

Key Points
Results based on our allogeneic model suggest genetic strategies aimed at correcting at least 20% of HSCs are necessary to reverse SCD.
A minority of donor myeloid cells is adequate because of the vast differences in RBC survival between donor and recipient.
Abstract
Novel curative therapies using genetic transfer of normal globin-producing genes into autologous hematopoietic stem cells (HSCs) are in clinical trials for patients with sickle cell disease (SCD). The percentage of transferred globin necessary to cure SCD is currently not known. In the setting of allogeneic nonmyeloablative HSC transplants (HSCTs), stable mixed chimerism is sufficient to reverse the disease. We regularly monitored 67 patients after HSCT. After initially robust engraftment, 3 of these patients experienced declining donor myeloid chimerism (DMC) levels with eventual return of disease. From this we discovered that 20% DMC is necessary to reverse the sickle phenotype. We subsequently developed a mathematical model to test the hypothesis that the percentage of DMC necessary is determined solely by differences between donor and recipient red blood cell (RBC) survival times. In our model, the required 20% DMC can be entirely explained by the large differences between donor and recipient RBC survival times. Our model predicts that the requisite DMC and therefore necessary level of transferred globin is lowest in patients with the highest reticulocyte counts and concomitantly shortened RBC lifespans.
Introduction
Hematopoietic stem cell (HSC) transplant (HSCT) is the only curative option for patients with sickle cell disease (SCD). HLA-matched sibling HSCT has become increasingly successful, and haploidentical approaches are being explored to increase the donor pool. We and others have shown that stable mixed chimerism is sufficient to reverse the sickle phenotype without graft-versus-host disease.1-5 Kean and colleagues showed in a murine SCD model that relatively low levels of donor white blood cell (WBC) chimerism (3%-18%) were associated with higher red blood cell (RBC) chimerism (84%-95%).6 Furthermore, 100% donor peripheral-blood RBC chimerism was necessary to achieve hematopoietic cure. Another murine study showed that 26% WBC chimerism improved organ pathology and RBC lifespan.7
Advances have been made in gene therapy such that clinical trials are now under way for the first time in patients with SCD. The question of what percentage of donor chimerism is necessary to cure SCD is critical in identifying the target percentage of the transferred globin gene needed to reverse SCD. A murine study in which various ratios of bone marrow from SCD mice were transplanted into normal mice showed that donor myeloid chimerism (DMC) levels exceeding 25% were associated with >90% normal hemoglobin, but 70% DMC was necessary to correct the anemia.8 On the other hand, 1 human study suggested that a whole-blood WBC donor chimerism level as low as 11% was sufficient to render patients free of SCD3; we note, however, that lineage-specific chimerism was not performed. A more recent study showed that 5% whole-blood WBC donor chimerism was associated with severe anemia and a sickle hemoglobin (HbS) of 60.8% (donor, 39.6%),9 therefore not sufficient to reverse the sickle phenotype. We have previously demonstrated that peripheral-blood DMC linearly reflects the HSC compartment.10 The observation that 1 patient with 40% to 50% donor WBC chimerism had 97% to 100% donor RBC chimerism, whereas another with 25% to 30% donor WBC chimerism had only up to 69% RBC chimerism, suggests that the relationship between donor HSC chimerism and RBC chimerism is nonlinear.11 In this brief report, we characterize this relationship mathematically and determine the percentage of DMC necessary to reverse the sickle phenotype in patients who underwent HSCT.
Study design
All patients with SCD transplanted at the National Institutes of Health between 2003 and 2015 were included in the analysis (clinicaltrials.gov identifiers NCT00061568, NCT02105766, and NCT00977691). All protocols were approved by the National Heart, Lung, and Blood Institute Institutional Review Board, and all patients signed informed consent. Patients who underwent HLA-matched sibling HSCT received alemtuzumab, 300 cGy total-body irradiation, and sirolimus. Patients who received haploidentical HSCT were treated with alemtuzumab, 400 cGy total-body irradiation, and a dose escalation of cyclophosphamide (0-100 mg/kg on days 3 and/or 4 posttransplant).12
We subsequently developed a mathematical model to test the hypothesis that the percentage of necessary DMC to reverse SCD is determined solely by the differences between donor and recipient RBC half-lives. We model the dynamics of hematopoietic cells in 2 compartments, early myeloid progenitor and mature erythroid, separately for donor and recipient cells. The model assumes first-order kinetics for erythropoiesis and RBC senescence/destruction. The kinetic constants of donor and recipient cells in the immature compartments (before globin gene expression) are assumed to be identical, whereas the kinetic constants of the mature compartments are assumed to be different. Data from patients at specified milestone dates were used to characterize the kinetic constants via nonlinear regression. The mathematical details of the model as well as the specifics of the nonlinear regression are presented in the supplemental Methods (available on the Blood Web site). Our model predicts that the fraction of mature donor RBCs in the periphery (fM) is a function of progenitor chimerism (fP) and donor and recipient mean erythrocyte lifetimes (tD and tR, respectively):
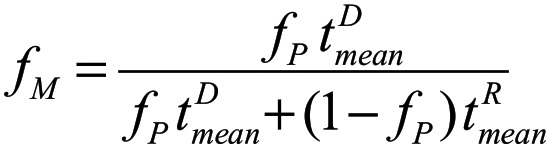 |
and that the fraction of HbS, fHbS is given by
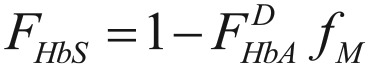 |
where 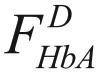 is the innate fraction of hemoglobin A in the donor.
is the innate fraction of hemoglobin A in the donor.
Results and discussion
In protocols testing both HLA-matched sibling and haploidentical nonmyeloablative HSCT, 67 SCD patients were transplanted followed by monitoring of myeloid (CD14/15) and lymphoid (CD3) chimerism, hemoglobin and HbS levels, and symptoms every 6 to 12 months. Three were identified who, despite robust initial engraftment, experienced declining DMC levels over prolonged follow-up, eventually associated with return of SCD. The patients’ ages were 24, 27, and 31 years; 2 were men (see supplemental Table 1). Indications for transplant were recurrent painful crises in 1, moyamoya syndrome in 1, and end-stage renal disease requiring hemodialysis, cardiomyopathy, and recurrent painful crises in 1 patient. One patient underwent HLA-matched sibling; 2 haploidentical HSCT patients enrolled in the second cohort of the study received 50 mg/kg cyclophosphamide on day 3 posttransplant. All 3 patients’ donors had sickle cell trait with HbS ranging from 36% to 40%. No patients received donor lymphocyte infusions or stem cell boosts during the period of follow-up, and the patients did not experience sepsis or viral reactivation peritransplant. The patients initially achieved nearly 100% DMC; however, their chimerism levels slowly decreased over time (Figure 1). As long as mean DMC was >20%, mean percentage of HbS remained below 50% (similar to their sickle cell trait donors) and the patients stayed free of SCD-related symptoms. However, mean DMC at 2, 3, 4, and 5 years posttransplant fell below 20%; they were 18%, 12%, 11%, and 10%, with corresponding mean percentage of HbS levels of 52.5%, 51.6%, 57.8%, and 64.2%. At these levels of HbS percentages, 2 patients had return of their recurrent painful crises, and the third patient with moyamoya syndrome developed severe anemia requiring reinitiation of chronic transfusion therapy.
Figure 1.

Percentage of DMC, measured HbS, and predicted HbS (Pred HbS) over time in 3 subjects with SCD who underwent allogeneic HSCT and experienced falling DMC.
Roberts and colleagues found, using a mathematical model in mice and humans, that a donor WBC chimerism as small as 13% was sufficient to cure thalassemia due to differences in donor and recipient RBC survival and ineffective erythropoiesis.13 Ineffective hematopoiesis is less significant in SCD; however, greatly shortened RBC lifespan occurs.14-17 A study recently reported, using a mathematical model utilizing differences in RBC survival between donors and recipients in a murine SCD model, that 16.2% non-S HSCs would lead to a mixture of 50% non-S RBCs in the periphery.18 They further predicted that in humans, 25% altered HSCs could result in about 65% nonsickling RBCs.
From our nonlinear regression to posttransplant data from our patients, we calculated an erythrocyte lifetime of 6.22 days (95% confidence interval, 1.22-29.21 days). Although we did not directly measure RBC lifetimes, we identified 4 patients with SCD in the literature in whom reticulocyte percentages ranged from 9.0% to 21.0% and corresponding RBC half-lives determined by radioactive sodium chromate were 1 to 11 days.19 We assumed that in erythropoietic homeostasis, RBC lifetime must be proportional to the inverse of the reticulocyte fraction; we confirmed this relationship using data from these patients with a good coefficient of determination (R2 = 0.91). Therefore, we reasoned that (at stable hemoglobin concentration) the reticulocyte fraction is a good proxy for RBC lifetime.
Figure 1 depicts a comparison between the measured and our model-predicted HbS fractions in our 3 patients transplanted using the kinetic parameters obtained via nonlinear regression. As observed in our 3 patients, our model predicts that DMC of at least 20% is necessary to reverse the sickle phenotype. Figure 2 shows that the fraction of peripheral-donor RBCs is very sensitive to RBC lifetimes: the shorter the RBC lifetime, the lower the percentage of DMC necessary to achieve an equivalent fraction of peripheral-donor RBCs. For example, if the recipient RBC lifetime is 6 days, 20% DMC would be necessary for expression of donor-type hemoglobin in our model. Whereas, if the recipient RBC lifetime is 1 day, 4% DMC would be sufficient to achieve donor-type hemoglobin (Figure 2).
Figure 2.

Fraction of HbS in 3 hypothetical homozygous sickle cell disease (HbSS) patients from a sickle cell trait donor for 3 different HbSS mean erythrocyte lifetimes.
We have shown for the first time how HbS levels and sickle phenotype are affected by percentage of DMC levels over time. Our model predicts that genetic strategies aimed at SCD must in general achieve correction levels of at least 20% to reverse the sickle phenotype. It should be noted, however, that the model might have limitations for broader application beyond nonmyeloablative allogeneic HSCT for SCD. Next, the mechanism explaining why a minority of donor myeloid cells is adequate is due solely to the vast differences in RBC survival between donor and recipient cells. Lastly, recipient RBC survival is lowest in patients with the highest reticulocyte counts, suggesting that gene therapy may be most efficacious in subjects with high reticulocyte percentages as our model predicts that lower levels of correction would be sufficient in this setting.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors acknowledge the patients and their families, Roger Kurlander and Jodie Keary for chimerism analysis, and the Department of Transfusion Medicine and clinical staff.
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.D.F., M.M.H., S.C., and J.F.T. designed the study; C.D.F., M.M.H., and S.C. performed data analysis; C.D.F., M.M.H., T.T., W.C., K.R., M.L., and J.F.T. provided protocol support; C.D.F., M.M.H., S.C., and J.F.T. wrote the manuscript; and C.D.F., M.M.H., S.C., T.T., W.C., K.R., M.L., and J.F.T. reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John F. Tisdale, Molecular and Clinical Hematology Branch, National Institute of Diabetes and Digestive and Kidney Diseases/National Heart, Lung, and Blood Institute, National Institutes of Health, 9000 Rockville Pike, Building 10, Room 9N-112, Bethesda, MD 20892; e-mail: johntis@nhlbi.nih.gov.
References
- 1.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreani M, Testi M, Gaziev J, et al. Quantitatively different red cell/nucleated cell chimerism in patients with long-term, persistent hematopoietic mixed chimerism after bone marrow transplantation for thalassemia major or sickle cell disease. Haematologica. 2011;96(1):128-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walters MC, Patience M, Leisenring W, et al. ; Multicenter Investigation of Bone Marrow Transplantation for Sickle Cell Disease. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7(12):665-673. [DOI] [PubMed] [Google Scholar]
- 4.Saraf SL, Oh AL, Patel PR, et al. Nonmyeloablative stem cell transplantation with alemtuzumab/low-dose irradiation to cure and improve the quality of life of adults with sickle cell disease. Biol Blood Marrow Transplant. 2016;22(3):441-448. [DOI] [PubMed] [Google Scholar]
- 5.King AA, Kamani N, Bunin N, et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am J Hematol. 2015;90(12):1093-1098. [DOI] [PubMed] [Google Scholar]
- 6.Kean LS, Manci EA, Perry J, et al. Chimerism and cure: hematologic and pathologic correction of murine sickle cell disease. Blood. 2003;102(13):4582-4593. [DOI] [PubMed] [Google Scholar]
- 7.Felfly H, Trudel M. Successful correction of murine sickle cell disease with reduced stem cell requirements reinforced by fractionated marrow infusions. Br J Haematol. 2010;148(4):646-658. [DOI] [PubMed] [Google Scholar]
- 8.Iannone R, Luznik L, Engstrom LW, et al. Effects of mixed hematopoietic chimerism in a mouse model of bone marrow transplantation for sickle cell anemia. Blood. 2001;97(12):3960-3965. [DOI] [PubMed] [Google Scholar]
- 9.Bolaños-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285-4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh MM, Wu CJ, Tisdale JF. In mixed hematopoietic chimerism, the donor red cells win. Haematologica. 2011;96(1):13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu CJ, Hochberg EP, Rogers SA, et al. Molecular assessment of erythroid lineage chimerism following nonmyeloablative allogeneic stem cell transplantation. Exp Hematol. 2003;31(10):924-933. [DOI] [PubMed] [Google Scholar]
- 12.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical peripheral blood stem cell transplantation. Blood Adv. 2017;1(11):652-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts C, Kean L, Archer D, Balkan C, Hsu LL. Murine and math models for the level of stable mixed chimerism to cure beta-thalassemia by nonmyeloablative bone marrow transplantation. Ann N Y Acad Sci. 2005;1054:423-428. [DOI] [PubMed] [Google Scholar]
- 14.Eadie GS, Brown IW Jr. Red blood cell survival studies. Blood. 1953;8(12):1110-1136. [PubMed] [Google Scholar]
- 15.Korell J, Coulter CV, Duffull SB. A statistical model for red blood cell survival. J Theor Biol. 2011;268(1):39-49. [DOI] [PubMed] [Google Scholar]
- 16.Shemin D, Rittenberg D. The life span of the human red blood cell. J Biol Chem. 1946;166(2):627-636. [PubMed] [Google Scholar]
- 17.Lindsell CJ, Franco RS, Smith EP, Joiner CH, Cohen RM. A method for the continuous calculation of the age of labeled red blood cells. Am J Hematol. 2008;83(6):454-457. [DOI] [PubMed] [Google Scholar]
- 18.Altrock PM, Brendel C, Renella R, Orkin SH, Williams DA, Michor F. Mathematical modeling of erythrocyte chimerism informs genetic intervention strategies for sickle cell disease. Am J Hematol. 2016;91(9):931-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinstein IM, Spurling CL, Klein H, Necheles TF. Radioactive sodium chromate for the study of survival of red blood cells. III. The abnormal hemoglobin syndromes. Blood. 1954;9(12):1155-1164. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.