase, LadA‐type alkane monooxygenase is more dominant in other Firmicutes, which possess only single or two copies of LadA. All three types of alkane monooxygenases found in the DR1 strain were found in only two other species, Nocardia jianxiensis (Actinobacteria) and Bacillus mycoides (Firmicutes). All examined Acinetobacter species (A. oleivorans DR1, A. baylyi and A. calcoaceticus) have six alkane monooxygenases, which indicates that the Acinetobacter species are specialists in degradation of alkane substrates. Results of distribution analysis of various alkane monooxygenase genes were found to be largely inconsistent with respect to results of the 16S rRNA gene phylogeny analysis; this also suggested that multiple copies of alkane monooxygenase genes occurred through horizontal gene transfer (HGT).
Figure 4.
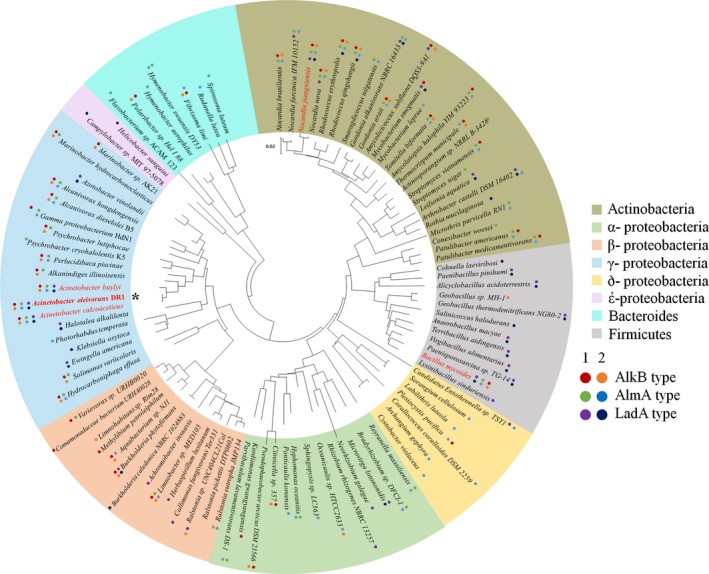
Neighbour‐joining phylogenetic tree of bacteria harbouring alkane monooxygenase homologs based on 16S rRNA. Each colour represents a phylum level of the bacterial community. The number on the right side indicates the type number of each alkane monooxygenase in the strain DR1. The scale bar represents the expected value of substitutions per point. Strain DR1 is highlighted and marked with an asterisk (*). Red letters indicate six alkane monooxygenase‐possessing bacteria.
Iron requirement during TRI metabolism
The ability to scavenge iron from the environment is an important characteristic of hydrocarbon‐degrading bacteria. Iron uptake is essential for synthesis of alkane monooxygenases because non‐haem di‐iron monooxygenase (AlkB) and cytochrome P450 monooxygenase superfamily proteins (AlmA and LadA) require iron. Genes related to siderophore biosynthesis and transportation in the DR1 strain were upregulated, proving that iron requirement is high when TRI is degraded (Table S2). In the genome of the DR1 strain, two clusters associated with siderophore synthesis are present: one cluster for acinetobactin synthesis (ent operon, homologous to enterobactin in Escherichia coli) and the other cluster for staphyloferrin synthesis (sbn operon). Interestingly, genes ranging from AOLE_RS07230 (sbnG) to AOLE_RS07255 (sbnA) showed over 38% amino acid sequence identity with the sbn operon of Staphylococcus aureus although two sbnE and sbnI genes are missing (Fig. 5A). Based on our RNA‐seq data, the ent operon was not significantly induced by TRI, but the sbn operon was induced. This upregulation was also checked using qRT‐PCR (Fig. S3). Comparison of the sbn operon among a number of Gram‐negative bacteria and Staphylococcus aureus using blastp demonstrated high variability between the species. Further genomic analysis showed much lower GC contents upstream of sbnA (14.2%) and downstream of sbnG (23.3%) than the average (38.6%) GC content. To confirm higher production of siderophore, the CAS assay was conducted in both exponential and stationary growth phases, which showed high siderophore production in TRI‐supplemented media (Fig. 5B). Higher CAS activity was observed than control (succinate). In addition, the expression of sbnA was also upregulated during C16 degradation (Fig. S4). Collectively, the expression of sbnA was linked to siderophore production when DR1 cells use C16 as well as TRI. Three ferric siderophore receptor proteins encoding genes (bfrZ) were upregulated by 2.1‐ to 17.1‐fold, which is also consistent with the qRT‐PCR data (2.2‐fold change), in the media supplemented with TRI (Table S2 and Fig. S3). Our data show that highly induced sbn operon affected the synthesis of siderophore during TRI degradation.
Figure 5.
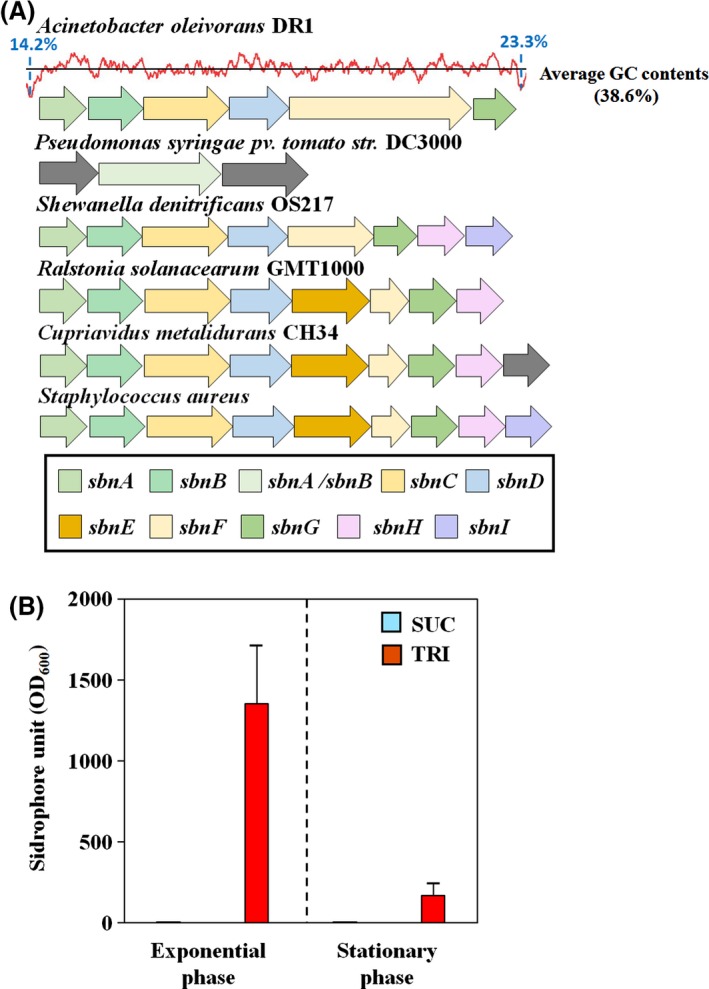
(A) AA alignment of sbn operon with other Gram‐negative strains and Gram‐positive Staphylococcus aureus. The red graph indicates GC contents in the sbn operon in DR1 genome, and the black line shows the average GC contents. Blue‐coloured GC contents were extremely lower than average. Different genes were coloured as shown in the boxes. (B) CAS assay for quantification of siderophore production under SUC and TRI supplementation. Siderophore production was not detected under SUC supplementation at both the exponential and stationary phase. Siderophore units were standardized by OD 600.
Trehalose biosynthesis during TRI degradation
The DR1 cells are known to display glucose metabolism disability due to the lack of glucokinase, which converts glucose to glucose‐6‐phosphate (Jung et al., 2011b). However, interestingly, trehalose (a disaccharide composed of two glucose molecules) metabolism‐related genes were highly upregulated during TRI metabolism (Table S2 and Fig. S3). In particular, the expression level of otsA and otsB genes, whose products are involved in synthesizing free trehalose from UDP‐glucose, was significantly increased (26.6‐ and 116.4‐fold respectively). Gluconeogenesis occurred during TRI degradation as indicated by high RPKM values of gluconeogenesis‐related genes (eno, gpmI, fda), which are associated with SUC metabolism (Table S2). Trehalose is also one of the representative alpha‐linked disaccharides synthesized intracellularly that helps cells to adapt to various stressors such as oxidative, heat, osmotic and desiccation stress (Benaroudj et al., 2001; Purvis et al., 2005; Doehlemann et al., 2006; Tapia et al., 2015). Trehalose measurement was performed using wild type and the otsA‐disrupted strain. Complete loss of trehalose accumulation was found only in the mutant (Fig. 6A), whereas wild‐type cells produced more trehalose in presence of TRI in both exponential and stationary growth phases. Sensitivity of the otsA mutant to salt stress (5% NaCl) was slightly greater than that of the wild type (Fig. 6B). In addition, growth defect of the otsA mutant was also observed in the presence of TRI (Fig. 6C). Collectively, these results suggest that TRI stimulates the synthesis of trehalose in the DR1 cells, which might enable cells to tolerate osmotic stress.
Figure 6.
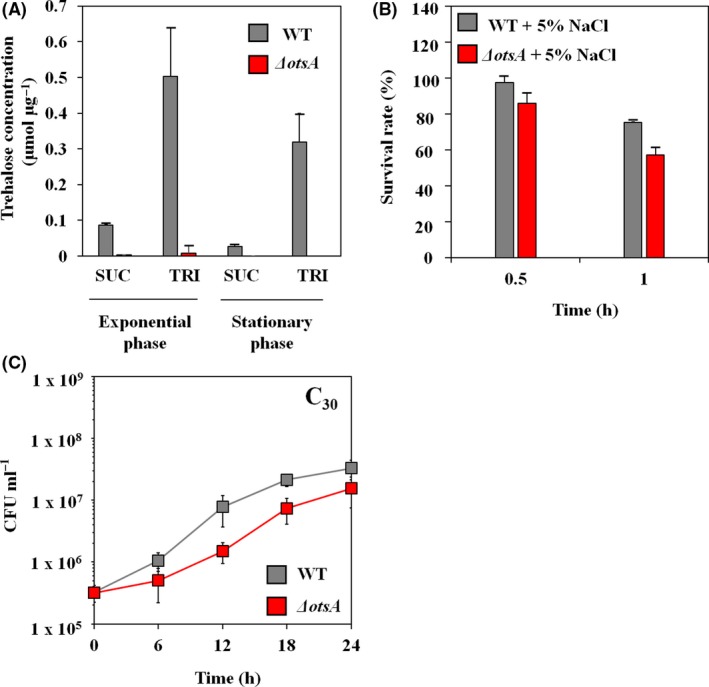
(A) Promoted trehalose production under TRI compared to SUC was confirmed using the trehalose assay. Scarcely produced trehalose was shown under SUC on the exponential and stationary phase. Trehalose was not detected on the ΔotsA (red bar) KO mutant even under TRI. (B) Survival rates of wild type (WT) and the ΔotsA mutant grown in TRI‐supplemented media were measured under PBS containing 5% NaCl for 1 h. Survival rates were calculated as follows: (CFU ml−1 at each time)/(CFU ml−1 at 0 h) × 100. (C) Growth assay of wild type (WT) and ΔotsA mutants in the TRI (C30) supplied MSB media.
Glyoxylate shunt and energy metabolism in the presence of TRI
The tricarboxylic acid (TCA) cycle is one of the main energy generating pathways involved in production of ATP, NADPH and FADH2. However, most TCA cycle‐related genes were downregulated during TRI assimilation. Instead, the aceA (encoding an isocitrate lyase) was highly upregulated in the presence of TRI (Table S2), although the glcB (encoding a malate synthase G) is not induced but constitutively expressed in both conditions (Table S2). The AckA (acetate kinase A) and Pta (phosphotransacetylase) genes are involved in producing ATP during conversion of acetyl‐CoA to acetate. The DR1 genome has AckA and Pta homologs, which have 45% and 49% amino acid similarities, respectively, with those of the E. coli K‐12 strain. These two ackA and pta genes were upregulated by 2.8‐ and 1.3‐fold with high RPKM values (Table S2 and Fig. S3). Growth defects of ΔaceA and ΔackA mutants were observed during TRI degradation, which indicated that both the glyoxylate shunt and Ack‐Pta pathway play important roles when DR cells grow on TRI (Fig. 7A). Growth defect of the ΔaceA mutant was observed under acetate condition, but the mutant cells could grow with longer lag phase under either hexadecane or hexadecanoic acid metabolism (Fig. S5). Furthermore, downregulation of F0F1 ATP synthase‐encoding genes (atp operon) also suggested the importance of the Ack‐Pta pathway in generation of ATP when the cells metabolize TRI. The results of the ATP assay supported the results of our expression and mutant analyses (Fig. 7B). Deletion of ackA resulted in less ATP production during TRI degradation. The ΔaceA mutant showed a lower survival rate than the wild type under H2O2‐treated conditions, suggesting that the glyoxylate bypass is essential not only for TRI degradation but also under oxidative stress conditions (Fig. 7C and D). Recently, we reported the importance of the glyoxylate shunt for defending cells against oxidative stress (Ahn et al., 2016). Other reports revealed that the ackA and pta genes might be important under reactive oxygen species (ROS) stress (Sadykov et al., 2013). Taken together, the glyoxylate shunt and Ack‐Pta pathway might be essential not only for metabolizing TRI but also for protecting cells under oxidative stress, which might be a consequence of TRI degradation.
Figure 7.
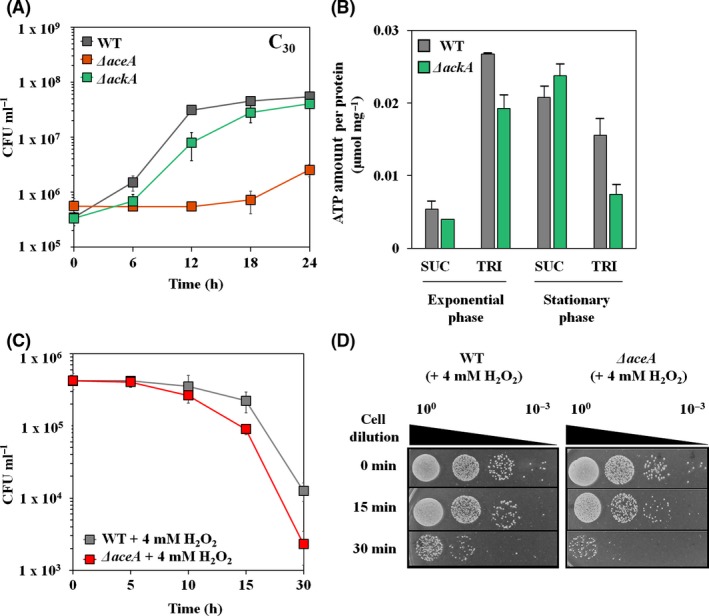
(A) Comparative growth assay of wild type (WT) and ΔaceA‐, ΔackA‐ mutants in the TRI‐supplemented media. (B) Quantification of intracellular ATP using Eliten ATP assay kit on both exponential and stationary phases. (C), (D) Comparative survival test of WT and ΔaceA mutant under 4 mm H2O2 (2MIC)‐treated conditions.
Discussion
Since its first isolation from soil, A. oleivorans DR1 has been a known alkane degrader (Kang et al., 2011). Simple growth measurements with different lengths of alkane chains suggested that DR1 cells could grow on media containing medium‐ and long‐chain alkanes (C12 to C30) but not short‐chain alkanes (C6, and C10) (Fig. 1A and Fig. S1C). This might result from the lack of a proper alkane monooxygenase for degrading short‐chain alkanes, because DR cells could utilize short‐chain alcohols and fatty acids (Fig. S1A). However, our data also suggested that decanol toxicity occurs for A. oleivorans (Figs S1D and S1E), which was also reported in other microorganisms such as Staphylococcus aureus (Togashi et al., 2007) and Mycobacteria (Mukherjee et al., 2013), although the toxicity mechanism remains unclear.
Phylogenetic analysis of alkane hydroxylases showed that three Acinetobacter species (A. oleivorans DR1, A. calcoaceticus and A. baylyi) and only two non‐Acinetobacter bacterial strains (Nocardia jiangxiensis and Bacillus mycoides) contain six alkane monooxygenases belonging to three different enzymes. The presence of alkane hydroxylase in multiple copies has been reported in many alkane degraders such as Alcanivorax borkumensis SK2 (Sabirova et al., 2006), Pseudomonas aeruginosa SJTD‐1 (Liu et al., 2014) and Geobacillus thermodenitrificans NG80‐2 (Liu et al., 2009). In addition, Alcanivorax dieselolei B‐5 possesses alkane hydroxylase of different systems (Liu et al., 2011). However, the Acinetobacter species genome is one of the rare cases in terms of alkane degradation repertoire as shown in Fig. 4.
The presence of mobile genetic elements adjacent to alkane hydroxylase in Rhodococcus sp. RHA1 and Azotobacter vinelandii may suggest that HGT would be one reason for the presence of multiple alkane hydroxylases in a bacterial genome, and this mobile element was also found near the ladA1 gene of the DR1 genome (Fig. S6). The existence of ladA located in the plasmid of G. thermodenitrificans NG80‐2 (pLW1071) also supports the occurrence of horizontal gene transfer. In addition, comparative genomic analysis revealed that DR1 possessed the largest genome among all Acinetobacter species probably due to not only gene recombination but also a relatively low rate of gene loss (Jung et al., 2011b). Possessing multiple alkane hydroxylase systems in multiple copies may confer ecological advantages for the colonization of niches rich in alkanes.
Increased expression of siderophore biosynthesis‐ and iron transportation‐related genes in DR1 grown on TRI was in concordance with what was observed in Rhodococcus erythropolis PR4 (Laczi et al., 2015). An increased need for iron might be attributable to high expression of alkane hydroxylase containing di‐iron clusters as reported from Pseudomonas oleovorans (Austin et al., 2000). In addition, the importance of iron metabolism might be valid especially in marine hydrocarbon‐degrading bacteria. A transposon mutagenesis study of Alkanivorax borkumenesis SK2 showed that a kinase sensor pfeS regulating iron acquisition by regulating synthesis and secretion of siderophore are essential under UV stress (Sabirova et al., 2008).
Several bacterial species, such as the Rhodococcus species, secrete various trehalose lipids as a biosurfactant during alkane degradation. A recent report proposed a hypothetical pathway for the synthesis of succinoyl trehalose lipid (STL) based on three essential genes (fda, tlsA and alkB) in Rhodococcus sp. SD‐74 during hexadecane degradation (Inaba et al., 2013). However, TLC analysis demonstrated similar patterns of glycolipid production in wild type and the ΔotsA mutant strain under TRI, indicating that trehalose synthesis is not necessary for glycolipid production in the DR1 strain (Fig. S7). Another possible hypothesis with respect to trehalose synthesis during TRI metabolism was that of water stress owing to hydrophobicity of the lipophilic substrate (Bhaganna et al., 2010). This is based on the highly upregulated kdp operon (Table S2), which is also induced under potassium‐limited conditions or osmotic stress (Asha and Gowrishankar, 1993).
The aceA gene encoding an isocitrate lyase in glyoxylate bypass was proven to be important for acetate, ethanol, poly‐3‐hydroxybutyrate, alkane metabolisms (Sabirova et al., 2006, 2011; Jung et al., 2011a; Zhang and Bryant, 2015; Ahn et al., 2016; Dunn et al., 2009). Interestingly, butanol metabolism appeared to occur through the glyoxylate bypass in Pseudomonas putida BIRD‐1 (Cuenca Mdel et al., 2016a,b) and loss of ethanol and acetate metabolisms was also observed in Candida albicans lacking the isocitrate lyase (Lorenz and Fink, 2001). Alkane degradation generates the TCA cycle intermediates from acetyl‐CoA via the glyoxylate bypass. To confirm the importance of the aceA gene during acetate and alkanes metabolisms, growth assays were conducted using wild type and mutant cells. Our data revealed that the aceA mutant could not grow on 1% sodium acetate (Fig. S6A); however, the aceA mutant could grow on hexadecane and hexadecanoic acid with longer lag phases (Figs S6B and S6C). This unexpected result suggests that DR1 cells might have additional alternative pathways to the glyoxylate shunt during alkane metabolism as seen in Rhodobacter spaeroedes and Methylobacterium extorquens AM1 (Ensign, 2006). Thus, growth defect of the ΔaceA mutant on TRI might be not due to the limitation in carbon, but due to the sensitivity to the stress generated by TRI. In addition, different pH changes during acetate‐ and hexadecane metabolism suggested that these two substrates are differently metabolized in DR1 cells (Fig. S6D).
It has been reported that outer membrane proteins, such as ompW, ompT, oprG and blc, might help to transport alkanes into periplasm of alkane‐degrading bacteria (Sabirova et al., 2006, 2011). In total, 42 annotated outer membrane proteins were identified in the DR1 genome (10 receptor proteins, 11 putative outer membrane proteins, 6 lipoproteins and 16 other outer membrane proteins). Both putative alkane transport system protein ompW and blc were upregulated in the presence of TRI (Table S2, Fig. 8, Fig. S3). Our RNA‐seq data also indicated that many oxidative stress defence genes were highly upregulated in TRI‐amended media (Table S2). All four catalase genes (katAc, katE, katP, katG) were upregulated, and the sodC gene encoding a superoxide dismutase was also induced (Table S2, Fig. 8). Other oxidative stress defence‐related genes (encoding a glutaredoxin, glutaredoxin and alkyl hydroperoxide reductase) were also upregulated by twofold with high RPKM values (> 500) in the presence of TRI (Table S2, Fig. 8). In addition, various general stress response genes (heat‐, cold‐shock protein coding genes, sigma factor, DNA‐repair related genes) were also induced (Table S2). The upregulation of three putative pha genes (phaA, phaB and phaC) participating in PHB synthesis led us to confirm the presence of PHB using microscopic analysis with Nile blue A (NBA). More PHB‐accumulated cells were observed with high intensity during TRI utilization (Fig. S8). A recent study revealed that a PHB‐intermediate (methyl esterified 3‐hydroxybutyrate) has hydroxyl radical scavenging activity to protect bacteria from ROS stress (Koskimäki et al., 2016). Therefore, upregulation of PHB synthesis genes might be involved in oxidative stress defence.
Figure 8.
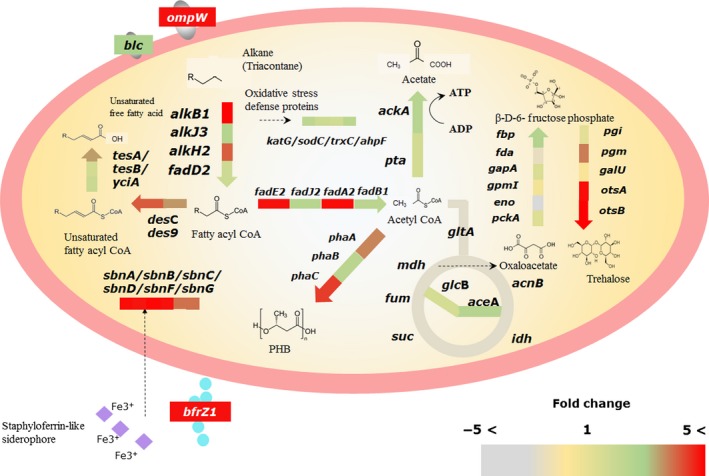
Schematic overview of gene expression within a DR1 cell in the presence of TRI. The colour spectrum indicates a fold change on TRI‐ compared to SUC‐supplemented media. Metabolic pathways were determined using the kegg pathway database and blast comparisons with proteins of experimentally proven function.
To our knowledge, this study is the first to evaluate an utilizable alkane range, expression patterns of alkane monooxygenases by chain length of the substrate, and physiological events in A. oleivorans DR1 grown on a long‐chain alkane. Notably, expression and mutant studies of alkane monooxygenases provided fundamental information related to the role of the multiple alkane hydroxylation system and regulation of alkane hydroxylases. In addition, RNA‐seq analysis provided valuable data regarding intracellular metabolic and physiological responses to a long‐chain alkane, such as synthesis of PHB, siderophore and trehalose, which were proven experimentally. These data could provide insights into the genotypic and phenotypic appearance of other alkane‐degrading bacteria during the metabolism of hydrophobic substances.
Experimental procedures
Bacterial strains, reagents and growth conditions
Acinetobacter oleivorans DR1, a diesel degrader, has been characterized in our previous studies (Kang and Park, 2010; Jung et al., 2011a; Kim and Park, 2013; Heo et al., 2014) and was used in this study. For the growth test of strain DR1, a seed culture was incubated in nutrient broth (Difco, Livonia, MI, USA) at 30°C and 220 r.p.m. overnight. Next, 1 ml of the seed culture was transferred to a 1.7‐ml microtube and washed twice with phosphate‐buffered saline (PBS, pH 7.4). Next, 1 × 105 to 1 × 106 CFU ml−1 of strain DR1 was transferred into 20 ml of minimal salt basal (MSB) medium (Hong et al., 2014) in a 50‐ml flask to observe the growth on different alkanes [0.1% (v/v for liquid alkanes C12–C22, w/v for solid alkanes C24–C30) alkanes]. However, M9 minimal media (Kang et al., 2011) was used to conduct only CAS assay for measurement of siderophore production during hexadecane metabolism. Growth curves were generated based on the colony‐forming unit (CFU) owing to water insolubility. All hydrocarbons (n‐alkanes) were purchased from Sigma‐Aldrich (St Louis, MO, USA), except for hexane (Junsei, Tokyo, Japan), decane (Wako, Osaka, Japan), decanol (Alfa Aesar, Haverhill, MA, USA), hexadecanol (Daejung, Gyeonggi‐do, Korea) and hexadecanoic acid (Junsei).
Survival and agar disc diffusion tests
To test the sensitivity of DR1 to decane and decanol, approximately 1 × 106 CFU ml−1 of PBS‐washed seed was inoculated into 50 ml fresh MSB media in a 100‐ml flask and the survival test was conducted. After the addition of 0.1% (v/v) decane or decanol, the mixture was shaken gently. Next, a 1 ml sample was transferred to a 1.7‐ml microtube and centrifuged at 1600 × g for 1 min. After washing the cell pellet twice, resuspended cells were serially diluted by 10−1 to 10−6. This procedure was repeated at each time point. Each resuspended sample (100 μl) was then spread onto a nutrient agar plate (NA). Colonies on all plates were counted using a colony doc‐it imaging station (UVP, Upland, CA, USA).
To demonstrate the susceptibility of the wild type and the otsA mutant against osmotic stress, a survival test was performed. Exponentially grown wild type and the otsA mutant in triacontane supplemented media (~18 h) were harvested and resuspended with 1 ml PBS. Each sample (500 μl) was inoculated into 20 ml PBS containing 5% NaCl. A 1‐ml sample was washed and then diluted with PBS to 10−4 at each time point. Diluted samples were spotted onto LB agar plates and the survival rate was calculated:
Survival rate = (CFU ml−1 at each time)/(CFU ml−1 at 0 h) × 100. For the H2O2‐sensitivity test of the wild type and ΔaceA mutant, overnight‐cultured strains in nutrient broth (NB) were diluted into 50 ml NB by 100‐fold. Exponential‐phase cells (OD600~0.6) were harvested and washed twice with PBS. Inoculation of approximately 106 cells per ml was conducted into fresh PBS (10 ml) containing 4 mm H2O2. At each time point, the cells were harvested and washed in PBS. Viable cells were quantified by counting the CFU. For the agar disc diffusion test, 50 ml MSB agar was autoclaved and cooled for 30 min. Next, 1–1.5 ml of inoculum was transferred into 50 ml MSB agar. The media was shaken gently and poured onto a plate and then dried for another 30 min. An 8‐mm paper disc (Advantec, Dublin, CA, USA) was used to absorb 20 μl of 50 and 100% (v/v) decane or decanol. The decane‐ or decanol‐soaked paper disc was dropped onto a dried MSB agar plate using sterilized forceps.
RNA isolation and transcriptomic analysis by RNA‐seq
Total RNA of DR1 cells was obtained from mid‐exponentially grown cells using an RNeasy kit (Qiagen, Hilden, Germany) per the manufacturer's instructions. All procedures for RNA sequencing and alignment were conducted by Chunlab (Seoul, South Korea). The Ribo‐Zero rRNA removal kit (Epicentre, Medison, WI, USA) was used for ribosomal RNA depletion according to the manufacturer's instructions. Libraries for Illumina sequencing were constructed with the TruSeq Stranded mRNA sample prep kit (Illumina, San Diego, CA, USA) following the manufacturer's protocol. RNA sequencing was performed on the Illumina HiSeq 2500 platform using single‐end 50‐bp sequencing. Sequence data for the reference genome were retrieved from the NCBI database. Quality‐filtered reads were aligned to the reference‐genome sequence using Bowtie2. The abundance of relative transcript was shown by reads per kilobase of the exon sequence per million mapped sequence reads (RPKM) defined as total exon reads/(mapped reads in millions × exon length in kilobases). Metabolic pathways were analysed based on the kegg pathway analysis and blast alignment with proteins (SRA accession number: SRS1307226).
Quantitative reverse transcription PCR (qRT‐PCR)
cDNA synthesized from 1 μg of each RNA was used as a template. The PCR mixture contained 1 μl of each primer (0.5 μm) and 2 μl 100‐fold diluted cDNA, 10 μl Power SYBR Green PCR Master Mix (Applied biosystems, Carlsbad, CA, USA) and 6 μl DW comprising a total volume of 20 μl. PCR conditions were set at 48°C for 30 min and 95°C for 10 min for the first holding stage, followed by 42 cycles of 15 s at 95°C for the cycling stage, finally, 15 s at 95°C, 1 min at 60°C and again 15 s at 95°C for the melt curve stage. The expression levels of each gene were normalized by 16S rDNA, as described previously (Watanabe et al., 2001). The quantification results were based on triplicate samples. Primer sequences are shown in Table S3.
Northern blot analysis
Northern blotting was conducted as described previously (Lee et al., 2006). Briefly, RNA concentration was measured at OD260 nm. Samples of total RNA (1 μl) were loaded onto an ethidium bromide‐stained agarose gel containing 0.25 m formaldehyde and electrophoresed. Separated RNA was transferred to a nylon membrane (Schleicher & Schuell BioScience, Dassel, Germany) using a TurboBlotter. mRNA was quantified by hybridizing the membrane with a P32‐labelled probe. Primers used for amplification are represented in Table S3. Autoradiography was conducted using an imaging plate (Fujifilm, Tokyo, Japan) and a multiplex Bio‐imaging system (Fujifilm).
Phylogenetic analyses of the 16 rRNA gene and alkane monooxygenases
AlkB‐, AlmA‐ and LadA‐type alkane monooxygenase homologs in other bacteria were collected using blastp (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequences with over 90% coverage and at least 30% similarity with the query were selected, and bacterial 16S rRNA sequences were searched for in the silva (http://www.arb-silva.de/) web browser. After trimming multiple aligned sequences by clustal w, neighbour‐joining (NJ) phylogenetic trees were drawn using mega 6.0 software. The same criteria were adapted for comparison of alkane homologs between strain DR1 and other bacteria using blastp. The insertion sequence (IS) and mobile genetic element (MGE) were searched using an IS finder (https://www-is.biotoul.fr/) and aclame (http://aclame.ulb.ac.be/).
Microscopic analysis
Dual staining with NBA and DAPI was performed as previously described (Oshiki et al., 2013). Briefly, heat‐fixed cells were treated with 1% ethanolic Nile blue A (NBA) for 30 min to visualize the intracellular granules accumulated by PHA. After washing with 100% ethanol, DR1 cells were stained with 2 μg ml−1 DAPI for 5 min. NBA‐stained granules and DAPI‐stained cells were detected as bright red and blue at 550 nm‐, 365 nm wavelength excitation filters respectively.
Chrome azurol S (CAS) assay
To quantify the production of siderophore, the CAS assay was conducted as previously described (Dale et al., 2004). Briefly, all 1/10 diluted supernatants and a blank were mixed with equal volumes of CAS shuttle solution and then allowed to stand for 30 min at room temperature. The absorbance at 630 nm was determined with MSB as the blank and distilled water (DW) as the reference standard. Siderophore units were measured as follows: (A630 of MSB−A630 of sample)/A630 of MSB × 100% and were standardized by cell density (OD600).
Trehalose assay
Supernatant and crude cells grown on 50 ml MSB media were separated by ultracentrifugation (3000 r.p.m., 20 min). Then, intracellular trehalose was extracted by boiling the cell pellet at 90°C for 20 min. Trehalose levels of supernatant and crude cells were measured using a Trehalose assay kit K‐TREH (Megazyme International Ireland, Bray, Ireland) according to the manufacturer's instructions. All samples were standardized by the Bradford assay, and triplicates were used.
ATP assay
Measurement of intracellular ATP concentration was conducted using the ENLITEN ATP Assay System Bioluminescence Detection Kit (Promega, Medison, WI, USA) in accordance with the manufacturer's instructions. Briefly, exponentially cultured DR1 and ΔackA KO mutant were harvested and resuspended in 1% trichloroacetic acid (TCA) buffer. Prior to the measurement, the samples were neutralized by sixfold dilution with 250 mm Tris–acetate buffer (pH 7.75). The luminescence was measured using a microplate reader (Hidex, Turku, Finland).
Construction of the single crossover knockout (KO) mutants
The alkR1 (AOLE_RS10595), alkR2 (AOLE_RS13405), alkB1 (AOLE_RS10590), alkB2 (AOLE_RS13400), otsA (AOLE_RS15640), ackA (AOLE_RS17025) and aceA (AOLE_RS14285) genes were amplified by PCR from genomic DNA and cloned into the pVIK 112 plasmid. The amplified fragments were digested with KpnI/XbaI for alkB1, alkB2 and aceA, and EcoRI/KpnI for otsA and ackA. Ligation into each of the restriction enzyme‐treated sites of pVIK112 was performed; the plasmids were subsequently transformed into the E. coli S17‐1λ pir strain. Recombined plasmids were extracted and then transformed to strain DR1. KO mutants were screened on NA containing 50 μg ml−1 kanamycin.
To produce an alkB double KO mutant, alkB2 amplicon and pEX18Gm vector were treated with the same restriction enzymes as above. After ligation, the recombined vector was transformed into the E. coli S17‐1λ pir strain. The vector obtained by extraction from E. coli was again transformed into strain alkB1 single KO strain, and then, the double KO mutant was selected on NA containing 50 μg ml−1 kanamycin and 15 μg ml−1 gentamicin.
To construct an aceA complemented strain, the amplified fragment of aceA and pRK415 vector was digested with BamHI/EcoRI. The constructed plasmid was transformed into the E. coli TOP10. Complementation was performed by transforming this constructed vector into aceA KO strain, which was screened on LB containing 50 μg ml−1 kanamycin and 20 μg ml−1 tetracycline. Gel electrophoresis and sequencing were conducted with KO_F, KO_R, and KO_C primers for verification (Table S3).
Thin‐layer chromatography (TLC) analysis
Glycolipid of the wild type and mutant strain was analysed by thin‐layer chromatography as previously described (Espuny et al., 1995). Briefly, mid‐exponentially grown cells in 50 ml MSB medium were harvested by ultracentrifugation. Cells collected were resuspended in 5 ml PBS and brought to pH 2 using H2SO4. Then, 5 ml of the mixture (chloroform: methanol = 2:1) was added and vortexed for 5 min. The organic phase was dried in a rotary evaporator and resuspended in 40 μl fresh chloroform/methanol mixture. Samples were plated on silica gel sheets G 60 (Merck, Kenilworth, NJ, USA) and developed with chloroform/methanol/water = 65:25:4). Visualization of developed samples was performed by treatment with TLC reagents: iodine vapour for lipid staining and 1‐naphthol reagent for hydrocarbon detection (Wang and Benning, 2011).
Conflict of interest
None declared.
Supporting information
Table S1. Information about the raw RNA‐seq used in this study.
Table S2. RPKM values of transcriptome in DR1 strain under SUC and TRI. Fold changes were determined by RPKM values of each gene under TRI compared to SUC.
Table S3. Information about primer sequences used in qRT‐PCR, Northern blotting, and construction of knock out mutants.
Fig. S1. Growth assays and survival tests on (A) hexane, (B) decane, and their derivatives. (C) Measurement of colony‐forming units (CFUs) for 24 h to verify the inability of cells to grow on 0.1% hexane and decane. All experiments were performed in triplicate and their means are represented. (D) Survival tests on 0.1% decanol within 15 min was performed for A. oleivorans DR1. (E) Verification of decanol‐high toxicity towards DR1 in a paper disk assay. The results of the paper disk assay on decane (top) and decanol (bottom) are shown as 50% (left) and 100% (right).
Fig. S2. The expression analysis of alkB in wild type strain and alkB single mutants using Northern blot hybridization. (A) alkB1 expression in wild type‐, and ΔalkB2 strain. (B) alkB2 expression in wild type‐, and ΔalkB1 strain.
Fig. S3. Validation of six‐upregulated genes in RNA‐seq profile using qRT‐PCR.
Fig. S4. (A) CAS activity and (B) relative expression level of sbnA in DR1 cells grown on 10 mm succinate (SUC, cyan), and 0.1% hexadecane (HEX, red) supplemented M9 medium.
Fig. S5. The scheme of MGE site in the upstream of ladA1 in DR1 strain and tnpA‐encoding genes in Azotobacter vinelandii.
Fig. S6. Growth assay of wild type‐, ΔaceA‐, ΔaceA(pRK415::aceA) strain on (A) 1% Sodium acetic acid, (B) 1% hexadecane, (C) 1% hexadecanoic acid. (D) pH measurement of wild type strain during sodium acetic acid (NaAc, red), and hexadecane (HEX, green) assimilation. Circle and square indicates OD600 and pH, respectively.
Fig. S7. Comparison of intracellular glycolipid between wild type and ΔotsA KO strain using thin‐layer chromatography (TLC). Numbers beside the column indicate the R f value of each band. Left side indicates detection of hydrocarbon in glycolipid using 1‐naphthol reagent. Right side indicates detection of lipid in glycolipid using iodine vapor.
Fig. S8. Microscopic analysis of NBA‐, DAPI‐ dual stained DR1 for PHB detection. Mid‐exponential cells were stained and observed by no filter (control), 365 nm‐ (DAPI), and 550 nm‐wavelength filters (NBA). Merged images of DAPI and NBA are shown on the right side (DAPI + NBA).
Microbial Biotechnology (2017) 10(6), 1809–1823
Funding information
This work was supported by the National Research Foundation of Korea (NRF) grant‐funded by the Korea government (MSIP) (No. NRF‐2017R1A2B4005838) WP was supported by a Korea University Grant.
References
- Ahn, S. , Jung, J. , Jang, I. A. , Madsen, E. L. , and Park, W. (2016) Role of glyoxylate shunt in oxidative stress response. J Biol Chem 291: 11928–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asha, H. , and Gowrishankar, J. (1993) Regulation of kdp operon expression in Escherichia coli: evidence against turgor as signal for transcriptional control. J Bacteriol 175: 4528–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin, R. N. , Chang, H. K. , Zylstra, G. J. , and Groves, J. T. (2000) The non‐heme diiron alkane monooxygenase of Pseudomonas oleovorans (AlkB) hydroxylates via a substrate radical intermediate. J Am Chem Soc 122: 11747–11748. [Google Scholar]
- Bach, H. , Berdichevsky, Y. , and Gutnick, D. (2003) An exocellular protein from the oil‐degrading microbe Acinetobacter venetianus RAG‐1 enhances the emulsifying activity of the polymeric bioemulsifier emulsan. Appl Environ Microbiol 69: 2608–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaroudj, N. , Lee, D. H. , and Goldberg, A. L. (2001) Trehalose accumulation during cellular stress protects cells and cellular proteins from damage by oxygen radicals. J Biol Chem 276: 24261–24267. [DOI] [PubMed] [Google Scholar]
- Bhaganna, P. , Volkers, R. J. , Bell, A. N. , Kluge, K. , Timson, D. J. , McGrath, J. W. , et al (2010) Hydrophobic substances induce water stress in microbial cells. Microb Biotechnol 3: 701–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca Mdel, S. , Molina‐Santiago, C. , Gómez‐García, M. R. , and Ramos, J. L. (2016a) A Pseudomonas putida double mutant deficient in butanol assimilation: a promising step for engineering a biological biofuel production platform. FEMS Microbiol Lett 363: fnw018. [DOI] [PubMed] [Google Scholar]
- Cuenca Mdel, S. , Roca, A. , Molina‐Santiago, C. , Duque, E. , Armengaud, J. , Gómez‐Garcia, M. R. , and Ramos, J. L. (2016b) Understanding butanol tolerance and assimilation in Pseudomonas putida BIRD‐1: an integrated omics approach. Microb Biotechnol 9: 100–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale, S. E. , Doherty‐Kirby, A. , Lajoie, G. , and Heinrichs, D. E. (2004) Role of siderophore biosynthesis in virulence of Staphylococcus aureus: identification and characterization of genes involved in production of a siderophore. Infect Immun 72: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehlemann, G. , Berndt, P. , and Hahn, M. (2006) Trehalose metabolism is important for heat stress tolerance and spore germination of Botrytis cinerea . Microbiology 152: 2625–2634. [DOI] [PubMed] [Google Scholar]
- Dunn, M. F. , Ramírez‐Trujillo, J. A. , and Hernández‐Lucas, I. (2009) Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology 155: 3166–3175. [DOI] [PubMed] [Google Scholar]
- Ensign, S. A. (2006) Revisiting the glyoxylate cycle: alternate pathways for microbial acetate assimilation. Mol Microbiol 61: 274–276. [DOI] [PubMed] [Google Scholar]
- Espuny, M. J. , Egido, S. , Mercade, M. E. , and Manresa, A. (1995) Characterization of trehalose tetraester produced by a waste lubricant oil degrader Rhodococcus sp. Toxicol Environ 48: 83–88. [Google Scholar]
- Feng, L. , Wang, W. , Cheng, J. , Ren, Y. , Zhao, G. , Gao, C. , et al (2007) Genome and proteome of long‐chain alkane degrading Geobacillus thermodenitrificans NG80‐2 isolated from a deep‐subsurface oil reservoir. Proc Natl Acad Sci USA 104: 5602–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo, A. , Jang, H. J. , Sung, J. S. , and Park, W. (2014) Global transcriptome and physiological responses of Acinetobacter oleivorans DR1 exposed to distinct classes of antibiotics. PLoS ONE 9: e110215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, H. , Ko, H. J. , Choi, I. G. , and Park, W. (2014) Previously undescribed plasmids recovered from activated sludge confer tetracycline resistance and phenotypic changes to Acinetobacter oleivorans DR1. Microb Ecol 67: 369–379. [DOI] [PubMed] [Google Scholar]
- Inaba, T. , Tokumoto, Y. , Miyazaki, Y. , Inoue, N. , Maseda, H. , Nakajima‐Kambe, T. , et al (2013) Analysis of genes for succinoyl trehalose lipid production and increasing production in Rhodococcus sp. strain SD‐74. Appl Environ Microbiol 79: 7082–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y. , Mao, G. , Wang, Y. , and Bartlam, M. (2013) Structural insights into diversity and n‐alkane biodegradation mechanisms of alkane hydroxylases. Front Microbiol 4: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, J. , and Park, W. (2015) Acinetobacter species as model microorganisms in environmental microbiology: current state and perspectives. Appl Microbiol Biotechnol 99: 2533–2548. [DOI] [PubMed] [Google Scholar]
- Jung, J. , Baek, J. H. , and Park, W. (2010) Complete genome sequence of the diesel‐degrading Acinetobacter sp. strain DR1. J Bacteriol 192: 4794–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, J. , Noh, J. , and Park, W. (2011a) Physiological and metabolic responses for hexadecane degradation in Acinetobacter oleivorans DR1. J Microbiol 49: 208–215. [DOI] [PubMed] [Google Scholar]
- Jung, J. , Madsen, E. L. , Jeon, C. O. , and Park, W. (2011b) Comparative genomic analysis of Acinetobacter oleivorans DR1 to determine strain‐specific genomic regions and gentisate biodegradation. Appl Environ Microbiol 77: 7418–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y. S. , and Park, W. (2009) Protection against diesel oil toxicity by sodium chloride‐induced exopolysaccharides in Acinetobacter sp. strain DR1. J Biosci Bioeng 109: 118–123. [DOI] [PubMed] [Google Scholar]
- Kang, Y. S. , and Park, W. (2010) Trade‐off between antibiotic resistance and biological fitness in Acinetobacter sp. strain DR1. Environ Microbiol 12: 1304–1318. [DOI] [PubMed] [Google Scholar]
- Kang, Y. S. , Jung, J. , Jeon, C. O. , and Park, W. (2011) Acinetobacter oleivorans sp. nov. is capable of adhering to and growing on diesel‐oil. J Microbiol 49: 29–34. [DOI] [PubMed] [Google Scholar]
- Kim, J. , and Park, W. (2013) Identification and characterization of genes regulated by AqsR, a LuxR‐type regulator in Acinetobacter oleivorans DR1. Appl Microbiol Biotechnol 97: 6967–6978. [DOI] [PubMed] [Google Scholar]
- Koskimäki, J. J. , Kajula, M. , Hokkanen, J. , Ihantola, E. L. , Kim, J. H. , Hautajärvi, H. , et al (2016) Methyl‐esterified 3‐hydroxybutyrate oligomers protect bacteria from hydroxyl radicals. Nat Chem Biol 12: 332–338. [DOI] [PubMed] [Google Scholar]
- Laczi, K. , Kis, Á. , Horváth, B. , Maróti, G. , Hegedüs, B. , Perei, K. , and Rákhely, G. (2015) Metabolic responses of Rhodococcus erythropolis PR4 grown on diesel oil and various hydrocarbons. Appl Microbiol Biotechnol 99: 9745–9759. [DOI] [PubMed] [Google Scholar]
- Lee, Y. , Peña‐Llopis, S. , Kang, Y. S. , Shin, H. D. , Demple, B. , Madsen, E. L. , et al (2006) Expression analysis of the fpr (ferredoxin‐NADP+ reductase) gene in Pseudomonas putida KT2440. Biochem Biophys Res Commun 339: 1246–1254. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Dong, Y. , Zhang, J. , Zhang, A. , Wang, L. , and Feng, L. (2009) Two novel metal‐independent long‐chain alkyl alcohol dehydrogenases from Geobacillus thermodenitrificans NG80‐2. Microbiology 155: 2078–2085. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Wang, W. , Wu, Y. , Zhou, Z. , Lai, Q. , and Shao, Z. (2011) Multiple alkane hydroxylase systems in a marine alkane degrader, Alcanivorax dieselolei B‐5. Environ Microbiol 13: 1168–1178. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Xu, J. , Liang, R. , and Liu, J. (2014) Characterization of the medium‐ and long‐chain n‐alkanes degrading Pseudomonas aeruginosa Strain SJTD‐1 and its alkane hydroxylase genes. PLoS ONE 9: e105506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Tang, J. , Bai, Z. , Hecker, M. , and Giesy, J. P. (2015) Distribution of petroleum degrading genes and factor analysis of petroleum contaminated soil from the Dagang Oilfield, China. Sci Rep 5: 11068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, M. C. , and Fink, G. R. (2001) The glyoxylate cycle is required for fungal virulence. Nature 412: 83–86. [DOI] [PubMed] [Google Scholar]
- Maeng, J. H. , Sakai, Y. , Tani, Y. , and Kato, N. (1996) Isolation and characterization of a novel oxygenase that catalyzes the first step of n‐alkane oxidation in Acinetobacter sp. strain M‐1. J Bacteriol 178: 3695–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, K. , Tribedi, P. , Mukhopadhyay, B. , and Sil, A. K. (2013) Antibacterial activity of long‐chain fatty alcohols against mycobacteria . FEMS Microbiol Lett 338: 177–183. [DOI] [PubMed] [Google Scholar]
- Oshiki, M. , Onuki, M. , Satoh, H. , and Mino, T. (2013) Microbial community composition of polyhydroxyalkanoate‐accumulating organisms in full‐scale wastewater treatment plants operated in fully aerobic mode. Microbes Environ 28: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis, J. E. , Yomano, L. P. , and Ingram, L. O. (2005) Enhanced trehalose production improves growth of Escherichia coli under osmotic stress. Appl Environ Microbiol 71: 3761–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak, A. , Geissdörfer, W. , and Hillen, W. (1998) Expression of alkane hydroxylase from Acinetobacter sp. Strain ADP1 is induced by a broad range of n‐alkanes and requires the transcriptional activator AlkR. J Bacteriol 180: 5822–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo, F. (2010) Enzymes for aerobic degradation of alkanes. In Handbook of Hydrocarbon and Lipid Microbiology. Timmis K. N., McGenity T. J., van der Meer J. R., and de Lorenzo V. (eds). Berlin: Springer, pp. 782–793. [Google Scholar]
- Rosenberg, M. , Bayer, E. A. , Delarea, J. , and Rosenberg, E. (1982) Role of thin fimbriae in adherence and growth of Acinetobacter calcoaceticus RAG‐1 on hexadecane. Appl Environ Microbiol 44: 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirova, J. S. , Ferrer, M. , Regenhardt, D. , Timmis, K. N. , and Golyshin, P. N. (2006) Proteomic insights into metabolic adaptations in Alcanivorax borkumensis induced by alkane utilization. J Bacteriol 188: 3763–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirova, J. S. , Chernikova, T. N. , Timmis, K. N. , and Golyshin, P. N. (2008) Niche‐specificity factors of a marine oil‐degrading bacterium Alcanivorax borkumensis SK2. FEMS Microbiol Lett 285: 89–96. [DOI] [PubMed] [Google Scholar]
- Sabirova, J. S. , Becker, A. , Lünsdorf, H. , Nicaud, J. M. , Timmis, K. N. , and Golyshin, P. N. (2011) Transcriptional profiling of the marine oil‐degrading bacterium Alcanivorax borkumensis during growth on n‐alkanes. FEMS Microbiol Lett 319: 160–168. [DOI] [PubMed] [Google Scholar]
- Sadykov, M. R. , Thomas, V. C. , Marshall, D. D. , Wenstrom, C. J. , Moormeier, D. E. , Widhelm, T. J. , et al (2013) Inactivation of the Pta‐AckA pathway causes cell death in Staphylococcus aureus . J Bacteriol 195: 3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia, H. , Young, L. , Fox, D. , Bertozzi, C. R. , and Koshland, D. (2015) Increasing intracellular trehalose is sufficient to confer desiccation tolerance to Saccharomyces cerevisiae . Proc Natl Acad Sci USA 112: 6122–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throne‐Holst, M. , Wentzel, A. , Ellingsen, T. E. , Kotlar, H. K. , and Zotchev, S. B. (2007) Identification of novel genes involved in long‐chain n‐alkane degradation by Acinetobacter sp. strain DSM 17874. Appl Environ Microbiol 73: 3327–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi, N. , Shiraishi, A. , Nishizaka, M. , Matsuoka, K. , Endo, K. , Hamashima, H. , and Inoue, Y. (2007) Antibacterial activity of long‐chain fatty alcohols against Staphylococcus aureus . Molecules 12: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Beilen, J. B. , and Funhoff, E. G. (2007) Alkane hydroxylases involved in microbial alkane degradation. Appl Microbiol Biotechnol 74: 13–21. [DOI] [PubMed] [Google Scholar]
- Van Beilen, J. B. , Li, Z. , Duetz, W. A. , Smits, T. H. M. , and Witholt, B. (2003) Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci Technol 58: 427–440. [Google Scholar]
- Wang, Z. and Benning, C. (2011) Arabidopsis thaliana polar glycerolipid profiling by thin layer chromatography (TLC) coupled with gas‐liquid chromatography (GLC) [WWW document]. URL http://www.jove.com/video/2518/arabidopsis-thaliana-polar-glycerolipid-profiling-thin-layer.html. [DOI] [PMC free article] [PubMed]
- Watanabe, K. , Kodama, Y. , and Harayama, S. (2001) Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J Microbiol Methods 44: 253–262. [DOI] [PubMed] [Google Scholar]
- Wentzel, A. , Ellingsen, T. E. , Kotlar, H. K. , Zotchev, S. B. , and Throne‐Holst, M. (2007) Bacterial metabolism of long‐chain n‐alkanes. Appl Microbiol Biotechnol 76: 1209–1221. [DOI] [PubMed] [Google Scholar]
- Zhang, S. , and Bryant, D. A. (2015) Biochemical validation of the glyoxylate cycle in the cyanobacterium Chlorogloeopsis fritschii strain PCC 9212. J Biol Chem 290: 14019–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information about the raw RNA‐seq used in this study.
Table S2. RPKM values of transcriptome in DR1 strain under SUC and TRI. Fold changes were determined by RPKM values of each gene under TRI compared to SUC.
Table S3. Information about primer sequences used in qRT‐PCR, Northern blotting, and construction of knock out mutants.
Fig. S1. Growth assays and survival tests on (A) hexane, (B) decane, and their derivatives. (C) Measurement of colony‐forming units (CFUs) for 24 h to verify the inability of cells to grow on 0.1% hexane and decane. All experiments were performed in triplicate and their means are represented. (D) Survival tests on 0.1% decanol within 15 min was performed for A. oleivorans DR1. (E) Verification of decanol‐high toxicity towards DR1 in a paper disk assay. The results of the paper disk assay on decane (top) and decanol (bottom) are shown as 50% (left) and 100% (right).
Fig. S2. The expression analysis of alkB in wild type strain and alkB single mutants using Northern blot hybridization. (A) alkB1 expression in wild type‐, and ΔalkB2 strain. (B) alkB2 expression in wild type‐, and ΔalkB1 strain.
Fig. S3. Validation of six‐upregulated genes in RNA‐seq profile using qRT‐PCR.
Fig. S4. (A) CAS activity and (B) relative expression level of sbnA in DR1 cells grown on 10 mm succinate (SUC, cyan), and 0.1% hexadecane (HEX, red) supplemented M9 medium.
Fig. S5. The scheme of MGE site in the upstream of ladA1 in DR1 strain and tnpA‐encoding genes in Azotobacter vinelandii.
Fig. S6. Growth assay of wild type‐, ΔaceA‐, ΔaceA(pRK415::aceA) strain on (A) 1% Sodium acetic acid, (B) 1% hexadecane, (C) 1% hexadecanoic acid. (D) pH measurement of wild type strain during sodium acetic acid (NaAc, red), and hexadecane (HEX, green) assimilation. Circle and square indicates OD600 and pH, respectively.
Fig. S7. Comparison of intracellular glycolipid between wild type and ΔotsA KO strain using thin‐layer chromatography (TLC). Numbers beside the column indicate the R f value of each band. Left side indicates detection of hydrocarbon in glycolipid using 1‐naphthol reagent. Right side indicates detection of lipid in glycolipid using iodine vapor.
Fig. S8. Microscopic analysis of NBA‐, DAPI‐ dual stained DR1 for PHB detection. Mid‐exponential cells were stained and observed by no filter (control), 365 nm‐ (DAPI), and 550 nm‐wavelength filters (NBA). Merged images of DAPI and NBA are shown on the right side (DAPI + NBA).