

Abstract
Ischemic heart disease is a major cause of mortality and disability worldwide. Timely reperfusion is currently the most effective method of treating ischemic heart disease; however, abrupt reperfusion may cause ischemia/reperfusion (I/R) injury. Apoptosis serves an important role in the progression of myocardial I/R injury and it has been demonstrated that the mitochondria are the center of regulation for apoptosis. Penehyclidine hydrochloride (PHC) is used during surgery and has recently been identified as a new type of anticholinergic drug. It has been demonstrated in vivo that pretreatment with PHC reduces myocardial apoptosis in rat hearts. The present study aimed to investigate the effects of PHC post-conditioning on myocardial cell apoptosis in a rat model of myocardial I/R and to determine whether the mitochondria-induced pathway was activated. Male Wistar rats were evenly and randomly categorized into 4 experimental groups as follows: i) Sham group; ii) I/R group; iii) PHC+sham group; and iv) PHC+I/R group. A PHC (1 mg/kg) post-conditioning approach (5 min before reperfusion) was used in addition to I/R in the PHC-treated groups. Following 3 h reperfusion, flow cytometry and terminal deoxynucleotidyl transferase dUTP nick end labeling staining were performed to measure myocardial cell apoptosis. A JC-1 staining method was performed to measure the mitochondrial membrane potential of myocardial cells. The expression of Bax, Bcl-2, voltage dependent anion-selective channel protein 1 (VDAC1), cytosol cytochrome c (cyt-c) and cleaved caspase-3 was analyzed using western blotting. PHC post-conditioning significantly reduced apoptosis in cardiomyocytes, significantly downregulated the expression of Bax, VDAC1, cytosol cytochrome c and cleaved caspase-3 but significantly upregulated the expression of Bcl-2. PHC post-conditioning also restored the mitochondrial membrane potential. Thus, the present study demonstrated that PHC post-conditioning protects cardiomyocytes against apoptosis in the rat model of myocardial I/R by inhibiting the mitochondria-induced intrinsic pathway.
Keywords: ischemia-reperfusion, apoptosis, mitochondrial, penehyclidine hydrochloride, post-conditioning
Introduction
Ischemic heart disease is a major cause of mortality and disability globally. According to the World Health Organization, it is predicted to become the primary global cause of mortality by the year 2020 (1). It accounts for 1.4 million cases of mortality in developed countries and 5.7 million in developing countries (2). Many risk factors, including smoking, obesity, hypertension, hyperlipidemia and hyperglycemia, may induce ischemic heart disease, which subsequently lead to congestive heart failure, myocardial infarction and even cardiac arrest (3,4). Timely reperfusion is currently the most effective method of treating ischemic heart disease (5). However, abrupt reperfusion causes ischemia/reperfusion (I/R) injury (6). Apoptosis serves an important role in the progression of myocardial I/R injury (7,8) and it has been demonstrated that the mitochondria are the center of regulation for apoptosis (9,10). As the primary apoptotic pathway, the mitochondria-induced intrinsic pathway is initiated by I/R and increases the size of mitochondrial permeability transition pores (MPTP), which in turn induces the release of apoptogenic factors and activation of the downstream apoptotic cascade reaction (11).
Penehyclidine hydrochloride (PHC) may be used during surgery; however, it has also recently been identified as a new type of anticholinergic drug (12). PHC selectively acts on nicotinic and muscarinic (muscarinic 1 and muscarinic 3 subtypes) receptors, which induces anticholinergic effects (13). The major benefit of PHC compared with other anticholinergic drugs is that it does not cause muscarinic 2 receptor-associated cardiovascular side effects (14). PHC stabilizes the heart rate without autonomic nervous modulation (15). Our previous study demonstrated that PHC pre-treatment reduces myocardial apoptosis in rat hearts in vivo (16); however, its mechanism of action remains unclear. Furthermore, ischemic heart disease is unpredictable in most cases; thus, post-conditioning treatment is clinically more significant than pre-conditioning treatment. Therefore, the present study aimed to investigate the effects of PHC post-conditioning on myocardial cell apoptosis in a rat model of myocardial I/R and to determine whether the mitochondrial-induced pathway is involved.
Materials and methods
Animal model
The present study was approved by the Ethics Committee of Capital Medical University (Beijing, China). The rats were cared for following the National Institutes of Health Guide for the Care and Use of Laboratory animals (17). A total of 24 8-week old Wistar rats (male; 220–250 g) were provided by Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Rats had free access to food and water and were housed in standard cages (22°C, 50% humidity, 12:12 h light/dark cycle). An animal model was induced following a previously described protocol (16). All rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (300 mg/kg; Yulong Algae Co., Ltd., Qingdao, China). Following thoracotomy, the rat heart was exposed. Myocardial I/R was induced by occlusion of the left anterior descending coronary artery and the coronary artery was subjected to 30 min ischemia followed by 3 h reperfusion. Rats in the sham operation group underwent the same procedure with no ligation. Rats were subsequently euthanized by injection of potassium chloride at the end of the experiment.
Experimental design
PHC injections were obtained from Chengdu List Pharmaceutical Co., Ltd. (Chengdu, China). They were sealed for preservation and stored in cool, dry place (20°C). PHC was prepared immediately prior to the start of the experiment. PHC (1 mg/ml) was diluted in normal saline. The 24 healthy male Wistar rats were randomly and evenly categorized into 4 experimental groups as follows: i) Sham group consisting of rats undergoing sham surgery; ii) I/R group consisting of rats subjected to 30 min myocardial ischemia followed by 3 h reperfusion; iii) PHC+sham group consisting of rats injected with PHC (1 mg/kg) via the tail vein, 25 min following sham surgery; and iv) PHC+I/R group consisting of rats subjected to 30 min myocardial ischemia, treated with PHC (1 mg/kg) via the tail vein 25 min following ischemia and then undergoing 3 h reperfusion. The experimental design is presented in Fig. 1.
Figure 1.
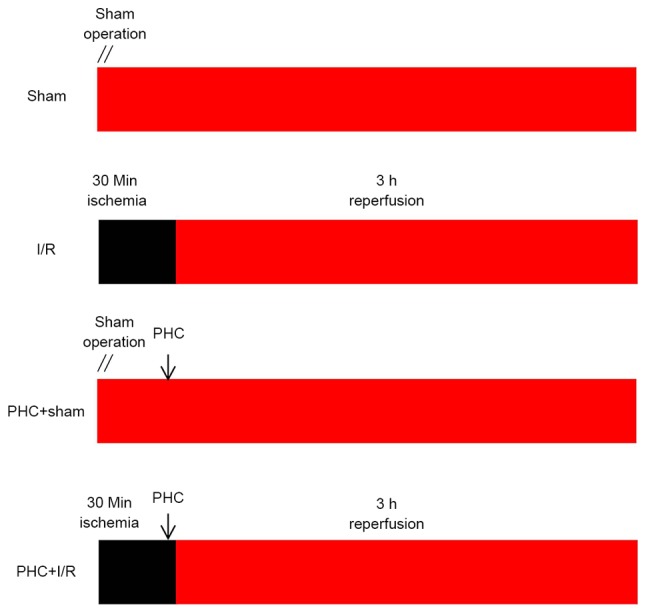
Experimental design. Rats were divided into: i) Sham group, in which rats underwent a sham operation; ii) I/R group, in which rats were subjected to 30 min myocardial ischemia followed by 3 h reperfusion; iii) PHC+sham group, in which rats were injected with PHC (1 mg/kg) via the tail vein 25 min following the sham operation; and iv) PHC+I/R group, in which rats were subjected to 30 min of myocardial ischemia and subsequently treated with PHC (1 mg/kg) via the tail vein 5 min prior to reperfusion, followed by 3 h reperfusion. The black box represents the period of ischemia. The red box represents the period of perfusion or reperfusion. I/R, ischemia/reperfusion; PHC, penehyclidine hydrochloride.
Flow cytometry
Apoptotic cells were analyzed by flow cytometry, as previously described (18). Following 3 h reperfusion, rat hearts were removed and myocardial tissue sections from frozen samples were shredded, digested and washed. The myocardial tissue was centrifuged for 10 min at 160 × g at 4°C and the precipitate was collected. The precipitate was washed twice with PBS and centrifuged for 10 min at 160 × g at 4°C. The precipitate was then resuspended in binding buffer containing 5 µl Annexin V-fluorescein isothiocyanate (FITC; BD Biosciences, Franklin Lakes, NJ, USA) and 5 µl propidium iodide staining solution (BD Biosciences). The solution was incubated for 15 min at 37°C in the dark. Apoptosis (%) was quantified using the Annexin V-FITC and propidium iodide apoptosis detection kit (BD Biosciences) by flow cytometry as previously described (BD Biosciences) (18). The result was analyzed with CellQuest Pro software version 5.2 (BD Biosciences).
Terminal deoxynucleotidyltransferase-mediated dUTP nick end labelling (TUNEL) staining
TUNEL staining was used to measure the rate of apoptosis, as previously described (16). Following 3 h reperfusion, rat hearts were removed. Myocardial tissue sections were dehydrated in ascending series of ethanol, cleared in xylene and embedded in paraffin. Paraffin-embedded myocardial tissues were cut into sections 5-µm thick, dewaxed with xylene and stained for TUNEL analysis. Sections were penetrated for 8 min with 0.1% Triton X-100 (50 µl; Beyotime Institute of Biotechnology, Haimen, China) and washed 3 times for 5 min each in PBS at 37°C. Sections were then blocked for 10 min by 3% H2O2 and washed 3 times for 5 min each in PBS at 37°C. TUNEL reaction mixture was added to the sections, which were then incubated for 60 min at 37°C in a dark in a humidified environment. Sections were washed 3 times in PBS for 5 min each at 37°C. Converter-POD (50 µl; Roche Diagnostics, Basel, Switzerland) was added to the sections and incubated for 30 min in a humid environment at 37°C. Sections were washed 3 times for 5 min each in PBS at 37°C. Sections were incubated with DAB substrate for 10 min at 15°C and washed 3 times for 5 min each in PBS at 37°C. Finally, sections were stained by hematoxylin for 3 min at 37°C and fixed by neutral balata. TUNEL-positive cells were counted using a fluorescent microscope (Olympus, Tokyo, Japan) at a magnification of ×400.
JC-1 staining
Following 3 h reperfusion, rat hearts were removed and myocardial tissue sections were shredded, centrifuged and stained using the JC-1 staining method, as previously described (19). Myocardial tissue sections were homogenized in mitochondrial isolation buffer and centrifuged at 1,000 × g for 5 min at 4°C. The supernatant was centrifuged at 11,000 × g for 10 min at 4°C and the precipitate was collected. The precipitate was resuspended in JC-1 solution (×200 JC-1 stock solution, X5 JC-1 staining buffer, ddH2O; Keygenbio, Nanjing, China) and incubated for 20 min at 37°C. A Lumat LB9507 chemic luminous instrument (Berthold Technologies, Bad Wildbad, Germany) was used to detect the absorbency of the samples. The ratio of red/green absorbency represents mitochondrial membrane potential (MMP).
Western blot analysis
Following 3 h reperfusion, rat hearts were removed. Apoptosis-related proteins were extracted and measured using western blot analysis, as previously described (20). Bax, Bcl-2 cytosol cytochrome c (cyt-c) and cleaved caspase-3 proteins were extracted by using a cytosol extraction kit (Wanleibio Co., Ltd., Shanghai, China). Voltage-dependent anion-selective channel protein 1 (VDAC1) protein was extracted by using a mitochondria extraction kit (Wanleibio Co., Ltd.). The expression of Bax, Bcl-2, VDAC1, cyt-c and cleaved caspase-3 was measured using a BCA Protein assay kit (Beyotime Institute of Biotechnology). The proteins (40 µg) were separated by 12% SDS-PAGE and transferred onto polyvinylidene fluoride membranes. Following blocking with 5% non-fat milk for 1 h and washing with TTBS (150 mM NaCl, 20 mM Tris, 0.1% Tween-20, pH 7.6) for 5 min, membranes were incubated with specific Bcl-2 antibody (1:500; cat. no/WL01556), Bax antibody (1:500; cat. no. WL01637; both from Wanleibio Co., Ltd.), VDAC1 antibody (1:500, cat. no. bs-1461R; Bioss, Beijing, China), β-actin antibody (1:1,000, WL01845), COX IV (1:500, WL01794) or cyt-c antibody (1:500, WL01571) (all from Wanleibio Co., Ltd.) overnight at 4°C and then with goat anti-rabbit immunoglobulin G conjugated to horseradish peroxidase (1:5,000, cat. no. WLA023a; Wanleibio Co., Ltd.) for 45 min at 37°C. An ECL western blotting detection system (Wanleibio Co., Ltd.) was used to determine protein bands. Image software GeneTools 4.3.7 (Syngene, Frederick, MD, USA) was used to quantify the band intensities. The band densities were normalized to β-actin or COX IV.
Statistical analysis
The results were obtained from 6 different samples and all data were expressed as the mean ± standard deviation. SPSS 13.0 (SPSS, Inc., Chicago, IL, USA) was used for analysis. The differences between data were evaluated using a two-way analysis of variance followed by Bonferroni post hoc testing. P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of PHC on cardiomyocyte apoptosis
Following 3 h reperfusion, apoptotic cells were analyzed using flow cytometry (Fig. 2A). There was a decreased proportion of apoptotic cells in the PHC+sham group compared with the Sham group; however, this difference was not significant (5.52±2.32% vs. 6.20±2.93%). The proportion of apoptotic cells in the I/R group was significantly increased compared with the Sham group (25.12±5.92% vs. 6.20±2.93%) and significantly decreased in the PHC+I/R group compared with the I/R group (7.35±3.42% vs. 25.12±5.92%, both P<0.001; all Fig. 2A).
Figure 2.

Effect of PHC on cardiomyocyte apoptosis. (A) Representative images of flow cytometry. Cell populations were regarded, respectively, as early-stage apoptotic in the LR quadrant, living in the LL quadrant, late-stage apoptotic in the UR quadrant and necrotic in the UL quadrant. (B) Representative images of myocardial TUNEL staining. TUNEL-positive cells were regarded as apoptotic and counted using a microscope at ×400 magnification. Cells in the nucleus contained brown granules suggesting positive staining and are indicated by red arrows. Normal cell nuclei were stained blue with hematoxylin. (C) The cardiomyocyte apoptotic rate was expressed as a ratio of TUNEL-positive cells (apoptotic cells) to the total number of cardiomyocytes. Data were expressed as the mean ± standard deviation (n=6 each group). Scale bar, 20 µm. ##P<0.01 vs. sham; **P<0.01 vs. I/R. ns, not significant (P>0.05); I/R, ischemia/reperfusion; PHC, penehyclidine hydrochloride; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labelling; LL, lower left; UL, upper left; LR, lower right; UR, upper right; FITC, fluorescein isothiocyanate.
Following 3 h reperfusion, TUNEL staining was performed (Fig. 2B) to measure the rate of apoptosis as the proportion of apoptotic cells in the sample. The difference in the apoptotic rate of cardiomyocytes in the PHC+sham group compared with the Sham group was not significant (3.62±1.5% vs. 2.95±1.4%; Fig. 2C). The apoptotic rate was significantly increased in the I/R group compared with the Sham group (58.09±6.2% vs. 2.95±1.4%) and significantly decreased in the PHC+I/R group compared with the I/R group (15.84±2.6% vs. 58.09±6.2%, both P<0.01; Fig. 2C).
Effect of PHC on the expression of apoptotic regulatory proteins
The expression of the apoptotic regulatory proteins Bax and Bcl-2 following 3 h reperfusion was measured using western blotting (Fig. 3A and B). The difference in the expression of Bax (0.92±0.03 vs. 1.00; Fig. 3A) and Bcl-2 (1.0±0.04 vs. 1.00; Fig. 3B) in the PHC+sham group compared with the Sham group were not significant. The expression of Bax was significantly increased in the I/R group compared with sham group (1.65±0.05 vs. 1.00) and significantly decreased in the PHC+I/R group compared with the I/R group (0.89±0.03 vs. 1.65±0.05, both P<0.01; Fig. 3A). The expression of Bcl-2 was significantly decreased in the I/R group compared with the Sham group (0.39±0.03 vs. 1.00) and significantly increased in the PHC+I/R group compared with the I/R group (1.01±0.03 vs. 0.39±0.03, both P<0.01; both Fig. 3B).
Figure 3.

Effect of PHC on the expression of apoptosis-related proteins. Following 3 h reperfusion, the expression of apoptosis-related proteins was analyzed using western blotting. Band densities were normalized to β-actin or COX IV. (A) Bax protein expression. (B) Bcl-2 protein expression. (C) VDAC1 protein expression. (D) Cytosol cyt-c protein expression. (E) Cleaved caspase-3 protein expression. Data are expressed as mean ± standard deviation (n=6). ##P<0.01 vs. sham; **P<0.01 vs. I/R. ns, not significant (P>0.05); I/R, ischemia/reperfusion; PHC, penehyclidine hydrochloride; COX IV, cytochrome c oxidase subunit 4; VDAC1, voltage dependent-selective anion channel protein 1; cyt-c, cytochrome-c.
Effect of PHC treatment on apoptosis-related protein expression
The expression of the apoptosis-related proteins VDAC1, cytosol cyt-c and cleaved caspase-3 was measured using western blotting following 3 h reperfusion.
The difference between the expression of VDAC1 in the PHC+sham group and the sham group was not significant (0.94±0.03 vs. 1.00). The expression of VDAC1 was significantly increased in the I/R group compared with the sham group (1.62±0.06 vs. 1.00) and significantly decreased in the PHC+I/R group compared with the I/R group (0.95±0.02 vs. 1.62±0.06, both P<0.01; Fig. 3C).
The difference between the expression of cytosol cyt-c in the PHC+sham group and the sham group was not significant (0.95±0.02 vs. 1.00). The expression of cytosol cyt-c was significantly increased in the I/R group compared with the Sham group (2.37±0.06 vs. 1.00) and significantly decreased in the PHC+I/R group compared with the I/R group (0.96±0.03 vs. 2.37±0.06, both P<0.01; Fig. 3D).
The difference between the expression of cleaved caspase-3 in the PHC+sham group and the Sham group was not significant (0.94±0.02 vs. 1.00). The expression of cleaved caspase-3 was significantly increased in the I/R group compared with the sham group (3.51±0.1 vs. 1.00) and significantly decreased in the PHC+I/R group compared with the I/R group (1.40±0.03 vs. 3.51±0.1, both P<0.01; all Fig. 3E).
Effect of PHC treatment on MMP
The cellular MMP was measured using a JC-1 staining method following 3 h reperfusion. The difference in MMP (0.99±0.03 vs. 1.00±0.03) between the PHC+sham group and the Sham group was not significant. The I/R group exhibited a significantly decreased MMP compared with the sham group, (0.36±0.01 vs. 1.00±0.03) and MMP was significantly increased in the PHC+I/R group compared with the I/R group (0.73±0.08 vs. 0.36±0.01, both P<0.01; all Fig. 4).
Figure 4.
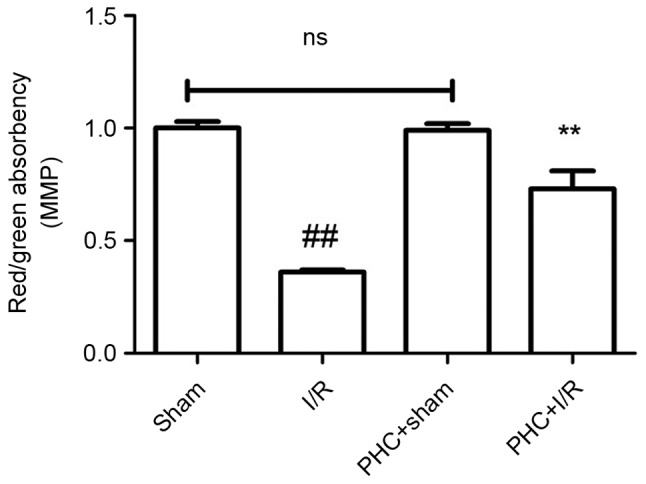
Effect of PHC on the MMP. Following 3 h reperfusion, a JC-1 staining method was performed to detect MMP. The ratio of red absorbency/green absorbency represents the MMP. Data are expressed as mean ± standard deviation (n=6). ##P<0.01 vs. sham; **P<0.01 vs. I/R. ns, not significant (P>0.05); I/R, ischemia/reperfusion; PHC, penehyclidine hydrochloride; MMP, myocardial cell mitochondrial membrane potential.
Discussion
During the course of reperfusion therapy in ischemic heart disease, there may be a rapid and severe onset of I/R injury. Thus, the focus of current studies in the field is to identify therapies that attenuate myocardial I/R injury.
The pathogenesis of I/R injury includes oxidative stress, cellular apoptosis, Ca2+ overload and an inflammatory response (21). Previous studies have demonstrated that myocardial apoptosis serves an important role in I/R injury (22–24). Apoptosis leads to a reduction in myocardial cells and cardiac function (25). Therapeutic strategies that inhibit the apoptosis of myocardial cells attenuate I/R injury (26). There are a number of apoptotic pathways, including the mitochondria-induced intrinsic pathway, the death receptor-induced extrinsic pathway and the endoplasmic reticulum stress-induced pathway (27–29). The initiation of the mitochondria-induced intrinsic pathway by pro-apoptotic signaling leads to the opening of MPTP, the release of apoptogenic factors and the activation of the apoptotic cascade reaction (11). Yu et al (30) demonstrated that PHC attenuates apoptosis in focal cerebral I/R and exhibits a neuroprotective effect. Lin et al (16) indicated that pretreatment with PHC reduces myocardial apoptosis in rat hearts in vivo. The results of the aforementioned studies demonstrate that I/R induces cell apoptosis, whereas PHC reduces cell apoptosis. Central components of the mitochondria-induced intrinsic pathway include Bcl-2 family members, MMP, MPTP, apoptogenic factor and caspase family members (9,11,31). The proportion of apoptotic cells, the apoptotic rate, the expression of apoptotic regulatory proteins Bax and Bcl-2, MMP, expression of MPTP via VDAC1, expression of the apoptogenic factor cytosol cyt-c and expression of the caspase family member cleaved caspase-3 were examined in the present study to determine whether PHC post-conditioning inhibits myocardial apoptosis via the mitochondria-induced intrinsic pathway. The present study demonstrated that rats undergoing I/R exhibit significantly increased numbers of apoptotic cardiomyocytes and an increased apoptotic rate, whereas PHC post-conditioning significantly decreased the number of apoptotic cardiomyocytes and the apoptotic rate. Thus, the results of the present study are consistent with the results of studies conducted by Yu et al (30) and Lin et al (16), as it was demonstrated that PHC treatment attenuates I/R injury by inhibiting apoptosis. Furthermore, the present study indicated that PHC post-conditioning effectively protects rats against I/R-induced myocardial apoptosis. The application of post-conditioning has more clinical significance than preconditioning, as ischemic heart disease is unpredictable in most cases (32).
The Bcl-2 family proteins pro-apoptotic Bax and anti-apoptotic Bcl-2 serve a key role in regulating the mitochondrial apoptotic process (33). It has been demonstrated that the balance between Bax and Bcl-2 is associated with apoptotic suppression or activation (34). Bax increases the size of MPTPs, whereas Bcl-2 decreases the size of MPTPs during apoptosis (35). The present study demonstrated that I/R significantly increased the expression of Bax but significantly decreased the expression of Bcl-2. Following treatment with PHC, the expression of Bax was significantly decreased, while the expression of Bcl-2 was significantly increased. These results indicate that PHC post-conditioning protects against I/R injury by regulating the balance between Bax and Bcl-2.
The MMP mediates mitochondrial activity and cellular survival (36) and I/R disrupts the MMP, which induces myocardial apoptosis (37). A decrease in the MMP is characterized as a key factor in the activation of apoptotic pathways. The present study demonstrated that myocardial I/R injury was accompanied by a decrease in the MMP. Treatment with PHC leads to a partial but significant recovery of the MMP. Thus, the results indicate that PHC post-conditioning maintains the MMP, which attenuates I/R-induced myocardial apoptosis.
MPTPs are complex protein channels in the inner mitochondrial membrane and their opening leads to a decrease in the MMP and the onset of mitochondrial swelling (38). VDAC is an important component of MPTPs located in the mitochondrial outer membrane (39). VDAC1 is the major form of VDAC and is a gatekeeper in MPTPs, and its expression serves an important role in mediating mitochondrial apoptosis (40). It has been previously demonstrated that an increase in VDAC1 expression leads to an increase in mitochondria membrane permeability (40). The present study demonstrated that there was a significant increase in VDAC1 expression following I/R, whereas PHC post-conditioning significantly decreased the expression of VDAC1. These results indicate that PHC may stabilize the mitochondrial membrane, inhibit the opening of MPTPs and protect mitochondrial activity against I/R-induced cardiomyocyte apoptosis.
The mitochondrial apoptogenic factor cyt-c is released from the mitochondrial inter-membrane space into the cytosol during reperfusion and induces the apoptotic process (41). Cytosol cyt-c combines with other apoptosis factors to form an apotosome, thus triggering a further apoptotic cascade (42). The present study demonstrated that I/R significantly increased the expression of cytosol cyt-c protein, whereas PHC post-conditioning significantly decreased the expression of cytosol cyt-c protein. These results indicate that PHC post-conditioning protects against cardiomyocyte apoptosis by inhibiting the release of cyt-c from the mitochondria into the cytosol.
Caspase-3 exhibits a high propensity and catalytic turnover to cleave substrates and is therefore a key executioner protease (43). Cleaved caspase-3 induces irreversible and intense apoptosis (44). The present study demonstrated that I/R significantly increased levels of cleaved caspase-3, whereas PHC post-conditioning significantly decreased cleaved caspase-3 levels in the cytosol. These results indicate that PHC post-conditioning decreases I/R-induced apoptosis by attenuating the activation of caspase-3.
In conclusion, the present study demonstrated that PHC post-conditioning protects cardiomyocytes against apoptosis in a rat model of myocardial I/R. PHC served an important role in inhibiting myocardial apoptosis through the mitochondria-induced intrinsic pathway. Its anti-apoptotic mechanisms consist of suppressing of Bax expression, activating Bcl-2, promoting the recovery of MMP and inhibiting VDAC1, cytosol cyt-c and cleaved caspase-3 in myocardial I/R. The present study indicated that a PHC post-conditioning approach may be developed as a novel therapeutic strategy for treating I/R-induced myocardial apoptosis. However, the anti-apoptotic mechanism of action of PHC post-conditioning remains unclear and requires further study.
Acknowledgements
The present study was supported by the National Nature Science Foundation of China (grant no. 81471902).
References
- 1.Ostadal B. The past, the present and the future of experimental research on myocardial ischemia and protection. Pharmacol Rep. 2009;61:3–12. doi: 10.1016/S1734-1140(09)70002-7. [DOI] [PubMed] [Google Scholar]
- 2.Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: A review of global methodologies of mortality measurement. Circulation. 2013;127:749–756. doi: 10.1161/CIRCULATIONAHA.112.128413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel AV, Bangalore S. Challenges with evidence-based management of stable ischemic heart disease. Curr Cardiol Rep. 2017;19:11. doi: 10.1007/s11886-017-0820-7. [DOI] [PubMed] [Google Scholar]
- 4.Cordero A, Galve E, Bertomeu-Martinez V, Bueno H, Fácila L, Alegria E, Cequier Á, Ruiz E, González-Juanatey JR. Trends in risk factors and treatments in patients with stable ischemic heart disease seen at cardiology clinics between 2006 and 2014. Rev Esp Cardiol (Engl Ed) 2016;69:401–407. doi: 10.1016/j.recesp.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Chi HJ, Chen ML, Yang XC, Lin XM, Sun H, Zhao WS, Qi D, Cai J, Dong JL. Progress in therapies for myocardial ischemia reperfusion injury. Curr Drug Targets. 2016 Apr 1; doi: 10.2174/1389450117666160401120308. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 6.Barrere-Lemaire S, Nargeot J, Piot C. Delayed postconditioning: Not too late? Trends Cardiovas Med. 2012;22:173–179. doi: 10.1016/j.tcm.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Qiao Y, Zhao Y, Liu Y, Ma N, Wang C, Zou J, Liu Z, Zhou Z, Han D, He J, et al. miR-483-3p regulates hyperglycaemia-induced cardiomyocyte apoptosis in transgenic mice. Biochem Biophys Res Commun. 2016;477:541–547. doi: 10.1016/j.bbrc.2016.06.051. [DOI] [PubMed] [Google Scholar]
- 8.Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, Lips DJ, Doevendans PA. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004;61:414–426. doi: 10.1016/j.cardiores.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 9.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/S0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 10.Teringova E, Tousek P. Apoptosis in ischemic heart disease. J Transl Med. 2017;15:87. doi: 10.1186/s12967-017-1191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 12.Zhan J, Xiao F, Li JJ, Zhang ZZ, Chen K, Wang YP, Wang YL. Penehyclidine hydrochloride decreases pulmonary microvascular permeability by upregulating beta arrestins in a murine cecal ligation and puncture model. J Surg Res. 2015;193:391–398. doi: 10.1016/j.jss.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Wang YA, Zhou WX, Li JX, Liu YQ, Yue YJ, Zheng JQ, Liu KL, Ruan JX. Anticonvulsant effects of phencynonate hydrochloride and other anticholinergic drugs in soman poisoning: Neurochemical mechanisms. Life Sci. 2005;78:210–223. doi: 10.1016/j.lfs.2005.04.071. [DOI] [PubMed] [Google Scholar]
- 14.Ma TF, Zhou L, Wang Y, Qin SJ, Zhang Y, Hu B, Yan JZ, Ma X, Zhou CH, Gu SL. A selective M1 and M3 receptor antagonist, penehyclidine hydrochloride, prevents postischemic LTP: Involvement of NMDA receptors. Synapse. 2013;67:865–874. doi: 10.1002/syn.21693. [DOI] [PubMed] [Google Scholar]
- 15.Liu XB, Pan S, Yang XG, Li ZW, Sun QS, Zhao Z, Ma HC, Cui CR. Effect of penehyclidine hydrochloride on heart rate variability in hysteroscopy. Exp Ther Med. 2015;10:181–186. doi: 10.3892/etm.2015.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin D, Ma J, Xue Y, Wang Z. Penehyclidine hydrochloride preconditioning provides cardioprotection in a rat model of myocardial Ischemia/Reperfusion injury. PLoS One. 2015;10:e138051. doi: 10.1371/journal.pone.0138051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Research NRCU. Guide for the Care and Use of Laboratory Animals. National Academies Press (US); Washington (DC): 1996. [PubMed] [Google Scholar]
- 18.Li T, Xu XH, Tang ZH, Wang YF, Leung CH, Ma DL, Chen XP, Wang YT, Chen Y, Lu JJ. Platycodin D induces apoptosis and triggers ERK- and JNK-mediated autophagy in human hepatocellular carcinoma BEL-7402 cells. Acta Pharmacol Sin. 2015;36:1503–1513. doi: 10.1038/aps.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koka S, Das A, Salloum FN, Kukreja RC. Phosphodiesterase-5 inhibitor tadalafil attenuates oxidative stress and protects against myocardial ischemia/reperfusion injury in type 2 diabetic mice. Free Radic Biol Med. 2013;60:80–88. doi: 10.1016/j.freeradbiomed.2013.01.031. [DOI] [PubMed] [Google Scholar]
- 20.Reifschneider NH, Goto S, Nakamoto H, Takahashi R, Sugawa M, Dencher NA, Krause F. Defining the mitochondrial proteomes from five rat organs in a physiologically significant context using 2D blue-native/SDS-PAGE. J Proteome Res. 2006;5:1117–1132. doi: 10.1021/pr0504440. [DOI] [PubMed] [Google Scholar]
- 21.Sharma V, Bell RM, Yellon DM. Targeting reperfusion injury in acute myocardial infarction: A review of reperfusion injury pharmacotherapy. Expert Opin Pharmaco. 2012;13:1153–1175. doi: 10.1517/14656566.2012.685163. [DOI] [PubMed] [Google Scholar]
- 22.Zhu T, Yao Q, Wang W, Yao H, Chao J. iNOS induces vascular endothelial cell migration and apoptosis via autophagy in ischemia/reperfusion injury. Cell Physiol Biochem. 2016;38:1575–1588. doi: 10.1159/000443098. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Cao Y, Shen M, Wang B, Zhang W, Liu Y, He X, Wang L, Xia Y, Ding M, et al. DIDS reduces ischemia/reperfusion-induced myocardial injury in rats. Cell Physiol Biochem. 2015;35:676–688. doi: 10.1159/000369728. [DOI] [PubMed] [Google Scholar]
- 24.Sima N, Lü W, Xie X. Early proteins E6 and E7 of human papillomavirus may attenuate ischemia-reperfusion injury. Med Hypotheses. 2011;76:607–609. doi: 10.1016/j.mehy.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, Li G, Fu X, Qi Y, Li M, Lu G, Hu J, Wang N, Chen Y, Bai Y, Cui M. PDCD5 protects against cardiac remodeling by regulating autophagy and apoptosis. Biochem Biophys Res Commun. 2015;461:321–328. doi: 10.1016/j.bbrc.2015.04.032. [DOI] [PubMed] [Google Scholar]
- 26.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.RES.79.5.949. [DOI] [PubMed] [Google Scholar]
- 27.Park YJ, Choi C, Chung KH, Kim KH. Pharbilignan C induces apoptosis through a mitochondria-mediated intrinsic pathway in human breast cancer cells. Bioorg Med Chem Lett. 2016;26:4645–4649. doi: 10.1016/j.bmcl.2016.08.054. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Wang QL, Nong XH, Zhang XY, Xu XY, Qi SH, Wang YF. Oxalicumone A, a new dihydrothiophene-condensed sulfur chromone induces apoptosis in leukemia cells through endoplasmic reticulum stress pathway. Eur J Pharmacol. 2016;783:47–55. doi: 10.1016/j.ejphar.2016.04.056. [DOI] [PubMed] [Google Scholar]
- 29.Lee HP, Li TM, Tsao JY, Fong YC, Tang CH. Curcumin induces cell apoptosis in human chondrosarcoma through extrinsic death receptor pathway. Int Immunopharmacol. 2012;13:163–169. doi: 10.1016/j.intimp.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Yu C, Wang J. Neuroprotective effect of penehyclidine hydrochloride on focal cerebral ischemia-reperfusion injury. Neural Regen Res. 2013;8:622–632. doi: 10.3969/j.issn.1673-5374.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palchaudhuri R, Lambrecht MJ, Botham RC, Partlow KC, van Ham TJ, Putt KS, Nguyen LT, Kim SH, Peterson RT, Fan TM, Hergenrother PJ. A small molecule that induces intrinsic pathway apoptosis with unparalleled speed. Cell Rep. 2015;13:2027–2036. doi: 10.1016/j.celrep.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weintraub WS, Boden WE. Reexamining the efficacy and value of percutaneous coronary intervention for patients with stable ischemic heart disease. JAMA Intern Med. 2016;176:1190–1194. doi: 10.1001/jamainternmed.2016.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009;21:871–877. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Xia A, Xue Z, Li Y, Wang W, Xia J, Wei T, Cao J, Zhou W. Cardioprotective effect of betulinic Acid on myocardial ischemia reperfusion injury in rats. Evid Based Complement Alternat Med. 2014;2014:573745. doi: 10.1155/2014/573745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria-specificity in membrane targeting for death. Biochim Biophys Acta. 2011;1813:532–539. doi: 10.1016/j.bbamcr.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 36.Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, O'Rourke B, Akar FG. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J Mol Cell Cardiol. 2010;49:565–575. doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadenbach B, Ramzan R, Moosdorf R, Vogt S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion. 2011;11:700–706. doi: 10.1016/j.mito.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Xiao-Feng L, Wen-Ting Z, Yuan-Yuan X, Chong-Fa L, Lu Z, Jin-Jun R, Wen-Ya W. Protective role of 6-Hydroxy-1-H-Indazole in an MPTP-induced mouse model of Parkinson's disease. Eur J Pharmacol. 2016;791:348–354. doi: 10.1016/j.ejphar.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Shoshan-Barmatz V, Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion. 2012;12:24–34. doi: 10.1016/j.mito.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Marenzi G, Giorgio M, Trinei M, Moltrasio M, Ravagnani P, Cardinale D, Ciceri F, Cavallero A, Veglia F, Fiorentini C, et al. Circulating cytochrome c as potential biomarker of impaired reperfusion in ST-segment elevation acute myocardial infarction. Am J Cardiol. 2010;106:1443–1449. doi: 10.1016/j.amjcard.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 42.Lee HJ, Lee HJ, Lee EO, Ko SG, Bae HS, Kim CH, Ahn KS, Lu J, Kim SH. Mitochondria-cytochrome c-caspase-9 cascade mediates isorhamnetin-induced apoptosis. Cancer Lett. 2008;270:342–353. doi: 10.1016/j.canlet.2008.05.040. [DOI] [PubMed] [Google Scholar]
- 43.Demon D, Van Damme P, Vanden Berghe T, Deceuninck A, Van Durme J, Verspurten J, Helsens K, Impens F, Wejda M, Schymkowitz J, et al. Proteome-wide substrate analysis indicates substrate exclusion as a mechanism to generate caspase-7 versus caspase-3 specificity. Mol Cell Proteomics. 2009;8:2700–2714. doi: 10.1074/mcp.M900310-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao L, Duanmu W, Yin Y, Zhou Z, Ge H, Chen T, Tan L, Yu A, Hu R, Fei L, Feng H. Dihydroartemisinin exhibits anti-glioma stem cell activity through inhibiting p-AKT and activating caspase-3. Pharmazie. 2014;69:752–758. [PubMed] [Google Scholar]