

Summary
In recent decades, considerable effort has been devoted to finding microbial source‐tracking (MST) markers that are suitable to assess the health risks of faecally polluted waters, with no universal marker reported so far. In this study, the abundance and prevalence of a crAssphage‐derived DNA marker in wastewaters of human and animal origins were studied by a new qPCR assay with the ultimate aim of assessing its potential as an MST marker. crAssphage showed up to 106 GC/ml in the sewage samples of human origin, in both the total DNA and the viral DNA fraction. In wastewaters containing animal faecal remains, 39% of the samples were negative for the presence of the crAssphage sequence, while those showing positive results (41% of the samples) were at least 1 log10 unit lower than the samples of human origin. Noteworthy, the log10 values of the ratio (R) crAssphage (GC/ml)/Escherichia coli (CFU/ml) varied significantly depending on the human or animal origin (R > 1.5 for human samples and R < −1.5 for animal wastewater samples. This study opens the way for further research to explore if different specific animal variants of crAssphage exist and whether other zones of the crAssphage genome are better suited to source discrimination.
Introduction
Waterborne diseases transmitted via the faecal–oral route make a significant contribution to the burden of diseases worldwide (WHO, 2008). Faecal pollution has several origins, human or diverse animals, and determination of the faecal sources accompanied by appropriate water resource management policies could contribute to improving the microbial quality of water (Blanch et al., 2006; Hagedorn et al., 2011). Source‐tracking methods allow the origin of faecal pollution in a particular body of water to be determined (Scott et al., 2002; U.S. EPA, 2005; Hagedorn et al., 2011; Harwood et al., 2014). A number of approaches have been developed to identify the source of faecal pollution, in attempts to associate various animals with faecal contamination of natural waters (Blanch et al., 2006; Harwood et al., 2014). These methods are in various stages of development and approval. Those based on microbiological determination are defined as microbial source‐tracking (MST) methods.
The analysis of gastrointestinal microorganisms indicates that about 400 different species of bacteria can be found in the animal intestine and that populations are of the order of 1011/g, although only a very small fraction of these bacteria have been cultured (Zoetendal et al., 2004). The intestinal microbiome has been characterized in detail in several animal hosts, including humans (Eckburg et al., 2005; Gill et al., 2006), pigs (Leser et al., 2002) and cattle (Ramsak et al., 2000). Culture‐independent methods suggest that numerically dominant bacteria in the colon of animals are anaerobic and belong to the bacterial phylum Cytophaga‐Flavobacter‐Bacteroides, with Bacteroides being one of the most common genera in the animal intestine (Matsuki et al., 2002; Eckburg et al., 2005; Ley et al., 2008; Bradford et al., 2013; Ishikawa et al., 2013; Wei et al., 2013). However, anaerobic bacteria (Bacteroides, Bifidobacterium) are not easily grown in the laboratory, which has limited their use as faecal indicators in the past, and for this reason molecular methods for their detection have been developed (Bernhard and Field, 2000; Haugland et al., 2010; Gómez‐Doñate et al., 2012; Green et al., 2014; Mayer et al., 2016).
In this context, the molecular detection of host‐specific Bacteroides species as well as bacteriophages infecting different Bacteroides species (Tartera and Jofre, 1987; Payan et al., 2005; Gómez‐Doñate et al., 2011; Jofre et al., 2014; McMinn et al., 2014; Venegas et al., 2015) have proved to be useful in MST studies. However, a drawback with both approaches is a certain geographical inconsistency of the results (Reischer et al., 2013; Jofre et al., 2014).
Recent metagenomic studies of human intestinal contents, and ‘in silico’ analyses have revealed DNA sequences present in the majority of published human faecal metagenomes (Dutilh et al., 2014). These sequences have been identified as belonging to a single, previously unidentified bacteriophage, named crAssphage after the cross assembly program used to generate it (Dutilh et al., 2014). This virtual bacteriophage was assigned to Bacteroides ssp in accordance with CRISPR analysis. However, the phage does not resemble previously reported Bacteroides phages like phage B‐40 (ATCC 51477‐B1 eB40‐8) infecting Bacteroides fragilis (Tartera and Jofre, 1987) and phage ΦB124‐14 which infects strain GB‐124 of B. fragilis (Ogilvie et al., 2012).
This study presents the molecular detection of a crAssphage‐derived DNA sequence (referred to herein as crAssphage) in samples with faecal pollution of known origin (human or animal) using a newly designed qPCR and evaluates the differential occurrence of crAssphage in human‐polluted samples and its potential as a molecular MST marker.
Results and discussion
Sequencing crAssphage amplimer from HM samples
In order to design a qPCR to detect the crAssphage genome first, a total of 10 samples of human (HM) sewage DNA samples were amplified and sequenced using primers reported in Fig. S1 targeting a region of 1331 bp. The amplicons generated revealed certain sequence variation, indicating that different crAssphage variants may exist. This is in accordance with previous results from Liang and coworkers that showed high genetic diversity in ORF00018 and ORF00039 of the crAssphage sequence available at GenBank (accession number NC_024711.1) in the faeces of Chinese patients (Liang et al., 2016). Nevertheless, it was possible to find a conserved region of 78 bp, which was chosen to develop a qPCR assay for the molecular quantification of crAssphage.
Validation of the crAssphage qPCR assay
The standard curves of the crAssphage assay were reproducible. The average standard curve is shown in Fig. 1 with the corresponding equation. The qPCR assay was shown to have an efficiency of 100.4% and showed a limit of quantification (LOQ) of 1.27 gene copies (GC)/μl of the analysed sample (7 ul) of the qPCR mixture. The LOQ is defined as the last value of the standard curve that showed consistent and reproducible results and that is used to calculate the efficiency the qPCR assay.
Figure 1.
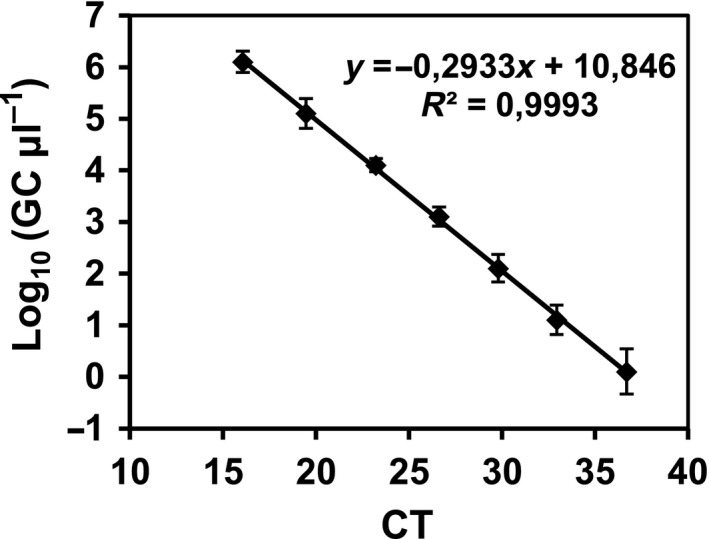
crAssphage qPCR assay. Standard curve obtained with pGEM plasmid containing the 1331‐bp sequence fragment obtained from a HM sewage sample DNA. CT, threshold cycle; GC, gene copies.
Quantification of crAssphage in sewage and wastewater samples
crAssphage was detected in the viral DNA and the total DNA fraction, although the concentration was around 1 log10 units lower than in total DNA fraction (data not shown). crAssphage has never been isolated, and we are not certain of their prevalence as a free virion in environmental settings and, therefore, we decided to use the total DNA fraction to analyse simultaneously free virions and prophages inserted within the Bacteroides genomes.
High concentrations of crAssphage (GC/ml) were detected in the total DNA fraction of 100% of the 23 samples of raw municipal sewage tested (Fig. 2A), with values ranging from 5.4 to 6.9 log10 GC/ml and an average value of 6.24 (standard error (SE) = 0.1). These values are higher than those reported for other microbial source markers found in high numbers such as norovirus (da Silva et al., 2007), adenovirus and polyomavirus (Bofill‐Mas et al., 2006), human bifidobacteria (Gómez‐Doñate et al., 2012) and human Bacteroidetes (Gómez‐Doñate et al., 2016; Mayer et al., 2016).
Figure 2.
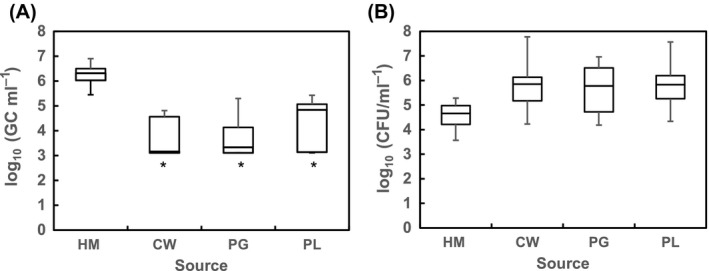
Densities (represented in box‐and‐whisker plot graphs) of (A) crAssphage (GC/ml) and (B) Escherichia coli (CFU/ml) in the different source samples: HM, human sewage (n = 23); PG, porcine wastewater (n = 15); CW, cow wastewater (n = 12); PL, poultry wastewater (n = 14). *, in these samples the lowest fence value in the graph corresponds to the limit of quantification of the qPCR as some of the samples were considered negative in the qPCR; therefore, the actual lowest value is expected to be lower than the plotted value.
However, crAssphage was also found in samples contaminated with faecal remains from certain animals, though in lower concentrations than in the human sources. Sixty‐one per cent of the samples (n = 41) collected from sources contaminated with faeces of different animals were positive for the presence of crAssphage. Logarithmic values presented as box‐and‐whisker plot graphs are shown in Fig. 2A. In this case, the values ranged from 3.2 to 4.8 log10 GC/ml in CW, 3.2 to 5.3 log10 GC/ml in PG and 3.2 to 5.4 in PL samples, with average values of 3.7 (SD = 0.75), 3.7 (SD = 0.67) and 4.1 (SD = 0.95) for the different animal wastewaters respectively. The concentration of the phage was significantly (p < 0.05) higher in human than in animal wastewaters.
The crAssphage sequence was originally identified from metagenomic analysis of human samples and was reported to be highly abundant in humans (Dutilh et al., 2014). To the best of our knowledge, there is only one published study of the prevalence of this phage in non‐human samples (Stachler and Bibby, 2014). Stachler and Bibby studied the presence of the phage in metagenomes from different animals including cow, pig and chicken, but the phage was not detected in any sample, except those from bats. In our study, crAssphage was detected in more than half of the animal samples analysed but in lower densities than in human samples. These differences between the results of the two studies may be due to two main reasons. First, the low number of samples analysed by Stachler and Bibby (n was between 1 and 4 for the genomes analysed); second, crAssphage may be not the dominant phage in animal samples and may not be detected in metagenomics analysis, but may be detected when targeting a specific sequence. In any case, the high abundance of this phage found in our study is important for developing an MST method, so that small amounts of faecal contamination can be detected. However, according to our results, the detection of its presence on its own may not be sufficient to track the origin of the contamination. Therefore, the use of the bacteriophage in combination with another faecal marker was studied.
crAssphage and Escherichia coli for tracking human faecal contamination
As determined by the concentration of the universal indicator E. coli, the different samples tested had quite different concentrations of faecal residues with the highest values corresponding to the samples of animal origin (Fig. 2B). To examine the potential effect of the concentration of faecal remains in the samples, the crAssphage results were considered versus the abundance of E. coli by calculating the log10 value of the ratio between crAssphage (GC/ml) and E. coli (CFU/ml) (i.e. log10 [crAssphage (GC/ml)/E. coli (CFU/ml)]). The arithmetic means (± standard error) of the log10 values of these ratios are plotted in Fig. 3.
Figure 3.
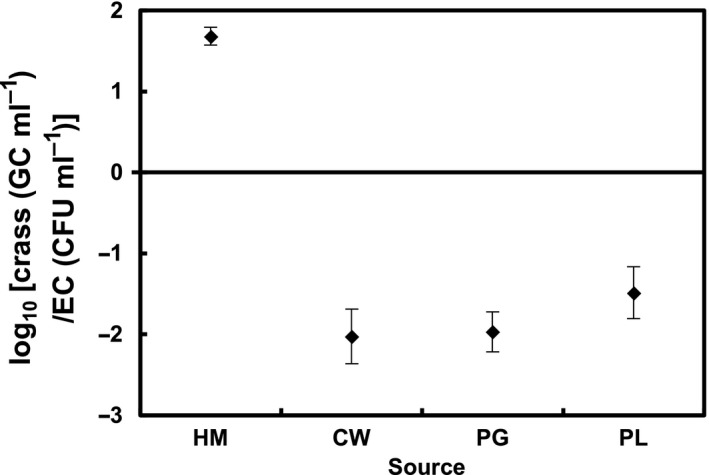
Observed log10 ratio [crAssphage (GC/ml)/Escherichia coli (CFU/ml)] for the different faecal pollution sources (mean ± standard error). crass: crAssphage; EC: E. coli.
The log10 values of the ratios differed significantly (Kruskal–Wallis, p < 0.05) between human (ratio average = 1.7, min = 0.6 and max = 2.3) and non‐human samples (ratio average < −1.5, max = −0.9 and min = −4.7 for CW, max = −0.7 and min = −3.4 for PG, and max = 0.33 and min = −3.9 for PL), and therefore, it was possible to distinguish human from non‐human pollution source, based on the use of only two markers.
Field studies using most of the numerous chemical and microbiological methods available to track sources of faecal contamination have shown that the existing methods are insufficient and that different markers are needed (Blanch et al., 2004; Muniesa et al., 2012). Other authors have also indicated that the abundance ratio between a discriminating and non‐discriminating marker may be a useful tool. For example, in the late 1960s, Geldreich and Kenner (Geldreich and Kenner, 1969) proposed the ratio faecal coliforms/Streptococcus fecalis, and more recently, Sauer et al. (2011) discussed the usefulness of the abundance ratio human Bacteroidales/total Bacteroidales and Newton et al. (2011) proposed the use of the abundance ratio among Lachno2, a human‐associated phylotype within the Lachnospiraceae family and quantitative values of enterococci obtained by qPCR. And more recently, Muniesa and coworkers reported the usefulness of the ratio somatic coliphages/phages infecting Bacteroides thetaiotaomicron GA17 strain to predict human faecal pollution sources (Muniesa et al., 2012).
In conclusion, a methodology was developed to detect human faecal polluted water based on the quantification of just two markers: the recently reported crAssphage and the worldwide used indicator E. coli. Accordingly, crAssphage was present in some animal samples, but the use of the log10 ratio obtained among crAssphage/E. coli was suitable for the significant differentiation of human versus diverse animal faecal pollution sources. Furthermore, the observed numbers of crAssphage are amongst the highest reported for any human marker so far and therefore the proposed method besides being easy to perform, and robust could be used in highly diluted samples. These results are quite promising and make further research worthwhile to establish if different specific animal variants of crAssphage exist, and whether other genome regions of crAssphage are better suited to discriminating the animal source.
Experimental procedures
Wastewater samples
A total of 64 wastewater samples were collected from various sources. Twenty‐three samples were obtained from different urban wastewater treatment plants (WWTPs) (HM samples) serving populations ranging from 5000 to 384 000 inhabitants in Catalonia (NE Spain). A total of 14 samples of poultry slurry (PL samples) were collected from two poultry slaughter houses that each slay 60 000 animals per week. A total of 15 samples of pig slurry (PG samples) were obtained from four pig abattoirs that slay 12 500, 15 000, 15 000 and 5000 pigs per week. Finally, a total of 12 samples of cattle slurry (CW samples) were collected from four slaughterhouses butchering weekly between 250 and 2000 calves.
Detection of E. coli
Escherichia coli was used as indicator of bacterial faecal pollution. Escherichia coli was enumerated by membrane filtration based on the ISO standard method 16649‐1:2001 with an initial resuscitation stage on MMGA (4 h at 37 °C) followed by incubation in chromogenic TBX agar at 44 °C (ISO, 2001).
Total DNA extraction
Total DNA was isolated from 0.2 ml samples with the QIAamp DNA blood minikit (Qiagen GmbH, Hilden, Germany), following the manufacturer's instructions. The DNA was suspended in a final volume of 200 μl of elution buffer. The integrity of the genomic DNA extracted was evaluated by 0.8% agarose gel electrophoresis and ethidium bromide staining.
Bacteriophage DNA extraction
The bacteriophage DNA fraction was extracted from 0.2 ml samples previously filtered through low‐protein‐binding 0.22 μm‐pore‐size membrane filters (Millex‐GP, Millipore) using the method described above.
End‐point PCR assay
DNA was amplified by PCR from 10 HM samples using primers derived from the crAssphage sequence available in GenBank (accession number NC_024711.1) (Fig. S1) targeting a 1331 bp fragment (crAss1 5′‐CTGATAGTATGATTGGTAATG‐3′ and crAss2 5′‐AAGATAGTTGGAGAACTTAT‐3′) and sequenced using the ABI Prism BigDye 3.1 terminator cycle sequencing ready reaction kit (Life Technologies, Madrid, Spain).
Quantitative PCR assay
A qPCR assay was developed based on the previously obtained consensus sequences from HM samples. crAss‐UP primer (5′‐AGGAGAAAGTGAACGTGGAAACA‐3′), crAss‐LP primer (5′‐TAAAGCTTAAAGTTGGTGCTCGTT‐3′) (which differed in a single base pair from the crAssphage sequence available at GenBank) and a Taqman probe with a 3′‐FAM carboxyfluorescein reporter and a 5′‐MGBNFQ (minor groove binding non‐fluorescent) quencher (FAM‐AGGATTTGGAGAAGGAA‐MGBNFQ) were designed using Primer Express 3.0.1 (Applied Biosystems) to amplify a 78‐bp fragment encoding the KP06_gp31 gene. The qPCR was performed using 7 μl of the DNA (total or phage DNA) extracted from each wastewater sample. The assay was performed under standard conditions as previously described (Gómez‐Doñate et al., 2012). All the samples were run in duplicate. Threshold cycle (Ct) data were expressed as the number of GC according to the values obtained with the standard for each qPCR reaction.
To generate standards for the qPCR assay, the 1331‐bp fragment was amplified from a HM sample. The amplified 1331‐bp fragment was cloned into the pGEM‐T Easy vector following the manufacturer's instructions (Promega Biotech Ibérica, Barcelona, Spain) and transformed by electroporation (2.5 kV, 25 F capacitance and 200 Ω resistance) into E. coli DH5α electrocompetent cells. The ampicillin‐resistant colonies containing the vector with the insert were selected, verified by PCR and used to purify the plasmid using a Qiagen Plasmid Midi purification kit (Qiagen, Valencia, CA). A NanoDrop ND‐1000 spectrophotometer (Thermoscientifics, Wilmington, DE) was used to evaluate the concentration and purity of the construct containing each band.
To calculate the number of gene copies (GC) in the prepared stock, the following equation was used as follows: [concentration of pGEM‐T Easy::insert (ng/μl)/molecular mass (ng/mol)] × 6.022 × 1023 molecules/mol = number of molecules of pGEM‐T Easy::insert/μl. Ten‐fold serial dilutions of the stock were performed with double‐distilled water and stored at −80 °C until used. The stocks were amplified in duplicate in five independent experiments, and the average of the Ct results was used to elaborate standard curves.
Statistical analyses
The data corresponding to the different sample types are presented as box plots of multiple variables. Comparison of data from all types of samples was conducted using the Kruskal–Wallis test (statgraphics plus software (version 5.1)). For ratio calculations, the limit of detection was used for samples which were below the limit of detection.
Conflict of Interest
None declared.
Supporting information
Fig. S1. CrAssphage qPCR assay. Location of primers and probes designed for the end‐point and qPCR assay.
Acknowledgements
We thank Prof. Rob Edwards at San Diego State University for the information on the primer sequences to conduct the End‐Point PCR.
Microbial Biotechnology (2017) 10(6), 1775–1780
Originality‐Significance Statement: In this manuscript, a qPCR was designed for the detection of crAssphage, which was later used to develop a method to predict human faecal pollution. High numbers of crAssphage were detected in wastewaters with human faecal pollution, although the crAssphage target sequence was detected in some animal wastewater samples (pig, cow and specially poultry) for the first time. Nevertheless, the log10 ratio crAssphage/E. coli allowed the precise detection of human faecal pollution.
Funding Information
This study was supported by the Generalitat de Catalunya [2014SGR007] and by the Xarxa de Referència en Biotecnologia (XRB).
References
- Bernhard, A. E. , and Field, K. G. (2000) Identification of nonpoint sources of fecal pollution in coastal waters by using host‐specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl Environ Microbiol 66: 1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanch, A. R. , Belanche‐Muñoz, L. , Bonjoch, X. , Ebdon, J. , Gantzer, C. , Lucena, F. , et al (2004) Tracking the origin of faecal pollution in surface water: an ongoing project within the European Union research programme. J Water Health 2: 249–260. [PubMed] [Google Scholar]
- Blanch, A. R. , Belanche‐Muñoz, L. , Bonjoch, X. , Ebdon, J. , Gantzer, C. , Lucena, F. , et al (2006) Integrated analysis of established and novel microbial and chemical methods for microbial source tracking. Appl Environ Microbiol 72: 5915–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill‐Mas, S. , Albinana‐Gimenez, N. , Clemente‐Casares, P. , Hundesa, A. , Rodriguez‐Manzano, J. , Allard, A. , et al (2006) Quantification and stability of human adenoviruses and polyomavirus JCPyV in wastewater matrices. Appl Environ Microbiol 72: 7894–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, S. A. , Morales, V. L. , Zhang, W. , Harvey, R. W. , Packman, A. I. , Mohanram, A. , and Welty, C. (2013) Transport and fate of microbial pathogens in agricultural settings. Crit Rev Environ Sci Technol 43: 775–893. [Google Scholar]
- Dutilh, B. E. , Cassman, N. , McNair, K. , Sanchez, S. E. , Silva, G. G. Z. , Boling, L. , et al (2014) A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun 5: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg, P. B. , Bik, E. M. , Bernstein, C. N. , Purdom, E. , Dethlefsen, L. , Sargent, M. , et al (2005) Diversity of the human intestinal microbial flora. Science 308: 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldreich, E. E. , and Kenner, B. A. (1969) Concepts of fecal streptococci in stream pollution. J Water Pollut Control Fed 41(Suppl): R336+. [PubMed] [Google Scholar]
- Gill, S. R. , Pop, M. , DeBoy, R. T. , Eckburg, P. B. , Turnbaugh, P. J. , Samuel, B. S. , et al (2006) Metagenomic analysis of the human distal gut microbiome. Science 312: 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Doñate, M. , Payán, A. , Cortés, I. , Blanch, A. R. , Lucena, F. , Jofre, J. , and Muniesa, M. (2011) Isolation of bacteriophage host strains of Bacteroides species suitable for tracking sources of animal faecal pollution in water. Environ Microbiol 13: 1622–1631. [DOI] [PubMed] [Google Scholar]
- Gómez‐Doñate, M. , Ballesté, E. , Muniesa, M. , and Blanch, A. R. (2012) New molecular quantitative PCR assay for detection of host‐specific Bifidobacteriaceae suitable for microbial source tracking. Appl Environ Microbiol 78: 5788–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Doñate, M. , Casanovas‐Massana, A. , Muniesa, M. , and Blanch, A. R. (2016) Development of new host‐specific Bacteroides qPCRs for the identification of fecal contamination sources in water. Microbiol Open 5: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, H. C. , Haugland, R. A. , Varma, M. , Millen, H. T. , Borchardt, M. A. , Field, K. G. , et al (2014) Improved HF183 quantitative real‐time PCR assay for characterization of human fecal pollution in ambient surface water samples. Appl Environ Microbiol 80: 3086–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn C., Blanch A. R., and Harwood V. J. (eds) (2011) Microbial Source Tracking: Methods, Applications, and Case Studies. New York, NY: Springer. [Google Scholar]
- Harwood, V. J. , Staley, C. , Badgley, B. D. , Borges, K. , and Korajkic, A. (2014) Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol Rev 38: 1–40. [DOI] [PubMed] [Google Scholar]
- Haugland, R. A. , Varma, M. , Sivaganesan, M. , Kelty, C. , Peed, L. , and Shanks, O. C. (2010) Evaluation of genetic markers from the 16S rRNA gene V2 region for use in quantitative detection of selected Bacteroidales species and human fecal waste by qPCR. Syst Appl Microbiol 33: 348–357. [DOI] [PubMed] [Google Scholar]
- Ishikawa, E. , Matsuki, T. , Kubota, H. , Makino, H. , Sakai, T. , Oishi, K. , et al (2013) Ethnic diversity of gut microbiota: species characterization of Bacteroides fragilis group and genus Bifidobacterium in healthy Belgian adults, and comparison with data from Japanese subjects. J Biosci Bioeng 116: 265–270. [DOI] [PubMed] [Google Scholar]
- ISO (2001) Microbiology of Food and Animal Feeding Stuffs – Horizontal Method for the Enumration of B‐glucoronidase‐positive Escherichia coli ‐ Part 1: Colony‐count Technique at 44°C using Membranes and 5‐Bromo‐4‐chloro‐3‐indolyl B‐glucuronide. ISO 16649‐1:2001 04. Geneva, Switzerland: International Organization of Standardization. [Google Scholar]
- Jofre, J. , Blanch, A. R. , Lucena, F. , and Muniesa, M. (2014) Bacteriophages infecting Bacteroides as a marker for microbial source tracking. Water Res 55: 1–11. [DOI] [PubMed] [Google Scholar]
- Leser, T. D. , Amenuvor, J. Z. , Jensen, T. K. , Lindecrona, R. H. , Boye, M. , and Møller, K. (2002) Culture‐independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl Environ Microbiol 68: 673–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Lozupone, C. A. , Hamady, M. , Knight, R. , and Gordon, J. I. (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6: 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Y. , Zhang, W. , Tong, Y. , and Chen, S. (2016) CrAssphage is not associated with diarrhoea and has high genetic diversity. Epidemiol Infect 144: 3549–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki, T. , Watanabe, K. , Fujimoto, J. , Miyamoto, Y. , Takada, T. , Matsumoto, K. , et al (2002) Development of 16S rRNA‐gene‐targeted group‐specific primers for the detection and identification of predominant bacteria in human feces. Appl Environ Microbiol 68: 5445–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, R. E. , Bofill‐Mas, S. , Egle, L. , Reischer, G. H. , Schade, M. , Fernandez‐Cassi, X. , et al (2016) Occurrence of human‐associated Bacteroidetes genetic source tracking markers in raw and treated wastewater of municipal and domestic origin and comparison to standard and alternative indicators of faecal pollution. Water Res 90: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMinn, B. R. , Korajkic, A. , and Ashbolt, N. J. (2014) Evaluation of Bacteroides fragilis GB‐124 bacteriophages as novel human‐associated faecal indicators in the United States. Lett Appl Microbiol 59: 115–121. [DOI] [PubMed] [Google Scholar]
- Muniesa, M. , Lucena, F. , Blanch, A. R. , Payán, A. , and Jofre, J. (2012) Use of abundance ratios of somatic coliphages and bacteriophages of Bacteroides thetaiotaomicron GA17 for microbial source identification. Water Res 46: 6410–6418. [DOI] [PubMed] [Google Scholar]
- Newton, R. J. , Vandewalle, J. L. , Borchardt, M. A. , Gorelick, M. H. , and McLellan, S. L. (2011) Lachnospiraceae and Bacteroidales alternative fecal indicators reveal chronic human sewage contamination in an urban harbor. Appl Environ Microbiol 77: 6972–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie, L. A. , Caplin, J. , Dedi, C. , Diston, D. , Cheek, E. , Bowler, L. , et al (2012) Comparative (meta)genomic analysis and ecological profiling of human gut‐specific bacteriophage φB124‐14. PLoS ONE 7: e35053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payan, A. , Ebdon, J. , Taylor, H. , Gantzer, C. , Ottoson, J. , Papageorgiou, G. T. , et al (2005) Method for isolation of Bacteroides bacteriophage host strains suitable for tracking sources of fecal pollution in water. Appl Environ Microbiol 71: 5659–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsak, A. , Peterka, M. , Tajima, K. , Martin, J. C. , Wood, J. , Johnston, M. E. A. , et al (2000) Unravelling the genetic diversity of ruminal bacteria belonging to the CFB phylum. FEMS Microbiol Ecol 33: 69–79. [DOI] [PubMed] [Google Scholar]
- Reischer, G. H. , Ebdon, J. E. , Bauer, J. M. , Schuster, N. , Ahmed, W. , Åström, J. , et al (2013) Performance characteristics of qPCR assays targeting human‐ and ruminant‐associated Bacteroidetes for microbial source tracking across sixteen countries on six continents. Environ Sci Technol 47: 8548–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, E. P. , VandeWalle, J. L. , Bootsma, M. J. , and McLellan, S. L. (2011) Detection of the human specific Bacteroides genetic marker provides evidence of widespread sewage contamination of stormwater in the urban environment. Water Res 45: 4081–4091. [DOI] [PubMed] [Google Scholar]
- Scott, T. M. , Rose, J. B. , Jenkins, T. M. , Farrah, S. R. , and Lukasik, J. (2002) Microbial source tracking: current methodology and future directions. Appl Environ Microbiol 68: 5796–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva, A. K. , Le Saux, J.‐C. , Parnaudeau, S. , Pommepuy, M. , Elimelech, M. , and Le Guyader, F. S. (2007) Evaluation of removal of noroviruses during wastewater treatment, using real‐time reverse transcription‐PCR: different behaviors of genogroups I and II. Appl Environ Microbiol 73: 7891–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachler, E. , and Bibby, K. (2014) Metagenomic evaluation of the highly abundant human gut bacteriophage CrAssphage for source tracking of human fecal pollution. Environ Sci Technol Lett 1: 405–409. [Google Scholar]
- Tartera, C. , and Jofre, J. (1987) Bacteriophages active against Bacteroides fragilis in sewage‐polluted waters. Appl Environ Microbiol 53: 1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA (2005) Microbial source tracking. EPA/ORD Source Tracking Group.
- Venegas, C. , Diez, H. , Blanch, A. R. , Jofre, J. , and Campos, C. (2015) Microbial source markers assessment in the Bogotá River basin (Colombia). J. Water Health 13: 801–810. [DOI] [PubMed] [Google Scholar]
- Wei, S. , Morrison, M. , and Yu, Z. (2013) Bacterial census of poultry intestinal microbiome. Poult Sci 92: 671–683. [DOI] [PubMed] [Google Scholar]
- WHO (2008) The global burden disease. 2004. Update.
- Zoetendal, E. G. , Collier, C. T. , Koike, S. , Mackie, R. I. , and Gaskins, H. R. (2004) Molecular ecological analysis of the gastrointestinal microbiota: a review. J Nutr 134: 465–472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CrAssphage qPCR assay. Location of primers and probes designed for the end‐point and qPCR assay.