

Abstract
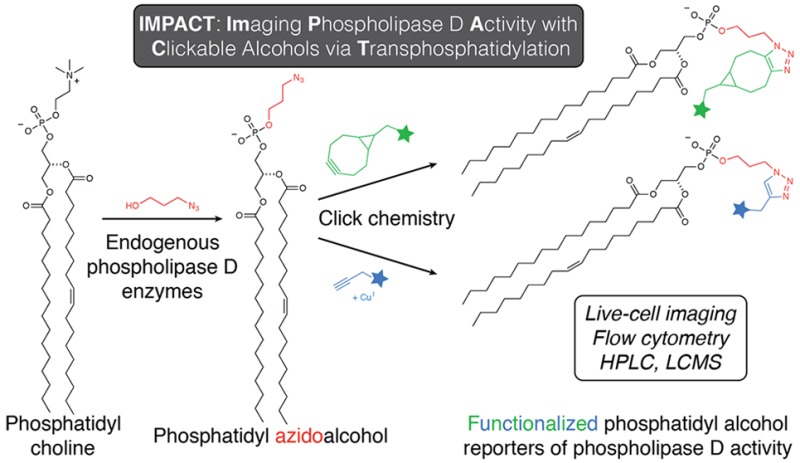
Chemical imaging techniques have played instrumental roles in dissecting the spatiotemporal regulation of signal transduction pathways. Phospholipase D (PLD) enzymes affect cell signaling by producing the pleiotropic lipid second messenger phosphatidic acid via hydrolysis of phosphatidylcholine. It remains a mystery how this one lipid signal can cause such diverse physiological and pathological signaling outcomes, due in large part to a lack of suitable tools for visualizing the spatial and temporal dynamics of its production within cells. Here, we report a chemical method for imaging phosphatidic acid synthesis by PLD enzymes in live cells. Our approach capitalizes upon the enzymatic promiscuity of PLDs, which we show can accept azidoalcohols as reporters in a transphosphatidylation reaction. The resultant azidolipids are then fluorescently tagged using the strain-promoted azide–alkyne cycloaddition, enabling visualization of cellular membranes bearing active PLD enzymes. Our method, termed IMPACT (Imaging Phospholipase D Activity with Clickable Alcohols via Transphosphatidylation), reveals pools of basal and stimulated PLD activities in expected and unexpected locations. As well, we reveal a striking heterogeneity in PLD activities at both the cellular and subcellular levels. Collectively, our studies highlight the importance of using chemical tools to directly visualize, with high spatial and temporal resolution, the subset of signaling enzymes that are active.
Short abstract
Simple azidoalcohols enable a click chemistry-based method for imaging the subcellular localizations of the active pools of phospholipase D signaling enzymes.
Signal transduction pathways allow cells to translate biochemical cues from the extracellular environment into changes in metabolism, gene expression, and behavior. Second messengers are key signaling intermediates in these pathways whose downstream effects depend greatly on cell type, physiological state, and, importantly, the intracellular location of their production. Because the spatial regulation of signaling is so critical for ensuring desired physiological outcomes, imaging-based tools have become indispensable for studying the dynamics of signaling events within live cells.
Phospholipase D (PLD) enzymes impact intracellular signaling by synthesizing the pleiotropic lipid second messenger phosphatidic acid (PA).1 PLD-mediated synthesis of PA leads to diverse physiological changes,2 including modifications to membrane curvature, vesicle trafficking, and the actin cytoskeleton as well as activation of protein kinases.3 These changes ultimately cause modulations in cell growth, division, migration, and other behaviors.4 To achieve such a diverse set of physiological outcomes from a sole signaling agent, cells use multiple upstream signals to selectively activate different PLD isozymes at specific locations to control PA production spatiotemporally.5
Several strategies exist to image PLD signaling, each with its strengths and drawbacks.6 Fluorescent protein fusions have revealed dynamic localizations of PLD1 and PLD2, the two isozymes responsible for PA generation via hydrolysis of phosphatidylcholine (PC).7−10 It is now well appreciated, however, that the localization of total enzyme pools often does not correlate well with the subpopulations that are active.11,12 Conversely, there exist several genetically encoded probes to directly visualize PA, consisting of positively charged, PA-binding peptides fused to fluorescent proteins; however, they cannot distinguish between different biosynthetic pools of PA originating from PLDs, diacylglycerol kinases, or lysophosphatidic acid acyltransferases.6,13 Furthermore, these probes can perturb signaling by masking the target lipid, and their binding often depends on additional ligands or membrane bilayer properties, leading to biased localizations.
We set out to develop a universal, unbiased imaging strategy to identify and track discrete pools of PLD-generated PA within live cells. We focused our efforts on PA generated by PLD for several reasons. First, PLDs are upregulated in several pathological scenarios, including cancer, neurodegeneration, autoimmunity, and infectious disease.10,14,15 As well, PLD1 has low basal but highly inducible activity and a localization that has been reported to change upon activation,7,16 suggesting differences between total and active pools of enzyme that may have major implications for signaling.
Using fluorescent protein fusions, localizations of PLD1 and PLD2 have been reported in the membranes of several organelles, most prominently the Golgi apparatus, endosomes, lysosomes, and the plasma membrane. Surprisingly, we reveal here, using a chemical imaging technique that can directly monitor PLD activity, that the bulk of endogenous PLD activity, both basal and stimulated, appears to occur at the endoplasmic reticulum (ER) and Golgi apparatus, as well as small but distinct endosomal and lysosomal pools. These results have important implications for understanding cellular control of PLD signaling. Furthermore, they highlight the importance of using approaches to visualize active populations of signaling enzymes rather than total pools of enzyme or signaling agents for dissecting metabolic and signaling pathways.
To image the dynamics of PLD-dependent PA synthesis, we capitalized on the ability of PLD enzymes, which normally hydrolyze PC to generate PA, to accept small primary alcohols in a transphosphatidylation reaction to produce phosphatidyl alcohols.1,17 Transphosphatidylation with ethanol or 1-butanol has been widely used to assay PLD activity in vitro by thin-layer chromatography or mass spectrometry.17,18 High concentrations of alcohols can be used (1%, or approximately 150 mM), to block the PLD-mediated production of PA, and lower concentrations may be used as tracers that do not substantially perturb rates of PA synthesis. However, these methods do not reveal the subcellular localization of where the PA is being generated. Antibodies to image phosphatidyl ethanol exist but have not been widely employed, perhaps due to issues of sensitivity and specificity. We recently reported that PLDs can accept alkynols as substrates, with detection of PLD activity enabled at much lower alcohol concentrations due to subsequent probe tagging using the copper-catalyzed azide–alkyne cycloaddition (CuAAC).19 While this approach could be used to image cellular PLD activity, due to the toxicity of copper, alkynols are not suitable PLD probes to follow the dynamics of PLD activity and signaling within live cells.
Here we present a chemical method termed IMPACT (Imaging Phospholipase D Activity with Clickable Alcohols via Transphosphatidylation) to image pools of PA produced by PLD. Our two-step strategy involves stimulation of endogenous PLD activity in the presence of an azidoalcohol to generate phosphatidyl azidoalcohols, followed by tagging using copper-free click chemistry to append imaging or other detection probes (Figure 1A). Importantly, our approach does not significantly perturb endogenous PA levels, and the cytocompatibility of copper-free click chemistry enabled the imaging of the subcellular locations of PLD activity within live cells, revealing unexpected localizations for PLD-mediated production of PA.
Figure 1.

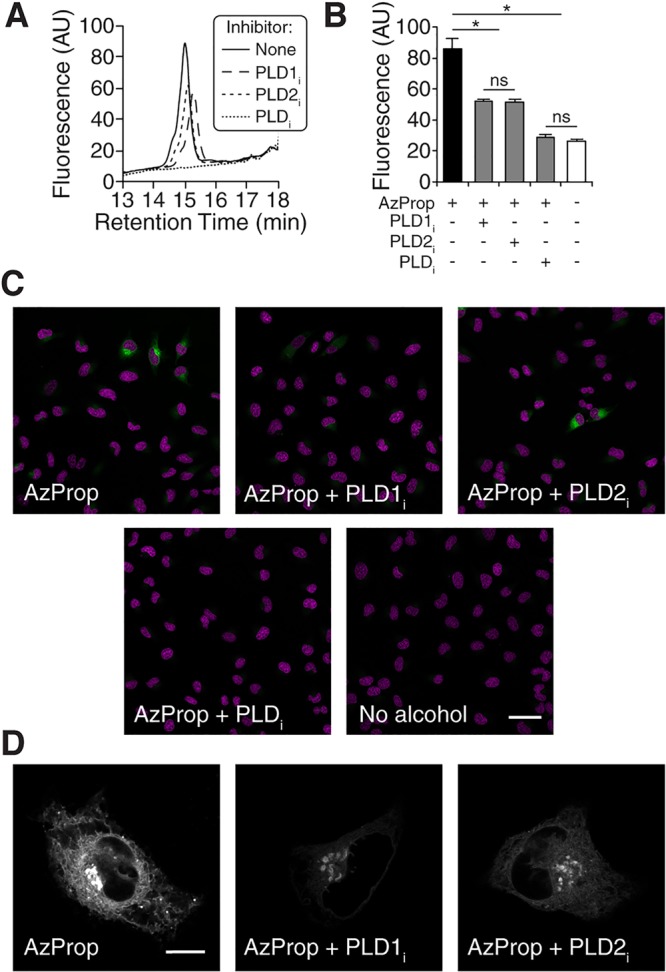
Azidoalcohols can be used as reporters of PMA-stimulated, endogenous PLD activity in HeLa cells. (A) Schematic of method to monitor PLD activity using azidoalcohols and click chemistry. (B, C) Azidoalcohols can report on PLD activity in mammalian cells. HeLa cells were first treated with DMSO (solid lines) or a PLD inhibitor (B, FIPI (750 nM, dashed lines); C, VU0359595 (250 nM, dashed lines) or VU0364739 (350 nM, dotted lines)) for 30 min, then with the indicated azidoalcohol (1 mM) or vehicle (B, dotted lines) for 20 min, and then stimulated with PMA (100 nM) for 20 min, followed by lipid extraction, SPAAC tagging with BODIPY-cyclooctyne 1, and analysis by fluorescence-coupled HPLC. (D–F) AzProp faithfully reports on PLD-mediated PA synthesis and does not inhibit its production. (D, E) HeLa cells were labeled with AzProp (1 mM) for 20 min (D and E, white lines) or no alcohol (E, black lines) and then stimulated with PMA (100 nM) for 20 min, followed by lipid extraction, CuAAC labeling with alkynyl ammonium salt 2a, and analysis by LC/ESI-TOF MS. Shown are relative amounts of the individual phosphatidyl alcohol (D) or endogenous phosphatidic acid (E) species, indicated as number of carbons:degree of unsaturation in lipid tails. (F) HeLa cells were first treated with FIPI (PLDi, 750 nM) or DMSO vehicle for 30 min, then with AzProp (1 mM), butanol (1% w/v), or no alcohol for 20 min, and then stimulated with PMA (100 nM) for 20 min, followed by lipid extraction, CuAAC labeling with 2a and analysis by LC/ESI-TOF MS. Shown are the relative total amounts of all phosphatidic acid species detected. *, p < 0.01; ns, not significant. For D–F, n = 3 (3 technical replicates each of 3 independent biological experiments), and the horizontal bar represents the mean.
We began by treating HeLa cells with a panel of azidoalcohols (2-azidoethanol (AzEt), 3-azido-1-propanol (AzProp), 4-azido-1-butanol (AzBut), and 5-azido-1-pentanol (AzPent)) and stimulating endogenous PLD activity using phorbol 12-myristate 13-acetate (PMA). Following lipid extraction and tagging by strain-promoted azide–alkyne cycloaddition (SPAAC) with BODIPY-cyclooctyne 1,20 HPLC analysis revealed the presence of fluorescent lipid species for all azidoalcohols tested (Figures 1B and S1). Importantly, treatment of cells during the labeling procedure with the pan-PLD inhibitor 5-fluoro-2-indolyl deschlorohalopemide (FIPI, or PLDi)21 led to complete loss of the fluorescent lipid species (Figure 1B), confirming that the fluorescently labeled lipids were derived exclusively from PLD activity. We then used isoform-selective PLD inhibitors to assess the relative contributions of PLD1 and PLD2. Treatment with isoform-selective PLD1 (VU0359595)22 or PLD2 (VU0364739)23 inhibitors led to decreases in approximately 75% and 25%, respectively, of the signal (Figure 1C). These data demonstrate that our method can report on both PLD1 and PLD2 activity and is consistent with the majority of PMA-stimulated PLD activity deriving from the PLD1 isozyme.24 In terms of efficacy, AzProp, AzBut, and AzPent all performed roughly equivalently, while AzEt was a poorer PLD substrate (Figure 1B). Given its commercial availability and synthetic tractability, as well as its superior performance in imaging experiments (vide infra), we elected to focus our efforts on AzProp.
We then turned to liquid chromatography–mass spectrometry (LCMS) based lipidomics analysis to unequivocally confirm the identity of the labeled lipids as phosphatidyl azidoalcohols. We were initially unable to detect either the underivatized or BODIPY-labeled phosphatidyl azidoalcohols from HeLa cell extracts, presumably due to a combination of poor ionization under electrospray conditions and, for the former, an overlap with the mass range of abundant cellular phospholipids. To overcome these issues, we tested a panel of clickable derivatization tags to enhance LCMS detection following electrospray ionization.
We first synthesized a phosphatidyl azidoalcohol lipid standard, phosphatidyl azidopropanol, via an in vitro chemoenzymatic reaction between 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and AzProp catalyzed by a commercially available PLD. We then subjected this azidolipid standard to copper-catalyzed azide–alkyne cycloaddition (CuAAC) labeling with a variety of alkynes bearing charged, polar, and nonpolar functional groups, followed by electrospray ionization time-of-flight (ESI-TOF) analysis (Figure S2A). We found that reagent 2a, which endowed the azidolipid with a quaternary ammonium group, a known enhancer of signal in ESI-MS,25 performed optimally (Figure S2B and Table S1).
LCMS analysis of lipidomes from cells treated with AzProp and stimulated with PMA, followed by CuAAC labeling with 2a, revealed labeling of several phosphatidyl alcohols differing in lipid tail length and degree of unsaturation. Importantly, the relative abundances of the different phosphatidyl alcohol species mirrored those of the natural PA species in the cell and, as expected for PLD-derived PA,18,26 were enriched in lipids with shorter lipid tail lengths and lower levels of unsaturation (Figure 1D and Table S2). As well, the lower alcohol concentrations enabled by our two-step labeling procedure resulted in no diminishment in the levels of natural PA species in the cell, confirming that our approach does not perturb endogenous PLD signaling (Figures 1E,F and Table S3).
Having established that azidoalcohols such as AzProp could effectively serve as faithful and nonperturbative reporters of endogenous PLD activity, we set out to image the localization of PLD-dependent PA synthesis in live cells. We first incubated cells with various azidoalcohols and stimulated PLD activity with PMA. Subsequently, we labeled cells for 10 min with 1, a cell-permeable, cyclooctyne–fluorophore conjugate that exhibits minimal nonspecific binding to cellular membranes.20 Following a brief rinse-out, we imaged the cells by confocal microscopy, observing strong fluorescence labeling of several intracellular compartments (Figures 2A and S3). Control experiments using FIPI, VU0359595, and VU0364739 again confirmed that the bulk of the labeling under PMA stimulation can be ascribed to PLD1 activity (Figures 2A,B). Among the azidoalcohols, the signal-to-background was highest for AzProp and AzEt, possibly because excess alcohol was more easily rinsed out of cells for these more hydrophilic alcohols (Figure S3). Among these two, we elected to proceed with AzProp due to its higher level of labeling in imaging and HPLC experiments (Figures S3 and 1B).
Figure 2.

Live-cell imaging of PMA-stimulated PLD activity reveals pools of active enzyme on ER, Golgi, lysosomal, and endosomal membranes. (A, B) HeLa cells were first treated with the indicated PLD inhibitor (PLDi (FIPI), 750 nM; PLD1i (VU0359595), 250 nM; PLD2i (VU0364739), 350 nM) or DMSO vehicle for 30 min, followed by AzProp (1 mM) for 20 min, and then stimulated with PMA (100 nM) for 20 min. Cells were then incubated with 1 (1 μM) for 10 min, rinsed for 15 min, and imaged by confocal microscopy. Arrowheads denote AzProp-positive puncta. (C–F) HeLa cells were transfected with the indicated plasmids (ER, STIM1-mRFP; Golgi, mCherry-P4M; Lysosomes (Lyso), LAMP1-mRFP; Endosomes (Endo), Rab5-mRFP; PLD1, mCherry-PLD1) and then labeled as in panel A with AzProp, PMA, and 1. Shown are single z-slices, with zoomed-in regions indicated by dashed outline shown in the upper right corner. For entire z-stack, see Movies S1 and S2. For D–F, frames from time-lapse movies of representative zoomed-in regions (indicated by dashed outline) are shown. Solid arrowheads denote LAMP1/Rab5/PLD1 puncta that colocalize with the AzProp label, and hollow arrowheads denote LAMP1/Rab5/PLD1 puncta that do not contain the AzProp label. In merged images, colocalization appears white. For C–F, colocalization is demonstrated by intensity plots along a one-dimensional profile corresponding to the dashed line in the merged image. Pearson correlation coefficients (R2) are provided to aid in interpreting colocalization of the two markers. Scale bars: 10 μm for all except 1 μm for zoomed-in regions in C, time-lapse images in D–F, and one-dimensional profiles in C–F.
Because there is a delay between PLD-mediated phosphatidyl azidoalcohol production and imaging following the click chemistry step, we performed additional control experiments to verify that the observed labeling pattern is reflective of PLD activity and not of phosphatidyl alcohol redistribution between different organelle membranes. To address potential diffusion or trafficking during the SPAAC reaction, we altered the labeling procedure by fixation immediately after the PMA stimulation, followed by CuAAC tagging with an alkyne–rhodamine 110 conjugate (Figure S4A). Second, to test for redistribution during the PMA stimulation, we shortened the PMA stimulation time from 20 to 5 min, using both the live-cell SPAAC and fixed-cell CuAAC detection protocols (Figure S4B). In both cases, the overall labeling pattern was largely similar to that in Figure 2A, suggesting that there is no observable change in localization due to the SPAAC labeling step.
To determine the subcellular localization of the fluorescent phosphatidyl alcohols, we performed colocalization experiments of IMPACT-derived fluorescence with various organelle markers by confocal microscopy and super-resolution structured illumination microscopy (SR-SIM). The majority of the labeling colocalized strongly with markers of the Golgi apparatus and ER (Figures 2C and S5A,B and Movies S1 and S2). To validate these colocalization analyses, we performed analogous colocalization experiments on fixed cells labeled either with SPAAC identically to the live-cell samples but fixed prior to imaging or with CuAAC following fixation as previously shown (Figure S6). We attribute any differences in relative fluorescence intensity of the IMPACT label in between various protocols (e.g., Figure S6 compared to Figure 2C) to the fixation step.
We then assessed, using fluorescence recovery after photobleaching (FRAP), the time scale on which the fluorescent lipid reporters of PLD activity diffused and trafficked around the cell. In these studies, we observed rapid (<5 s) FRAP in the ER, suggesting that, as expected, the fluorescently labeled lipids diffused very rapidly within the lipid bilayer of an individual organelle (Figure S7A and Movie S3). However, we observed minimal FRAP in a region of the Golgi apparatus over 20 min, suggesting that trafficking of the fluorescent phosphatidyl alcohols occurs much more slowly (Figure S7B and Movie S4). These FRAP studies support the idea that our labeling protocol, including the SPAAC and associated rinse steps, has sufficient temporal resolution to report on the localizations of PLD activity in live cells.
While PLD-dependent PA production at the Golgi apparatus is well documented for many physiological processes occurring on Golgi membranes,1 we were surprised to observe such strong and striking fluorescent phosphatidyl alcohol labeling of the ER. The ER is the principal cellular site of de novo phospholipid biosynthesis, in which PA is a central intermediate.27 However, PA pools in the ER are generally thought to be produced from glycerol 3-phosphate via acyltransferase activities,27 though a single study has proposed functions for PLD-generated PA in promoting vesicle trafficking from the ER to the Golgi via the Sar1 GTPase.28 Our data suggest, however, that a substantial fraction of inducible PLD activity may occur at ER membranes, which is surprising given limited evidence for functions and localizations of PLDs at ER membranes.
Additionally and importantly, we noticed a small number of IMPACT-derived bright puncta in each cell. These puncta exhibited partial colocalization by confocal microscopy and SR-SIM with markers of both lysosomes (LAMP1) and endosomes (Rab5), consistent with known roles for PLD1-generated PA on these organelles, including in mTOR activation and macroautophagy (Figures 2D,E and S5C,D and Movies S5 and S6).29−31 Interestingly, only a subset of these organelles contained the labeled phosphatidyl alcohols.
We reasoned that if the fluorescent phosphatidyl alcohols are indeed faithful reporters of PLD activity, then performing the AzProp/SPAAC labeling in cells overexpressing a fluorescently tagged PLD1 construct should result in increased IMPACT labeling on PLD1-positive structures. Therefore, we generated an mCherry-tagged PLD1 and found that, as expected for an overexpressed, tagged PLD1, it localized predominantly to puncta corresponding to lysosomes and endosomes (Figure S8 and Movies S7 and S8).1
When we incubated mCherry-PLD1-expressing cells with AzProp, followed by PMA stimulation and SPAAC labeling with 1, we observed an increase in IMPACT fluorescence in bright puncta that indeed colocalized with mCherry-PLD1 (Figure 2F and Movie S7). Strikingly, while virtually every fluorescent phosphatidyl alcohol spot was positive for mCherry-PLD1, only a small subset of mCherry-PLD1-positive puncta were positive for fluorescent phosphatidyl alcohol (Figure 2F and Movie S7). These results suggest an unappreciated spatial heterogeneity in PLD activation at the subcellular level, wherein, even under strong stimulation with PMA, only a subset of PLD enzymes are activated.
Up to this point, we had focused our efforts on monitoring acute PLD activation in response to a stimulus, using PMA as a model pharmacological agent to mimic activation of several signal transduction pathways. In the absence of a stimulus, however, PLD enzymes do display a much lower but appreciable level of basal activity.9,16,32 The ability to detect this much lower level of basal PLD activity could enable the study of the consequences of aberrant PLD levels that occur in disease, notably in several cancers.14,15 Thus, we then set out to determine whether IMPACT using AzProp displayed sufficient sensitivity to detect basal, endogenous PLD activity.
To accomplish this, we treated HeLa cells with 1 mM AzProp, followed by lipid extraction, SPAAC labeling with 1, and HPLC analysis. While short incubations of AzProp (e.g., 20 min) did not result in appreciable labeling, slightly longer labeling times of 2 h led to detection of fluorescent phosphatidyl alcohols (Figure 3A). Treatment with FIPI prevented phosphatidyl alcohol production, and use of the isoform-selective inhibitors VU0359595 and VU0364739 revealed that roughly half of the unstimulated PLD activity in HeLa cells can be attributed to each of PLD1 and PLD2 (Figure 3A).
Figure 3.
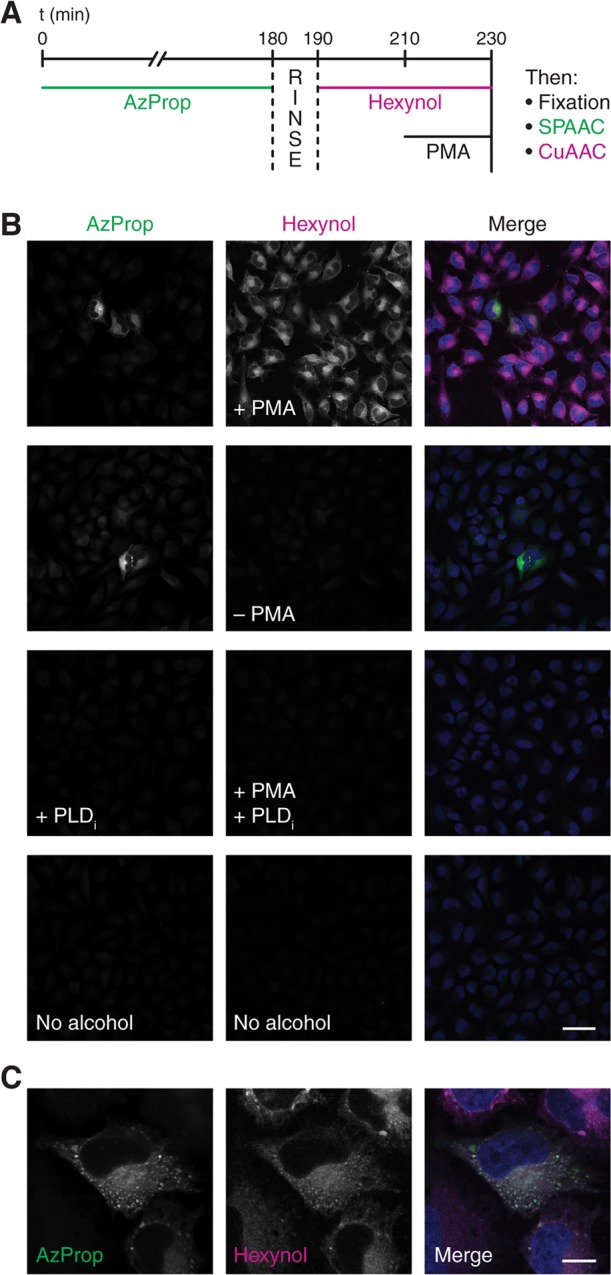
AzProp labeling reveals heterogeneity of unstimulated PLD activity at the cellular level. HeLa cells were first treated with the indicated PLD inhibitor (PLDi (FIPI), 750 nM; PLD1i (VU0359595), 250 nM; PLD2i (VU0364739), 350 nM) or DMSO vehicle for 30 min, followed by AzProp (1 mM) for 120 min (or no alcohol for 120 min as indicated in panels B and C) and then further processed for analysis as described below. (A) For HPLC analysis, lipids were extracted, extracts were tagged by SPAAC with 1 and then analyzed by fluorescence-coupled HPLC. (B–D) For live-cell analysis, cells were then incubated with 1 (1 μM) for 10 min, rinsed for 15 min. (B) Cells were then analyzed by flow cytometry. Shown are mean fluorescence intensities of the cell populations. Black bars: AzProp + no inhibitor. Gray bars: AzProp + indicated PLD inhibitor. White bars: no alcohol. Error bars represent SEM. *, p < 0.01; n = 3 biological replicates. (C, D) Cells were further stained with Hoechst 33342 to mark nuclei and imaged by confocal microscopy. Single z-slices are shown at low (C) and high (D) magnification, and in panel C, AzProp fluorescence is in green and Hoechst 33342 is in magenta. Scale bars: 50 μm (C), 10 μm (D).
To image sites of basal PLD activity within live cells, we treated cells with 1 mM AzProp for 2 h, rinsed the cells, and then performed the SPAAC reaction with 1. Analysis of populations of labeled cells by flow cytometry revealed a modest amount of fluorescence that was 4-fold higher in cells treated with AzProp than in cells treated with no alcohol (Figure 3B). Importantly, the use of FIPI and the isoform-selective inhibitors in these flow cytometry experiments gave results that were consistent with the in vitro lipid analysis shown in Figure 3A, indicating that the AzProp-dependent cellular fluorescence was due entirely to PLD enzymes and split evenly between the activities of PLD1 and PLD2 (Figure 3B).
We then examined the distribution of basal PLD activity at both the cellular and subcellular levels. At the cellular level, we observed a striking heterogeneity, wherein a small subset of cells exhibited high IMPACT-derived fluorescence (Figure 3C), while the majority of cells exhibited much lower fluorescence (Figure S9). While an examination of the flow cytometry data did not reveal a discernible, separate IMPACThigh population, we did notice an asymmetry in the histogram of IMPACT fluorescence, with a larger tail at higher fluorescence values. Skewness is a statistical measure for quantitation of this asymmetry, with positive skewness reflecting an increase at the high end of the distribution and negative skewness reflecting an increase at the low end. The flow cytometry data indeed exhibited a positive skewness value, potentially reflecting the contributions of the IMPACThigh cells to the overall population distribution (Figure S10).
When we performed similar IMPACT labeling of basal PLD activity in the presence of the PLD1 isoform-selective inhibitor VU0359595, we observed the disappearance of the high-fluorescence cells by confocal microscopy (Figure 3C), while treatment with the PLD2 inhibitor VU0364739 did not eliminate the high-fluorescence population (Figure 3C). Thus, at the cellular level, we attribute the high fluorescence of a minority of cells to PLD1 activity and the moderate fluorescence in the majority of cells to PLD2. These data are consistent with the constitutive but modest activity of PLD2 compared to the highly inducible activity of PLD1.9 Further, these conclusions are supported by a decrease in population skewness observed by flow cytometry analysis upon treatment with VU0359595 but an increase in skewness upon treatment with VU0364739 (Figure S10). Notably, the heterogeneity of PLD1 at the cellular level is unexpected and quite different from the case where PLD1 activity is stimulated by PMA, where virtually all cells exhibited high and roughly equivalent levels of fluorescence (e.g., Figure 2B).
The subcellular localization of AzProp-marked basal PLD activity appeared to be similar to that of AzProp-marked, PMA-stimulated PLD enzymes, that is localized to a mixture of ER, Golgi, endosomal, and lysosomal membranes (Figure 3D). We used isoform-selective inhibitors to probe the relative contributions of each PLD isoform to PA biosynthetic activity on different organelle membranes. Use of the PLD1-selective inhibitor VU0359595 led to selective disappearance of most of the ER-derived fluorescence but only a portion of the Golgi-derived fluorescence (Figure 3D). By contrast, the PLD2-selective inhibitor VU0364739 caused a decrease in Golgi-derived fluorescence but had minimal effect on ER-derived fluorescence (Figure 3D). Thus, we conclude that, in HeLa cells, the bulk of ER-localized, basal PLD activity is due to PLD1, whereas PLD activity on Golgi membranes is due to a roughly equal mix of PLD1 and PLD2.
Our observation of largely similar labeling patterns between acutely stimulated and tonic PLD activity in separate experiments raised the question of whether we could, within the same cell, tag membranes orthogonally to distinguish these distinct biochemical activities temporally and spatially. To accomplish this, we developed a sequential, two-color labeling protocol to mark basal PLD activity with one fluorophore and stimulated PLD activity with a second fluorophore (Figure 4A). We first treated cells with AzProp for 3 h in the absence of a PLD stimulus to generate phosphatidyl azidoalcohols. Followed by a brief rinse, we then added 5-hexyn-1-ol (hexynol) in the presence of a PMA stimulus to mark membranes bearing acutely stimulated PLD enzymes with phosphatidyl alkynols.19 After fixation, we performed sequential SPAAC and CuAAC reactions with 1 and then a tetramethylrhodamine–azide conjugate, respectively, followed by imaging by confocal microscopy.
Figure 4.
Sequential, two-color imaging protocol using AzProp and hexynol enables visualization of basal and PMA-stimulated PLD activity. (A) Schematic of experimental setup. HeLa cells were treated with AzProp (1 mM) for 180 min, rinsed for 10 min, and then treated with hexynol (1 mM) for 20 min. Where indicated, cells were then stimulated with PMA (100 nM) in the presence of hexynol for 20 min. Cells were then fixed with paraformaldehyde, sequentially labeled first by SPAAC with 1 and then by CuAAC with an azido tetramethylrhodamine conjugate, mounted in medium containing DAPI, and imaged by confocal microscopy. Where PLDi is indicated in panel B, cells were incubated with FIPI (750 nM) for 30 min prior to AzProp labeling and throughout both alcohol labeling steps. (B, C) Confocal microscopy images of cells labeled as described above. In merged images, AzProp-derived fluorescence is in green, hexynol-derived fluorescence is magenta, and DAPI is blue; colocalization of AzProp and hexynol appears as white. Shown are single z-slices at low (B) and high (C) magnification. Scale bars: 50 μm (B), 10 μm (C).
Consistent with our live-cell imaging (Figure 3C), in this experiment the nonstimulated PLD displayed high AzProp-derived fluorescence only in a minority of cells, whereas hexynol marked equally high PMA-stimulated PLD activity in all cells (Figure 4B). Various negative controls, including the omission of PMA and the addition of FIPI, validated the specificity of each alcohol probe in this sequential labeling protocol (Figure 4B). At the subcellular level, the two different phosphatidyl alcohol populations exhibited a substantial colocalization, suggesting that basal and active PLD pools occupy similar membrane compartments (Figure 4C). Given that both AzProp and hexynol are expected to report on PLD1 and PLD2 activities under our labeling conditions, we were pleased to observe such strong colocalization. As an important control, when we switched the order of AzProp and hexynol labeling, i.e., using a 3-h hexynol treatment to mark unstimulated PLD activity and a 20 min AzProp incubation in the presence of PMA to report on stimulated PLD, we observed identical results (Figure S11).
Collectively, these IMPACT imaging experiments offer several potential insights regarding the localization and activity of PLD enzymes. First, the localization of endogenous PLD activity, both basal and PMA-stimulated, appears different from the localization of fluorescently tagged PLD enzymes. Second, our data suggest that a major pool of PLD activity may reside in the ER, indicating underappreciated and potentially novel functions for PLD-dependent PA production in this organelle. Third, we observe a striking heterogeneity in IMPACT labeling at both the cellular and subcellular levels. At the cellular level, basal PLD1 activity appears low, but a minority of cells within a given population exhibit what appears to be stochastically high PLD1 levels. At the subcellular level, even under strong PMA stimulation, only a subset of overexpressed, endosomally and lysosomally localized PLD1 enzymes colocalize with IMPACT labeling, suggesting that endogenous PLD1 enzymes may have highly variable activity across these compartments as well.
In sum, we have developed a nonperturbative, chemical approach for imaging and profiling endogenous cellular PLD activity within live cells termed IMPACT that both recapitulated known localizations of PLD-mediated PA production and also highlighted the existence of underappreciated cellular pools of active PLD. We anticipate that this method, which is specific to PLD-generated PA, will be an important part of the growing toolset for studying PA, which includes complementary genetically encoded sensors that bind to total cellular PA independent of its biosynthetic origins. Collectively, our study underscores the importance of using chemical imaging tools that can directly report on enzymatic activity to study dynamic lipid signaling events.
Acknowledgments
This work was supported in part by the National Institutes of Health (R00GM110121 to J.M.B.), a Beckman Young Investigator Award from the Arnold and Mabel Beckman Foundation to J.M.B., and a National Science Foundation Graduate Research Fellowship to T.W.B. (NSF DGE-165-0441). We thank the Cornell University NMR facility (NSF MRI: CHE-1531632), the Cornell Biotechnology Resource Center Imaging Facility (NSF MRI: DBI-1428922), H. Alex Brown (Vanderbilt University) for providing VU0359595 and VU0364739, Robert A. DiStasio Jr. and Megan Gelsinger for assistance with statistical analysis, the Baird, Mao, and Yu laboratories (Cornell University) for providing plasmids, and Michael Frohman (Stony Brook University) and Jeremy Thorner (University of California, Berkeley) for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00222.
Author Contributions
T.W.B. performed all experiments. T.W.B. and J.M.B. conceived of experiments, interpreted data, and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Selvy P. E.; Lavieri R. R.; Lindsley C. W.; Brown H. A. Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 2011, 111, 6064–6119. 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Su Y.; Wang X.. Phosphatidic Acid-Mediated Signaling. In Lipid-mediated Protein Signaling; Springer: Netherlands, Dordrecht, 2013; pp 159–176. [DOI] [PubMed] [Google Scholar]
- Foster D. A.; Salloum D.; Menon D.; Frias M. A. Phospholipase D and the Maintenance of Phosphatidic Acid Levels for Regulation of Mammalian Target of Rapamycin (mTOR). J. Biol. Chem. 2014, 289, 22583–22588. 10.1074/jbc.R114.566091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cambronero J. Phospholipase D in Cell Signaling: From a Myriad of Cell Functions to Cancer Growth and Metastasis. J. Biol. Chem. 2014, 289, 22557–22566. 10.1074/jbc.R114.574152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J.-H.; Lee C. S.; Hwang D.; Ryu S. H. Understanding of the roles of phospholipase D and phosphatidic acid through their binding partners. Prog. Lipid Res. 2012, 51, 71–81. 10.1016/j.plipres.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Kassas N.; Tanguy E.; Thahouly T.; Fouillen L.; Heintz D.; Chasserot-Golaz S.; Bader M.-F.; Grant N. J.; Vitale N. Comparative Characterization of Phosphatidic Acid Sensors and Their Localization during Frustrated Phagocytosis. J. Biol. Chem. 2017, 292, 4266–4279. 10.1074/jbc.M116.742346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G.; Altshuller Y. M.; Vitale N.; Huang P.; Chasserot-Golaz S.; Morris A. J.; Bader M.-F.; Frohman M. A. Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J. Cell Biol. 2003, 162, 305–315. 10.1083/jcb.200302033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes W. E.; Parker P. J. Endosomal localization of phospholipase D 1a and 1b is defined by the C-termini of the proteins, and is independent of activity. Biochem. J. 2001, 356, 727–736. 10.1042/bj3560727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley W. C.; Sung T. C.; Roll R.; Jenco J.; Hammond S. M.; Altshuller Y.; Bar-Sagi D.; Morris A. J.; Frohman M. A. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 1997, 7, 191–201. 10.1016/S0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- Nelson R. K.; Frohman M. A. Physiological and pathophysiological roles for phospholipase D. J. Lipid Res. 2015, 56, 2229–2237. 10.1194/jlr.R059220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. W.; Moellering R. E.; Cravatt B. F. An Activity-Based Imaging Probe for the Integral Membrane Hydrolase KIAA1363. Angew. Chem., Int. Ed. 2012, 51, 966–970. 10.1002/anie.201107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonović M.; Bogyo M. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev. Proteomics 2008, 5, 721–730. 10.1586/14789450.5.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Wang Z.; Lu M.; Yonekubo Y.; Liang X.; Zhang Y.; Wu P.; Zhou Y.; Grinstein S.; Hancock J. F.; Du G. Temporal Production of the Signaling Lipid Phosphatidic Acid by Phospholipase D2 Determines the Output of Extracellular Signal-Regulated Kinase Signaling in Cancer Cells. Mol. Cell. Biol. 2014, 34, 84–95. 10.1128/MCB.00987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Frohman M. A. Cellular and physiological roles for phospholipase D1 in cancer. J. Biol. Chem. 2014, 289, 22567–22574. 10.1074/jbc.R114.576876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H. A.; Thomas P. G.; Lindsley C. W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discovery 2017, 16, 351–367. 10.1038/nrd.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond S. M.; Jenco J. M.; Nakashima S.; Cadwallader K.; Gu Q.; Cook S.; Nozawa Y.; Prestwich G. D.; Frohman M. A.; Morris A. J. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J. Biol. Chem. 1997, 272, 3860–3868. 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- Brown H. A.; Henage L. G.; Preininger A. M.; Xiang Y.; Exton J. H. Biochemical analysis of phospholipase D. Methods Enzymol. 2007, 434, 49–87. 10.1016/S0076-6879(07)34004-4. [DOI] [PubMed] [Google Scholar]
- Pettitt T. R.; McDermott M.; Saqib K. M.; Shimwell N.; Wakelam M. J. Phospholipase D1b and D2a generate structurally identical phosphatidic acid species in mammalian cells. Biochem. J. 2001, 360, 707–715. 10.1042/bj3600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumpus T. W.; Baskin J. M. A Chemoenzymatic Strategy for Imaging Cellular Phosphatidic Acid Synthesis. Angew. Chem., Int. Ed. 2016, 55, 13155–13158. 10.1002/anie.201607443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamudi S. H.; Satapathy R.; Kim J.; Su D.; Ren H.; Das R.; Hu L.; Alvarado-Martínez E.; Lee J. Y.; Hoppmann C.; Peña-Cabrera E.; Ha H.-H.; Park H.-S.; Wang L.; Chang Y.-T. Development of background-free tame fluorescent probes for intracellular live cell imaging. Nat. Commun. 2016, 7, 1–9. 10.1038/ncomms11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W.; Yeku O.; Olepu S.; Genna A.; Park J.-S.; Ren H.; Du G.; Gelb M. H.; Morris A. J.; Frohman M. A. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 2009, 75, 437–446. 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. A.; Scott S. A.; Lavieri R.; Buck J. R.; Selvy P. E.; Stoops S. L.; Armstrong M. D.; Brown H. A.; Lindsley C. W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 2009, 19, 1916–1920. 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieri R. R.; Scott S. A.; Selvy P. E.; Kim K.; Jadhav S.; Morrison R. D.; Daniels J. S.; Brown H. A.; Lindsley C. W. Design, synthesis, and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 inhibitor. J. Med. Chem. 2010, 53, 6706–6719. 10.1021/jm100814g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exton J. H. Regulation of phospholipase D. FEBS Lett. 2002, 531, 58–61. 10.1016/S0014-5793(02)03405-1. [DOI] [PubMed] [Google Scholar]
- Santa T.; Al-Dirbashi O. Y.; Fukushima T. Derivatization reagents in liquid chromatography/electrospray ionization tandem mass spectrometry for biomedical analysis. Drug Discoveries Ther. 2007, 1 (2), 108–118. [PubMed] [Google Scholar]
- Pettitt T. R.; Martin A.; Horton T.; Liossis C.; Lord J. M.; Wakelam M. J. Diacylglycerol and phosphatidate generated by phospholipases C and D, respectively, have distinct fatty acid compositions and functions. Phospholipase D-derived diacylglycerol does not activate protein kinase C in porcine aortic endothelial cells. J. Biol. Chem. 1997, 272, 17354–17359. 10.1074/jbc.272.28.17354. [DOI] [PubMed] [Google Scholar]
- Vance J. E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. 10.1111/tra.12230. [DOI] [PubMed] [Google Scholar]
- Pathre P.; Shome K.; Blumental-Perry A.; Bielli A.; Haney C. J.; Alber S.; Watkins S. C.; Romero G.; Aridor M. Activation of phospholipase D by the small GTPase Sar1p is required to support COPII assembly and ER export. EMBO J. 2003, 22, 4059–4069. 10.1093/emboj/cdg390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M.-S. Vps34 and PLD1 take center stage in nutrient signaling: their dual roles in regulating autophagy. Cell Commun. Signaling 2015, 13, 44. 10.1186/s12964-015-0122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall’Armi C.; Hurtado-Lorenzo A.; Tian H.; Morel E.; Nezu A.; Chan R. B.; Yu W. H.; Robinson K. S.; Yeku O.; Small S. A.; Duff K.; Frohman M. A.; Wenk M. R.; Yamamoto A.; Di Paolo G. The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 2010, 1, 142. 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idkowiak-Baldys J.; Baldys A.; Raymond J. R.; Hannun Y. A. Sustained receptor stimulation leads to sequestration of recycling endosomes in a classical protein kinase C- and phospholipase D-dependent manner. J. Biol. Chem. 2009, 284, 22322–22331. 10.1074/jbc.M109.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G.; Altshuller Y. M.; Kim Y.; Han J. M.; Ryu S. H.; Morris A. J.; Frohman M. A. Dual requirement for rho and protein kinase C in direct activation of phospholipase D1 through G protein-coupled receptor signaling. Mol. Biol. Cell 2000, 11, 4359–4368. 10.1091/mbc.11.12.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.