

Abstract
Previous studies have demonstrated that nicotinamide phosphoribosyltransferase (NAMPT) promoted inflammation and permeability of vascular endothelial cells following cardiopulmonary bypass (CPB). In addition, mitogen-activated protein kinase (MAPK) signaling was activated and contributed to these cell responses. However, the mechanism by which NAMPT regulates cellular inflammation and permeability remains unknown, and whether NAMPT regulates MAPK signaling during this process is also not clear. The present study established an anoxia-reoxygenation (A-R) model using human umbilical vein endothelial cells (HUVECs) and investigated the regulation of MAPK signaling by NAMPT by using small RNA transfection, ELISA and western blot analysis. The results demonstrated that A-R significantly induced the expression levels of NAMPT and cellular permeability-associated proteins, and the release of several inflammatory factors. Furthermore, calcium and MAPK signaling were evidently increased. When the A-R cells were transfected with NAMPT small interfering RNA, the expression of cellular permeability-associated proteins was downregulated, the release of inflammatory factors was decreased, and calcium and MAPK signaling was blocked. These data suggest that NAMPT may activate MAPK signaling to promote A-R-induced inflammation and permeability enhancement of HUVECs. Therefore, the current study indicates that NAMPT may be a potential drug target for A-R-induced endothelial cell injury subsequent to CPB.
Keywords: nicotinamide phosphoribosyltransferase, anoxia-reoxygenation, inflammation, cellular permeability, mitogen-activated protein kinase signaling
Introduction
Cardiopulmonary bypass (CPB) is a necessary procedure during open-heart surgery, and its use in clinical applications has been expanding. However, CPB may induce postoperative pulmonary dysfunction (1,2). During CPB, oxygen is supplied to the lung only through the bronchus, thus, ischemia and hypoxia may occur in the lung. Therefore, the lung is often reperfused, resulting in anoxia-reoxygenation (A-R)-induced acute injury (3). A-R has been reported to induce inflammation and enhance the cellular permeability of pulmonary vascular endothelial cells, which then led to pneumonedema, inducing hyoxemia, acute respiratory distress syndrome and even mortality (4). Thus, inhibiting inflammation and cellular permeability may improve the prognosis following CPB (5).
Pre-B-cell colony-enhancing factor was first identified and cloned from peripheral blood lymphocytes in 1994 by Samal et al (6). Subsequently, this protein was also detected in adipose cells, and named visfatin or nicotinamide phosphoribosyltransferase (NAMPT) (7). Studies have demonstrated that NAMPT serves important roles in lung injury following CPB (8,9). NAMPT is often highly expressed during acute lung injury, and exhibits a close correlation with the levels of inflammatory factors, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and IL-6 (8). In addition, a previous report has also identified that CPB promoted the release of inflammatory factors and enhanced the cellular permeability of pulmonary vascular endothelial cells, while upregulation of NAMPT contributed to A-R-induced cellular permeability enhancement (9), suggesting that NAMPT may facilitate CPB-triggered inflammation and the changes in cellular permeability. However, the mechanism through which NAMPT regulates these cell responses remains unclear.
Mitogen-activated protein kinase (MAPK) signaling serves important roles in cell inflammation, apoptosis, proliferation and differentiation (10). MAPK signaling includes three pathways: Extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 MAPK pathways. Furthermore, MAPK signaling is activated by post-translational phosphorylation mediated by various kinases (11,12). It has been demonstrated that MAPK signaling was activated in the rat lung tissue following CPB (13). However, this signaling was blocked following MAPK signaling inhibitor treatment, and simultaneously, the release of inflammatory factors and infiltration were restrained (14). Therefore, MAPK signaling may participate in CPB-induced lung injury, although whether NAMPT regulates CPB-triggered inflammation and cellular permeability enhancement of lung tissue through MAPK signaling remains unknown.
In the present study, an A-R model was established using human umbilical vein endothelial cells (HUVECs) to investigate the regulation of MAPK signaling by NAMPT during CPB-induced A-R. The present data demonstrated that NAMPT was upregulated by A-R, and subsequently induced the release of inflammatory factors and the expression levels of cellular permeability-associated proteins. Additionally, calcium and MAPK signaling were activated; however, NAMPT knockdown blocked A-R-induced MAPK signaling and the cell responses, including inflammation and cellular permeability enhancement. The present study also indicated that NAMPT may promote A-R-induced inflammation and cellular permeability enhancement through activating MAPK signaling.
Materials and methods
Chemicals
Chemicals were purchased commercially as follows: Bicinchoninic acid (BCA) kit, enhanced chemiluminescence (ECL) reagent, Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from GE Healthcare Life Sciences (Hyclone; Logan, UT, USA); horseradish peroxidase (HRP)-labeled goat anti-Rabbit IgG (H+L) (cat. no. A0208) and glyceraldehyde phosphate dehydrogenase (GAPDH) mouse monoclonal antibodies (cat. no. AF0006) were purchased from Beyotime Institute of Biotechnology (Shanghai, China); enzyme-linked immunosorbent assay (ELISA) kits for TNF-α (cat. no. MBS2502004), IL-1β (cat. no. MBS175901) and IL-6 (cat. no. MBS021993; all from MyBioSource, Inc., San Diego, CA, USA); rabbit polyclonal antibodies against myosin light-chain (MLC; cat. no. ab79935), phosphorylated (p)-MLC (cat. no. ab2480), vascular endothelial (VE)-cadherin (cat. no. ab33168), p-VE-cadherin (cat. no. ab119785), β-catenin (cat. no. ab6302), focal adhesion kinase (FAK; cat. no. ab61113), p-FAK (cat. no. ab39967), paxillin (cat. no. ab2264), p38 MAPK (cat. no. ab197348), p-p38 (cat. no. ab47363), ERK (cat. no. ab17942) and p-ERK (cat. no. ab24157) were from Abcam (Cambridge, MA, USA); and Lipofectamine 2000 was from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Cell culture, establishment of A-R model and small interfering RNA (siRNA) transfection
HUVECs (cat. no. CRL1730) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and maintained in DMEM supplemented with 10% (v/v) FBS. The A-R model was established as described in a previous study (9). Briefly, HUVECs in the logarithmic growth phase were arranged into the sterilized hypoxic box. The box was filled with 5% CO2 and 94% N2, and the oxygen concentration decreased to 1% within 30 min. After 20-h hypoxic treatment, the cells were maintained in a normal incubator with 5% CO2 at 37°C to establish the A-R model.
After an additional 9 h, A-R-treated HUVECs were used in subsequent siRNA transfection. For NAMPT knockdown, NAMPT siRNA and negative control siRNA were synthesized by Invitrogen; Thermo Fisher Scientific, Inc. according to the reported sequences: NAMPT siRNA, 5′-CCACCCAACACAAGCAAAGUUUAUU-3′; and negative control siRNA, 5′-UUCUCCGAACGUGUCACGUTT-3′. These siRNAs were then transfected into the A-R-treated HUVECs in FBS-free DMEM by using Lipofectamine 2000 following the manufacturer's protocol (Invitrogen; Thermo Fisher Scientific, Inc.). After 6-h transfection, the cells were cultured in DMEM with 10% FBS for an additional 48 h.
Detection of the TNF-α, IL-1β and IL-6 content by ELISA and of the Ca2+ levels by Fura-2-acetoxymethyl ester (Fura-2-AM)
The four treatment groups included the normal control cells (without A-R induction), A-R model cells, A-R cells transfected with negative control siRNA and A-R cells transfected with NAMPT siRNA transfection. Following treatment, the cells were centrifuged at 1,000 × g at 4°C for 5 min to harvest the supernatant. According to the ELISA kit instructions, the contents of TNF-α, IL-1β and IL-6 in each group were detected based on three independent replicates using a microplate reader (Infinite F50; Tecan Trading AG, Männedorf, Switzerland).
For detection of Ca2+ levels, the cells were treated with 1 µM Fura-2-AM (Invitrogen; Thermo Fisher Scientific, Inc.) and 2 mM calcium chloride (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 30 min at 37°C. Subsequently, cells were washed three times with phosphate-buffered saline, and fluorescence was detected at 555 nm using a laser scan confocal microscope (Carl Zeiss LSM 700; Carl Zeiss, Thornwood, NY, USA).
Western blot analysis of protein expression levels
The cells were centrifuged at 1,000 × g at 4°C for 5 min, and the precipitates were lysed in radioimmunoprecipitation assay buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0, 5.0 mM EDTA, pH 8.0, 0.5 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride) containing protease inhibitors for 30 min. The cell lysates were then centrifuged at 12,000 × g at 4°C for 10 min, and protein concentrations in the supernatants were detected using the BCA kit according to the manufacturer's instructions. Next, the proteins were treated with SDS sample buffer in boiling water for 10 min, then separated using 12.5% SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The membranes were incubated with blocking buffer (5% bovine serum albumin (Sigma-Aldrich; Merck KGaA), 50 mM Tris, 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature, and subsequently incubated with the corresponding primary antibodies against MLC, p-MLC, VE-cadherin, p-VE-cadherin, β-catenin, FAK, p-FAK, paxillin, p38 MAPK, p-p38, ERK and p-ERK at a 1:1,000 dilution at 4°C overnight. After being washed with wash buffer (50 mM Tris, 150 mM NaCl, 0.1% Tween-20) three times for 5 min each time, the membranes were incubated with the HRP-labeled goat anti-rabbit IgG (H+L) secondary antibody at room temperature for 2 h. Following treatment with the ECL regent, the target protein bands were observed by Molecular Image ChemiDoc XRS system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The protein expression was quantitatively analyzed by Quantity One software (version 4.62; Bio-Rad Laboratories, Inc.).
Statistical analysis
All the data were collected from three independent replicates and analyzed with SPSS version 17.0 software package (SPSS, Inc., Chicago, IL, USA). Statistically significant differences were analyzed by Student's t-test and considered when P<0.05.
Results
NAMPT knockdown by siRNA transfection inhibits A-R-induced release of inflammatory factors in HUVECs
In the present study, an A-R model was established in HUVECs in order to investigate the mechanisms underlying A-R-induced inflammation. Compared with the normal control, A-R clearly upregulated NAMPT protein expression (Fig. 1A). Following transfection of the cells with NAMPT siRNA, NAMPT expression was markedly downregulated, even under A-R treatment, as compared with the negative control siRNA transfection group (Fig. 1A). Next, the contents of three inflammatory factors in the culture medium of HUVECs were detected using ELISA subsequent to NAMPT knockdown. As shown in Fig. 1B-D, TNF-α, IL-1β and IL-6 contents were significantly increased following A-R treatment along with the NAMPT upregulation (P<0.01). However, NAMPT knockdown by siRNA transfection significantly inhibited the A-R-upregulated inflammatory factor contents (P<0.01; Fig. 1B-D). Therefore, the results suggest that A-R induces inflammation in HUVECs by upregulating NAMPT expression.
Figure 1.

NAMPT knockdown by siRNA transfection inhibits A-R-induced release of inflammatory factors in HUVECs. (A) Western blots demonstrating the expression of NAMPT in the four groups of treated cells, including normal cells, A-R-induced cells, A-R-induced cells transfected with negative control siRNA, and A-R-induced cells transfected with NAMPT siRNA. Enzyme-linked immunosorbent assay was used to analyze the contents of (B) TNF-α, (C) IL-1β and (D) IL-6 in the four treated cell groups. Normal and negative control siRNA-transfected cells were used as the controls corresponding to the A-R-induced cells and A-R+NAMPT siRNA-transfected cells, respectively. Values are expressed as the mean ± standard deviation based on three independent replicates. Statistical significance was calculated with the Student's t-test. **P<0.01. NAMPT, nicotinamide phosphoribosyltransferase; A-R, anoxia-reoxygenation; siRNA, small interfering RNA; TNF-α, tumor necrosis factor-α; IL, interleukin.
NAMPT knockdown blocks A-R-induced expression of cellular permeability-associated proteins and Ca2+ increase
A previous study has demonstrated that the cellular permeability of endothelial cells is mediated by intercellular adherens and the junction between cell and extracellular matrix (15). VE-cadherin interacts with β-catenin to form intercellular adherens junctions, and is then connected to actin cytoskeleton to regulate cellular permeability (16). In addition, FAK interacts with paxillin and vinculin to form cell-extracellular matrix adhesions, and regulates cellular permeability through actin cytoskeleton (16). Calcium signaling activated MLC kinase (MLCK) to phosphorylate MLC and promote the connection between MLC and actin for the regulation of cell contraction (17). Therefore, calcium signaling is able to regulate intercellular adherens junctions and cell-extracellular matrix adhesions for cellular permeability changes through adjusting actin. Previous studies observed that NAMPT induced the enhancement of cellular permeability of pulmonary vascular endothelial cell during A-R-induced acute lung injury (18,19), and the aforementioned results of the present study suggested that NAMPT was upregulated in HUVECs under A-R treatment. Therefore, the current study attempted to clarify whether NAMPT serves a role in the A-R-induced cellular permeability enhancement of HUVECs.
The calcium-mediated phosphorylation of MLC (p-MLC protein) was first detected to investigate the regulation of actin cytoskeleton by NAMPT under A-R induction. The data revealed that A-R significantly upregulated (P<0.01) the Ca2+ level and the phosphorylation of MLC compared with the normal control group (Fig. 2). When NAMPT was depleted by siRNA transfection, the levels of Ca2+ and p-MLC were significantly downregulated (P<0.01) under A-R treatment. These results suggested that NAMPT may promote A-R-induced actin cytoskeleton contraction by activating the Ca2+-MLC pathway.
Figure 2.

NAMPT increases Ca2+ levels and MLC phosphorylation. (A) Ca2+ levels detected by Fura-2-acetoxymethyl ester assay, and (B) protein levels of MLC and p-MLC detected by western blot analysis in the four groups of treated cells. MLC was used as the internal control of p-MLC. Bar graph shows the mean ratios of the abundance of p-MLC to that of MLC in each sample. All the values are reported as the mean ± standard deviation based on three independent replicates. Statistically significant differences were calculated with the Student's t-test. **P<0.01. NAMPT, nicotinamide phosphoribosyltransferase; A-R, anoxia-reoxygenation; siRNA, small interfering RNA; MLC, myosin light-chain; p-MLC, phosphorylated MLC.
Subsequently, the levels of VE-cadherin phosphorylation (p-VE-cadherin protein) and β-catenin expression were examined to determine whether NAMPT is able to regulate the A-R-induced derangement of intercellular adherens junctions. Compared with the normal control, A-R significantly upregulated the levels of p-VE-cadherin and β-catenin (P<0.01; Fig. 3). However, the increased expression was significantly blocked subsequent to NAMPT knockdown (P<0.01; Fig. 3). The data indicated that NAMPT may regulate intercellular adherens junctions during A-R.
Figure 3.

NAMPT regulates intercellular adherens by promoting the phosphorylation of VE-cadherin and expression of β-catenin, analyzed by western blot analysis in the four groups of treated cells. GAPDH was used for internal standardization. Bar graph demonstrates the mean ratios of the abundance of p-VE-cadherin to that of VE-cadherin, or β-catenin relative to GAPDH in each sample. All the values are reported as the mean ± standard deviation based on three independent replicates. Statistically significant differences were calculated with the Student's t-test. **P<0.01. NAMPT, nicotinamide phosphoribosyltransferase; A-R, anoxia-reoxygenation; siRNA, small interfering RNA; VE, vascular endothelial; p-, phosphorylated.
The study also examined the levels of FAK phosphorylation (p-FAK protein) and paxillin expression to determine the effect of cell-extracellular matrix adhesions by NAMPT under A-R treatment. Clearly, the levels of p-FAK and paxillin significantly increased following A-R induction compared with the normal control levels (P<0.01; Fig. 4). By contrast, NAMPT knockdown significantly suppressed this A-R-induced upregulation (P<0.01; Fig. 4). The results suggested that NAMPT may regulate cell-extracellular matrix adhesions during A-R. Collectively, the data indicate that NAMPT may mediate cellular permeability by affecting the intercellular adherens and the junction between cell and extracellular matrix.
Figure 4.
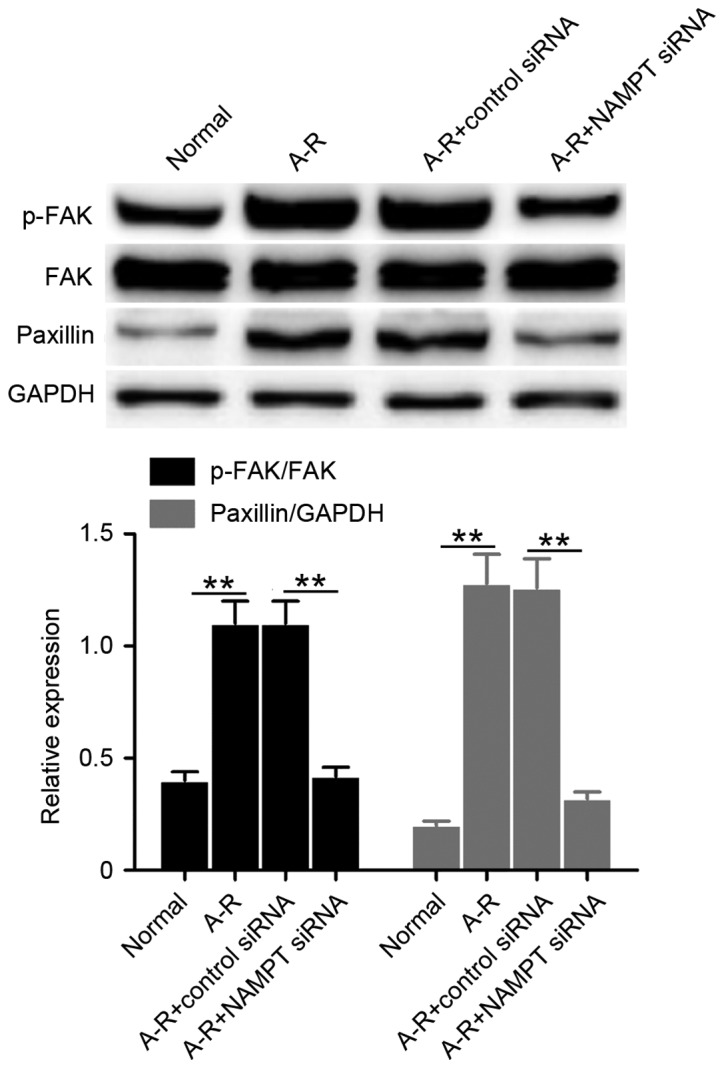
NAMPT regulates cell-extracellular matrix adhesions by promoting FAK phosphorylation and paxillin expression. The expression levels of p-FAK and paxillin were analyzed by western blot analysis in the four groups of treated cells. GAPDH was used for internal standardization. Bar graph reports the mean ratios of the abundance of p-FAK to that of FAK, or paxillin relative to GAPDH in each sample. All the values are reported as the mean ± standard deviation based on three independent replicates. **P<0.01. NAMPT, nicotinamide phosphoribosyltransferase; A-R, anoxia-reoxygenation; siRNA, small interfering RNA; FAK, focal adhesion kinase; p-, phosphorylated.
NAMPT knockdown restrains A-R-activated MAPK signaling
It has been demonstrated that MAPK signaling is activated during CPB-triggered acute lung injury, and mediates cellular inflammation and permeability (20). Thus, the present study tested the phosphorylation of p38 MAPK and ERK to investigate the regulation of MAPK signaling by NAMPT under A-R treatment. Compared with the normal control, p38 and ERK phosphorylation significantly increased following A-R treatment (P<0.01; Fig. 5). However, siRNA-mediated NAMPT knockdown significantly decreased the A-R-induced phosphorylation of p38 and ERK (P<0.01; Fig. 5). These results suggested that NAMPT positively regulates the A-R-activated MAPK signaling.
Figure 5.
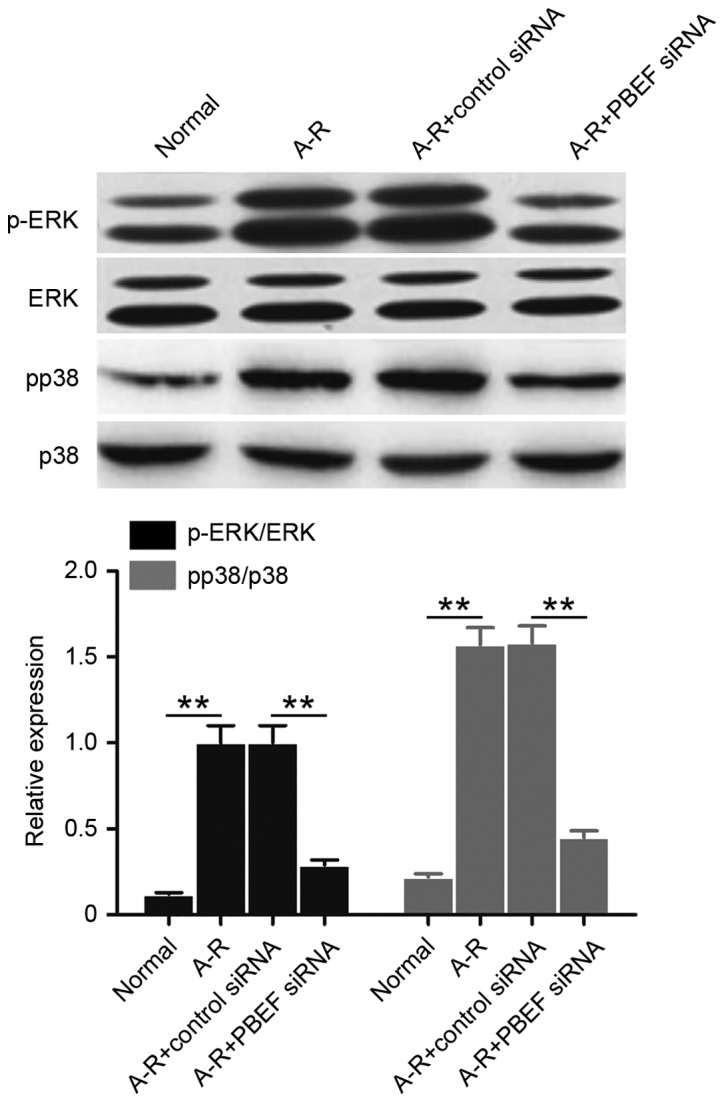
NAMPT promotes A-R-induced activation of MAPK signaling. The phosphorylated expression levels of ERK and p38 were analyzed by western blot analysis in the four treated cell groups. Bar graph demonstrates the mean ratios of the abundance of phosphorylated protein to that of the corresponding protein in each sample. All the values are reported as the mean ± standard deviation based on three independent replicates. **P<0.01. NAMPT, nicotinamide phosphoribosyltransferase; A-R, anoxia-reoxygenation; siRNA, small interfering RNA; MAPK, mitogen-activated protein kinase; p-, phosphorylated.
Discussion
Previous studies revealed that NAMPT serves as a growth factor and cytokine (18). In particular, NAMPT has been identified as an inflammatory factor and a potential novel biomarker in acute lung injury (21). Further research demonstrated that NAMPT was expressed in lung microvascular endothelium (21). In mice, NAMPT regulated ventilator-induced lung injury through mediating the release of inflammatory factors, including IL-6 and TNF-α (22). Previous studies have also observed that NAMPT expression was upregulated in pulmonary cells, and that it may contribute to inflammatory factor expression (23). Therefore, NAMPT is considered to participate in the inflammation during acute lung injury (18). In the present study, an A-R-induced model was established using HUVECs to investigate how NAMPT regulates inflammation. It was observed that A-R significantly induced the expression of NAMPT and the release of TNF-α, IL-1β and IL-6, suggesting that NAMPT may promote A-R-induced inflammation by facilitating the release of inflammatory factors. Next, NAMPT knockdown was performed via siRNA transfection, and it was demonstrated that the release of these three inflammatory factors was controlled by the high expression of NAMPT. Thus, these results indicated that NAMPT promotes A-R-induced inflammation by facilitating the release of TNF-α, IL-1β and IL-6. Similar results were also observed in a previous study during rat acute lung injury, in which NAMPT was highly expressed, and NAMPT inhibitor decreased TNF-α and IL-1β contents (24). Notably, inhibition of NAMPT was able to attenuate the pandemic H1N1 2009 virus-induced inflammation in the lung endothelium, for example, by decreasing the expression levels of IL-6, IL-8, and TNF-α (25). Therefore, NAMPT may be an important upstream factor that regulates inflammation.
Cellular permeability alterations in endothelial cells participate in acute lung injury, while the cellular permeability in these cells has been demonstrated to be mediated by intercellular and cell-extracellular matrix adherens (15). VE-cadherin interacts with β-catenin to form intercellular adherens junctions, and then targets the actin cytoskeleton to regulate cellular permeability (16). Furthermore, FAK interacts with paxillin and vinculin to form cell-extracellular matrix adhesions, and subsequently regulates cellular permeability through the actin cytoskeleton (16). Calcium signaling regulates intercellular adherens junctions and cell-extracellular matrix adhesions for cellular permeability changes through activating MLCK-mediated MLC phosphorylation. Phosphorylated MLC then promotes the connection between MLC and actin to regulate cell contraction (17). In human pulmonary artery endothelial cells, high expression of NAMPT has been reported to regulate thrombin-induced alterations of cellular permeability by promoting Ca2+ entry, MLC phosphorylation and actin polymerization (26). The present study also identified that NAMPT regulated the A-R-induced cellular permeability changes in HUVECs by mediating actin function, including Ca2+ signaling and MLC phosphorylation. Furthermore, it was demonstrated that NAMPT regulated VE-cadherin phosphorylation and β-catenin expression, suggesting the regulation of intercellular adherens junctions by NAMPT. Meanwhile, NAMPT also mediated cell-extracellular matrix adhesions by regulating FAK phosphorylation and paxillin expression. Hence, NAMPT serves a vital role in the regulation of cellular permeability under A-R conditions. Combined with previous research, the current study suggests that NAMPT regulates cellular permeability by mediating intercellular and cell-extracellular matrix adherens.
NAMPT promotes acute lung injury through regulating inflammation and cellular permeability, while MAPK signaling may also be involved in this process (18). MAPK signaling includes the ERK, p38 MAPK and JNK pathways, and serves an important role in cell inflammation, apoptosis, proliferation and differentiation. MAPK signaling is activated by post-translational phosphorylation mediated by various kinases (11,12). Studies have shown that p38 MAPK and ERK pathways participate in the inflammation of endothelial cells, while JNK pathway does not function in this process (27). In pig lungs following CPB, ERK and p38 MAPK were detected to be significantly activated (28). Furthermore, in reperfused rat lungs, the phosphorylation of p38 MAPK and ERK was also upregulated (13). Our previous study demonstrated that p38 MAPK was activated in rat lungs following CPB and that the inhibitor of p38 MAPK restrained CPB-triggered inflammation (14). Therefore, MAPK signaling serves an important role in acute lung injury and the associated inflammation. In addition, MAPK signaling also regulates the cellular permeability of pulmonary endothelial cells (29,30). Phosphorylated p38 MAPK was able to activate heat shock protein 27 in order to regulate the cellular permeability of rat pulmonary microvascular endothelial cells (31). p38 MAPK also regulated TGF-β-mediated MLC phosphorylation to affect permeability of endothelial cells (32). During vascular endothelial growth factor-induced cellular permeability changes, ERK was identified as an important regulator (33). Although NAMPT promoted inflammation in human pulmonary microvascular endothelial cells, inhibition of MAPK blocked NAMPT function, suggesting that NAMPT may regulate inflammation via MAPK signaling (34). These studies indicated that NAMPT-mediated lung injury may depend on MAPK signaling.
In the present study, it was observed that A-R significantly induced the expression of NAMPT and the phosphorylation of p38 MAPK and ERK in HUVECs. When NAMPT was depleted by siRNA transfection, the phosphorylation levels decreased. Clearly, NAMPT controls the A-R-induced activation of MAPK signaling. Combined with previous research, it can be concluded that NAMPT promotes A-R-induced inflammation and cellular permeability through MAPK signaling. The current study indicates that NAMPT may be a potential drug target for CPB-triggered acute injury of endothelial cells.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 81260054).
References
- 1.Huffmyer JL, Groves DS. Pulmonary complications of cardiopulmonary bypass. Best Pract Res Clin Anaesthesiol. 2015;29:163–175. doi: 10.1016/j.bpa.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiessen S, Vanhorebeek I, Van den Berghe G. Glycemic control and outcome related to cardiopulmonary bypass. Best Pract Res Clin Anaesthesiol. 2015;29:177–187. doi: 10.1016/j.bpa.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Durandy Y. Minimizing systemic inflammation during cardiopulmonary bypass in the pediatric population. Artif Organs. 2014;38:11–18. doi: 10.1111/aor.12195. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari RS, Andrade CF. Oxidative stress and lung ischemia-reperfusion injury. Oxid Med Cell Longe. 2015;2015:590987. doi: 10.1155/2015/590987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chao YK, Wu YC, Yang KJ, Chiang LL, Liu HP, Lin PJ, Chu Y. Pulmonary perfusion with L-arginine ameliorates post-cardiopulmonary bypass lung injury in a rabbit model. J Surg Res. 2011;167:e77–e83. doi: 10.1016/j.jss.2009.10.041. [DOI] [PubMed] [Google Scholar]
- 6.Samal B, Sun YH, Stearns G, Xie CS, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437. doi: 10.1128/MCB.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang YC, Chang TJ, Lee WJ, Chuang LM. The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism. 2010;59:93–99. doi: 10.1016/j.metabol.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Bajwa EK, Yu CL, Gong MN, Thompson BT, Christiani DC. Pre-B-cell colony-enhancing factor gene polymorphisms and risk of acute respiratory distress syndrome. Crit Care Med. 2007;35:1290–1295. doi: 10.1097/01.CCM.0000260243.22758.4F. [DOI] [PubMed] [Google Scholar]
- 9.Yang W, Zeng Y, Li B, Zhou J, Gong Y, Xu J, Dong X. Pre-B-cell colony enhancing factor (PBEF) increases endothelial permeability in hypoxia/re-oxygenation model. Int J Clin Exp Med. 2015;8:8842–8847. [PMC free article] [PubMed] [Google Scholar]
- 10.Mordret G. MAP kinase kinase: A node connecting multiple pathways. Biol Cell. 1993;79:193–207. doi: 10.1016/0248-4900(93)90138-5. [DOI] [PubMed] [Google Scholar]
- 11.L'Allemain G. Deciphering the MAP kinase pathway. Prog Growth Factor Res. 1994;5:291–334. doi: 10.1016/0955-2235(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 12.Munshi A, Ramesh R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer. 2013;4:401–408. doi: 10.1177/1947601913485414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markou T, Chambers DJ. Lung injury after simulated cardiopulmonary bypass in an isolated perfused rat lung preparation: Role of mitogen-activated protein kinase/Akt signaling and the effects of theophylline. J Thorac Cardiovasc Surg. 2014;148:2335–2344. doi: 10.1016/j.jtcvs.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong X, Liu Y, Du M, Wang Q, Yu CT, Fan X. P38 mitogen-activated protein kinase inhibition attenuates pulmonary inflammatory response in a rat cardiopulmonary bypass model. Eur J Cardiothorac Surg. 2006;30:77–84. doi: 10.1016/j.ejcts.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 15.Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994;267:L223–L241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- 16.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 17.Van Lierop JE, Wilson DP, Davis JP, Tikunova S, Sutherland C, Walsh MP, Johnson JD. Activation of smooth muscle myosin light chain kinase by calmodulin. Role of LYS(30) and GLY(40) J Biol Chem. 2002;277:6550–6558. doi: 10.1074/jbc.M111404200. [DOI] [PubMed] [Google Scholar]
- 18.Sun ZJ, Lei H, Zhang Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013;24:433–442. doi: 10.1016/j.cytogfr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahl TB, Yndestad A, Skjelland M, Øie E, Dahl A, Michelsen A, Damås JK, Tunheim SH, Ueland T, Smith C, et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: Possible role in inflammation and plaque destabilization. Circulation. 2007;115:972–980. doi: 10.1161/CIRCULATIONAHA.106.665893. [DOI] [PubMed] [Google Scholar]
- 20.Zakkar M, Guida G, Suleiman M, Angelini GD. Cardiopul-monary bypass and oxidative stress. Oxid Med Cell Longe. 2015;2015:189863. doi: 10.1155/2015/189863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye SQ, Simon BA, Maloney JP, Zambelli-Weiner A, Gao L, Grant A, Easley RB, McVerry BJ, Tuder RM, Standiford T, et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med. 2005;171:361–370. doi: 10.1164/rccm.200404-563OC. [DOI] [PubMed] [Google Scholar]
- 22.Hong SB, Huang Y, Moreno-Vinasco L, Sammani S, Moitra J, Barnard JW, Ma SF, Mirzapoiazova T, Evenoski C, Reeves RR, et al. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med. 2008;178:605–617. doi: 10.1164/rccm.200712-1822OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu P, Li H, Cepeda J, Zhang LQ, Cui X, Garcia JG, Ye SQ. Critical role of PBEF expression in pulmonary cell inflammation and permeability. Cell Biol Int. 2009;33:19–30. doi: 10.1016/j.cellbi.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, Zhang H, Cheng PY, Zhou FC. The influence of pre-B-cell colony enhancing factor on adhesive molecule in pulmonary cells in rats with acute lung injury/acute respiratory distress syndrome. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2013;25:159–163. doi: 10.3760/cma.j.issn.2095-4352.2013.03.010. (In Chinese) [DOI] [PubMed] [Google Scholar]
- 25.Gao W, Mao Q, Feng AW, Sun HM, Sun WK, Lu X, Su X, Shi Y. Inhibition of pre-B cell colony-enhancing factor attenuates inflammation and apoptosis induced by pandemic H1N1 2009 in lung endothelium. Respir Physiol Neurobiol. 2011;178:235–241. doi: 10.1016/j.resp.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 26.Ye SQ, Zhang LQ, Adyshev D, Usatyuk PV, Garcia AN, Lavoie TL, Verin AD, Natarajan V, Garcia JG. Pre-B-cell-colony-enhancing factor is critically involved in thrombin-induced lung endothelial cell barrier dysregulation. Microvasc Res. 2005;70:142–151. doi: 10.1016/j.mvr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Liu SW, Qiao SB, Yuan JS, Liu DQ. Visfatin stimulates production of monocyte chemotactic protein-1 and interleukin-6 in human vein umbilical endothelial cells. Horm Metab Res. 2009;41:281–286. doi: 10.1055/s-0028-1102914. [DOI] [PubMed] [Google Scholar]
- 28.Khan TA, Bianchi C, Araujo EG, Ruel M, Voisine P, Sellke FW. Activation of pulmonary mitogen-activated protein kinases during cardiopulmonary bypass. J Surg Res. 2003;115:56–62. doi: 10.1016/S0022-4804(03)00236-1. [DOI] [PubMed] [Google Scholar]
- 29.Yang D, Xie P, Guo S, Li H. Induction of MAPK phosphatase-1 by hypothermia inhibits TNF-alpha-induced endothelial barrier dysfunction and apoptosis. Cardiovasc Res. 2010;85:520–529. doi: 10.1093/cvr/cvp323. [DOI] [PubMed] [Google Scholar]
- 30.Sun HM, Hong LZ, Shen XK, Lin XQ, Song Y, Shi Y. Antithrombin-III without concomitant heparin improves endotoxin-induced acute lung injury rats by inhibiting the activation of mitogen-activated protein kinase. Chin Med J (Engl) 2009;122:2466–2471. [PubMed] [Google Scholar]
- 31.Liu T, Milia E, Warburton RR, Hill NS, Gaestel M, Kayyali US. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J Cell Physiol. 2012;227:1438–1445. doi: 10.1002/jcp.22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg PL, MacNaughton DE, Clements RT, Minnear FL, Vincent PA. p38 MAPK activation by TGF-beta1 increases MLC phosphorylation and endothelial monolayer permeability. Am J Physiol Lung Cell Mol Physiol. 2002;282:L146–L154. doi: 10.1152/ajplung.2002.282.1.L146. [DOI] [PubMed] [Google Scholar]
- 33.Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, II, Durán WN. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–H100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 34.Ming GF, Ma XH, Xu DM, Liu ZY, Ai YH, Liu HX, Shi ZH. PBEF promotes the apoptosis of pulmonary microvascular endothelial cells and regulates the expression of inflammatory factors and AQP1 through the MAPK pathways. Int J Mol Med. 2015;36:890–896. doi: 10.3892/ijmm.2015.2283. [DOI] [PubMed] [Google Scholar]