

Abstract
L-asparaginase is a critical chemotherapeutic agent for acute lymphoblastic leukemia (ALL). It hydrolyzes plasma asparagine into aspartate and NH3, causing asparagine deficit and inhibition of protein synthesis and eventually, leukemic cell death. However, patient relapse often occurs due to development of resistance. The molecular mechanism by which ALL cells acquire resistance to L-asparaginase is unknown. Therefore, we sought to identify genes that are involved in L-asparaginase resistance in primary leukemic cells. By unbiased genome-wide RNAi screening, we found that among 10 resistant ALL clones, six hits were for opioid receptor mu 1 (oprm1), two hits were for carbonic anhydrase 1 (ca1) and another two hits were for ubiquitin-conjugating enzyme E2C (ube2c). We also found that OPRM1 is expressed in all leukemic cells tested. Specific knockdown of OPRM1 confers L-asparaginase resistance, validating our genome-wide retroviral shRNA library screening data. Methadone, an agonist of OPRM1, enhances the sensitivity of parental leukemic cells, but not OPRM1-depleted cells, to L-asparaginase treatment, indicating that OPRM1 is required for the synergistic action of L-asparaginase and methadone, and that OPRM1 loss promotes leukemic cell survival likely through downregulation of the OPRM1-mediated apoptotic pathway. Consistent with this premise, patient leukemic cells with relatively high levels of OPRM1 are more sensitive to L-asparaginase treatment compared to OPRM1-depleted leukemic cells, further indicating that OPRM1 loss has a crucial role in L-asparaginase resistance in leukemic patients. Thus, our study demonstrates for the first time, a novel OPRM1-mediated mechanism for L-asparaginase resistance in ALL, and identifies OPRM1 as a functional biomarker for defining high-risk subpopulations and for the detection of evolving resistant clones. Oprm1 may also be utilized for effective treatment of L-asparaginase-resistant ALL.
Introduction
The 2013 SEER Stat Fact Sheets from the National Cancer Institute1 indicate that there were ~78 000 people with acute lymphoblastic leukemia (ALL) in the United States alone, roughly 66% of those affected are youths, making ALL the most widespread pediatric cancer. L-asparaginase is one of the central components of current combination chemotherapy against ALL. Normally, L-asparagine is synthesized from L-aspartic acid and NH3 by L-asparagine synthetase whose level is critically downregulated in leukemic cells.2, 3 Thus, leukemic cells heavily depend on the availability of plasma asparagine for their protein synthesis and survival. L-asparaginase in the ALL treatment regimen depletes plasma asparagine by hydrolyzing the latter into aspartic acid and NH3, thereby causing cellular asparagine deficit, inhibition of protein synthesis and eventually, leukemic cell death. Although the outcome in childhood leukemia has improved significantly in the recent past, challenges arise as ALL patients treated with L-asparaginase relapse due to development of resistance, resulting in refractory disease. It would stand to reason that L-asparaginase-resistant cells have generated a mechanism that provides access to asparagine. One proposed mechanism involves the upregulation of L-asparagine synthetase activity, as elevated L-asparagine synthetase level was detected in resistant primary samples and cell lines.4 Indeed, cellular L-asparagine synthetase level correlates with the half-maximal inhibitory concentration (IC50) obtained following asparaginase treatment.5 However, there have also been reports where no correlation between L-asparagine synthetase level and L-asparaginase resistance was observed,6, 7 suggesting that other unknown factors may contribute to L-asparaginase resistance. To elucidate the molecular mechanism by which ALL cells acquire resistance to L-asparaginase, we sought to identify genes that are involved in L-asparaginase resistance. In this study, we employed an unbiased genome-wide loss-of-function screening against L-asparaginase-sensitive pediatric ALL cells. We found that among randomly selected resistant clones, a significant majority has shown loss of the opioid receptor mu 1 (OPRM1). We present further analyses of this finding and demonstrate that, indeed, L-asparaginase refractory ALL coincides with downregulation of OPRM1.
Results and discussion
To identify genes that are involved in L-asparaginase resistance, we utilized an unbiased genome-wide RNAi screening8 that targets 24 000 unique shRNAs covering 8 000 vital genes in continuously growing leukemia cells (POETIC2) established from a pediatric leukemia patient. These cells were prepared by high-density culture of blast cells of a 14-year-old patient diagnosed with pre-B ALL with p16 deletion that maintained growth and survival in vitro. Initially, cells were infected with retrovirus carrying pRS-shRNA library and infected cells were selected by treatment with puromycin (2 μg/ml) for 3 weeks. Cells infected with pRS-shRNA were then cultured in the presence of L-asparaginase (10 mIU/ml) for an additional 2 weeks. To isolate individual resistant colonies in soft agar, an additional 1-week culture in the presence of puromycin and L-asparaginase was performed and a total of 10 single colonies were formed and isolated. Isolated clones were amplified individually and as a pool of the 10 clones. Cell were fed with fresh media containing L-asparaginase (10 mIU/ml) every 3 days. As shown in Figure 1a, parental (designated as *) and control retroviral vector-infected cells are sensitive (IC50=50.7 mIU/ml) to L-asparaginase treatment. In contrast, the pooled shRNA library-infected cells are resistant to L-asparaginase treatment. To test whether cell survival after puromycin and L-asparaginase treatments is due to infection of retrovirus carrying shRNAs, genomic DNA isolated from the pooled resistant cells was isolated and used to perform PCR using primers that flank the shRNA inserts. The presence of the 642 bp PCR product after the first (puromycin) and the second (L-asparaginase) screening of pRS-shRNA library-infected cells indicate that these cells, indeed, harbor the retroviral shRNA insert (Figure 1b). Sequencing of bar codes for each individual clone was then performed to identify the target genes that account for L-asparaginase resistance. Our data show that among the 10 resistant clones, six hits were for opioid receptor mu 1 gene (oprm1) and two hits each for carbonic anhydrase 1 (ca1)9 and ubiquitin-conjugating enzyme E2C (ube2c)10 genes (Figure 1c).
Figure 1.

Genome-wide RNAi screening identifies loss of OPRM1 in the development of L-asparaginase resistance in leukemic cells. (a) Reduced L-asparaginase sensitivity in shRNA library-infected cells (•) compared to parental cells (○) and cells infected with an empty pRS vector (▪). Cells were treated with increasing concentrations of L-asparaginase for 4 days and viability was measured. Values are means±s.d. of three independent experiments. (b) Presence of shRNA inserts in cells infected with retrovirus carrying shRNA library. PCR was performed using gDNAs as template and pRS forward (5'-CCCTTGAACCTCCTCGTTCGACC-3') and reverse (5'-GAGACGTGCTACTTCCATTTGTC-3') primers. (c) Identification of genes responsible for L-asparaginase resistance. Ten L-asparaginase-resistant clones isolated by soft agar colony formation assay were subjected to gDNA isolation and PCR using the primers described in b. 642 bp PCR products resolved in 1% agarose gel were cut, extracted and sequenced using pRS-sequence primer (5'-GCTGACGTCATCAACCCGCT-3').
Next, we tested whether OPRM1 is expressed in our model cell system and other leukemic cells. As shown in Figure 2, OPRM1 is expressed in our model cell system (*) and all four other leukemic cells tested. To validate our data from the genome-wide retroviral shRNA library screening, we infected the leukemic cells with retrovirus carrying shOPRM1 and tested whether specific knockdown of OPRM1 confers L-asparaginase resistance. Indeed, shOPRM1-infected cells that show reduced levels of OPRM1 mRNA (Figure 3a, left panels) and protein (Figure 3a, right panels) are resistant to L-asparaginase treatment (Figure 3b).
Figure 2.
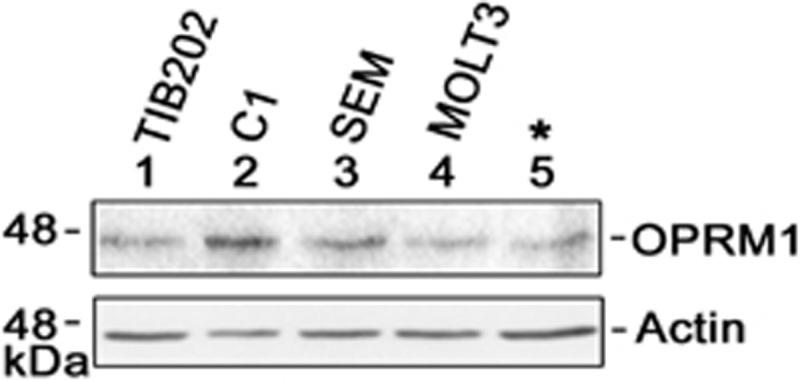
OPRM1 levels in leukemic cells. Cell lysates (30 μg for actin blot; 80 μg for OPRM1 blot) were resolved in SDS–PAGE and subjected to immunoblotting with OPRM1 (Pierce, Waltham, MA, USA) or actin (Santa Cruz Biotech, Dallas, TX, USA) antibody. SDS–PAGE, SDS–polyacrylamide gel electrophoresis.
Figure 3.

Specific knockdown of OPRM1 confers L-asparaginase resistance. (a) OPRM1 mRNA (left) and protein (right) are significantly reduced in cells infected with retrovirus carrying pRS-shOPRM1. Representative data from three independent experiments are shown. (b) Cells infected with retrovirus carrying pRS-shOPRM1 confers L-asparaginase resistance. Values are means±s.d. of three independent experiments.
Interestingly, a previous study11 showed that stimulation of the opioid receptor leads to activation of inhibitory Gi-proteins which block adenylyl cyclase activity, which in turn, reduces cellular cAMP level and subsequently induces apoptosis by caspase activation in leukemic cells. A separate study12 also showed that activation of opioid receptor reduces cAMP level and increases sensitivity of glioblastoma cells to doxorubicin. Together, these findings suggest that leukemic cells utilize the OPRM1-cAMP-caspase pathway to acquire L-asparaginase resistance. Since methadone, an μ-opioid receptor agonist, was found to sensitize leukemic cells to doxorubicin treatment,13 and the OPRM1-cAMP-caspase pathway may be utilized by leukemic cells for both methadone sensitization and L-asparaginase resistance, we investigated L-asparaginase sensitivity in both parental and OPRM1-depleted cells with and without methadone treatment. If the OPRM1-cAMP-caspase pathway is utilized by leukemic cells for both methadone sensitization and L-asparaginase resistance, parental cells will show synergistic sensitivity when the cells are treated with L-asparaginase+methadone compared to cells treated with L-asparaginase or methadone alone. However, leukemic cells resistant to L-asparaginase due to OPRM1 loss will be insensitive to L-asparaginase+methadone treatment. As shown in Figure 4a, methadone enhances the sensitivity of parental cells, but not OPRM1-depleted cells, to L-asparaginase treatment, indicating that OPRM1 is required for the synergistic action of L-asparaginase and methadone. Our findings also suggest that depletion of OPRM1 promotes leukemic cell survival through downregulation of the OPRM1-mediated apoptotic pathway. Thus, our data demonstrate that the OPRM1-mediated pathway may have a critical role in L-asparaginase sensitivity and development of resistance. Consistent with this finding, all leukemic cells with relatively high level of OPRM1 are significantly (P<0.05) sensitive to L-asparaginase treatment compared to our model leukemic cells depleted of OPRM1 (Figure 4b).
Figure 4.

(a) Cells depleted of OPRM1 are insensitive to methadone addition. Cells infected with pRS and pRS-shOPRM1 were treated with methadone and/or L-asparaginase for 4 days before cell viability assay. **P<0.05. (b) Increased sensitivity of TIB202 (▪), SEM (◊), MOLT3 (♦), C1 (□) and model leukemic (○) cells compared to model leukemic cells depleted of OPRM1 (•). Cells were treated with increasing concentrations of L-asparaginase for 4 days before cell viability assay. (c) Reduced level of OPRM1 is linked to L-asparaginase resistance in leukemic cells isolated from ALL patients. Peripheral blood samples were obtained from children diagnosed with leukemia at the Alberta Children’s Hospital following local ethics board approval and parental consent (REB815-1383-REN1). A panel of primary leukemia cells were treated with increasing concentrations of L-asparaginase before cell viability assay. The ratios of the OPRM1 vs actin band intensities were determined following densitometric scanning of bands using the NIH Image J 1.61 software (Bethesda, MD, USA). Values in A and B are means±s.d. of three independent experiments.
To investigate the clinical relevance of our findings, a panel of additional leukemic cells isolated from ALL patients were assessed to determine whether reduced level of OPRM1 is linked to L-asparaginase resistance. To do so, primary leukemic cells from five ALL patients were analyzed for OPRM1 levels and L-asparaginase sensitivity. As shown in Figure 4c, leukemic cells with higher levels of OPRM1 are more sensitive to L-asparaginase treatment compared to those with lower levels of OPRM1. This finding further demonstrates the link between loss of OPRM1 and the development of L-asparaginase resistance.
In accord with our observations, further examination of the data at the cBioportal for Cancer Genomics,14 that offers large-scale cancer genomics data set analysis, revealed the connection of oprm1 deletions and mutations to hematopoietic and other cancers. OPRM1 is deleted in 10% of adenoid cystic carcinoma and 6.3% of diffuse large B-cell lymphoma. OPRM1 is mutated in 15% of desmoplastic melanoma, 2.1% of diffuse large B-cell lymphoma, 1.7% of adenoid cystic carcinoma and 0.6% of chronic lymphocytic leukemia. Thus, our study demonstrates for the first time a novel molecular apparatus for L-asparaginase resistance in ALL, and identifies OPRM1 as a functional biomarker for defining high-risk subpopulations and for the detection of evolving resistant clones. Absence of sufficient data sets in publicly available databases (for example, cBioportal and Oncomine) prevented us from further acquiring a significant relationship between OPRM1 and ALL. Nonetheless, we propose that oprm1 can be targeted for effective treatment of L-asparaginase-resistant ALL. Characterization of L-asparaginase resistance due to loss of carbonic anhydrase 1 or ubiquitin-conjugating enzyme E2C in ALL is underway.
Acknowledgments
This work was supported in part by grants from the CIHR (MOP-123400) and NSERC (RGPIN/312985-2011) to KYL.
Author contributions
SK performed most of the experiments and wrote a draft. VMS isolated primary leukemic cells from patients and contributed to revision. JLR, KL, SK and AN conceived the idea and JLR, KL and AN contributed to revision.
Footnotes
The authors declare no conflict of interest.
References
- Howlader NNA, Krapcho M, Miller D, Bishop K, Altekruse SF, Kosary CL et al. SEER Cancer Statistics Review. National Cancer Institute: Bethesda, MD, USA, 2016. [Google Scholar]
- Killander D, Dohlwitz A, Engstedt L, Franzen S, Gahrton G, Gullbring B et al. Hypersensitive reactions and antibody formation during L-asparaginase treatment of children and adults with acute leukemia. Cancer 1976; 37: 220–228. [DOI] [PubMed] [Google Scholar]
- Li BS, Gu LJ, Luo CY, Li WS, Jiang LM, Shen SH et al. The downregulation of asparagine synthetase expression can increase the sensitivity of cells resistant to l-asparaginase. Leukemia 2006; 20: 2199–2201. [DOI] [PubMed] [Google Scholar]
- Chen SH. Asparaginase therapy in pediatric acute lymphoblastic leukemia: a focus on the mode of drug resistance. Pediatr Neonatol 2015; 56: 287–293. [DOI] [PubMed] [Google Scholar]
- Su N, Pan YX, Zhou M, Harvey RC, Hunger SP, Kilberg MS. Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatr Blood Cancer 2008; 50: 274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci O, Starkova J, Otova B, Madzo J, Kalinova M, Hrusak O et al. Upregulation of asparagine synthetase fails to avert cell cycle arrest induced by L-asparaginase in TEL/AML1-positive leukaemic cells. Leukemia 2004; 18: 434–441. [DOI] [PubMed] [Google Scholar]
- Stams WA, den Boer ML, Beverloo HB, Meijerink JP, Stigter RL, van Wering ER et al. Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood 2003; 101: 2743–2747. [DOI] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004; 428: 431–437. [DOI] [PubMed] [Google Scholar]
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017; 12: 61–88. [DOI] [PubMed] [Google Scholar]
- Xie C, Powell C, Yao M, Wu J, Dong Q. Ubiquitin-conjugating enzyme E2C: a potential cancer biomarker. Int J Biochem Cell Biol 2014; 47: 113–117. [DOI] [PubMed] [Google Scholar]
- Friesen C, Roscher M, Hormann I, Fichtner I, Alt A, Hilger RA et al. Cell death sensitization of leukemia cells by opioid receptor activation. Oncotarget 2013; 4: 677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen C, Hormann I, Roscher M, Fichtner I, Alt A, Hilger R et al. Opioid receptor activation triggering downregulation of cAMP improves effectiveness of anti-cancer drugs in treatment of glioblastoma. Cell Cycle 2014; 13: 1560–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen C, Roscher M, Alt A, Miltner E. Methadone, commonly used as maintenance medication for outpatient treatment of opioid dependence, kills leukemia cells and overcomes chemoresistance. Cancer Res 2008; 68: 6059–6064. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]