

Muscular dystrophies are a clinically and genetically heterogeneous group of skeletal muscle‐wasting diseases. Even for experts in the field of neuromuscular diseases, it is becoming increasingly difficult to distinguish and accurately diagnose all forms of muscular dystrophy on clinical grounds alone, as there is currently a still growing number of different genetic loci for muscular dystrophies.1 At present, therapy of muscular dystrophies is predominantly based on symptomatic treatment and supportive care. However, within the last years, promising new molecular therapies have been developed facilitating causative therapy in the near future.2, 3, 4
Duchenne muscular dystrophy (DMD) represents the most frequent hereditary childhood myopathy, leading to progressive muscle atrophy and weakness and premature death; interestingly, the underlying genetic defect in the dystrophin gene with an X‐linked trait has been described more than 25 years ago. The worldwide incidence is estimated with 1:5000 male newborns.5, 6 First symptoms are usually noted between age 3 and 5, while loss of ambulation occurs around age 12, along with scoliosis, contractures, respiratory and cardiac impairment requiring early ventilatory support and heart medication7 (Table 1). Life expectancy is reduced to 30–40 years of age although multidisciplinary symptomatic and surgical treatment has considerably improved survival within the last two decades.8, 9
Table 1.
Typical course and transition in DMD; life expectancy is around 30 years and can be prolonged by invasive ventilation therapy
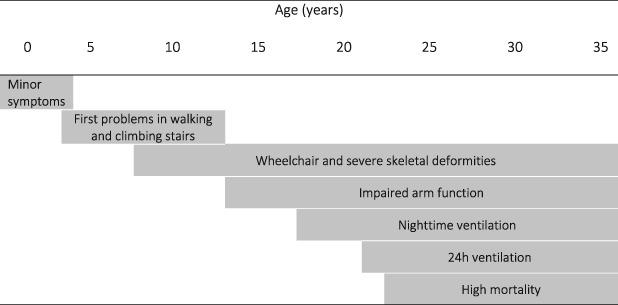
|
Becker muscular dystrophy (BMD) is the allelic phenotype of DMD with wider variability of clinical symptoms but usually milder impairment and slower progression with an estimated incidence of 1:20 000.6 The mutation in the dystrophin gene does not cause a reading frame interruption but a so called in frame mutation that leads to translation of different amounts of a truncated Dystrophin protein that can maintain a low protein function level within the muscle cells of these patients.10, 11
Similar to leading the way towards genetic diagnostics back then, DMD today is within the main focus of new gene‐based therapies.
Over the last decades, the identification of the dystrophin gene has led to a deeper understanding of the muscle cell membrane and the proteins responsible for membrane stability. Animal models, mainly the mdx mouse and the golden retriever muscular dystrophy (GRMD) dog—with natural mutations or specifically developed by gene targeting—have strongly contributed to enhance knowledge on disease progression, underlying pathology and therapeutic development.12 Recently, a promising large animal model for studying disease mechanisms and for the development and evaluation of targeted therapies of Duchenne muscular dystrophy, the DMDpig, had been successfully established.13, 14 Pigs are particularly suitable animal models for translational biomedical research as they reflect many anatomical and physiological characteristics of humans. This animal model reflects a frequent mutation of human DMD patients, a deletion of exon 52; therefore, the DMD pig represents a promising model for testing targeted genetic treatments.
Two predominant therapeutic strategies can be differentiated—dystrophin‐based therapies, e.g. stem cell therapy, virus‐based gene therapy, exon skipping by antisense oligonucleotides or morpholinos, e.g. Eteplirsen/Exondys51®, approved in the US in 2016 for DMD with deletions amendable to exon 51 skipping, and stop codon read‐through by Ataluren/Translarna®, which had been conditionally approved as the first drug in DMD in 2014 in Europe for treatment of nonsense‐mutation DMD (nmDMD), and dystrophin‐independent treatments such as utrophin modulation, alpha‐7‐integrin up‐regulation, myostatin inhibition, follistatin gene therapy, along with other strategies for muscle growth.15 Unfortunately, until now, the large size of the dystrophin gene hinders direct replacement through viral gene therapy.
Nonsense stop codon read‐through therapy16, 17, 18, 19 works by selectively inducing ribosomal read‐through of premature stop codons but not normal stop codons. DMD patients harbouring specific nonsense mutations would potentially benefit from this therapy (11% of German DMD patients). Exon skipping aims to moderate disease progression by restoring the open reading frame of dystrophin transcripts resulting in the production of partly functional dystrophin protein by AONs.20, 21 Since 60% to 65% of all DMD patients carry a deletion of one or more exons, the skipping of certain exons would be beneficial to a relatively large numbers of patients.22
Recently, the CRISPR (Clustered Regularly Interspaced Palindromic Repeat)/Cas9 system has been identified as a revolutionary disease‐modifying technology. The versatility of CRISPR/Cas9‐based platforms makes them promising tools for the correction of monogenetic diseases.23 In 2016, local and systemic delivery of Cas9 and gRNAs with AAV vectors was shown in the mdx mouse, the most frequently used DMD animal model, leading to expression of a truncated but functional dystrophin protein in skeletal and cardiac muscle.24, 25, 26 Young et al.27 deleted exons 45–55 in induced pluripotent stem cells (iPSCs) of DMD patients and differentiated them into cardiomyocytes or skeletal muscle cells. The large deletion restored the reading frame of the DMD gene and also resulted in a truncated but functional protein. Genome editing with CRISPR/Cas 9 creates a permanent modification of the gene; therefore, once‐in‐a‐lifetime might be sufficient for a stable dystrophin expression. However, it is still a challenge to sufficiently reach every muscle in the body, including the heart. Cell therapy has the limitation that it is currently not possible to deliver enough cells to all muscles in the body,27 while the genome‐editing design may offer a therapeutic approach with a reasonable clinical applicability, reaching out to approximately 60% of DMD patients.28
Gene therapeutic methods such as stop codon read‐through, exon skipping, and gene editing by exon snipping are promising future disease‐modifying or even curative treatments for DMD. Until now, corticosteroids, heart medication, non‐invasive and invasive ventilation, physiotherapy, and supportive care represent the gold standard of therapy; still, loss of ambulation, respiratory, and cardiac decompensation cannot be prevented.29, 30
TREAT‐NMD (www.TREAT‐NMD.eu) is a worldwide network for neuromuscular diseases that provides an infrastructure to support the delivery of promising new therapies for patients. Within TREAT‐NMD patient registries for DMD were initially established in five European partner countries (Germany, UK, Hungary, France, and Italy). This project was highly successful with by now >13 500 patients worldwide from 31 European countries, demonstrating the urgent need of patient registries in rare diseases.22, 23, 24, 25, 26, 27, 28, 29, 30, 31 The impact of the national registries is further enhanced through the continued inclusion in the global DMD registry.
The main objectives of the German DMD registry (www.dmd‐register.de) are to assess the feasibility, facilitate the planning of appropriate clinical trials, and support the enrolment of patients, as new therapeutic strategies and clinical trials are in preparation. When a clinical trial is being planned, it is very important that patients suitable for that trial can be found and contacted quickly to facilitate enrolment in the trial. The best way of ensuring this is to collect relevant data about patients in a single registry containing the information that researchers will need, including each patient's particular genetic defect and other key information about their disease. Since DMD is a rare disorder, clinical trials are likely to recruit from several countries to reach statistical significance. It is therefore of high importance to harmonize trial infrastructures, standards of care, outcome measures, and patient registries internationally. The national and international registries give a unique opportunity not only to effectively recruit patients for international clinical trials but also to learn more about prevalence, standards of care, and the natural history of the disease. Furthermore, analysing the type and frequency of DMD patient specific mutations is an invaluable tool for diagnostics, basic scientific research, trial planning, trial readiness, and improved clinical care. Trial readiness implies standardized patient registries with many benefits to registered patients, such as feedback on standards of care and new research developments. For industry, the benefits are easy access to the patient community, a clear concept of the target market, feasibility and planning of clinical trials, and recruitment of patients into clinical trials.
We analysed genetic data for 724 DMD mutations held within the German DMD database; our findings mirrored the numbers identified by Bladen et al.22 Within our registry, a total of 557 large mutations were observed (77% of total mutations), of which 481 (86%) were deletions (1 exon or larger) and 76 (14%) were duplications (1 exon or larger).
There were 167 small mutations (smaller than 1 exon, 23% of all mutations), of which 43 (26%) were small deletions, 11 (7%) small insertions and 21 (13%) affected the splice sites; 83 (50%) were stop‐codon‐mutations. Seventy‐eight mutations were identified within the database that would potentially benefit from novel genetic therapies for DMD such as stop codon read‐through therapies (11% of total mutations).
Additionally, 391 mutations would potentially benefit from exon skipping therapy (84% of out of frame deletions and 54% of total mutations). The top ten exon skips within the database and the percentage of mutations that would be rescued by such exon skips were deletion of exon 51 (14% of total mutations/22% of deletions), 53 (9%/14%), 45 (8%/13%), 44 (9%/14%),43 (3%/5%), 46(4%/7%), 50(3%/5%), 52(3%/5%), 55(6%/9%), and 8(1%/2%).
Regarding emerging new therapies, it becomes more and more important to address health economic questions; recently, we assessed the cost of illness (COI) of Duchenne and the milder allelic Becker muscular dystrophy (DMD/BMD) from a socio‐economic and clinical perspective in the background of emerging new innovative (curative) DMD therapies.32
Three hundred and sixty‐three patients with genetically confirmed DMD and BMD were enrolled; patients were divided into corresponding severity stages according to disease progression and motor function (Table 2) for analysis of differences between the consumption of resources of direct medical services, indirect and informal care cost and health‐related quality of life (HRQOL) between DMD and BMD, but also within the course of the disease. Estimated annual total disease burden including direct medical/non‐medical, indirect and informal care costs of DMD totaled €65263, which was nearly twice as high when compared with €36651 in BMD. Informal care cost, indirect cost caused by productivity loss and absenteeism of patients and caregivers, and non‐medical cost were identified as highly important cost drivers. Total cost increased significantly with disease progression and consistent with the clinical severity of the distinct phenotype regarding all cost items included in the study whereas patients' HRQOL declined with disease progression.
Table 2.
Definition of patient/parent self‐evaluated clinical severity stages (Schreiber et al.32)
| Stage | Clinical characteristics of DMD patients |
|---|---|
| I | Early ambulatory with mild impairment: Gower's manoeuvre, waddling gait, walking on toes, and problems with climbing stairs. |
| II | Late ambulatory with high impairment: walking becomes increasingly difficult, more problems climbing stairs and getting up from the floor, and part‐time wheelchair use. |
| III | Early non‐ambulatory: loss of ambulation, active manual wheelchair use still possible, independent standing, and sitting still possible for some time. |
| IV | Late non‐ambulatory: independent electric wheelchair use but decline of upper limb function and ability to sit independently. |
| V | Non‐ambulatory with confinement to bed: loss of independent mobility and hand function preserved on a low level. |
Estimating the worldwide prevalence of DMD to be 4.78 and for BMD 1.53 per 100 000 males as recently published,33 and according to recent calculations of the German population,34 about 1 900 DMD patients and 600 BMD patients are supposed to live in Germany. Altogether, this implicates a yearly disease burden of €123 million for DMD, which showed to be more than five times higher than for BMD with estimated €22 million without even including additional cost of respiratory management, medical aids, and extra direct cost per patient.32
These health economic assessments are crucial for the funding of development programs for rare diseases since early benefit assessments are required for reimbursement of therapies, contributing to the facilitation of an efficient translation of innovations from clinical research over marketing authorization to patient access to a new therapy.32
Conclusions
New developments of personalized gene therapy aim at genetically defined disease subgroups in DMD, based on the underlying molecular mechanism and the resulting phenotype, and set an example for other hereditary diseases. We have learned tremendously within the last decade; however, there is still a long way to go until these therapeutic strategies will be able to finally cure—and not only modify—pathology and phenotype of DMD patients.
Conflict of Interest
None declared.
Acknowledgements
This article is based on a previously published paper (Schreiber et al., 2014), updated regarding factual contents and modified for style, and on current data from the German DMD patient registry (www.dmd‐register.de). The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle.35 We thank H. Saodiy for scientific assistance and database research.
Walter, M. C. , and Reilich, P. (2017) Recent developments in Duchenne muscular dystrophy: facts and numbers. Journal of Cachexia, Sarcopenia and Muscle, 8: 681–685. doi: 10.1002/jcsm.12245.
References
- 1. Thompson R, Straub V. Limb‐girdle muscular dystrophies—international collaborations for translational research. Nat Rev Neurol 2016;12:294–309. [DOI] [PubMed] [Google Scholar]
- 2. Seto JT, Bengtsson NE, Chamberlain JS. Therapy of genetic disorders‐novel therapies for Duchenne muscular dystrophy. Curr Pediatr Rep 2014;2:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guiraud S, Aartsma‐Rus A, Vieira NM, Davies KE, van Ommen GJ, Kunkel LM. The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet 2015;16:281–308. [DOI] [PubMed] [Google Scholar]
- 4. Gee P, Xu H, Hotta A. Cellular reprogramming, genome editing, and alternative CRISPR Cas9 technologies for precise gene therapy of Duchenne muscular dystrophy. Stem Cells Int 2017;2017: 8765154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mendell JR, Lloyd‐Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013;48:21–26. [DOI] [PubMed] [Google Scholar]
- 6. Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years' experience in Wales (UK). Eur J Hum Genet 2013;21:1049–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 2002;12:926–929. [DOI] [PubMed] [Google Scholar]
- 8. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. D. M. D. care considerations working group: diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93. [DOI] [PubMed] [Google Scholar]
- 9. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. D. M. D. care considerations working group: diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9:177–189. [DOI] [PubMed] [Google Scholar]
- 10. Bushby KM, Gardner‐Medwin D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I Natural history J Neurol 1993;240:98–104. [DOI] [PubMed] [Google Scholar]
- 11. Comi GP, Prelle A, Bresolin N, Moggio M, Bardoni A, Gallanti A, et al. Clinical variability in Becker muscular dystrophy. Genetic, biochemical and immunohistochemical correlates. Brain 1994;117:1–14. [DOI] [PubMed] [Google Scholar]
- 12. Yu X, Bao B, Echigoya Y, Yokota T. Dystrophin‐deficient large animal models: translational research and exon skipping. Am J Transl Res 2015;7:1314–1331. [PMC free article] [PubMed] [Google Scholar]
- 13. Klymiuk N, Blutke A, Graf A, Krause S, Burkhardt K, Wuensch A, et al. Dystrophin‐deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum Mol Genet 2013;22:4368–4382. [DOI] [PubMed] [Google Scholar]
- 14. Fröhlich T, Kemter E, Flenkenthaler F, Klymiuk N, Otte KA, Blutke A, et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci Rep 2016;6:33362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walter MC. Update therapie bei muskeldystrophien. Drug Res 2015;65:S24. [DOI] [PubMed] [Google Scholar]
- 16. Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside‐induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol 2000;48:164–169. [PubMed] [Google Scholar]
- 17. Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 2001;49:706–711. [PubMed] [Google Scholar]
- 18. Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following singleand multiple‐dose administration to healthy male and female adult volunteers. J Clin Pharmacol 2007;47:430–444. [DOI] [PubMed] [Google Scholar]
- 19. Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447:87–91. [DOI] [PubMed] [Google Scholar]
- 20. van Ommen GJ, van Deutekom J, Aartsma‐Rus A. The therapeutic potential of antisense‐mediated exon skipping. Curr Opin Mol Ther 2008;10:140–149. [PubMed] [Google Scholar]
- 21. Aartsma‐Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, et al. Theoretic applicability of antisense‐mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 2009;30:293–299. [DOI] [PubMed] [Google Scholar]
- 22. Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT‐NMD DMD Global database: analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum Mutat 2015;36:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phimister EG. The CRIDSPR way to think about Duchenne's. NEJM 2016;374:1684–1686. [DOI] [PubMed] [Google Scholar]
- 24. Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez‐Ortiz E, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016;351:400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016;351:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016;351:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, et al. A single CRISPR‐Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC‐derived muscle cells. Cell Stem Cell 2016;18:533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Breitbart A, Murry CE. Imprecision medicine: a one‐size‐fits‐many approach for muscle dystrophy. Cell Stem Cell 2016;18:423–424. [DOI] [PubMed] [Google Scholar]
- 29. Sejerson T, Bushby K, T‐NENo E. Standards of care for Duchenne muscular dystrophy: brief TREAT‐NMD recommendations. Adv Exp Med Biol 2009;652:13–21. [DOI] [PubMed] [Google Scholar]
- 30. Hoffman EP, Reeves E, Damsker J, Nagaraju K, McCall JM, Connor EM, et al. Novel approaches to corticosteroid treatment in Duchenne muscular dystrophy. Phys Med Rehabil Clin N Am 2012;23:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bladen CL, Rafferty K, Straub V, Monges S, Moresco A, Dawkins H, et al. The TREAT‐NMD Duchenne muscular dystrophy registries: conception, design and utilisation by industry and academia. Hum Mutat 2013;34:1449–1457. [DOI] [PubMed] [Google Scholar]
- 32. Schreiber‐Katz O, Klug C, Thiele S, Schorling E, Zowe J, Reilich P, et al. Comparative cost of illness analysis and assessment of health care burden of Duchenne and Becker muscular dystrophies. OJRD 2014;9:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta‐analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014;24:482–491. [DOI] [PubMed] [Google Scholar]
- 34. Statistisches Bundesamt [https://www.destatis.de/DE/ZahlenFakten/GesellschaftStaat/Bevoelkerung/Bevoelkerung.html]
- 35. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]