

Abstract
Background
Chronic kidney disease (CKD) patients experience skeletal muscle wasting and decreased exercise endurance. Our previous study demonstrated that indoxyl sulfate (IS), a uremic toxin, accelerates skeletal muscle atrophy. The purpose of this study was to examine the issue of whether IS causes mitochondria dysfunction and IS‐targeted intervention using AST‐120, which inhibits IS accumulation, or mitochondria‐targeted intervention using L‐carnitine or teneligliptin, a dipeptidyl peptidase‐4 inhibitor which retains mitochondria function and alleviates skeletal muscle atrophy and muscle endurance in chronic kidney disease mice.
Methods
The in vitro effect of IS on mitochondrial status was evaluated using mouse myofibroblast cells (C2C12 cell). The mice were divided into sham or 5/6‐nephrectomized (CKD) mice group. Chronic kidney disease mice were also randomly assigned to non‐treatment group and AST‐120, L‐carnitine, or teneligliptin treatment groups.
Results
In C2C12 cells, IS induced mitochondrial dysfunction by decreasing the expression of PGC‐1α and inducing autophagy in addition to decreasing mitochondrial membrane potential. Co‐incubation with an anti‐oxidant, ascorbic acid, L‐carnitine, or teneligliptine restored the values to their original state. In CKD mice, the body and skeletal muscle weights were decreased compared with sham mice. Compared with sham mice, the expression of interleukin‐6 and atrophy‐related factors such as myostatin and atrogin‐1 was increased in the skeletal muscle of CKD mice, whereas muscular Akt phosphorylation was decreased. In addition, a reduced exercise capacity was observed for the CKD mice, which was accompanied by a decreased expression of muscular PCG‐1α and increased muscular autophagy, as reflected by decreased mitochondria‐rich type I fibres. An AST‐120 treatment significantly restored these changes including skeletal muscle weight observed in CKD mice to the sham levels accompanied by a reduction in IS levels. An L‐carnitine or teneligliptin treatment also restored them to the sham levels without changing IS level.
Conclusions
Our results indicate that IS induces mitochondrial dysfunction in skeletal muscle cells and provides a potential therapeutic strategy such as IS‐targeted and mitochondria‐targeted interventions for treating CKD‐induced muscle atrophy and decreased exercise endurance.
Keywords: Chronic kidney disease, Indoxyl sulfate, Muscle atrophy, Mitochondrial function, L‐carnitine, Dipeptidyl peptidase‐4 inhibitor
Introduction
Patients with chronic kidney disease (CKD) experience skeletal muscle wasting and decreased exercise endurance.1 An impaired physical performance was observed from an early stage of CKD.2 Physical inactivity derived from skeletal muscle atrophy, referred to as sarcopenia, seriously influences the prognosis of CKD patients because their physical inactivity is correlated with renal prognosis and mortality.3, 4 Therefore, maintaining physical performance is considered to be an essential factor for improving the prognosis of CKD patients. However, robust evidence regarding effective therapeutics for CKD‐induced physical inactivity is currently lacking.
So far, several molecular mechanisms have been proposed to explain CKD‐induced skeletal muscle atrophy.1, 5 Oxidative stress and inflammatory cytokines are thought to play an important role in CKD‐induced skeletal muscle atrophy and decreasing of exercise capacity.1, 6 In a recent study, using indoxyl sulfate (IS)‐loaded half‐nephromized mice and a mouse myofibroblast (C2C12) cell system, we reported that, among the various uremic toxins, IS strongly induced skeletal muscle atrophy by increasing the production of atrogin‐1, a member of the muscle specific ubiquitin ligase family, and myostatin, a negative regulator of muscle growth, via inducing muscular oxidative stress‐mediated inflammation.7 In addition, Nishikawa et al. also showed that IS might be involved in a decrease in exercise capacity via inducing muscular oxidative stress in CKD model mice and that IS enhanced the production of reactive oxygen species (ROS) in C2C12 cells.8 These findings suggest that IS has a critical role in the development of CKD‐induced skeletal muscle atrophy.
It is well known that exercise capacity is strongly related to mitochondrial function in skeletal muscle. The amount of mitochondria is regulated by both mitochondria biosynthesis and its degradation.9, 10 Tamaki et al. recently reported that the amount of muscular mitochondria was decreased in the early stage of CKD mice and that it was associated with increased oxidative stress and inflammatory cytokines, such as tumor necrosis factor (TNF)‐α and interleukin (IL)‐6.6 In fact, oxidative stress and inflammation cause the expression of the peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α), a master regulator for mitochondrial biosynthesis, to be reduced and to an increase in autophagy, a mitochondria degradation system. Interestingly, Brault et al. demonstrated that the overexpression of PGC‐1α caused a resistance to muscle atrophy that was induced by denervation or fasting.11 Similarly, Wenz et al. also showed that the overexpression of PGC‐1α in mice prevented muscle atrophy and hence extended their life span.12 These findings led us to hypothesize that IS causes mitochondria dysfunction in skeletal muscle cells. If this is correct, therapeutic strategies designed to inhibit IS accumulation or to retain muscular mitochondria function would be effective for the prevention and treatment of CKD‐induced muscle atrophy.
The purpose of this study was to reveal whether (i) IS causes mitochondrial dysfunction and (ii) whether IS‐ and mitochondria‐targeted therapeutics alleviate CKD‐induced muscle atrophy and the accompanying impaired exercise endurance. AST‐120, a charcoal absorbent, is used in IS‐targeted therapeutics, because it absorbs indole, a precursor of IS, in the gut flora, resulting in a reduced level of IS synthesis in the liver.13 For mitochondrial‐targeted therapeutics, we choose L‐carnitine because it transports free‐fatty acids to mitochondria to maintain mitochondrial function, and CKD patients are occasionally given L‐carnitine supplementation due to a L‐carnitine deficiency. We also used teneligliptin, a dipeptidyl peptidase‐4 (DPP‐4) inhibitor that can be safely administered to patients with reduced renal function, because glucagon‐like peptide‐1 (GLP‐1), a major target of DPP‐4, increased mitochondrial membrane potential and oxygen consumption in addition to increasing the expression of PGC‐1α.14
Methods
Chemicals and materials
Indoxyl sulfate, dihydroethidium, and rabbit polyclonal anti‐mouse glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) antibody were purchased from Sigma‐Aldrich (St Louis, MO). Rabbit polyclonal anti‐myostatin antibody was purchased from proteintech (Chicago, USA). AST‐120 was kindly provided from Kureha Corporation (Tokyo, Japan). The antibiotic and antimycotic mixture (10 000 U/mL penicillin, 10 000 μg/mL streptomycin, 25 μg/mL amphotericin B), ascorbic acid was purchased from Nacalai Tesque (Kyoto, Japan). Dulbecco's modified eagle medium and Dulbecco's phosphate‐buffered saline (D‐PBS) were purchased from Gibco (Invitrogen, Grand Island, NY). L‐carnitine was purchased from Otsuka Phamaceutical Co., Ltd. (Tokyo, Japan). Teneligliptin was generously provided from Mitsubishi Tanabe Pharma Co., Ltd. (Osaka, Japan). All methods were carried out in accordance with approved guidelines. All experimental protocols were approved by Kumamoto University.
Cell culture
Mouse C2C12 myoblast cells were purchased from the RIKEN Bioresource Center Cell Bank (Ibaraki, Japan). C2C12 myoblast cells were cultured in Dulbecco's modified eagle medium supplemented with 10% fetal bovine serum (Hyclone Laboratories, Logan UT, USA), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B, and maintained under 37°C and 5% CO2.
Real‐time reverse transcription polymerase chain reaction (RT‐PCR) analysis
Real‐time RT‐PCR analysis was performed as described in a previous report.7 In a typical run, total RNA was extracted using RNAiso PLUS (TaKaRa Bio Inc., Shiga, Japan) according to the manufacturer's protocol. The concentration and the purity of the RNA extract were determined by the absorbance at 260 and 280 nm. The cDNA was synthesized using the PrimeScript® RT master mix (TaKaRa Bio Inc.). Quantitative real‐time RT‐PCR analysis of PGC‐1α, myostatin, atrogin‐1, and GAPDH was performed in an iCycler thermal cycler (Bio‐Rad) with an iQ5 qRT‐PCR detection system attached (Bio‐Rad) using SYBR® Premix Ex TaqII (TaKaRa Bio Inc.). Polymerase chain reaction amplifications were performed under the following conditions: 95°C for 3 min, for 40 cycles at 95°C for 10 s (denaturation step), at 60°C for 1 min (annealing/extension steps). The primers used are shown in Supporting Information Table 1. The threshold cycle (Ct) values for each gene amplification were normalized by subtracting the Ct value calculated for GAPDH.
Table 1.
Renal function, body weight, and muscle weight profile for sham, 5/6‐nephrectomized (CKD) and AST‐120, L‐carnitine, or teneligliptin‐treated CKD mice
| CKD | |||||
|---|---|---|---|---|---|
| Sham | Control | AST‐120 | Carnitine | Teneligliptin | |
| BUN (mg/dL) | 21.1 ± 1.2 | 56.7 ± 9.4b | 45.5 ± 6.0a | 53.9 ± 9.4b | 66.9 ± 12.8b |
| SCr (mg/dL) | 0.2 ± 0.02 | 0.8 ± 0.31a | 0.4 ± 0.13 | 1.4 ± 0.42a | 1.0 ± 0.05b |
| Final BW (g) (at 28 weeks) | 49.1 ± 2.7 | 39.3 ± 1.4a | 44.2 ± 0.5 | 45.4 ± 1.4 | 43.4 ± 0.9 |
| Tibialis anterior (mg) | 74.6 ± 3.3 | 70.0 ± 2.6 | 71.5 ± 1.7 | 74.0 ± 2.7 | 75.0 ± 3.7 |
| Soleus (mg) | 12.5 ± 0.6 | 10.3 ± 0.3a | 10.7 ± 0.6 | 11.5 ± 0.5 | 11.3 ± 0.4 |
| Gastrocnemius (mg) | 216.0 ± 6.6 | 188.9 ± 9.1a | 212.0 ± 5.5 | 211.0 ± 6.2 | 207.6 ± 6.2 |
Data are expressed as the mean ± standard error of the mean.
p < 0.05.
p < 0.01 compared with sham.
CKD, chronic kidney disease; BUN, blood urea nitrogen; SCr, serum creatinine; BW, body weight.
Mitochondrial staining
C2C12 myoblast cells were seeded at six well‐plate sets with flame sterilized‐cover glasses and cultured overnight. After cell adhesion, cells were starved for 2 h with serum free medium and then treated with or without IS. The effect of ascorbic acid, L‐carnitine, or teneligliptin was determined after a simultaneous treatment with IS. After a 3 h treatment, the cells were washed with PBS and then incubated with MitoRed (Dojin Chemical Co., Ltd, Kumamoto, Japan), MitoGreen (Invitrogen, Grand Island, NY), and Hoechist33342 (Dojin chemical Co., Ltd, Kumamoto, Japan) in D‐PBS for 20 min. After incubation, cells were washed with D‐PBS and observed by microscopy.
Animal experiments
All animal experiments were carried out in accordance with the approved guidelines of Kumamoto University for the care and use of laboratory animals. All animal experiments and procedures were approved by Kumamoto University. sea:ICR mice (5 weeks, male) were purchased from Kudo Co., Ltd (Saga, Japan) and bred on a 12 h day/night cycle. 5/6‐nephrectomized model mice were produced in a two‐step surgery procedure according to previous reports.15 In brief, two‐thirds of the right kidney was removed, and 1 week later, the left kidney was removed. At 4 weeks after the final surgery, the mice were randomized by blood urea nitrogen (BUN) and body weight, and were assigned to AST‐120 (charcoal oral absorbent, 8 w/w% in powder diet), L‐carnitine (560 mg/kg, drinking water), or teneligliptin (60 mg/kg, drinking water) treatment groups. Sham‐operated mice and control mice (CKD‐operated mice) received a normal diet and water. At 24 weeks after the final surgery, treadmill experiments were performed. At 4 weeks after the treadmill experiment, the mice were sacrificed, and blood or skeletal muscle tissue was collected.
Blood urea nitrogen and creatinine
The plasma levels for BUN and creatinine were measured by a Fujidrychem7000 and drychem slide system (FUJIFILM, Kanagawa, Japan) following the manufacturer's protocol.
High‐performance liquid chromatography (HPLC) analysis
Indoxyl sulfate levels in the plasma and gastrocnemius were measured by an HPLC method, as described previously.7, 16 In brief, plasma or a gastrocnemius homogenate extracted with radioimmunoprecipitation assay buffer containing 150 mM NaCl, 1% nonidet P‐40, 10 mM Tris–HCl (pH 7.4), and 1% protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) was mixed with acetonitrile (1:9, v/v for the plasma sample or 1:3, v/v for the tissue homogenate sample) and centrifuged at 12 000 g for 10 min. The supernatant was collected and mixed with ultrapure water (1:1, v/v for a plasma sample or 3:2, v/v for a tissue homogenate sample). The sample was loaded to HPLC with 20 μL for the plasma sample and 50 μL for the tissue homogenate. The HPLC system consisted of an Agilent 1100 series intelligent pump and a fluorescence spectrophotometer. A LiChro‐sorb RP‐18 column (Cica Merk, Tokyo, Japan) was used as the stationary phase. The mobile phase consisted of 0.2 M acetate buffer (pH 4.0)–acetonitrile (3:1, v/v) for IS. The flow rate was 1.0 mL/min. Indoxyl sulfate was detected by means of a fluorescence monitor with excitation/emission wavelengths set to 280 and 375 nm, respectively.
Western blotting analysis
Western blotting analysis was performed as described in a previous report.7 Total protein was extracted with radioimmunoprecipitation assay buffer containing 150 mM NaCl, 1% nonidet P‐40, 10 mM Tris–HCl (pH 7.4), 1% protease inhibitor cocktail, and 1% phosphatase inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). A 30 μg sample of protein was mixed with sample buffer containing 50 mM dithiothreitol, heated 100°C, and separated by 10% sodium dodecyl sulfate–poly‐acrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene fluoride membrane and then immunoblotted with antibodies against myostatin (Cell Signaling Technology, MA, USA), Akt (Cell Signaling Technology, MA, USA), p‐Akt (Cell Signaling Technology, MA, USA), and LC3 (Sigma‐Aldrich, St. Louis, MO) under the room temperature for 1 h. The sample was then immunoblotted with horseradish peroxidase conjugated secondary antibody at room temperature for 1 h. The intensity of each band was detected using LAS4000mini (GE Healthcare, UK Ltd, Backinghamshire, England) and quantified using ImageJ software. The densitometric intensity was normalized with GAPDH expression.
Enzyme‐linked immunosorbent assay (ELISA)
Interleukin‐6 and TNF‐α in gastrocnemius were measured using ELISA MAX™ Deluxe set (Biolegend, San Diego, USA) following the manufacturer's protocol.
Treadmill experiments
The running distance for the mice was measured using a treadmill (MK‐680S, Muromachi kikai, Tokyo, Japan), in accordance with a previous report.9 At 3 days before the experiment, we acclimated the mice to a motor‐driven treadmill at 14 m/min and a 0% incline. Mice were forced to run until they were exhausted (they remained on the electrical shocker plate). Electrical stimulation was set at the recommended intensity described in the instructions. The detailed treadmill conditions are described in Supporting Information Table S2.
Succinate dehydrogenase (SDH) stain
Succinate dehydrogenase staining was performed as described in previous reports.17, 18 In brief, 10 μm thick of frozen sections of skeletal muscle was dried by a dryer at room temperature for 30 min. Tissue sections were reacted with reaction solution (100 mM phosphate buffer, 1.5 mM nitroblue tetrazolium, 48 mM disodium succinate) for 45 min at 37°C. After the reaction, the sections were washed 3 times with ion‐exchanged water and mounted with mount‐quick aqueous (Daido Sangyo Co., Ltd, Saitama, Japan). The resulting sections were observed by microscopy (Keyence, BZ‐9000 microscope, Osaka, Japan).
Statistical analyses
The means for the two groups of data were compared by the unpaired t‐test. The means for the groups were compared by analysis of variance followed by Tukey's multiple comparison. A probability value of P < 0.05 was considered to be significant.
Results
The effect of indoxyl sulfate on mitochondria and the effect of anti‐oxidant, L‐carnitine, and teneligliptin on indoxyl sulfate‐induced mitochondrial dysfunction in C2C12 cells
We investigated the effect of IS on mitochondrial biosynthesis‐ or degradation‐related factors such as PGC‐1α, a master regulator for mitochondrial biosynthesis or autophagy (LC3II/LC3I ratio), one of the key systems for mitochondrial degradation, using C2C12 cells. As shown in Figure 1A, IS significantly decreased the mRNA expression of PGC‐1α and increased the ratio of LC3II/LC3I. Moreover, MitoRed staining clearly indicated that incubation with IS for 3 h caused a decrease in mitochondrial membrane potential in C2C12 cells compared with untreated controls, while IS had no effect on the amount of mitochondria, as evidenced by MitoGreen staining.
Figure 1.
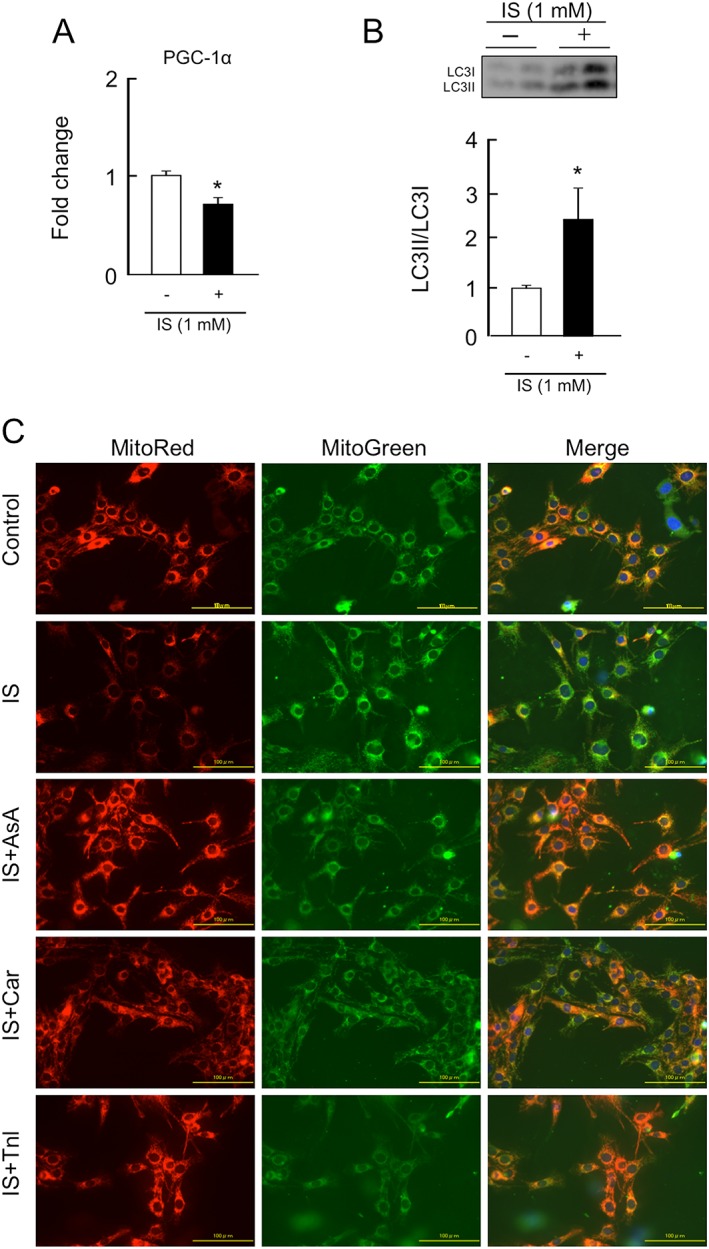
Effect of indoxyl sulfate on peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha expression, autophagy, or mitochondrial membrane potential, and the effect of anti‐oxidant, L‐carnitine, and teneligliptin on indoxyl sulfate‐induced mitochondrial dysfunction in C2C12 cells. (A) mRNA expression of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha at 3 h after a 1 mM indoxyl sulfate treatment was determined by real‐time reverse transcription polymerase chain reaction. (B) Protein expression of LC3II/LC3I at 3 h after 1 mM indoxyl sulfate treatment was determined by western blot. (C) Mitochondrial membrane potential was determined by MitoRed staining (the interaction of MitoRed with mitochondria depends of the membrane potential of the mitochondria) and MitoGreen staining (MitoGreen appears to localize to mitochondria regardless of mitochondrial membrane potential), the mitochondria selective fluorescent probe at 3 h after a 1 mM indoxyl sulfate treatment. Effect of ascorbic acid (an anti‐oxidant, 200 μμ; AsA), L‐carnitine (5 mM; Car), or teneligliptin (1 μM; Tnl) on indoxyl sulfate‐induced mitochondrial decline of membrane potential at 3 h after 1 mM indoxyl sulfate treatment. AsA, Car, or Tnl was co‐incubated with indoxyl sulfate. Data are expressed the mean ± standard error of the mean (n = 3–4). * p < 0.05 compared with control.
Co‐incubation with ascorbic acid, an anti‐oxidant, restored the IS‐induced decrease in mitochondrial membrane potential, suggesting the importance of oxidative stress in IS‐induced mitochondrial dysfunction (Figure 1C). Similar to ascorbic acid, L‐carnitine or teneligliptine also inhibited the development of impaired mitochondrial functions in the presence of IS.
The effect of AST‐120, L‐carnitine, and teneligliptin on chronic kidney disease‐induced muscle atrophy using 5/6‐nephrectomized mice
To confirm the findings obtained using C2C12 cells, we examined the effect of therapeutic intervention on CKD‐induced muscle atrophy and muscle endurance using 5/6‐nephrectomized mice. The experimental scheme for evaluating the 24 week administration of AST‐120, L‐carnitine, and teneligliptin to CKD mice is shown in Figure 2. Blood urea nitrogen (BUN) and serum creatinine levels in the CKD mice (control) were significantly increased compared with those in sham (healthy) mice (Table 1). However, no significant differences in renal function were observed between the CKD groups used in this study (Table 1), indicating that the therapeutic effect of each intervention was not due to an amelioration of the renal function. The body weights of the CKD mice were significantly decreased as compared with that of sham mice, while this reduction was suppressed by the administration of AST‐120, L‐carnitine, or teneligliptin (Table 1). We also measured the weights of skeletal muscle of the mice. As a result, the weights of soleus and gastrocnemius were significantly decreased in CKD mice as compared with those of sham mice. Similar to body weight, these muscular weight changes were suppressed by treatment with AST‐120, L‐carnitine, or teneligliptin (Table 1). There were no significant changes in the weight of the tibialis anterior among the groups.
Figure 2.

Experimental scheme for evaluating AST‐120, L‐carnitine, and teneligliptin on chronic kidney disease mice. sea:ICR mice (♂, 5 weeks) were used in the experiment. 5/6‐nephrectomized mice (chronic kidney disease mice) were produced in two‐step surgery according to the previous reports [Miyamoto. KI. 2013]. At 4 weeks after final surgery, mice were randomized by blood urea nitrogen and body weight, and were assigned to AST‐120 (oral charcoal absorbent, 8 w/w% in powder diet), L‐carnitine (560 mg/kg, drinking water), or teneligliptin (60 mg/kg, drinking water)‐treated group. Sham‐operated mice and control mice (chronic kidney disease‐operated mice) were received normal diet and water. At the 24 weeks after the final surgery, treadmill experiment was performed. Four weeks after treadmill experiment, mice were sacrificed, and blood or skeletal muscle tissue was collected.
The effect of AST‐120, L‐carnitine, and teneligliptin on plasma and muscular indoxyl sulfate levels in chronic kidney disease mice
To clarify whether AST‐120, L‐carnitine, and teneligliptin influence the accumulation of IS, the IS levels in plasma and skeletal muscle (gastrocnemius) of CKD mice were measured. As shown in Figure 3A, the plasma concentration of IS in the CKD mice was eight‐fold higher than that of sham mice (4.7 ± 0.4 μM for sham mice; 39.2 ± 7.7 μM for CKD mice). As expected, the elevated IS level in plasma was decreased to the levels of the sham mice by the administration of AST‐120 (6.4 ± 0.6 μM for CKD + AST mice). In contrast, L‐carnitine or teneligliptin had no effect on plasma IS levels in CKD mice. We also measured IS levels in skeletal muscle (gastrocnemius) (Figure 3B). The IS level in skeletal muscle was increased by three‐fold in the case of the CKD mice compared with the sham mice. Similar to the changes in the plasma IS level, IS levels in skeletal muscle were significantly reduced by AST‐120, while the administration of L‐carnitine or teneligliptin had no effect on the IS levels in muscles of CKD mice (13.4 ± 1.2 pmol/mg protein for sham mice; 38.3 ± 6.0 pmol/mg protein for CKD mice; 13.9 ± 1.5 pmol/mg protein for CKD + AST‐120; 35.9 ± 5.5 pmol/mg protein for CKD + Car; 39.6 ± 8.5 pmol/mg protein for CKD + Tnl) (Figure 3B).
Figure 3.
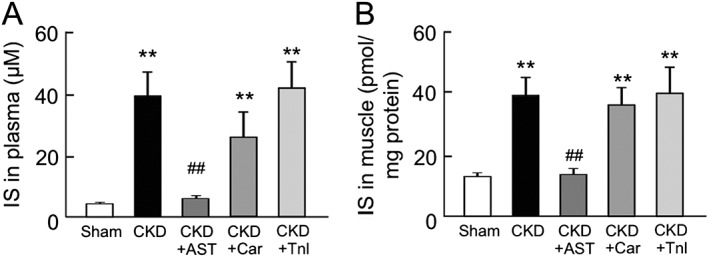
Plasma and skeletal muscle concentration of indoxyl sulfate in chronic kidney disease and AST‐120 (AST), L‐carnitine (Car), or teneligliptin (Tnl)‐treated chronic kidney disease mice. The concentration of indoxyl sulfate in (A) plasma and (B) gastrocnemius was determined by high‐performance liquid chromatography. Data are expressed the mean ± standard error of the mean (n = 5–9). ** p < 0.01 compared with sham. ## p < 0.01 compared with chronic kidney disease.
The effect of AST‐120, L‐carnitine, and teneligliptin on the expression of inflammatory cytokines in chronic kidney disease mice
We previously reported that the expression of inflammatory cytokine genes is increased in skeletal muscles of IS‐loaded half‐nephromized mice.7 Here, we evaluated the effect of AST‐120, L‐carnitine, and teneligliptin on the expression of inflammatory cytokines in CKD mice. As shown in Figure 4A, the IL‐6 levels in skeletal muscle (gastrocnemius) were significantly increased in the CKD mice. This increase tended to be suppressed by the AST‐120 treatment (Figure 4A). The same tendency was also observed in the case of TNF‐α levels (Figure 4B). On the other hand, the administration of L‐carnitine and teneligliptin had no effect on IL‐6 and TNF‐α levels in skeletal muscle.
Figure 4.
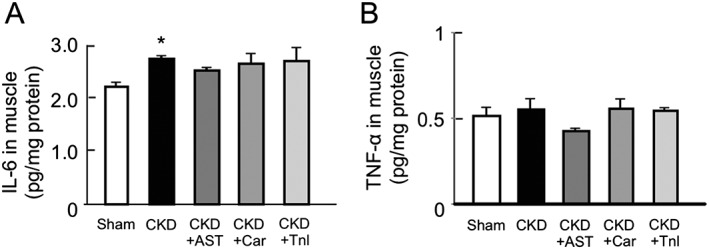
Effect of AST‐120 (AST), L‐carnitine (Car), or teneligliptin (Tnl) on protein expression of interleukin‐6 and tumor necrosis factor‐α in skeletal muscle of chronic kidney disease mice. (A) Interleukin‐6 and (B) tumor necrosis factor‐α expression in gastrocnemius were determined by enzyme‐linked immunosorbent assay. Data are expressed the means ± standard error of the mean (n = 5–9). * p < 0.05 compared with sham.
The effect of AST‐120, L‐carnitine, and teneligliptin on the expression of myostatin, atrogin‐1, and Akt phosphorylation in chronic kidney disease mice
We next evaluated the expression of muscle atrophy‐related genes such as myostatin and atrogin‐1 in the gastrocnemius of CKD mice. Both the expression of mRNA and the corresponding protein, myostatin, were increased in the CKD mice (Figure 5A and B). Similarly, the mRNA expression of atrogin‐1 was also increased in the CKD mice (Figure 5C). Such inductions of myostatin and atrogin‐1 expression in CKD mice were significantly inhibited by the administration of AST‐120, L‐carnitine, and teneligliptin (Figure 5A–C). On the other hand, the Akt phosphorylation level in the gastrocnemius was significantly decreased in CKD mice but was recovered by the administration of AST‐120, L‐carnitine, and teneligliptin (Figure 5D).
Figure 5.
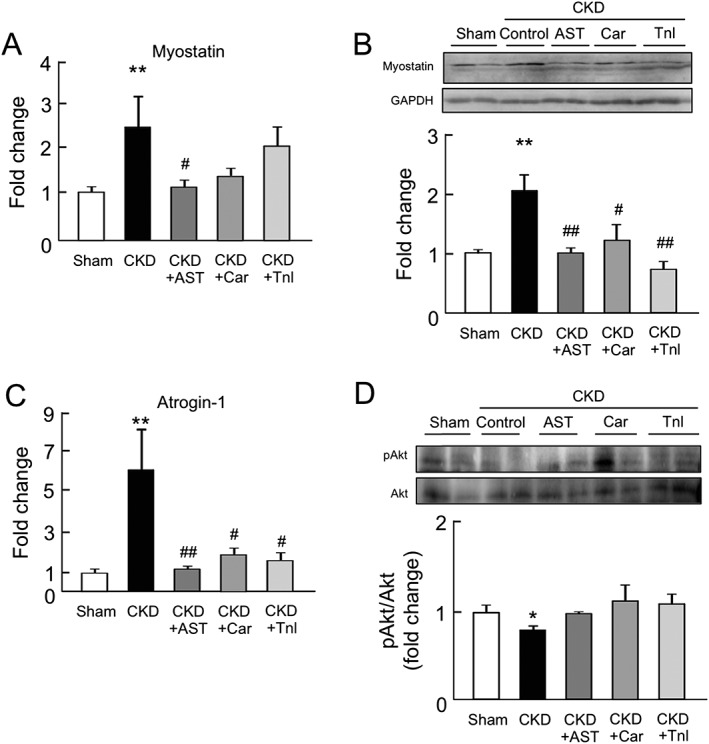
Effect of AST‐120 (AST), L‐carnitine (Car), or teneligliptin (Tnl) on myostatin, atrogin‐1 expression, and Akt phosphorylation in skeletal muscle of chronic kidney disease mice. (A) mRNA and (B) protein expressions of myostatin in gastrocnemius were determined by real‐time reverse transcription polymerase chain reaction and western blots. mRNA expression of (C) atrogin‐1 expression in the gastrocnemius was determined by real‐time reverse transcription polymerase chain reaction. (D) Akt phosphorylation in the gastrocnemius was determined by western blots. Relative intensity of pAkt/Akt was quantified using the ImageJ software. Data are expressed as the mean ± standard error of the mean (n = 4–9). * p < 0.05, ** p < 0.01 compared with sham. # p < 0.05, ## p < 0.01 compared with chronic kidney disease.
The effect of AST‐120, L‐carnitine, and teneligliptin on exercise capacity in chronic kidney disease mice
Because the capacity of CKD mice for exercise is known to be decreased in the early stages of CKD, we evaluated the effect of AST‐120, L‐carnitine, and teneligliptin on the exercise capacity (running distance) of CKD mice. As expected, the exercise capacity in CKD mice was lower than that of the sham mice. Interestingly, the administration of AST‐120, L‐carnitine, and teneligliptin significantly suppressed this impaired exercise capacity, i.e. exercise capacity was increased (Figure 6A).
Figure 6.
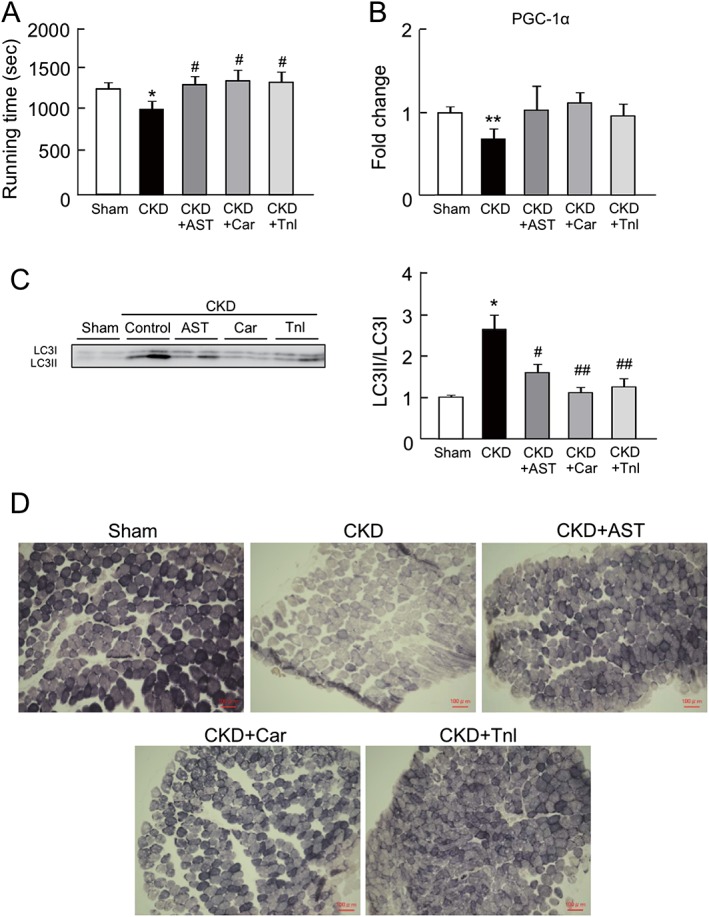
Effect of AST‐120 (AST), L‐carnitine (Car), or teneligliptin (Tnl) on mice exercise capacity, peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha expression, autophagy, and skeletal muscle fibre type in chronic kidney disease mice. (A) Mice exercise capacity was determined treadmill experiment. (B) mRNA expression of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha in gastrocnemius was determined by real‐time reverse transcription polymerase chain reaction. (C) Protein expression of LC3II/LC3I in gastrocnemius was determined by western blot. (D) Succinate dehydrogenase staining of soleus muscle was determined by succinate dehydrogenase staining. Data are expressed the mean ± standard error of the mean (n = 4–9). * p < 0.05, ** p < 0.01 compared with control. # p < 0.05, ## p < 0.01 compared with chronic kidney disease.
Previous reports and our in vitro experiments (Figure 1) demonstrated that there is mutual relationship between exercise capacity in treadmill experiments and muscular mitochondrial function.6, 9, 10, 19 We therefore further evaluated the mitochondrial status in skeletal muscle of CKD mice. The mRNA expression of PGC‐1α in the gastrocnemius was significantly decreased in CKD mice (Figure 6B), while the administration of AST‐120, L‐carnitine, and teneligliptin restored the change of PGC‐1α expression (Figure 6B). We also evaluated whether autophagy was induced in the gastrocnemius of the CKD mice. As shown in Figure 6C, the LC3II/LC3I ratio was significantly increased in the CKD mice, suggesting that autophagy was induced. The changes in the LC3II/LC3I ratio were inhibited by the administration of AST‐120, L‐carnitine, and teneligliptin (Figure 6C).
Muscle fibre distribution is altered by certain disease conditions due to the induction of fibre transition from type I fibres to type II fibres.20, 21, 22 The resulting reduced type I fibre content is associated with mitochondrial dysfunction, because type I fibres are mitochondria‐rich, slow twitch fibres. Therefore, we further investigated the muscle fibre distribution by SDH staining. This method utilizes the activity of SDH, a mitochondrial respiratory chain enzyme, to determine the myofibre type.18, 21 As shown in Figure 6D, the number of type I fibres was decreased in CKD mice, whereas these alterations were inhibited by the administration of AST‐120, L‐carnitine, and teneligliptin.
Discussion
Impaired physical performance is not only correlated with a renal prognosis and mortality but also constitutes an independent cardiovascular risk in patients with CKD.2, 4 However, evidence regarding a therapeutic strategy for treating CKD‐induced muscle atrophy is limited due to a lack of information concerning the molecular mechanism responsible. It is currently thought that the enhancement in oxidative stress and inflammatory cytokines is closely associated with CKD‐induced muscle atrophy.1, 23, 24 Interestingly, feeding a high protein diet not only reduces exercise endurance despite an increased muscle mass and muscle power in CKD mice6 but also exacerbates impaired renal function that is accompanied by an increased production of uremic toxins.25 This body of experimental evidence led us to hypothesize that uremic toxins play an important role in the mutual relationship between the aggravation of renal function, exercise capacity, and a high protein diet. In fact, our previous study, showing that among uremic toxins, IS strongly accelerates skeletal muscle atrophy via the induction of oxidative stress‐mediated inflammation using IS‐loaded half‐nephromized mice and a C2C12 cell system confirmed this assumption.7 The present study further demonstrated that IS also accelerates mitochondrial dysfunction via the induction of oxidative stress. Based on these findings, we explored the therapeutic impact of inhibiting the accumulation of IS and mitochondrial dysfunction on CKD‐induced muscle atrophy and decreased muscle endurance, and the findings showed that AST‐120, L‐carnitine, and teneligliptin function to potentiate the prevention of CKD‐induced physical inactivity mainly via maintaining mitochondrial function, suppressing atrogin‐1/myostatin expression, and recovering Akt phosphorylation in skeletal muscle.
It has been reported that a reduction in exercise capacity was observed even in an early stage of CKD.2, 6 Recent studies further indicated that such an impaired exercise capacity was induced prior to the development of muscle atrophy, and this was mainly caused by mitochondrial dysfunction in skeletal muscle.6 Similar results were also reported for early stage of CKD mice (16–20 weeks aged mice, 9–13 weeks after 5/6 Nx operation), and their exercise capacities were decreased accompanied by a reduction in mitochondrial function without muscle atrophy. On the other hand, the late stage of CKD mice (48 weeks aged mice, 41 weeks after 5/6 Nx operation) showed a marked decrease in both body weight and skeletal muscle weight with the impaired exercise capacity and mitochondrial dysfunction. In this study, we used an intermediate stage of CKD mice (34 weeks aged mice, 28 weeks after 5/6 Nx operation), and their characteristics as compared with sham mice were as follows1: The body weight and weights of the soleus or gastrocnemius were significantly decreased in CKD mice,2 plasma and muscular IS levels were significantly increased in CKD mice,3 IL‐6 and atrophy‐related factors (myostatin and atrogin‐1) in skeletal muscle were increased whereas muscular Akt phosphorylation was decreased in CKD mice,4 and exercise capacity was decreased in CKD mice and was accompanied by a reduction in muscular PCG‐1α expression and increased muscular autophagy as reflected by a decrease in type I fibres (mitochondria rich fibres). These features are well consistent with IS‐loaded half‐nephromized mice as shown previously.7 The administration of AST‐120 to the CKD mice significantly decreased the plasma and muscular IS levels and, hence, restored exercise capacity, muscle weight, and the number of type I slow twitch fibres and suppressed mitochondrial dysfunction. These findings further support the conclusion that IS is one of the humoral factors responsible for muscle atrophy in CKD.
Increased oxidative stress and inflammatory cytokines decrease mitochondria function.6 The present study revealed that IS decreases the expression of PGC‐1α (mitochondrial biosynthesis‐related factor) and increases autophagy (mitochondrial degradation‐related factor) in skeletal muscle in both in vivo and in vitro experiments. Interestingly, IS reduced membrane potential in C2C12 cells, and these events were markedly ameliorated by the treatment of an anti‐oxidant, ascorbic acid. Therefore, IS‐induced intracellular ROS production contributes to the induction of mitochondria dysfunction observed in CKD conditions. We previously proposed a mechanism for the IS‐induced production of ROS in skeletal muscle cells in which IS, which accumulates intracellularly via organic anion transporters, results in increased ROS production by the activation of NADPH oxidase and the aryl hydrocarbon receptor. Such enhanced ROS production might be involved in the IS‐induced mitochondria dysfunction reported herein. Further experiments will clearly be needed to clarify the molecular mechanism responsible for IS‐induced mitochondria dysfunction.
In CKD patients, a restriction of protein intake, a decrease in L‐carnitine biosynthesis, and the easy removal of L‐carnitine by dialysis cause a L‐carnitine deficiency. Such a L‐carnitine deficiency results in a decline in muscle power, fatigue, non‐ketotic hypoglycemia, or myocardial myopathy, while L‐carnitine supplementation is effective for myopathy or for a decrease in muscle mass and power in elderly people.26, 27 The findings reported herein indicate that an L‐carnitine treatment ameliorates the muscle atrophy and exercise capacity in CKD mice without affecting their renal function or the IS levels in both plasma and muscle. This could be due to the inhibition of mitochondrial dysfunction and decreased numbers of type I slow twitch fibres. Several reports have indicated that the L‐carnitine can exert a pleiotropic effect. For example, the administration of L‐carnitine to exercise‐loaded mice increased their exercise endurance via the up‐regulation of mitochondrial biosynthesis factors such as PGC‐1α.28 L‐carnitine also decreased the levels of markers for oxidative stress, such as 8‐OHdG and 4‐hydroxynonenal and TNF‐α in livers of non‐alcoholic steatohepatitis model mice,29 suggesting that L‐carnitine exhibits anti‐oxidative and anti‐inflammatory actions. In mice that are fed a high‐fructose diet which is known as an acquired model of insulin resistance, the presence of L‐carnitine reduced protein oxidation in the lens, and the decreased anti‐oxidative enzymes were recovered, thus preserving mitochondrial function.30 Moreover, the presence of L‐carnitine increases Akt phosphorylation and accelerates myotube formation in C2C12 cells31 and in the muscle of rats.32 These pleiotropic effects of L‐carnitine, including anti‐oxidative and anti‐inflammatory activities and maintaining Akt signaling, could play an important role in the protection of mitochondrial function in CKD conditions, especially in cases of higher IS accumulation, such as uremia. However, recently, Koeth et al. reported that metabolism by intestinal microbiota of dietary L‐carnitine produces trimethylamine‐N‐oxide (TMAO) and accelerates atherosclerosis in mice.33 In addition, they also showed that higher TMAO level predicted increase risks for prevalent cardiovascular disease and incident adverse cardiac events. On the other hand, other reports demonstrated that oral L‐carnitine supplementation increases TMAO but reduces markers of vascular injury in hemodialysis patients34 or L‐carnitine intake and high TMAO plasma levels correlate with low aortic lesions.35 Therefore, the link between gut microbiota metabolism of L‐carnitine and cardiovascular disease risk has generated much debate over the utility of supplemental intake of L‐carnitine.
We also found, for the first time, that teneligliptin has a therapeutic potential against CKD‐induced muscular dysfunction without causing changes in IS accumulation. A DPP‐4 inhibitor is used for treating patients with type 2 diabetes. The main mechanism for its anti‐diabetic effect is to inhibit DPP‐4, a degradative enzyme of incretin hormones such as GLP‐1 and glucose‐dependent insulinotropic polypeptide.36 Incretin has been known to exert pleiotropic effects on the central nervous system, the liver, cardiovascular, lung, kidney, or skeletal muscle. Kang et al. recently reported that GLP‐1 increased the mitochondrial membrane potential and oxygen consumption in addition to increasing of PGC‐1α expression.14 Thus, DPP‐4 inhibitors also exert pleiotropic effects via their ability to enhance the action of GLP‐1.37 In fact, Fukuda‐Tsuru et al. reported that a teneligliptin treatment suppressed mitochondrial dysfunction and lipid accumulation in livers in mice that had been fed a high‐fat diet.38 It is well known that GLP‐1 ameliorates insulin resistance via the activation of the PI3K‐Akt signal pathway in skeletal muscle.39 In addition, Kimura et al. reported that teneligliptin acts as a hydroxyl radical scavenger.40 Wang et al., using human proximal tubular cells, also reported that diportin, another DPP‐4 inhibitor, inhibited cell injury via the inhibition of IS‐induced NF‐κB signaling, ROS/p38MAPK/ERK, and recovering the PI3K‐Akt signaling pathway without involving the action of GLP‐1. This suggests that DPP‐4 inhibitors could have a direct cytoprotective function as well.41 Taking these findings into consideration, it would appear that teneligliptin may exert cytoprotective activities not only via GLP‐1 indirectly but also its direct action against CKD‐induced muscle atrophy.
In clinical settings, AST‐120 is used to suppress the progression of renal failure in CKD patients via inhibiting the accumulation of uremic toxins. On the other hand, L‐carnitine injections are occasionally administered to CKD patients, especially those who are receiving dialysis therapy because of the high incident of complications with L‐carnitine deficiency. In the case of teneligliptin, it can be used safely in diabetes patients with decreased renal function because it is excreted via both liver and kidney. Accordingly, AST‐120, L‐carnitine, and teneligliptin can be used to treat CKD patients with muscle atrophy and impaired muscle exercise endurance. Although AST‐120, L‐carnitine, and teneligliptin exhibit anti‐muscle atrophy actions, their mechanisms are different from each other. Thus, it may be expected that a combination of AST‐120 and L‐carnitine or AST‐120 and teneligliptin therapy or a combination of all may exert an additive or synergic effect against CKD‐induced muscle dysfunction.
In this in vitro study, C2C12 cells were incubated with 1 mM IS for 3 h in the absence of albumin, which is the highest individual total plasma IS concentration ever reported.42 It is important to consider the concentration of free uremic toxins on their biological activity, which means that the experiments need to be conducted using the correct albumin concentrations or with the correct free concentration. Vanholder et al. previously pointed out that the use of unrealistically high free concentrations of uremic toxins compared with the concentrations observed in human CKD and/or inappropriately low albumin concentrations may blur the interpretation of the biochemical effects of uremic toxins.43 On this point, the IS concentration used in this cell culture study may have been unusually high and the free concentration irrelevant. On the other hand, in clinical situations, uremic patients are routinely exposed to pathological IS concentrations for long periods of time (for many years). In the cell culture study, however, the cells were exposed to a high IS concentration only for several hours. High concentrations in in vitro experiments with s short exposure (~several hours) have been used in a number of previous studies, where the short exposure might compensate for the high concentration, in contrast to actual exposure periods, where uremic toxins would have more time to reach the intracellular compartments and to exert their biological activity.
Our experiments using mice are not necessarily equivalent to the human condition, and other presumed or unexpected actions of IS, AST‐120, L‐carnitine, and teneligliptin may contribute to the results on this study. Recently, Sato et al. also demonstrated that IS potentiated mitochondrial dysfunction as well as our study. In addition, a significant inverse association between plasma IS levels and skeletal muscle mass in patients with CKD was observed.44 The data suggest that IS is a pathogenic factor for muscle atrophy in the patients with CKD. However, the clinical data reported by Sato et al. were a correlation study and, as a result, would not have the same relevance as would be obtained in a large randomized study. In addition, correlating the concentration of IS with muscular atrophy cannot be considered to be proof of causation. Here, IS can be considered only as a marker of kidney failure, and there is a plethora of other retained compounds that could have played a role in this process, either directly or indirectly. Further clinical randomized studies will be needed to clarify whether IS is solely a marker of renal function or a pathogenic factor related to muscle atrophy and to confirm the anti‐muscle atrophy action of the above interventions in CKD patients in the future.
Conclusions
Together with our previous report,7 we demonstrated the existence of a mutual relationship between IS and skeletal muscle atrophy/exercise endurance in CKD conditions. Additionally, IS was found to induce mitochondrial dysfunction via increasing oxidative stress. Based on these molecular mechanisms, we provide two therapeutic strategies that are IS‐targeted (AST‐120) and mitochondria‐targeted (L‐carnitine and teneligliptin) interventions for CKD‐induced muscle atrophy and decreased exercise endurance. Thus, this study provides new beneficial evidence for IS‐ and mitochondria‐targeted intervention against CKD‐induced muscle atrophy and decreased exercise endurance.
Conflict of interest
H.W. received a grant from Mitsubishi Tanabe Pharma. All the other authors declare no competing financial interests.
Supporting information
Supplemental table 1. Primers used in real‐time RT‐PCR
Supplemental table 2. Protocol for the treadmill experiment
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia, and Muscle: update 2015.45
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (KAKENHI 16H05114) and the Nakatomi Foundation, Japan.
Enoki, Y. , Watanabe, H. , Arake, R. , Fujimura, R. , Ishiodori, K. , Imafuku, T. , Nishida, K. , Sugimoto, R. , Nagao, S. , Miyamura, S. , Ishima, Y. , Tanaka, M. , Matsushita, K. , Komaba, H. , Fukagawa, M. , Otagiri, M. , and Maruyama, T. (2017) Potential therapeutic interventions for chronic kidney disease‐associated sarcopenia via indoxyl sulfate‐induced mitochondrial dysfunction. Journal of Cachexia, Sarcopenia and Muscle, 8: 735–747. doi: 10.1002/jcsm.12202.
References
- 1. Wang XH, Mitch WE. Mechanisms of muscle wasting in chronic kidney disease. Nat Rev Nephrol 2014;10:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fried LF, Lee JS, Shlipak M, Chertow GM, Green C, Ding J, et al. Chronic kidney disease and functional limitation in older people: health, aging and body composition study. J Am Geriatr Soc 2006;54:750–756. [DOI] [PubMed] [Google Scholar]
- 3. Carrero JJ, Chmielewski M, Axelsson J, Snaedal S, Heimbürger O, Bárány P, et al. Muscle atrophy, inflammation and clinical outcome in incident and prevalent dialysis patients. Clin Nutr 2008;27:557–564. [DOI] [PubMed] [Google Scholar]
- 4. Beddhu S, Wei G, Marcus RL, Chonchol M, Greene T. Light‐intensity physical activities and mortality in the United States general population and CKD subpopulation. Clin J Am Soc Nephrol 2015;10:1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 6. Tamaki M, Miyashita K, Wakino S, Mitsuishi M, Hayashi K, Itoh H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int 2014;85:1330–1339. [DOI] [PubMed] [Google Scholar]
- 7. Enoki Y, Watanabe H, Arake R, Sugimoto R, Imafuku T, Tominaga Y, et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress‐mediated expression of myostatin and atrogin‐1. Sci Rep 2016;6:32084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishikawa M, Ishimori N, Takada S, Saito A, Kadoguchi T, Furihata T, et al. AST‐120 ameliorates lowered exercise capacity and mitochondrial biogenesis in the skeletal muscle from mice with chronic kidney disease via reducing oxidative stress. Nephrol Dial Transplant 2015;30:934–942. [DOI] [PubMed] [Google Scholar]
- 9. Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, et al. The transcriptional coactivator PGC‐1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab 2007;5:35–46. [DOI] [PubMed] [Google Scholar]
- 10. Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, et al. Rev‐erb‐α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med 2013;19:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator‐activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem 2010;285:19460–19471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC‐1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 2009;106:20405–20410. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med 1994;124:96–104. [PubMed] [Google Scholar]
- 14. Kang MY, Oh TJ, Cho YM. Glucagon‐like peptide‐1 increases mitochondrial biogenesis and function in INS‐1 rat insulinoma cells. Endocrinol Metab (Seoul) 2015;30:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe H, Miyamoto Y, Honda D, Tanaka H, Wu Q, Endo M, et al. p‐Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int 2013;83:582–592. [DOI] [PubMed] [Google Scholar]
- 16. Watanabe H, Noguchi T, Miyamoto Y, Kadowaki D, Kotani S, Nakajima M, et al. Interaction between two sulfate‐conjugated uremic toxins, p‐cresyl sulfate and indoxyl sulfate, during binding with human serum albumin. Drug Metab Dispos 2012;40:1423–1428. [DOI] [PubMed] [Google Scholar]
- 17. Nakatani T, Nakashima T, Kita T, Hirofuji C, Itoh K, Itoh M, et al. Succinate dehydrogenase activities of fibers in the rat extensor digitorum longus, soleus, and cardiac muscles. Arch Histol Cytol 1999;62:393–399. [DOI] [PubMed] [Google Scholar]
- 18. Lee CM, Lopez ME, Weindruch R, Aiken JM. Association of age‐related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic Biol Med 1998;25:964–972. [DOI] [PubMed] [Google Scholar]
- 19. Arany Z. PGC‐1 coactivators and skeletal muscle adaptations in health and disease. Curr Opin Genet Dev 2008;18:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Couturier A, Ringseis R, Mooren FC, Krüger K, Most E, Eder K. Carnitine supplementation to obese Zucker rats prevents obesity‐induced type II to type I muscle fiber transition and favors an oxidative phenotype of skeletal muscle. Nutr Metab (Lond) 2013;10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujita N, Nagatomo F, Murakami S, Kondo H, Ishihara A, Fujino H. Effects of hyperbaric oxygen on metabolic capacity of the skeletal muscle in type 2 diabetic rats with obesity. ScientificWorldJournal 2012;2012:637978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagatomo F, Fujino H, Kondo H, Gu N, Takeda I, Ishioka N, et al. PGC‐1α mRNA level and oxidative capacity of the plantaris muscle in rats with metabolic syndrome, hypertension, and type 2 diabetes. Acta Histochem Cytochem 2011;44:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 2011;25:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sriram S, Subramanian S, Sathiakumar D, Venkatesh R, Salerno MS, McFarlane CD, et al. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF‐κB. Aging Cell 2011;10:931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poesen R, Mutsaers HA, Windey K, van den Broek PH, Verweij V, Augustijns P, et al. The influence of dietary protein intake on mammalian tryptophan and phenolic metabolites. PLoS One 2015;10: e0140820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levitan MD, Murphy JT, Sherwood WG, Deck J, Sawa GM. Adult onset systemic carnitine deficiency: favorable response to L‐carnitine supplementation. Can J Neurol Sci 1987;14:50–54. [DOI] [PubMed] [Google Scholar]
- 27. Pistone G, Marino A, Leotta C, Dell'Arte S, Finocchiaro G, Malaguarnera M. Levocarnitine administration in elderly subjects with rapid muscle fatigue: effect on body composition, lipid profile and fatigue. Drugs Aging 2003;20:761–767. [DOI] [PubMed] [Google Scholar]
- 28. Kim JH, Pan JH, Lee ES, Kim YJ. L‐Carnitine enhances exercise endurance capacity by promoting muscle oxidative metabolism in mice. Biochem Biophys Res Commun 2015;464:568–573. [DOI] [PubMed] [Google Scholar]
- 29. Ishikawa H, Takaki A, Tsuzaki R, Yasunaka T, Koike K, Shimomura Y, et al. L‐carnitine prevents progression of non‐alcoholic steatohepatitis in a mouse model with upregulation of mitochondrial pathway. PLoS One 2014;9: e100627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Balasaraswathi K, Rajasekar P, Anuradha CV. Changes in redox ratio and protein glycation in precataractous lens from fructose‐fed rats: effects of exogenous L‐carnitine. Clin Exp Pharmacol Physiol 2008;35:168–173. [DOI] [PubMed] [Google Scholar]
- 31. Montesano A, Senesi P, Luzi L, Benedini S, Terruzzi I. Potential therapeutic role of L‐carnitine in skeletal muscle oxidative stress and atrophy conditions. Oxid Med Cell Longev 2015;2015:646171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Keller J, Couturier A, Haferkamp M, Most E, Eder K. Supplementation of carnitine leads to an activation of the IGF‐1/PI3K/Akt signalling pathway and down regulates the E3 ligase MuRF1 in skeletal muscle of rats. Nutr Metab (Lond) 2013;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fukami K, Yamagishi S, Sakai K, Kaida Y, Yokoro M, Ueda S, et al. Oral L‐carnitine supplementation increases trimethylamine‐N‐oxide but reduces markers of vascular injury in hemodialysis patients. J Cardiovasc Pharmacol 2015;65:289–295. [DOI] [PubMed] [Google Scholar]
- 35. Collins HL, Drazul‐Schrader D, Sulpizio AC, Koster PD, Williamson Y, Adelman SJ, et al. L‐Carnitine intake and high trimethylamine N‐oxide plasma levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP. Atherosclerosis 2016;244:29–37. [DOI] [PubMed] [Google Scholar]
- 36. Hansotia T, Maida A, Flock G, Yamada Y, Tsukiyama K, Seino Y, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 2007;117:143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007;132:2131–2157. [DOI] [PubMed] [Google Scholar]
- 38. Fukuda‐Tsuru S, Kakimoto T, Utsumi H, Kiuchi S, Ishii S. The novel dipeptidyl peptidase‐4 inhibitor teneligliptin prevents high‐fat diet‐induced obesity accompanied with increased energy expenditure in mice. Eur J Pharmacol 2014;723:207–215. [DOI] [PubMed] [Google Scholar]
- 39. Rupprecht LE, Mietlicki‐Baase EG, Zimmer DJ, McGrath LE, Olivos DR, Hayes MR. Hindbrain GLP‐1 receptor‐mediated suppression of food intake requires a PI3K‐dependent decrease in phosphorylation of membrane‐bound Akt. Am J Physiol Endocrinol Metab 2013;305:E751–E759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimura S, Inoguchi T, Yamasaki T, Yamato M, Ide M, Sonoda N, et al. A novel DPP‐4 inhibitor teneligliptin scavenges hydroxyl radicals: in vitro study evaluated by electron spin resonance spectroscopy and in vivo study using DPP‐4 deficient rats. Metabolism 2016;65:138–145. [DOI] [PubMed] [Google Scholar]
- 41. Wang WJ, Chang CH, Sun MF, Hsu SF, Weng CS. DPP‐4 inhibitor attenuates toxic effects of indoxyl sulfate on kidney tubular cells. PLoS One 2014;9: e93447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vanholder R, De Smet R, Glorieux G, Argilés A, Baurmeister U, Brunet P, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int 2003;63:1934–1943. [DOI] [PubMed] [Google Scholar]
- 43. Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p‐cresyl sulfate: a systematic review. J Am Soc Nephrol 2014;25:1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sato E, Mori T, Mishima E, Suzuki A, Sugawara S, Kurasawa N, et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci Rep 2016;6:36618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental table 1. Primers used in real‐time RT‐PCR
Supplemental table 2. Protocol for the treadmill experiment