

Abstract
Fundamental mechanisms governing the perpetuation of atrial fibrillation (AF), the most common arrhythmia seen in clinical practice, are poorly understood, which explains in part why AF prevention and treatment remain suboptimal. Although some clinical parameters have been identified as predicting a transition from paroxysmal to persistent AF in some patients, the molecular, electrophysiological and structural changes leading to such a progression have not been described in detail. Oxidative stress, atrial dilatation, calcium overload, inflammation, microRNAs and myofibroblast activation are all thought to be involved in AF-induced atrial remodeling. However, it is unknown to what extent and at which time points such alterations influence the remodeling process that perpetuates AF. Here we postulate a comprehensive model that might open new pathways for future investigation into mechanisms of AF perpetuation. We start from the premise that the progression to AF perpetuation is the result of an interplay among manifold signaling pathways with differing kinetics. Some such pathways have relatively fast kinetics (e.g., oxidative stress mediated shortening of refractory period); others likely depend on molecular processes with slower kinetics (e.g., transcriptional changes in myocyte ion channel protein expression mediated through inflammation and fibroblast activation). We stress the need to fully understand the relationships among such pathways should one hope to identify novel, truly effective targets for AF therapy and prevention.
Introduction
Atrial Fibrillation (AF) affects over 2.5 million Americans,1 and is the major cause of embolic stroke.2 In the USA and Europe, overall prevalence of AF is 0.9% and the number of people affected is projected to more than double over the next 2 decades.3–5 In fact, the projected rise in AF incidence is approaching epidemic proportions.1, 6 Yet despite its importance and more than 100 years of basic and clinical research, we still do not fully understand its fundamental mechanisms and have not learned how to treat it effectively. Some patients suffer relatively short (<7days) self-terminating episodes (i.e., paroxysmal) indefinitely, but a large proportion progress to long-lasting forms of AF.7 When AF lasts continuously for more than 7 days it is considered persistent AF.8 Spontaneous, pharmacological or ablative resumption of sinus rhythm is infrequent in persistent AF, with prompt recurrences or commonly failed cardioversions. AF lasting more than one year is termed “long-term persistent AF”.7 Persistent AF leads to electrical remodeling and fibrosis of the atria but the mechanism(s) remain poorly understood. Experimental and clinical data collected to date point to a very complex pathophysiology involving a large number of significant players, including oxidative stress, calcium overload, atrial dilatation, microRNAs, inflammation and myofibroblast activation (Figure 1), all of which are likely to be involved one way or another in AF-induced atrial extracellular matrix (ECM) and electrical remodeling.9, 10 The complexity of disease progression is further amplified by the positive feedback loops that can establish among many of such players. However, it is unknown to what extent and at which time points such interactions influence the remodeling process that perpetuates AF. This brief review addresses some of the most important factors underlying ECM remodeling, with particular attention paid to the role of oxidative stress and cardiac fibroblast-to-myofibroblast differentiation in the mechanisms of atrial remodeling associated with AF. We also briefly address other potentially important contributors, such as calcium overload, transient receptor potential channels (TRP channels) and microRNAs to the mechanisms of fibrosis and AF maintenance.
Figure 1.
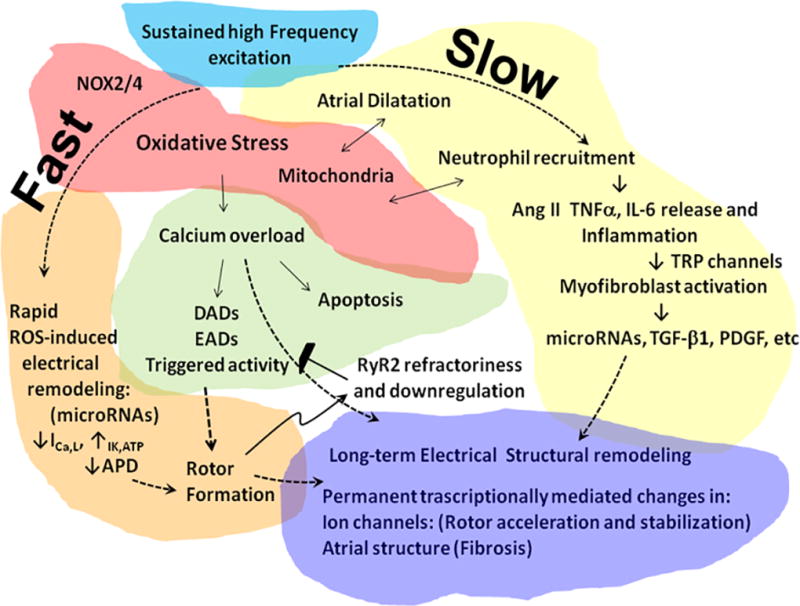
Working model for AF-induced remodeling and the substrate for AF perpetuation. Sustained high frequency excitation of the atria results in a complex series of pathophysiological events involving a large number of significant players. These include oxidative stress, calcium overload, atrial dilatation, inflammation and myofibroblast activation (Figure 1), all of which are likely to be involved in AF-induced atrial extracellular matrix (ECM) and electrical remodeling through transcriptionally mediated changes in both cardiac myocytes and fibroblasts.
A working Model of AF Perpetuation
We start from the premise that the progression to persistent AF is the result of interplay among multiple signaling pathways with differing kinetics,11 some fast and other slow, as illustrated diagrammatically in Figure 1. The role of many of such signaling pathways remains speculative. Yet fragmented evidence extracted from the literature, together with recent experimental results obtained in a clinically relevant model of persistent AF,12, 13 allows us to create a cohesive picture that might open new pathways for future investigation and hopefully the identification of novel targets for AF treatment and prevention. The model put forth in the scheme of Figure 1 predicts that, once AF is initiated, whether by premature triggered discharges from a pulmonary vein, by rapid electrical pacing of the atria, or by a simple wave break, the first consequence of the sustained high-frequency excitation would be the promotion of oxidative stress. The reactive oxygen species (ROS) released by nicotinamide adenine dinucleotide phosphate oxidases (NOX)2/4 would result in rapid (i.e., within hours or days) L-type Ca2+ current (ICa,L) reduction and inward rectifier K+ current (IK1) increase, leading to shortening of the atrial action potential duration (APD) and refractory period promoting the formation and stabilization of rotors. This would be followed by intracellular Ca2+ overload, promoting triggered activity, and apoptosis.14, 15 However, during sustained AF, the exceedingly high frequency of electrical excitation generated by the sustained rotor(s) should lead to refractoriness of the cardiac ryanodine receptor (RyR2) of the sarcoplasmic reticulum (SR)16 as well as downregulation of Ca2+ handling proteins,13, 17, 18 acting to prevent triggered activity. Nevertheless, Ca2+ overload, together with atrial dilatation, mitochondrial ROS and activation of inflammatory and pro-fibrotic pathways19 progressively alters gene expression. As demonstrated recently in a clinically relevant sheep model of persistent AF, the consequences of the above changes would be myocyte hypertrophy, interstitial fibrosis and ion channel remodeling, all of which would occur relatively slowly but reach critical levels when AF becomes persistent at a median time of about 2 months.13 As time progresses and remodeling continues to occur, enduring transcriptionally mediated changes in ion channel expression and atrial structure persistently work together to preserve sustained high electrical frequency excitation in a 'vicious cycle' that further promotes rotor stabilization, fibrosis and AF perpetuation (Figure 1).
Oxidative stress, inflammation and atrial fibrillation
The role of ROS in mediating changes in atrial ionic remodeling in AF is not well understood.20 Markers of oxidative stress have been documented in AF, and antioxidants have been shown to partly arrest electrical remodeling in animal models. Studies investigating the sources of ROS in AF have found an important role for NOX2/4 activity, which is increased in the fibrillating atria.21 Mitochondrial dysfunction and swelling are also known to occur in AF and mitochondrial ROS is potentially another important source of oxidative stress in AF22 NOX activity was shown to be increased in the goat after 2 weeks of atrial tachypacing and in human atrial samples obtained after post-operative AF.23 In contrast, for long-standing persistent AF in humans, and for animals whose atria were tachypaced for 6 months, the major source of ROS was attributed to mitochondrial oxidase and uncoupled nitric oxide synthase (NOS) activity.23 Previously unpublished experiments from our laboratory (Figure 2) indicate that NOX-derived ROS might be implicated in the electrical alterations seen in myocytes as early as 1 day after sustained tachypacing. In addition, a yet unpublished proteomic analysis during the transition to persistent AF (i.e., 58±21 days after the onset of atrial tachypacing) suggests that the elevated ROS that is observed in atrial myocytes from persistent AF animals may correlate with NOS reduction, xanthine oxidase elevation and/or mitochondrial dysfunction. Thus it is likely that these time-dependent changes in ROS also underlie the temporal changes in atrial remodeling (AF dominant frequency increase, atrial dilatation and fibrosis) known to occur in AF, as shown by our recent study13 and also by others.24 However, whether atrial oxidative stress directly affects atrial APD and refractoriness and thus contributes to rotor acceleration and stability in AF remains largely unknown. Several sarcolemmal ionic currents have been shown to be directly or indirectly modulated by ROS,25 but the relevance of these mechanisms in the context of human AF has not been demonstrated. Understanding this causal link will be important for the design of more rationale and effective upstream therapies that will help overcome the relatively limited success achieved in the clinical trials with systemic anti-oxidant therapy for AF.26
Figure 2.
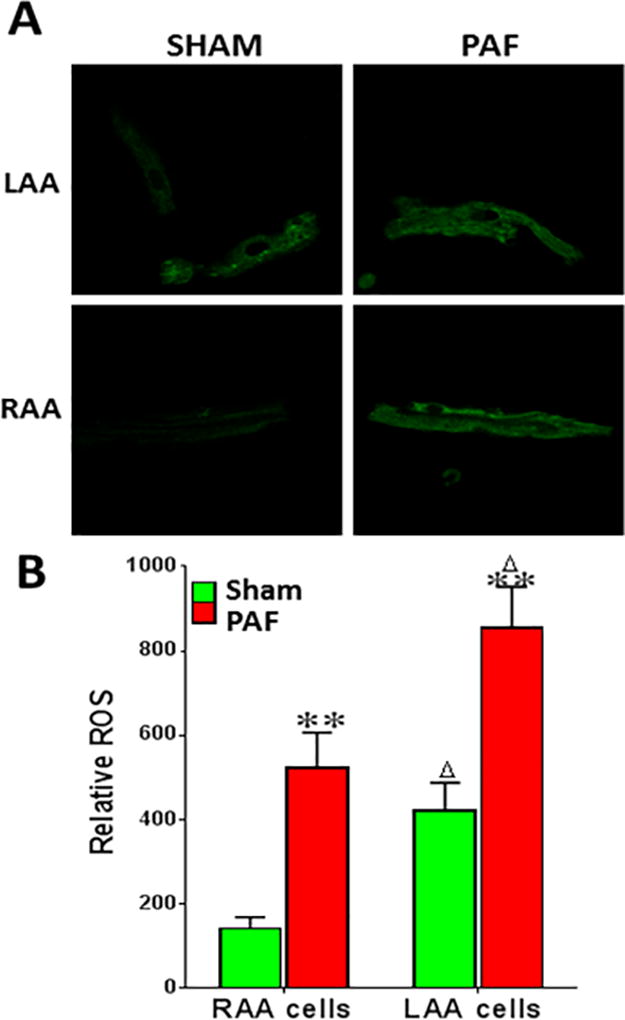
ROS is elevated in the atria of sheep with PAF. A. ROS fluorescence in freshly dissociated sheep myocytes from left atrial appendage (LAA) and right atrial appendage (RAA), (40×). B. Relative ROS levels in sheep atrial myocytes from sham and PAF sheep (**p<0.01) and in LAA versus RAA (in both sham and AF; Δp<0.05). ROS levels were measured using confocal fluorescence microscopy (excitation, 480 nm; emission, 510 nm) in atrial cells incubated in the dark with 10 µM dye dihydro-dichlorofluorescein-diacetate (H2DCF-DA, Sigma) for 20 minutes. Data were analyzed using Image J.
Whether spontaneous or pacing-induced, sustained AF is likely to lead to the release of proinflammatory cytokines and hormones related to cardiovascular disease and tissue injury, including angiotensin-II (Ang-II), tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-8.27 Both injury and mechanical stretch promote activation of leukocytes with subsequent release of inflammatory stimuli such as NOX–derived ROS, growth factors, and other hormones. Multiple studies have demonstrated a role for NOX in Ang-II function.28 However, more work is necessary to identify the precise molecular modifications of the putative signaling targets of ROS after Ang-II stimulation. Understanding which NOXs are activated by Ang-II in the atria, in normal physiology is also important, and may help us to define better interventions aimed at preventing the pro-fibrillatory effects of Ang-II activation. These are well-known triggers of fibroblast differentiation into myofibroblasts (see below), which are critical players in the development of fibrosis. Inflammatory cascades also lead to ion channel dysfunction, which along with myocyte apoptosis and matrix generation and turnover, likely contributes to both electrical and structural remodeling and predispose patients to AF.29
Role of calcium overload in AF perpetuation
Spontaneous calcium release promoting triggered activity is likely to be an important mechanism of AF initiation (Figure 1).30 However, whether RyR2 leakiness contributes to persistent AF is now disputed13, 17 A popular concept that had been promoted by some investigators over the last several years was that both initiation and maintenance of AF could be related to increased activity of protein kinase A (PKA) and/or calcium/calmodulin-dependent protein kinase II (CaMKII), with subsequent uncontrolled diastolic Ca2+ release from the SR. The idea is that Ca2+ released from the “leaky” RyR2 receptors in the sarcoplasmic reticulum would overactivate the Na+-Ca2+ exchanger (NCX) to extrude Ca2+ and produce an arrhythmogenic depolarizing current, thereby explaining both the contractile dysfunction and the high recurrence rate of the arrhythmia.31, 32 However, in a recent study in isolated rabbit atrial myocytes remodeling in response to sustained tachycardia for up to 5 days was shown to silence Ca2+ signaling through a failure of subcellular propagated Ca2+ release.17 Accordingly, Ca2+ silencing might be a protective mechanism against the massive Ca2+ overload that occurs during chronic AF.33 In another study in human atrial myocytes, while CaMKII appeared to facilitate catecholamine-evoked arrhythmias in atrial myocardium of patients with sinus rhythm, the same agonists failed to elicit arrhythmias in atrial myocardium of patients with chronic AF, likely related to atrial remodeling, which included decreases in CaMKII-mediated processes.18
The above results in patients are consistent with data derived from western blot analyses in sheep, designed to test whether remodeling was related to altered intracellular calcium dysfunction.13 Although the Na+-Ca2+ exchanger was increased in the left atrial appendage of persistent AF animals, both total RyR2 and phosphorylated RyR2 proteins were decreased, and the ratio of phosphorylated RyR2 to total RyR2 phosphorylation was unaffected. Hence, the transition from paroxysmal to persistent AF in the sheep model of atrial tachypacing did not seem to depend on Ca2+ leak or delayed after depolarizations (DADs).13 Altogether the above three studies challenge the idea that Ca2+ release dysfunction underlies AF maintenance. They all agree in that, during long-term sustained AF one should not expect an increase in the spontaneous release of Ca2+ from the SR, or that DADs/triggered activity is involved in AF maintenance or in the progression to stable forms of the arrhythmia. Thus as suggested recently,18 because of pathophysiological adaptation related to remodeling of calcium regulatory proteins, treatment with CaMKII inhibitors would be unlikely to affect persistent AF.
Cardiac Fibroblasts
The normal heart is comprised of four major cell types; myocytes, endothelial cells, smooth muscle cells (within vessels) and fibroblasts. The proportion of each cell type varies with species but altogether myocytes make up less than half of the cellular population of the mammalian heart.34 Pioneering studies in the 1970’s and 1980’s suggested that the heart consists of approximately 70% nonmyocytes and 30% cardiac myocytes.35 Importantly, fibroblasts make up the largest portion of the cellular number in the ventricles, ranging from 40 to over 60% of the total cell number35
The core functions of cardiac fibroblasts is to maintain the cardiac ECM homeostasis and to provide structural and mechanical support to the myocytes.34 The ECM is a network of fibers that in addition to providing structural and functional integrity to the heart also contains a number of cytokines and growth factors that can influence cardiac function.36 The cardiac ECM is a key mediator of the mechanical integration of cardiomyocytes, fibroblasts and blood vessels within the myocardium. The ECM also transmits extracellular mechanical signals to the cardiomyocytes. The major components of the ECM are fibrillar collagen types I and III; less abundant are collagen types IV, V and VI.37 The ECM also includes fibronectin, laminin, elastin and fibrillin, proteoglycans and glycoproteins, all of which are produced by the fibroblasts, and can be induced by a number of growth factors such as platelet-derived growth factor (PDGF),38 basic fibroblast growth factor (BFGF) and TGFβ during development and disease.39
Cardiac fibroblasts are elongated cells that contain a branched cytoplasm which surrounds an elliptical shaped nucleus.40 They have a large Golgi apparatus and no basement membrane. They can form monolayers in vitro and couple to one another via gap junctions, primarily made of Connexin43 (Cx43) and Connexin45 (Cx45).41 Fibroblasts contain proteins which aid in their identification. Such proteins are vimentin, which is present in the intermediate filaments of fibroblasts, and the more selective collagen receptor Discoidin Domain Receptor 2 (DDR2).42 DDR2 is not present in cardiac myocytes or cardiac endothelial cells but has been found in leukocytes and in tumors.43, 44 Recent studies suggest that periostin might be a selective marker for fibroblasts.45 Its expression in the heart is restricted to fibroblasts and is not expressed in cardiomyocytes, endothelial cells, or vascular smooth muscle cells in normal or injured hearts.45 Periostin expression is very low or absent in the healthy adult heart, but it increases substantially, and specifically, in myofibroblasts after damage. Thus periostin has been used to induce fibroblast specific knockout of Glycogen Synthase Kinase-3β (GSK-3β) by means of recombinase driven by the periostin promoter, strongly supporting the idea that periostin may be a useful specific marker for fibroblasts.46 In the healthy postnatal heart, interstitial cardiac fibroblasts do not express α-smooth muscle actin (α-SMA). In cardiac fibrosis, α-SMA expression is considered a sign of fibroblast activation and differentiation into myofibroblast.43
Cardiac fibroblasts are not electrically excitable and cannot maintain action potential propagation on their own although they may act as passive electrical conduits when connected through gap junctions with neonatal myocytes in vitro.47 Yet they do have distinct electrical properties, including a resting membrane potential between −30 mV and −37 mM.48 They also express several types of potassium channels (KCa1.1, KV1.5 and 1.6, KV4.2 and 4.3, Kir2.1, and Kir2.3), sodium channels (NaV1.2, 1.3, 1.5 and 1.7) and the Clnc3 channel.49 Recently, fibroblasts were shown to contain transient receptor potential (TRP) channels, which have been identified as another important activator of myofibroblast differentiation (see below).43, 50
Fibroblasts-to-myofibroblasts transdifferentiation
The origin of myofibroblasts remains controversial.51 Myofibroblasts have been thought to be a heterogeneous population derived from resident fibroblasts in the perivascular area, epithelial cells, endothelial cells, mesothelial cells, and circulating fibrocytes derived from bone marrow.43,52
Several cellular mechanisms have been proposed to explain the fibroblast-to-myofibroblast transdifferentiation. As discussed above, inflammation as seen in various cardiac pathologies, results in the recruitment of immune cells that release cytokines leading to fibroblast-to-myofibroblasts transformation.53 A recent study suggested that fibroblasts themselves can release proinflammatory cytokines and contribute to attract additional immune cells to further exacerbate the inflammatory response to injury. The latter suggests that under mechanical stress activated fibroblasts can lead to an inflammatory response.54
A variety of pro-fibrotic signaling pathways have been proposed to be changed during the initiation and progression of AF and other cardiac diseases.19, 36, 55 Such pathways can affect cardiac fibroblast proliferation and differentiation thus causing pathophysiological alterations via their effect on myofibroblasts. Stress related cytokines like IL-1 and TNF-α can activate stress activated mitogen-activated protein kinases (MAPKs) and their downstream partners (e.g., STAT1) to cause cardiac hypertrophy and/or apoptosis.36 Cultured human cardiac myofibroblasts show significant expression of MAPKs, including p38-α and p38-γ along with lower levels of p38-δ.56 Treatment of cardiac fibroblasts with TNF-α or IL-1 increases phosphorylation of p38.56 Activation of IL-1α induced phosphorylation of p38 can lead to IL-6 and matrix metalloproteinase-3 (MMP-3) mRNA and protein expression in these cells.56 Thus activation of p38 via MAPK pathway activation can exacerbate the release of proinflammatory cytokines like IL-6 and cause increased fibrosis via activation of cardiac fibroblasts56. Similar to the MAPK pathway, activation of AKT/GSK-3β, pathway can induce its effect via acting on the fibroblasts.46 The AKT/GSK-3β, pathway is activated by number of signaling molecules, including TGF-β.57 Activation of AKT by TGF-β or Ang-II can inhibit GSK-3β by increasing its phosphorylation and lead to hypertrophy and/or fibrosis. Fibroblasts specific deletion of GSK-3β in mice led to hypertrophy and increased deterioration of cardiac function after MI.46 Increased phosphorylation of GSK-3β was associated with increased α-SMA and thus increased transformation of fibroblasts into myofibroblasts.
In addition to acting through the canonical SMAD pathways TGF-β1 increases protein kinase C alpha (PKCα) activity in cultured rat aortic fibroblasts. TGF-β1-induced increased expression of α-SMA has been shown to be PKCα activity dependent.58 In addition, the increased fibroblast-myofibroblast transdifferentiation via the TGF-β1 PKCα pathway also involved an increased expression of phosphodiesterase 1A (PDE1A) in a dose-and-time dependent manner.58 Treatment of rat aortic advential fibroblasts with TGF-β1 caused nuclear translocation of the protein.59 Pharmacological inhibition of PDE1A reduced the TGF-β1-induced increased expression of α-SMA, whereas phorbol-12-myristate-13-acetate (a PKC activator) increased it. Finally, the upregulation of PKCα expression by TGF-β1 was also inhibited by PDE1A inhibition. Thus TGF-β1 can increase α-SMA expression and myofibroblast formation via a PDE1A-PKCα-dependent mechanism.59
Regardless of their origin or the pathway that activates them, cardiac fibroblasts are critical players in normal myocardial function and active contributors to ECM remodeling in response to cardiac injury.52 The link between ECM remodeling and AF maintenance is now universally recognized. Cardiac fibroblast proliferation and concomitant collagenous matrix accumulation (fibrosis) has been implicated in arrhythmia initiation and maintenance. Fibrosis affects electrical propagation through slow, discontinuous conduction with "zigzag" propagation, due to reduced regional coupling, abrupt changes in fibrotic bundle size and micro-anatomical reentry.60 In the case of AF, it has been suggested that sustained rapid atrial activation may further influence structural remodeling by directly altering fibroblast function, and may contribute to the upregulation of ECM genes in response to high-frequency excitation.29 The profibrotic effect was confirmed in vitro by decreased thymidine incorporation and increased α-SMA and collagen expression by fibroblasts cultured in the conditioned media of HL-1 cells that underwent tachypacing at 5 Hz for 24 hrs. The presence of the electromechanical uncoupler blebbistatin in the conditioned media allowed the demonstration that electrical rather than mechanical signaling was responsible for release of factors from HL-1 cells.61
TRP channels and atrial fibrillation
Increased intracellular calcium involving TRP channels in the plasma membrane of fibroblasts can led to activation of a wide variety of signaling pathways.43 Thus TRP channels have been identified as another important activator of myofibroblast differentiation.62 There are at least 7 members of the superfamily of cation specific TRP channels: TRPC (canonical), TRPM (melastatin), TRPV (vanilloid), TRPP (polycystin), TRPA (ankyrin), and TRPML (mucolipin), TRPN (NOMPC-like).43, 63, 64 Most TRP channels are activated by a number of ligands and by mechanical stretch, and allow Ca2+ and Na+ entry.
One of the canonical channels, TRPC3, is highly expressed in cardiac fibroblasts.65 TRPC3 increases extracellular-signal regulated kinase (ERK) activation and contributes to increased fibrosis. Atrial samples from AF patients and from tachypacing induced AF goats showed increased mRNA expression of TRPC3 as compared to levels in respective control samples.50 Cultured left atrial fibroblasts from dogs maintained in AF for one week showed increased TRPC3 with increased ERK phosphorylation and ECM gene expression.50 Inhibition of TRPC3 channels by pyrazole-3 suppressed AF dog atrial fibroblast proliferation rates, α-smooth muscle actin expression, and ERK phosphorylation, suggesting a role for increased Ca2+ influx via TRPC3 channels in AF associated atrial fibrosis.50 Of interest, atrial fibroblasts from chronic AF patients exhibit increased TRPM7 current and calcium influx.66 Silencing TRPM7 in atrial fibroblasts decreased TRPM7 current and calcium influx, and reduced fibroblast-to-myofibroblast differentiation and TGF-β mediated fibrosis. On the other hand, TGF-β treatment increased TRPM7 expression in atrial cells.66. TGF-β has been shown to involve TRPV4 in its effect on fibrosis markers in fibroblasts. TGF-β treatment also increases TRPV4 expression with increased calcium influx67
Role of microRNAs in atrial fibrillation
MicroRNAs (miRNAs) are a group of naturally occurring small non-coding RNA molecules that are partially complementary to one or more messenger RNAs.68 The main role of miRNAs is to control transcriptional expression of specific proteins by degrading specific mRNA. miRNAs are transcribed by RNA polymerase II as large RNA precursors called pri-miRNAs.69 Once in mature form, miRNAs form the RNA-induced silencing complex (RISC) responsible for the gene silencing observed due to miRNA expression and RNA interference.70 Since 2001, when the term miRNA was coined by Lee and colleagues,71 research in various areas of biology has suggested a strong role of these small RNA molecules in both normal physiology and pathophysiology.71, 72 Research in isolated cells and whole animals indicated that small RNA molecules play important roles in controlling processes involved in early development, lineage commitment, growth, differentiation, cell death, and metabolic control. A number of studies have suggested a role of miRNAs in cardiovascular diseases, including AF.73, 74
miR-21 in fibrosis and electrical remodeling
Cardiac fibroblasts have been shown to release miRNAs in special vesicles called exosomes which enables them to act as paracrine molecules. Relevant to AF, miR-21 has been shown to activate ERK and MAPK pathway.75. Therefore, by producing and releasing miR-21 miRNA, myofibroblasts can lead to increased hypertrophy and fibrosis.76 Transcriptomic analysis of left atrial samples from dogs with AF induced by right atrial tachypacing for 8 weeks and human atrial samples from AF patients with rheumatic heart disease AF showed that miR-223, miR-328, and miR-664 were upregulated >2 fold. However miR-101, miR-320, and miR-499 were downregulated by at least 50%.77 In the same study, adenoviral transfection in canine atrium and transgenic overexpression in mice of miR-328 increased AF vulnerability, diminished ICaL, and shortened APD, suggesting that miR-328 contributed to atrial electrical remodeling in AF through targeting L-type Ca2+ channel genes (CACNA1C;α1c) and CACNB1; β1).77 In a more recent study patients with AF expressed enhanced levels of miR-21 in cardiac myocytes.78 Myocytes also showed a reduced expression of CaV1.2, and a luciferase assay showed that CACNA1C and β2 (CACNB2) were targets for degradation by miR-21. miR-21 transfected in HL-1 cells showed both reduced ICaL and protein expression but there was no change in the t-type Ca2+ current ICaT.78 Upregulation of miR-22 increased migratory properties of fibroblasts suggesting that miR-22 can lead to increased fibrosis as well.79 Overexpression of miR-29 in isolated atrial fibroblasts decreased collagen and fibronectin gene expression, whereas knock down of miR-29 had the opposite effect.80 In atrial samples from patients with AF the level of miR-29 was significantly lower than sinus rhythm patients. Moreover circulating levels of miR-29 were also decreased in patients with CHF or AF. The decrease was further amplified if patients had AF along with CHF.81 Thus miR-29 may play an important role in the molecular mechanisms of lone AF and AF secondary to CHF.81
Targeted deletion of miR-1 has shown that it has an important role in cardiac conduction.82 On the other hand, it is well-known that up-regulation of the inward-rectifier channel Kir2.1 responsible for IK1 is important for AF maintenance,13, 83 and it has been shown that miR-1 reciprocally regulates Kir2.1 expression in coronary disease, contributing to arrhythmogenesis.84 In a study in 62 patients undergoing cardiac surgery, it was demonstrated that miR-1 levels were greatly reduced in 31 patients with AF, possibly contributing to up-regulation of Kir2.1, leading to increased IK1.85
Thus, a number of ubiquitously distributed miRs have been shown to be involved in structural or electrical remodeling, and several have been suggested to exhibit a strong arrhythmogenic potential.77 It is highly probable that multiple miRs contribute to controlling cardiac arrhythmogenesis and that different miRs are involved in different types of arrhythmias under different pathophysiological conditions, including AF.32, 86, 87
Interactions of (myo)fibroblasts with cardiomyocytes
Investigators have used co-cultures of cardiac myocytes and fibroblasts to investigate the role played by fibroblasts in the regulation of myocyte function. For example, using transwell culture plates it was shown that cardiac fibroblasts increased viability and decreased troponin-I expression in cardiac myocytes upon oxygen reperfusion after hypoxia.88 The increased viability was associated with activation of the ERK pathway but not the PI3K/AKT pathway. Analysis of the fibroblast secretome suggested that the increased myocyte survival involved ERK activation of tissue inhibitor of metalloproteinases-1 (TIMPs-1).88
In addition to paracrine interactions, mechanical coupling between fibroblasts and myocytes is likely to play important roles in myofibroblast-myocyte communication. Experiments by several laboratories demonstrated that co-culturing adult cardiac myocytes with cardiac myofibroblasts accelerates remodeling of the myocytes with morphological adaptation and de-differentiation.89–91 Figure 3 shows how upon spontaneous contact with a cardiomyocyte (CM) a myofibroblast (MF) was able to induce a vigorous stretch onto the membrane of the CMs. Rapid morphological change of the CM followed soon after heterocellular apposition leading to a dedifferentiated CM phenotype. As described by Driesen et al,90 time-lapse microscopy revealed that myocyte structural adaptation started at the distal end of the myocyte through disassembly of the intercalated disc and formation of cytoplasmic processes.90 It subsequently extended over the whole plasma membrane, changing the ultrastructure of the myocyte from a highly organized cylinder to a flat rounded myocyte, with extensive myofibrillar and mitochondrial rearrangement.90
Figure 3.

Myofibroblasts induce structural remodeling of cardiomyocytes. Fluorescence microscopy by confocal laser scanning. Protein organization at the myofibroblast (MF)–cardiomyocyte (CM). CM contact sites of vinculin. In the CM, vinculin is reorganized in strands parallel to the direction of the strain imposed by the MF. Modified by permission from Driesen RB, Verheyen FK, Dispersyn GD, Thone F, Lenders MH, Ramaekers FC, Borgers M. Structural adaptation in adult rabbit ventricular myocytes: Influence of dynamic physical interaction with fibroblasts. Cell Biochem Biophys. 2006;44:119–128.
Cardiac fibroblasts interact with myocytes through cell surface proteins of the α-integrin and cadherin families.92, 93 In neonatal cell co-cultures OB-cadherin was shown to express at myofibroblast-myofibroblast junctions but not at fibroblasts-myocyte junctions94 On the other hand significant expression of N-cadherin is seen between myocytes and fibroblasts.94 In a recent study using myofibroblasts with genetically modified expression of cadherins or mutant RhoA proteins in a co-culture model, N-cadherin-mediated mechanical coupling was shown to be responsible for slowing of conduction velocity. Silencing N-cadherin prevented the myofibroblast-dependent slowing of cardiac conduction.94
Electrotonic coupling of cardiac myocytes and nonmyocytes in vitro was first demonstrated in the early 1970s.95, 96 Subsequently, Rook et al showed that neonatal rat cardiac fibroblasts could connect electrically with other fibroblasts and with myocytes with single channel conductance values of 21pS and 32pS, respectively, suggesting the existence of two gap junction proteins between neonatal rat cardiocytes, Cx43 and another connexin that was not yet identified at the time.97 Gaudesius et al41 and Miragoli et al98 were first to demonstrate Cx43 and Cx45 expression at fibroblast–to-myocyte junctions. When inserts of fibroblasts were positioned in the middle of a myocyte strand, bidirectional electrotonic propagation was recorded, and impulse transmission from one side to the other was achieved for fibroblast inserts of up to 300 µm. Similarly, when fibroblasts were placed over myocyte strands, conduction velocity and depolarization upstroke velocity correlated with the fibroblasts/myocytes ratio in bimodal manner; i.e., increasing for low ratios and decreasing otherwise. Moreover, optical mapping studies from our laboratory have demonstrated that heterocellular coupling between cultured rat neonatal myocytes and myofibroblasts alters conduction velocity, reentry stability and complexity of wave propagation.47
However, despite accumulating evidence of potential heterocellular electrical coupling in vitro, electrical coupling between fully differentiated myocytes and fibroblasts has never been demonstrated in normal atrial or ventricular muscle. Camelliti et al suggested that fibroblast-myocyte coupling occurs in regions of isolated rabbit sinoatrial node preparations. They maintained that 10% of the total Cx45 of the region was located at membrane contacts between myocytes and fibroblasts and that Lucifer yellow dye transfer confirmed the functionality of heterogeneous gap junction coupling.99 Such data have yet to be confirmed by other investigators. In fact, whether in the normal, fully differentiated adult heart fibroblast-myocyte interactions also involve electrical connections through gap junctions continues to be a matter of debate. We hypothesize that myocyte electrical remodeling secondary to heterocellular gap junction-mediated interactions is a component of myocyte de-differentiation induced by the mechanical attachment to a myofibroblast. We surmise also that whether associated with cardiac injury or sustained AF, heterocellular electrical communication is a late process that occurs after fibroblasts switch phenotypically to myofibroblasts (see Figure 1), become contractile, express adhesive proteins and cytokines and attach to neighboring myocytes to force them to de-differentiate. This idea is based on recent experiments (discussed above) demonstrating that co-culturing adult cardiac myocytes with cardiac fibroblasts induces an accelerated remodeling of the myocytes with morphological adaptation and de-differentiation.89, 90
Concluding Remarks
The projected rise in AF incidence is approaching epidemic proportions.6 While AF usually starts with paroxysmal episodes, in a significant number of patients it evolves relentlessly to persistent and permanent forms. This likely reflects progressive electrophysiological and structural remodeling in both atria, making sources of the arrhythmia more stable and long-lasting. Yet the precise mechanisms have not been established. Oxidative stress, atrial dilatation, calcium overload, inflammation, TRPC channel mediated myofibroblast activation, epigenetic factors and microRNAs are all thought to be involved in AF-induced atrial remodeling. However, it is unknown to what extent and at which time points such alterations influence the remodeling process that perpetuates AF. Future research should be anchored by fundamental basic and translational science discoveries in genomics, proteomics and metabolomics and on the use of clinically relevant animal models and patient studies that should dramatically improve our current conceptualization, treatment and/or prevention of AF perpetuation.
Acknowledgments
Supported in part by the Leducq Foundation and by a research grant from University of Michigan Health Science-Peking University Health Science Center Joint Institute.
Footnotes
Disclosures: NONE
Literature Cited
- 1.McManus DD, Rienstra M, Benjamin EJ. An update on the prognosis of patients with atrial fibrillation. Circulation. 2012;126:e143–146. doi: 10.1161/CIRCULATIONAHA.112.129759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vergara P, Della Bella P. Management of atrial fibrillation. F1000Prime Rep. 2014;6:22. doi: 10.12703/P6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majeed A, Moser K, Carroll K. Trends in the prevalence and management of atrial fibrillation in general practice in england and wales, 1994–1998: Analysis of data from the general practice research database. Heart. 2001;86:284–288. doi: 10.1136/heart.86.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Andres N, Rossignol P, Iraqi W, Fay R, Nuee J, Ghio S, Cleland JG, Zannad F, Lacolley P. Association of galectin-3 and fibrosis markers with long-term cardiovascular outcomes in patients with heart failure, left ventricular dysfunction, and dyssynchrony: Insights from the care-hf (cardiac resynchronization in heart failure) trial. Eur J Heart Fail. 2012;14:74–81. doi: 10.1093/eurjhf/hfr151. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–473. [PubMed] [Google Scholar]
- 6.Miyasaka Y, Barnes M, Bailey K, Cha S, Gersh B, Seward J, Tsang T. Mortality trends in patients diagnosed with first atrial fibrillation a 21-year community-based study. J Am Coll Cardiol. 2007;49:986–992. doi: 10.1016/j.jacc.2006.10.062. [DOI] [PubMed] [Google Scholar]
- 7.Kerr CR, Humphries KH, Talajic M, Klein GJ, Connolly SJ, Green M, Boone J, Sheldon R, Dorian P, Newman D. Progression to chronic atrial fibrillation after the initial diagnosis of paroxysmal atrial fibrillation: Results from the canadian registry of atrial fibrillation. Am Heart J. 2005;149:489–496. doi: 10.1016/j.ahj.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 8.Saksena S, Hettrick DA, Koehler JL, Grammatico A, Padeletti L. Progression of paroxysmal atrial fibrillation to persistent atrial fibrillation in patients with bradyarrhythmias. Am Heart J. 2007;154:884–892. doi: 10.1016/j.ahj.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 9.Corradi D. Atrial fibrillation from the pathologist's perspective. Cardiovasc Pathol. 2014;23:71–84. doi: 10.1016/j.carpath.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Pinho-Gomes AC, Reilly S, Brandes RP, Casadei B. Targeting inflammation and oxidative stress in atrial fibrillation: Role of 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibition with statins. Antioxid Redox Signal. 2013;20:1268–1285. doi: 10.1089/ars.2013.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nattel S. Atrial electrophysiological remodeling caused by rapid atrial activation: Underlying mechanisms and clinical relevance to atrial fibrillation. Cardiovasc Res. 1999;42:298–308. doi: 10.1016/s0008-6363(99)00022-x. [DOI] [PubMed] [Google Scholar]
- 12.Filgueiras-Rama D, Martins RP, Ennis SR, Mironov S, Jiang J, Yamazaki M, Kalifa JR, Jalife J, Berenfeld O. High-resolution endocardial and epicardial optical mapping in a sheep model of stretch-induced atrial fibrillation. J. Vis. Exp. 2011 doi: 10.3791/3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martins RP, Kaur K, Hwang E, Ramirez RJ, Willis BC, Filgueiras-Rama D, Ennis SR, Takemoto Y, Ponce-Balbuena D, Zarzoso M, O'Connell RP, Musa H, Guerrero-Serna G, Avula UM, Swartz MF, Bhushal S, Deo M, Pandit SV, Berenfeld O, Jalife J. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation. 2014 doi: 10.1161/CIRCULATIONAHA.113.004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrev D, Nattel S. New insights into the molecular basis of atrial fibrillation: Mechanistic and therapeutic implications. Cardiovasc Res. 2011;89:689–691. doi: 10.1093/cvr/cvr021. [DOI] [PubMed] [Google Scholar]
- 15.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM, Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: Ryanodine receptor refractoriness during alternans and fibrillation. Circ Res. 2014;114:1410–1421. doi: 10.1161/CIRCRESAHA.114.302505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D, Schotten U. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest. 2014;124:4759–4772. doi: 10.1172/JCI70102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christ T, Rozmaritsa N, Engel A, Berk E, Knaut M, Metzner K, Canteras M, Ravens U, Kaumann A. Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc Natl Acad Sci U S A. 2014;111:11193–11198. doi: 10.1073/pnas.1324132111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suarez G, Meyerrose G. Heart failure and galectin 3. Ann Transl Med. 2014;2:86. doi: 10.3978/j.issn.2305-5839.2014.09.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang KC, Dudley SC., Jr Oxidative stress and atrial fibrillation: Finding a missing piece to the puzzle. Circulation. 2013;128:1724–1726. doi: 10.1161/CIRCULATIONAHA.113.005837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YM, Kattach H, Ratnatunga C, Pillai R, Channon KM, Casadei B. Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J Am Coll Cardiol. 2008;51:68–74. doi: 10.1016/j.jacc.2007.07.085. [DOI] [PubMed] [Google Scholar]
- 22.Van Wagoner DR. Oxidative stress and inflammation in atrial fibrillation: Role in pathogenesis and potential as a therapeutic target. J Cardiovasc Pharmacol. 2008;52:306–313. doi: 10.1097/FJC.0b013e31817f9398. [DOI] [PubMed] [Google Scholar]
- 23.Reilly SN, Jayaram R, Nahar K, Antoniades C, Verheule S, Channon KM, Alp NJ, Schotten U, Casadei B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation. 2011;124:1107–1117. doi: 10.1161/CIRCULATIONAHA.111.029223. [DOI] [PubMed] [Google Scholar]
- 24.Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–246. doi: 10.1016/s0008-6363(02)00258-4. [DOI] [PubMed] [Google Scholar]
- 25.Wagner S, Rokita AG, Anderson ME, Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal. 2013;18:1063–1077. doi: 10.1089/ars.2012.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Negi S, Shukrullah I, Veledar E, Bloom HL, Jones DP, Dudley SC. Statin therapy for the prevention of atrial fibrillation trial (stop af trial) J Cardiovasc Electrophysiol. 2010;22:414–419. doi: 10.1111/j.1540-8167.2010.01925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012;60:2263–2270. doi: 10.1016/j.jacc.2012.04.063. [DOI] [PubMed] [Google Scholar]
- 28.Dikalov SI, Nazarewicz RR. Angiotensin ii-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. 2013;19:1085–1094. doi: 10.1089/ars.2012.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ Res. 2014;114:1453–1468. doi: 10.1161/CIRCRESAHA.114.303211. [DOI] [PubMed] [Google Scholar]
- 30.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 31.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N. Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol. 2014 doi: 10.1161/CIRCEP.114.001973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jalife J, Delmar M, Anumonwo J, Berenfeld O, Kalifa J. Basic cardiac electrophysiology for the clinician. Blackwell Publishing; 2009. [Google Scholar]
- 34.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: The renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nag AC. Study of non-muscle cells of the adult mammalian heart: A fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 36.Pellman J, Lyon RC, Sheikh F. Extracellular matrix remodeling in atrial fibrosis: Mechanisms and implications in atrial fibrillation. J Mol Cell Cardiol. 2010;48:461–467. doi: 10.1016/j.yjmcc.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iraqi W, Rossignol P, Angioi M, Fay R, Nuee J, Ketelslegers JM, Vincent J, Pitt B, Zannad F. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: Insights from the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (ephesus) study. Circulation. 2009;119:2471–2479. doi: 10.1161/CIRCULATIONAHA.108.809194. [DOI] [PubMed] [Google Scholar]
- 38.Musa H, Kaur K, O'Connell R, Klos M, Guerrero-Serna G, Avula UM, Herron TJ, Kalifa J, Anumonwo JM, Jalife J. Inhibition of platelet-derived growth factor-ab signaling prevents electromechanical remodeling of adult atrial myocytes that contact myofibroblasts. Heart Rhythm. 2013;10:1044–1051. doi: 10.1016/j.hrthm.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiller M, Javelaud D, Mauviel A. Tgf-beta-induced smad signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 40.Porter KE, Turner NA. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 41.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 42.Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Heart Circ Physiol. 2013;305:H1363–1372. doi: 10.1152/ajpheart.00395.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis J, Molkentin JD. Myofibroblasts: Trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baum J, Duffy HS. Fibroblasts and myofibroblasts: What are we talking about? J Cardiovasc Pharmacol. 2011;57:376–379. doi: 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105:934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, Force T. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. 2014;130:419–430. doi: 10.1161/CIRCULATIONAHA.113.008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zlochiver S, Munoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamkin A, Kiseleva I, Wagner KD, Lammerich A, Bohm J, Persson PB, Gunther J. Mechanically induced potentials in fibroblasts from human right atrium. Exp Physiol. 1999;84:347–356. [PubMed] [Google Scholar]
- 49.Li GR, Sun HY, Chen JB, Zhou Y, Tse HF, Lau CP. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE. 2009;4:e7307. doi: 10.1371/journal.pone.0007307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif JC, Schotten U, Van Wagoner DR, Dobrev D, Nattel S. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2014;126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore-Morris T, Tallquist MD, Evans SM. Sorting out where fibroblasts come from. Circ Res. 2014;115:602–604. doi: 10.1161/CIRCRESAHA.114.304854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindner D, Zietsch C, Tank J, Sossalla S, Fluschnik N, Hinrichs S, Maier L, Poller W, Blankenberg S, Schultheiss HP, Tschope C, Westermann D. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res Cardiol. 2014;109:428. doi: 10.1007/s00395-014-0428-7. [DOI] [PubMed] [Google Scholar]
- 55.Lin CS, Pan CH. Regulatory mechanisms of atrial fibrotic remodeling in atrial fibrillation. Cell Mol Life Sci. 2008;65:1489–1508. doi: 10.1007/s00018-008-7408-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinfield JK, Das A, O'Regan DJ, Ball SG, Porter KE, Turner NA. P38 mapk alpha mediates cytokine-induced il-6 and mmp-3 expression in human cardiac fibroblasts. Biochem Biophys Res Commun. 2012;430:419–424. doi: 10.1016/j.bbrc.2012.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaur K, Zarzoso M, Ponce-Balbuena D, Guerrero-Serna G, Hou L, Musa H, Jalife J. Tgf-beta1, released by myofibroblasts, differentially regulates transcription and function of sodium and potassium channels in adult rat ventricular myocytes. PLoS One. 2013;8:e55391. doi: 10.1371/journal.pone.0055391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao PJ, Li Y, Sun AJ, Liu JJ, Ji KD, Zhang YZ, Sun WL, Marche P, Zhu DL. Differentiation of vascular myofibroblasts induced by transforming growth factor-beta1 requires the involvement of protein kinase calpha. J Mol Cell Cardiol. 2003;35:1105–1112. doi: 10.1016/s0022-2828(03)00207-4. [DOI] [PubMed] [Google Scholar]
- 59.Zhou HY, Chen WD, Zhu DL, Wu LY, Zhang J, Han WQ, Li JD, Yan C, Gao PJ. The pde1a-pkcalpha signaling pathway is involved in the upregulation of alpha-smooth muscle actin by tgf-beta1 in adventitial fibroblasts. J Vasc Res. 2010;47:9–15. doi: 10.1159/000231716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Bakker JM, van Capelle FJ, Janse MJ, Tasseron S, Vermeulen JT, de Jonge N, Lahpor JR. Slow conduction in the infarcted human heart. 'Zigzag' course of activation. Circulation. 1993;88:915–926. doi: 10.1161/01.cir.88.3.915. [DOI] [PubMed] [Google Scholar]
- 61.Burstein B, Qi XY, Yeh YH, Calderone A, Nattel S. Atrial cardiomyocyte tachycardia alters cardiac fibroblast function: A novel consideration in atrial remodeling. Cardiovasc Res. 2007;76:442–452. doi: 10.1016/j.cardiores.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 62.Yue Z, Xie J, Yu A, Stock J, Du J, Yue L. Role of TRP channels in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2014 doi: 10.1152/ajpheart.00457.2014. ajpheart 00457 02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 64.Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome biology. 2011;12:218. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yue Z, Zhang Y, Xie J, Jiang J, Yue L. Transient Receptor Potential (TRP) channels and cardiac fibrosis. Curr Top Med Chem. 2013;13:270–282. doi: 10.2174/1568026611313030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK. TRPv4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. doi: 10.1016/j.yjmcc.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ambros V. MicroRNA pathways in flies and worms: Growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 69.Cullen BR. Transcription and processing of human microRNA precursors. Mol Cell. 2004;16:861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 70.Papait R, Greco C, Kunderfranco P, Latronico MV, Condorelli G. Epigenetics: A new mechanism of regulation of heart failure? Basic Res Cardiol. 2013;108:361. doi: 10.1007/s00395-013-0361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee RC, Ambros V. An extensive class of small rnas in caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 72.Catalucci D, Latronico MV, Condorelli G. MicroRNAs control gene expression: Importance for cardiac development and pathophysiology. Ann N Y Acad Sci. 2008;1123:20–29. doi: 10.1196/annals.1420.004. [DOI] [PubMed] [Google Scholar]
- 73.Marian AJ. Recent developments in cardiovascular genetics and genomics. Circ Res. 2014;115:e11–17. doi: 10.1161/CIRCRESAHA.114.305054. [DOI] [PubMed] [Google Scholar]
- 74.Luo X, Yang B, Nattel S. MicroRNAs and atrial fibrillation: Mechanisms and translational potential. Nat Rev Cardiol. 2014 doi: 10.1038/nrcardio.2014.178. [DOI] [PubMed] [Google Scholar]
- 75.Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dong S, Ma W, Hao B, Hu F, Yan L, Yan X, Wang Y, Chen Z, Wang Z. MicroRNA-21 promotes cardiac fibrosis and development of heart failure with preserved left ventricular ejection fraction by up-regulating bcl-2. Int J Clin Exp Pathol. 2014;7:565–574. [PMC free article] [PubMed] [Google Scholar]
- 77.Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–2387. doi: 10.1161/CIRCULATIONAHA.110.958967. [DOI] [PubMed] [Google Scholar]
- 78.Barana A, Matamoros M, Dolz-Gaiton P, Perez-Hernandez M, Amoros I, Nunez M, Sacristan S, Pedraz A, Pinto A, Fernandez-Aviles F, Tamargo J, Delpon E, Caballero R. Chronic atrial fibrillation increases microRNA-21 in human atrial myocytes decreasing l-type calcium current. Circ Arrhythm Electrophysiol. 2014;7:861–868. doi: 10.1161/CIRCEP.114.001709. [DOI] [PubMed] [Google Scholar]
- 79.Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, Bauersachs J, Thum T. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age. 2013;35:747–762. doi: 10.1007/s11357-012-9407-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. Mir-29b as a therapeutic agent for angiotensin ii-induced cardiac fibrosis by targeting tgf-beta/smad3 signaling. Mol Ther. 2014;22:974–985. doi: 10.1038/mt.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dawson K, Wakili R, Ordog B, Clauss S, Chen Y, Iwasaki Y, Voigt N, Qi XY, Sinner MF, Dobrev D, Kaab S, Nattel S. MicroRNA29: A mechanistic contributor and potential biomarker in atrial fibrillation. Circulation. 2013;127:1466–1475. doi: 10.1161/CIRCULATIONAHA.112.001207. 1475e1461-1428. [DOI] [PubMed] [Google Scholar]
- 82.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking mirna-1–2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 83.Voigt N, Trausch A, Knaut M, Matschke K, Varro A, Van Wagoner DR, Nattel S, Ravens U, Dobrev D. Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:472–480. doi: 10.1161/CIRCEP.110.954636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA mir-1 regulates cardiac arrhythmogenic potential by targeting gja1 and kcnj2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 85.Girmatsion Z, Biliczki P, Bonauer A, Wimmer-Greinecker G, Scherer M, Moritz A, Bukowska A, Goette A, Nattel S, Hohnloser SH, Ehrlich JR. Changes in microRNA-1 expression and ik1 up-regulation in human atrial fibrillation. Heart Rhythm. 2009;6:1802–1809. doi: 10.1016/j.hrthm.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 86.Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, Horiguchi M, Nakamura T, Chonabayashi K, Hishizawa M, Hasegawa K, Kume N, Yokode M, Kita T, Kimura T, Ono K. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in apoe−/− mice. Journal of the American Heart Association. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McManus DD, Tanriverdi K, Lin H, Esa N, Kinno M, Mandapati D, Tam S, Okike ON, Ellinor PT, Keaney JF, Jr, Kevin Donahue J, Benjamin EJ, Freedman JE. Plasma microRNAs are associated with atrial fibrillation and change after catheter-ablation (the mirhythm study) Heart Rhythm. 2014 doi: 10.1016/j.hrthm.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abrial M, Da Silva CC, Pillot B, Augeul L, Ivanes F, Teixeira G, Cartier R, Angoulvant D, Ovize M, Ferrera R. Cardiac fibroblasts protect cardiomyocytes against lethal ischemia-reperfusion injury. J Mol Cell Cardiol. 2014;68:56–65. doi: 10.1016/j.yjmcc.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 89.Driesen RB, Dispersyn GD, Verheyen FK, van den Eijnde SM, Hofstra L, Thone F, Dijkstra P, Debie W, Borgers M, Ramaekers FC. Partial cell fusion: A newly recognized type of communication between dedifferentiating cardiomyocytes and fibroblasts. Cardiovasc Res. 2005;68:37–46. doi: 10.1016/j.cardiores.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 90.Driesen RB, Verheyen FK, Dispersyn GD, Thone F, Lenders MH, Ramaekers FC, Borgers M. Structural adaptation in adult rabbit ventricular myocytes: Influence of dynamic physical interaction with fibroblasts. Cell Biochem Biophys. 2006;44:119–128. doi: 10.1385/CBB:44:1:119. [DOI] [PubMed] [Google Scholar]
- 91.Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007;583:225–236. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hsueh WA, Law RE, Do YS. Integrins, adhesion, and cardiac remodeling. Hypertension. 1998;31:176–180. doi: 10.1161/01.hyp.31.1.176. [DOI] [PubMed] [Google Scholar]
- 93.Sepulveda JL, Gkretsi V, Wu C. Assembly and signaling of adhesion complexes. Curr Top Dev Biol. 2005;68:183–225. doi: 10.1016/S0070-2153(05)68007-6. [DOI] [PubMed] [Google Scholar]
- 94.Thompson SA, Blazeski A, Copeland CR, Cohen DM, Chen CS, Reich DM, Tung L. Acute slowing of cardiac conduction in response to myofibroblast coupling to cardiomyocytes through n-cadherin. J Mol Cell Cardiol. 2014;68:29–37. doi: 10.1016/j.yjmcc.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goshima K. Formation of nexuses and electrotonic transmission between myocardial and fl cells in monolayer culture. Exp Cell Res. 1970;63:124–130. doi: 10.1016/0014-4827(70)90339-3. [DOI] [PubMed] [Google Scholar]
- 96.Goshima K. Synchronized beating of myocardial cells mediated by fl cells in monolayer culture and its inhibition by trypsin-treated fl cells. Exp Cell Res. 1971;65:161–169. doi: 10.1016/s0014-4827(71)80062-9. [DOI] [PubMed] [Google Scholar]
- 97.Rook MB, van Ginneken AC, de Jonge B, el Aoumari A, Gros D, Jongsma HJ. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am J Physiol. 1992;263:C959–977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 98.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 99.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004;94:828–835. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]