

Abstract
Congenital myasthenic syndromes (CMS) are neuromuscular transmission disorders caused by mutations in genes encoding neuromuscular junction proteins. A 61-year-old female and her older sister showed bilateral ptosis, facial and proximal limb weakness, and scoliosis since childhood. Another female sibling had milder signs, while other family members were asymptomatic. Facial nerve repetitive stimulation in the proband showed decrement of muscle responses. Single fiber EMG revealed increased jitter and blocking. Muscle biopsy showed type 2-fiber atrophy, without tubular aggregates. Mutational analysis in the three affected siblings revealed two compound heterozygous mutations in DOK7: c.1457delC, that predicts p.Pro486Argfs*13 and truncates the protein C-terminal domain, and c.473G>A, that predicts p.Arg158Gln and disruption of the dok7-MuSK interaction in the phosphotyrosine binding (PTB) domain. Unaffected family members carried only one or neither mutation. Discussion. Two of the affected sisters showed marked improvement with salbutamol treatment, which illustrates the benefits of a correct diagnosis and treatment of DOK7-CMS.
Key Words: Myasthenic syndromes, congenital, dok-7, Electrophysiology, salbutamol, MRI
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Conflict of Interest
None of the authors have conflicts of interests.
List of acronyms
AChRs - acetylcholine receptors
BiPAP - Bilevel or two-level positive airway pressure
CK - muscle phosphocreatine kinase
CMSs - congenital myasthenic syndromes
DAP - 3,4 diaminopyridine
DOK7 – a gene encoding the intracellular protein dok-7
DTR - deep tendon reflexes
EDC - extensor digitorum communis muscle
HCUCH - University of Chile Clinical Hospital
LRP4 - LDL receptor related protein 4
MG - myasthenia gravis
MGC - Myasthenia Gravis Composite score
MRC - Medical Research Council muscle scale
MuSK - muscle specific kinase
NMJ - neuromuscular junction
PTB - phosphotyrosine-binding domain
tid - dosing three times a day
Congenital myasthenic syndromes (CMSs) comprise a group of neuromuscular transmission disorders caused by mutations in genes encoding neuromuscular junction (NMJ) proteins.1, 2 CMSs are often classified according with the primary site of neuromuscular transmission failure in presynaptic, synaptic, and postsynaptic.3 A recessive CMS, characterized by limb-girdle weakness and fatigability is due to mutations in DOK7, which encodes the intracellular protein dok-7.4 In turn Dok-7 binds muscle specific kinase (MuSK) at the phosphotyrosine-binding (PTB) domain. Dok-7 links the agrin-LRP4-MuSK signaling pathway with a poorly understood cascade of intracellular events leading to a dense aggregation of acetylcholine receptors (AChRs) at the crests of the postsynaptic folds.5 The CMS due to DOK7 mutations was first described in 2006, in the United Kingdom, where it represents around 20% of post-synaptic CMSs.6 DOK7-CMS genotype7 and phenotype8-11 have been extensively described. Identification of patients with DOK7-CMS is important since these patients may benefit from treatment with salbutamol12-15 or ephedrine,16 while acetylcholinesterase inhibitors and 3,4 diaminopyridine (3,4 DAP) may worsen symptoms. Here, we describe a four-generation Chilean family affected by a DOK7-CMS. To date, no patients with DOK7-CMS have ever been reported in Chile.
This report has been presented at the Annual Meeting of the American Academy of Neurology held in Washington DC, April 18-25, 2015.
Materials, Methods and Results
Case 1.
Case 1 is a 61-year-old female with a history of normal developmental milestones with scoliosis since childhood, and progressive bilateral ptosis since adolescence (Figure 1). During her first pregnancy at the age of 23, ptosis worsened and fluctuating limb-girdle, facial and neck weakness appeared. She was diagnosed with myasthenia gravis (MG) and treated with pyridostigmine, prednisone and azathioprine, without response. In her fifties (Figure 1A), she presented ptosis, and bilateral facial and proximal limb weakness (MRC3 = M3) in shoulder and pelvic girdles, with M4+ in distal muscles. Neck flexion scored M4. Barre and Mingazzini’s maneuvers scored both 30 seconds (N=130 and 75 seconds respectively). Deep tendon reflexes (DTR) and serum CK levels were in the normal range. Repetitive nerve stimulation of the facial nerve at 3 Hz showed 15% decrement of compound muscle action potential amplitudes and single fiber EMG in the EDC muscle showed increased jitter and blocking (Figure 2A and B). She continued pyridostigmine 60 mg/tid, and was adapted to daily life activities. At the age of 57, weakness worsened and she was hospitalized with pneumonia. She received rituximab (Mabthera®) without improvement. She started with nocturnal BiPAP, but did not tolerate the lie-down position. After recovery, she had dysphagia, drop-head syndrome, facial weakness and the ptosis was aggravated (Figure 1B). Barre and Mingazzini’s maneuvers scored 19 and 10 seconds respectively. Proximal limb weakness was scored at M3, with symmetrical distal involvement of upper and lower limbs (M4), requiring a wheelchair. Myasthenia Gravis Composite (MGC)16 score was 43 (normal 0, maximum score, 50). After unremarkable cardiologic assessment she started salbutamol 4 mg four times daily. Six months later, the facial weakness and ptosis persisted, but the head support was normal and the dysphagia was only occasional. Proximal limb weakness was in M4, and M5 distally. She walked with a cane, mainly due to the scoliosis. Barre and Mingazzini’s maneuvers scored 85 and 50 seconds respectively. MGC score was 24 (i.e. 19 points improvement). She maintains BiPAP use and continues treatment, and she is currently stable in her condition. A muscle biopsy and whole body muscle MRI were performed (Figure 2).
Fig 1.
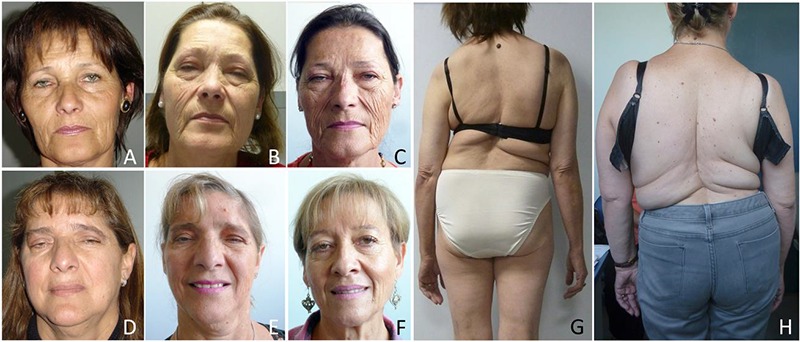
Clinical Features. Progression of facial signs, A to C. Index Case 1 at 52 years (A), 57 years (B) and at 59 years-old (C) after 1 year on salbutamol 4 mg/tid. D and E, Case 2, at 55 years (D) after blepharoplasty and at 60 years old (E); F, Case 3 at 57 years old. Note the mild ptosis on the right eyelid. Scoliosis. Case 1 (G) and Case 2 (H) presented scoliosis since childhood. Retrospectively, this was the first sign of the syndrome.
Fig 2.
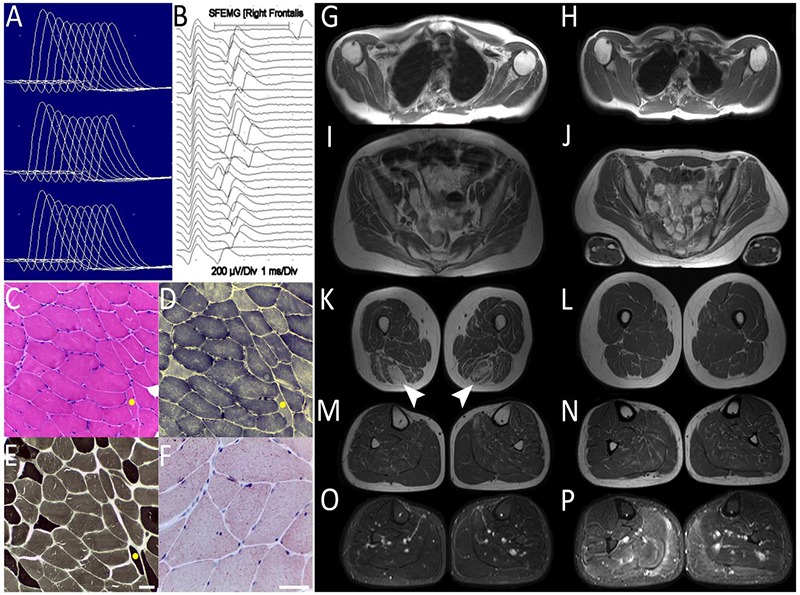
Results of Ancillary Diagnostic Tests. Case 1 Electrophysiological assessment A. Facial nerve repetitive stimulation test. Decremental responses (>15%) of nasalis muscle, at baseline, 1 and 2 min. post activation. B. Frontalis muscle Single Fiber Electromyography. Jitter increase and blocking. Muscle Biopsy. Case 1 left deltoid biopsy. C. HE, fiber size variability, angulated fibers and internalized nuclei. D. NADH-TR, Absence of tubular aggregates. E. Myosin ATPase pH 9.4 preserved fiber type proportion. F, Oil Red O, Diffuse lipid increase. The yellow dot in C, D, F, indicates the same fiber. Scale bars in E and F = 50 µm. Muscle MRI. Panels G to N, T1W and O.P, STIR sequences. Left panels G, I, C, M, O. Case 1 (60 years). G. Shoulder girdle. Diffuse mild fatty infiltration, greater in the subscapularis. I. Pelvic girdle, Gluteus minimus moderate fatty replacement. K. Thighs. Semimembranosus, biceps and sartorius moderate fatty infiltration. Sparing of adductors and quadriceps. Selective severe semitendinosus fatty replacement (arrowheads). M-O. Legs. Mild diffuse fatty replacement. Edema on the right medial gastrocnemius, soleus and lateral peroneus (O). Upper trunk evaluation was difficult due to severe dorsal scoliosis. Right panels H, J, N, L, P. Case 3 (59 years). Absence of fatty infiltration. Bilateral edema of gastrocnemius, soleus, central leg compartments and lateral peroneus, relatively greater on the right side (P).
Patients have given written consent to allow the publication of their face as part of this figure.
Case 2.
62-year-old sister of Case 1, has similar medical history as Case 1. She presented with ptosis since childhood and developed scoliosis during adolescence (Figure 1). The disease onset was during her first pregnancy at 24. She was diagnosed with MG and started on pyridostigmine without effect and poor tolerance. At the age of 25 she underwent thymectomy. She remained stable thereafter receiving pyridostigmine 60 mg/tid until the age of 40. During her second pregnancy evidenced severe worsening requiring mechanical ventilation. After recovery, she started with azathioprine 150 mg/day without improvement, remaining with fluctuating weakness but able to perform daily life activities. She underwent bilateral blepharoplasty twice, at the ages of 54 and 56. At her first visit, she presented neck flexion weakness (M3), proximal bilateral asymmetrical shoulder and pelvic girdles weakness (M3), with lesser impairment of distal upper and lower limb muscles (M4+). DTR and serum CK were in the normal range. Despite eyelid interventions, ptosis was severe (Figure 1D, E). Barre and Mingazzini’s maneuvers scored 45 and 27 seconds respectively. MGC score was 29. Salbutamol, 4 mg every 6 hrs. and nighttime BiPAP were initiated . One year later, ptosis was unchanged; Barre and Mingazzini’s maneuvers scored 116 and 45 seconds respectively and MGC score was 14 (i.e.15 score points improvement). No muscle biopsy or muscle MRI were performed in this patient.
Case 3.
This is a 59-year-old younger sister of Cases 1 and 2. She is asymptomatic but shows isolated mild right eyelid ptosis since she was 50 years old (Figure 1F). She works normally and practices gymnastics 2-3 times a week. She was never pregnant. A whole body muscle MRI showed edema in the gastrocnemius muscles (Figure 2). A single fiber EMG showed increased jitter in 5/10 fibers explored. No muscle biopsy was performed in this patient.
Demography and main clinical and laboratory features of the Dok-7 Chilean Family with c.473G>A [p.Arg158Gln] and c.1457delC [p.Pro486Arg] mutations are summarized in Table 1.
Table 1.
Main Clinical and Laboratory Features of Dok-7 Chilean Family with c.473G>A [p.Arg158Gln] and c.1457delC [p.Pro486Arg] mutations.
| Case | 1 | 2 | 3 |
|---|---|---|---|
| Clinical History | |||
| Age/Sex | 61/F | 62/f | 59/F |
| Age Onset | 18 | 19 | 57/F |
| Onset Symp. | Ptosis | Ptosis1/MSd | Ptosis |
| Ptosis | +++ | +++ | + |
| Face | +++ | ++ | N |
| Neck | +++ | +++ | N |
| Weakness Pattern (at first visit) | |||
| Limb Prox. | +++ | ++ | N |
| Limb Dist. | + | ++ | N |
| Trunk | +++ | ++ | N |
| MGC1 | 43 | 29 | - |
| MGC2 | 24 | 14 | - |
| OTR | ↓ | ↓ | ¯ |
| VC | ↓ | ↓ | N |
| Resp Treat | Pirid. → | Pirid. ↓ | - |
| Salb. ↑ | Salb. ↑ | ||
| Other | Scoliosis / | Scoliosis | Sportive/ |
| ↑pregn./ | ↑pregn./ | Never pregn. | |
| DHS/ | Thymect./ | ||
| BiPAP/ | blepharop. | ||
| CK=200 UI/L |
Abbreviations: MSd= myasthenic syndrome, 1. Ptosis always present. Muscle weakness: N, normal/no weakness. Weakness: +, mild; ++, moderate; +++, severe ↑pregn. = worsening during pregnancy. ↓=reduced; →= stable; ↑=improvement. CK [N]= 30-135 UI/L; DHS= dropped head syndrome. MGC1= before salbutamol treatment, MGC2= after salbutamol treatment.
Other family members.
One of us (JAB) assessed the parents, brothers, sisters and children of the affected women. All presented normal neurological examination and underwent genetic testing (Figure 3).
Fig 3.
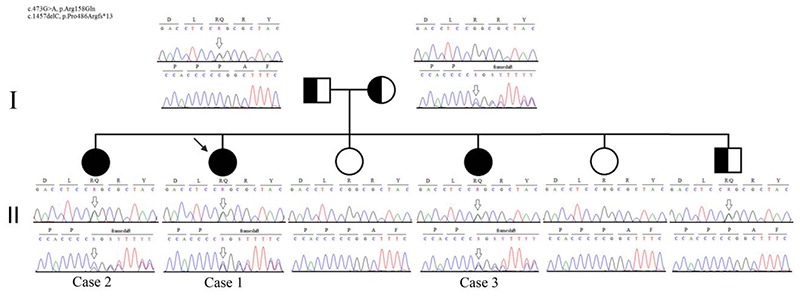
Genogram of the Affected Family. Two generation genetic tree of the Dok-7 Chilean family. The novel frameshift mutation variant c.1457delC p.Pro486Argfs*13 from the mother is present in the three affected sisters and the mutation c.473G>A, p.Arg158Gln from the father is also present at heterozygous state in one asymptomatic brother.
Muscle Biopsy.
Left deltoid muscle biopsy of Case 1 showed fiber size variability, angulated fibers, type 2-fiber atrophy and multiple internalized nuclei in some fibers. Most fibers had lipid accumulation. No tubular aggregates were seen. Electron microscopy was not done.
Muscle MRI Imaging.
Images were acquired using a 1.5 Tesla (T) system (Magnetom Symphony Maestro-Class, Siemens, Erlangen, Germany) and analyzed as described elsewere.18
Case 1 MRI performed at 60 years old (Figure 2 G, I, K, M, O), showed fatty infiltration and edema in most muscles with a selective severe asymmetric impairment of the semitendinosus muscle on the thighs as the most remarkable finding. Muscle MRI of Case 3 at 59 years, (Figure 2H, J, L, N, P), showed no fatty infiltration, but asymmetric bilateral edema on the gastrocnemius-soleus, central compartments of the legs and lateral peroneus.
Mutational Analysis
DNA amplification and sequencing were performed as described previously.19 The institutional review board of the University of California Davis approved the study and all participating individuals signed a consent form. There were no pathogenic mutations in the coding regions and flanking introns of CHRNA1 (MIM:100690); CHRNB1 (MIM:100710); CHRND (MIM:100720); CHRNE (MIM:100725) and RAPSN (MIM:601592). The amplification and sequencing of all exons and flanking introns of DOK7 (MIM:610285) in the proband and in her two affected sisters revealed two pathogenic sequence variants: c.473G>A in exon 4, which results in p.Arg158Gln; and c.1457delC in exon 7, which predicts p.Pro486Argfs*13. Unaffected family members carried only one or neither mutation.
The sequence variant c.473G>A, p.Arg158Gln (rs754633490) is rare (maximum alternate allele frequency in any subpopulation in ExAC = 0.00002) and has been previously reported as pathogenic6. The variant c.1457delC, p.Pro486Argfs*13, has never been reported.
Discussion
To date, no patients with CMS due to DOK7 mutations have ever been reported in Chile. Here we report a family harboring the combination of the novel DOK7 variant c.1457delC (p.Pro486Argfs*13) that truncates the protein at the C terminal domain, with the mutation c.473G>A (p.Arg158Gln) that disrupts the interaction of dok-7 with MuSK at the phosphotyrosine binding (PTB) domain4 and is determinant of pathogenicity. The combined effect of a PTB and a C terminal domain mutation in dok7 results in a recessively transmitted late onset CMS in this Chilean family, which is consistent with previously reported Dok-7 deficient patients.8
Other Latin American patients suffering from CMS have been previously included in some reports,8,20-22 involving a low number of cases. In the report of Mihaylova et al.21, DOK7 mutations were found in 4/25 patients with CMS (17%), representing the second most frequent cause of CMS in the southern Brazilian state of Parana. All patients in the report harbored the recurrent c.1124_1127dupTGCC DOK7 mutation, in combination with a novel aminoacid exchange DOK7 p.G54R in two patients with a mixed Portuguese-Amerindian-African origin, thus suggesting that this latter mutation comes from Africa or is Amerindian. Although no conclusion can be drawn concerning the putative origin of the novel DOK7 mutation c.1457delC reported here, identification of more cases of CMS in Chile may allow establishing its origin and significance. The phenotype of the affected patients consisted of limb girdle myasthenia with facial involvement, without ophthalmoplegia, but with high intrafamilial variability. It comprised a severe form in two sisters with onset in their twenties and particular aggravation by pregnancy; and a mild form in a third sister, who has never been pregnant and has only shown mild unilateral ptosis in her late fifties. The clinical and complementary testing findings in the Chilean family fully agree with CMS due to DOK7 mutations previously reported in the literature,8-11,21 including the worsening during pregnancies,23 and positive response to salbutamol.24 In fact, COLQ mutations would be less likely given the lack of a double motor response on the electrophysiological testing in two of our patients.20 The lack of response to esterase inhibitors, the myopathic features and the absence of arthrogryposis, allows distinction from AChR deficiency and RAPSN mutations respectively.8,10,25 Finally, the absence of tubular aggregates on the biopsy and lipidosis differentiates our patients from those with CMS related to congenital defects of glycosylation (GFPT1, DPAGT1 and ALG2 genes).8,25 Interestingly, pregnancy has been previously reported as a cause of deterioration on CMS patients,23 as it was observed in two of the siblings of this Chilean family. Although the underlying mechanism for this finding remains elusive, it is intriguing whether pregnancy (or its absence), could be another modifying factor of the clinical phenotype of DOK-CMS. Based on our observations, it seems advisable that patients affected by CMS be closely followed up during delivery and the immediate postpartum period. Muscle MRI in the index case showed a definite but non-specific involvement, similar to findings reported in a large MRI series of CMS.26 The most conspicuous MRI feature was the selective involvement of the semitendinosus at the posterior compartment of the thighs, a finding that has been previously described in desminopathy,27 and is therefore no specific. Nevertheless, MRI findings correlated with clinical severity, since minimal MRI changes were observed in the least affected sister. Muscle biopsy of the index case was non-specific either. As in previously reported in DOK7-related CMS,6,8,10 tubular aggregates were not observed. Recognition of DOK7-CMS is important as affected patients often respond poorly to anticholinesterase drugs, but well to sympathomimetics such as ephedrine and salbutamol. Both most affected sisters in this family greatly improved with salbutamol and experienced no significant side effects from this medication. However, improvement was not so evident for facial or respiratory muscles, but was clearly demonstrated by the improvement of muscle strength grading. Similarly to findings recently reported in a review article,24 no adverse reactions were observed in our patients. Unfortunately we cannot ascertain if an earlier treatment with salbutamol would have prevented the progressive motor impairment of our patients or adverted the worsening observed during pregnancy; however, we believe that at least it would have improved their quality of life.
In summary, in this report we aim at increasing awareness of timeliness in diagnosis of DOK-CMS, which would benefit patients with an early treatment, preventing complications including aggravation during pregnancy and puerperium, and avoiding unnecessary medical interventions.
Acknowledgments
Study Supported by NIH Grant 5R01NS049117 to RAM; Grant FONDECYT#1151383 and Grant VID ENL15/14 VID Universidad de Chile to JAB. We thank the patients for their generous participation. Thanks to Ms. Alejandra Trangulao MT, from the ICBM, Facultad de Medicina, Universidad de Chile for expert technical advice; Ms. Marcela Ramírez and Ms. Carolina Donoso from the Departamento de Neurología y Neurocirugía, HCUCH for their assistance and coordination of the study. We also thank Dr. Yamile Corredoira for reviewing the English version of the manuscript.
References
- 1.Rodriguez-Cruz PM, Palace J, Beeson D. Congenital myasthenic syndromes and the neuromuscular junction. Curr Opin Neurol 2014;27:566-75. [DOI] [PubMed] [Google Scholar]
- 2.Beeson D. Congenital myasthenic syndromes: recent advances. Curr Opin Neurol 2016; 29: 565-71. [DOI] [PubMed] [Google Scholar]
- 3.Engel AG, Shen XN, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 2015; 14: 420-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006,313:1975-8. [DOI] [PubMed] [Google Scholar]
- 5.Okada K, Inoue A, Okada M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006;312:1802-5. [DOI] [PubMed] [Google Scholar]
- 6.Palace J, Lashley D, Newsom-Davis J, et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain 2007;130:1507-15. [DOI] [PubMed] [Google Scholar]
- 7.Coussins J, Liu WW, Belaya K, et al. The spectrum of mutations that underlie the neuromuscular junction synaptopathy in DOK7 congenital myasthenic syndrome. Hum Mol Genet 2012;21,3765-75. [DOI] [PubMed] [Google Scholar]
- 8.Muller J, Herczegfalvi A, Vilchez JJ, et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain 2007;130:1497-506 [DOI] [PubMed] [Google Scholar]
- 9.Anderson JA, Jarae J, Bowe C, et al. Variable phenotypes associated with mutations in DOK7. Muscle Nerve 2008;37:448-56. [DOI] [PubMed] [Google Scholar]
- 10.Ben Ammar A, Petit F, Alexandri N, et al. Phenotype genotype analysis in 15 patients presenting a congenital myasthenic syndrome due to mutations in DOK7. J Neurol. 2010;257:754-66 [DOI] [PubMed] [Google Scholar]
- 11.Klein A, Pitt MC, McHugh JC, et al. DOK7 congenital myasthenic syndrome in childhood: early diagnostic clues in 23 children. Neuromuscul Disord 2013;23:883-91. [DOI] [PubMed] [Google Scholar]
- 12.Schara U, Barisic N, Deschauer M, et al. Neuromuscul Disord 2009;19:828-32. [DOI] [PubMed] [Google Scholar]
- 13.Burke G, Hiscock A, Klein A, et al. Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul Disord 2013;23:170-5. [DOI] [PubMed] [Google Scholar]
- 14.Lorenzoni PJ, Scola RH, Kay CSK, et al. Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J Neurol Sci 2013;331:155-7. [DOI] [PubMed] [Google Scholar]
- 15.Tsao Ch. Effective Treatment With Albuterol in DOK7 Congenital Myasthenic Syndrome in Children. Pediatr Neurol 2016;54:85-7. [DOI] [PubMed] [Google Scholar]
- 16.Lashley D, Palace J, Jayawant S, et al. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology 2010;74:1517-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns TM, Conaway M, Sanders DB. The MG Composite: A valid and reliable outcome measure for myasthenia gravis. Neurology 2010;74:1434-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Díaz J, Woudt L, Suazo L, et al. Broadening the imaging phenotype of dysferlinopathy at different disease stages. Muscle Nerve 2016;54:203-10 [DOI] [PubMed] [Google Scholar]
- 19.Maselli RA Fernandez JM, Arredondoet al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 2012;131:1123-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mihaylova V, Müller JS, Vilchez JJ, et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain. 2008. 131:747-59. [DOI] [PubMed] [Google Scholar]
- 21.Mihaylova V, Scola RH, Gervini B, et al. Molecular characterisation of congenital myasthenic syndromes in Southern Brazil. J Neurol Neurosurg Psychiatry. 2010. 81:973-7. [DOI] [PubMed] [Google Scholar]
- 22.Abicht A, Dusl M, Gallenmüller C, et al. Congenital myasthenic syndromes: achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: a study of 680 patients. Hum Mutat. 2012. 33:1474-84. [DOI] [PubMed] [Google Scholar]
- 23.Servais L, Baudoin H, Zehrouni K, et al. Pregnancy in congenital myasthenic syndrome. J Neurol. 2013;260:815-9. [DOI] [PubMed] [Google Scholar]
- 24.Witting N, Vissing J. Pharmacologic treatment of downstream of tyrosine kinase 7 congenital myasthenic syndrome. JAMA Neurol. 2014;71: 350-4. [DOI] [PubMed] [Google Scholar]
- 25.Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015,14:420-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fynlayson S, Morrow JM, Rodriguez-Cruz PM, et al. Muscle Nerve 2016;54:211-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 2008;71:758-65. [DOI] [PMC free article] [PubMed] [Google Scholar]