

Abstract
Serine resolvases are an interesting group of site-specific recombinases that, in their native contexts, resolve large fused replicons into smaller separated ones. Some resolvases are encoded by replicative transposons and resolve the transposition product, in which the donor and recipient molecules are fused, into separate replicons. Other resolvases are encoded by plasmids and function to resolve plasmid dimers into monomers. Both types are therefore involved in the spread and maintenance of antibiotic-resistance genes. Resolvases and the closely related invertases were the first serine recombinases to be studied in detail, and much of our understanding of the unusual strand exchange mechanism of serine recombinases is owed to those early studies. Resolvases and invertases have also served as paradigms for understanding how DNA topology can be harnessed to regulate enzyme activity. Finally, their relatively modular structure, combined with a wealth of structural and biochemical data, has made them good choices for engineering chimeric recombinases with designer specificity. This chapter focuses on the current understanding of serine resolvases, with a focus on the contributions of structural studies.
INTRODUCTION
The serine resolvases are a group of recombinases that, in their native contexts, resolve large fused replicons into smaller separated ones (1). Serine resolvases and the closely related invertases were the first serine recombinases to be studied in detail, and much of our understanding of the serine recombinase mechanism is owed to those early studies. Resolvases and invertases have also served as paradigms for understanding how DNA topology can be harnessed to regulate recombination reactions (2–5). Like other serine recombinases, the resolvases have a largely modular structure. In the resolvase case, the conserved catalytic domain is followed by a DNA-binding domain that is a simple helix-turn-helix similar to that found in many prokaryotic repressors (6). This modularity, combined with a wealth of structural and biochemical data, has made them good targets for engineering chimeric recombinases with designer sequence specificity (7, 8).
This chapter will focus on the current understanding of the mechanism of serine resolvases, with a focus on how structural studies have informed (and sometimes confounded) that understanding. For a broader view of serine recombinases, including an in-depth discussion of the large body of data addressing the mechanism of strand exchange, the reader is referred to the chapter by W.M. Stark. Additionally, the chapter by R.C. Johnson describes the invertases, which are quite closely related to the resolvases, and that by M.C.M. Smith describes the large serine recombinases, which have a different and much larger DNA-binding domain—also reviewed in reference (9). Although different groups of serine recombinases have different biological roles and regulatory properties, the strong conservation of the catalytic domain means that many of the lessons learned from the resolvase group can be directly applied to the rest of the serine recombinase family.
The generally accepted and well-supported mechanism of serine-recombinase-mediated strand exchange involves an unusual rotation of half of an entire complex relative to the other half. Briefly, formation of an active tetramer brings together the two dimer-bound crossover site DNAs (Figure 1). Formation of this tetramer is a key step that is regulated in different ways in different serine recombinase systems: see below for resolvases and chapters by RC Johnson and MCM Smith for other systems. Within the tetramer, the hydroxyl group of each subunit’s active site serine then attacks a particular DNA phosphate group, displacing the 3′ hydroxyl and creating a covalent protein–DNA linkage. Once double-strand breaks are formed, one half of the DNA-bound tetramer can rotate relative to the other. Although thermal energy is sufficient to drive rotation within an active tetramer, DNA supercoiling can provide an additional driving force and can favor rotation in one direction over the other. Once rotation has aligned the broken DNA ends with new partners, the free 3′ hydroxyls attack the phosphoserine linkages to reseal the DNA. The DNA can be re-ligated without the use of high-energy cofactors such as ATP because the chemical energy of the broken phosphodiester bond is stored in the phosphoserine linkage. However, as the net change in bond energy between substrate and product is zero, other factors are needed to tip the equilibrium towards product.
FIGURE 1.
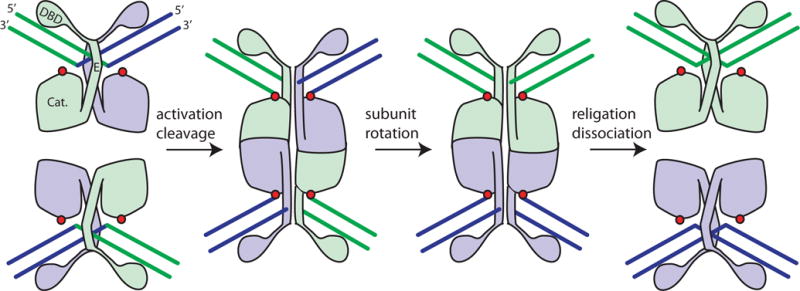
Cartoon of serine resolvase-mediated strand exchange. The wild-type protein initially binds crossover sites as an inactive dimer. Upon activation (see text), the catalytic domains (labeled “Cat.”) form a tetramer that synapses the two partner sites. Within the tetramer, the active site serines (red dots) attack the DNA, creating double strand breaks with 5′ phosphoserine linkages and 2-nucleotide 3′ overhangs. Two subunits and the DNA segments covalently linked to them can then rotate relative to the other two. A 180° rotation aligns the broken ends for re-ligation in the recombinant configuration. Much of both the dimer and tetramer interface is contributed by helix E. doi:10.1128/microbiolspec.MDNA3-0045-2014.f1
DNA resolvases play at least two biological roles. First, the transposition pathway of certain replicative transposons such as Tn3, γδ, and Tn4430 (see chapters by F. Dyda & A. B. Hickman and B. Hallet) leads to a “cointegrate” product in which the donor and target DNA replicons are fused, with a copy of the transposon at each junction (Figure 2A) (10). These elements generally encode site-specific recombinases, usually but not always of the serine family, that “resolve” the cointegrate product into two replicons, each bearing a copy of the transposon. Second, circular replicons (plasmids or entire bacterial chromosomes) can become dimerized as an accident of replication. When a collapsed replication fork is re-assembled in a RecA-dependent pathway, there are two possibilities for resolving the ensuing Holliday junction. In one case, the final product of replication will be two daughter circles, and in the other case the product will be one large fused circle (Figure 2B) (11). Although bacterial chromosomes (and certain plasmids) use tyrosine recombinases to resolve these dimers (see chapter by FX Barre), some plasmids encode their own serine resolvase systems. The functionally best-characterized of these is the β recombinase of Streptococcus pyogenes (12–14), whereas the closely related Sin recombinase, which is encoded by many staphylococcal plasmids, has been more extensively characterized biochemically and structurally (15, 16). Both transposon and plasmid resolvases act at sites termed “res” which include regulatory elements as well as the crossover sites.
FIGURE 2.
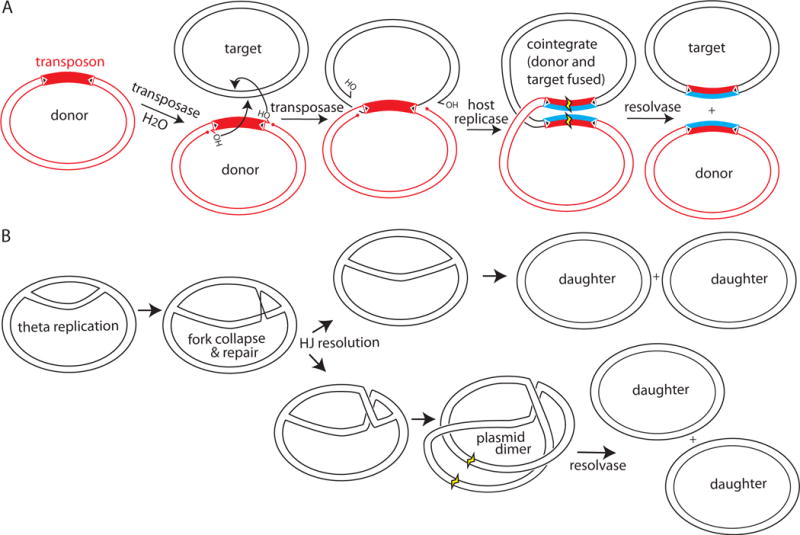
Biological roles for resolvases. (A) Some resolvases (e.g., γδ and Tn3) are encoded by replicative transposons. Their transposition creates a branched intermediate (center panel) that is processed by the host replication and repair machinery (new DNA strands are in blue) to yield a “cointegrate” (fourth panel) in which both the donor and recipient replicons are fused. Resolvase action at a res site within the transposon (yellow) resolves the co-integrate into the original donor and the recipient that now carries a copy of the transposon. (B) Some resolvases (e.g., β and Sin) are encoded by plasmids. Rescue of a stalled replication fork by a homologous recombination-mediated pathway can lead to a Holliday Junction (HJ) behind the rescued fork (second panel). Depending on which pair of strands is cut to resolve the Holliday Junction, replication results in two daughter circles (upper branch) or in a plasmid dimer (lower branch). Action of the plasmid-encoded resolvase at a res site (yellow) converts this dimer into two daughter circles. In both examples, the product circles are initially linked as catenanes (not shown for simplicity; see Figure 3) that are later separated by a host type II topoisomerase. doi:10.1128/microbiolspec.MDNA3-0045-2014.f2
The regulatory mechanism of serine resolvases exploits DNA topology to sense the relative location and orientation of the two partner res sites. Efficient recombination by wild-type (WT) resolvases occurs only between res sites that lie within the same circular DNA molecule and are in direct rather than inverted orientation. This requirement ensures that resolvases only catalyze the resolution of cointegrates (or dimers) and not inversion of the DNA segment between their cognate sites, or even intermolecular reactions that would create larger fused replicons. Formation of an active tetramer that synapses the two crossover sites occurs only within a larger complex termed a “synaptosome” that contains two copies of the full cognate res site for that resolvase, each one bound by multiple proteins. Although res sites for different serine resolvases vary in detail (Figure 3A), the topology of the DNA (when tested) is constant: the protein–protein and protein–DNA interactions within the synaptosome trap 3 supercoiling nodes (Figure 3B) reviewed in references (1, 17). Supercoiling therefore stabilizes the complex, and in fact the protein–protein interactions appear to be tuned to render stable synaptosome formation dependent on supercoiling (18). Requiring synaptosome formation before catalysis accomplishes several things, reviewed in reference (2). First, it greatly favors intramolecular over intermolecular recombination, because the energy barrier to trapping three DNA–DNA crossings between two res sites on the same supercoiled plasmid is much lower than that for trapping three crossings between two separate plasmids. Second, the synaptosome aligns the two crossover sites properly such that recombination will produce resolved rather than inverted products. Third, the topology of the synaptosome is such that the 180° rotation accompanying strand exchange causes a change in linking number of +4. This prediction was verified by careful experimental measurements (19). (For a useful primer on DNA topology and the changes predicted by different recombination mechanisms see (20), and for a more advanced mathematical treatment, see (21).) Assuming a physiological supercoiling density of roughly −0.025 and a 20-kb substrate, relaxing four superhelical turns corresponds to a ΔG of about −9 kcal/mol (22). For comparison, that is slightly more than the standard free energy of ATP hydrolysis, and so a considerable, although unconventional, driving force. The product circles are still topologically linked as a two-noded catenane, but that is a problem that any type II topoisomerase can handle. Additional rotations after the first 180° are possible, but create knots (23, 24). Finally, within the product catenane the architecture of the synaptosome is no longer stabilized by supercoiling as it was in the substrate, and thus the complex is more likely to dissociate. Regulation of the serine invertases is conceptually similar, although the details differ (see the chapter by RC Johnson).
FIGURE 3.
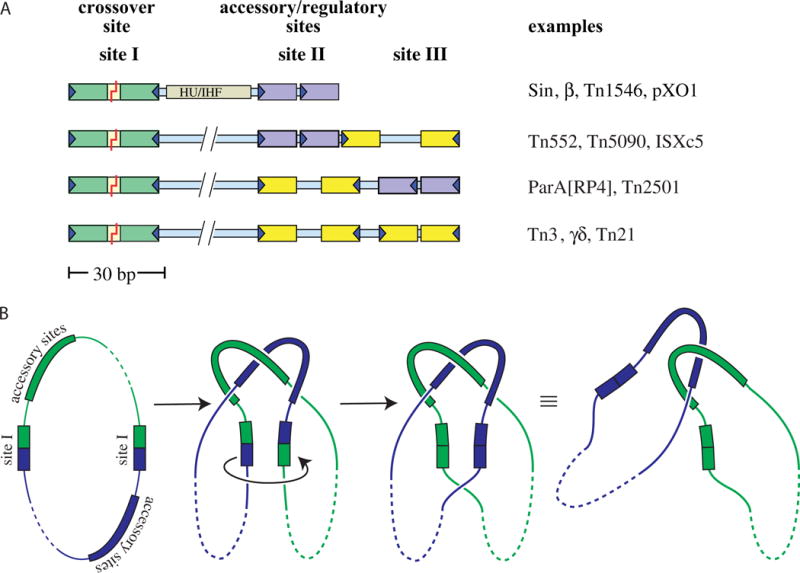
Res sites and the topology of the synaptosome. (A) Examples of serine resolvase res sites. Specific recognition sequences (~12 bp each) for individual resolvase subunits are shown as colored boxes: green for the crossover site (always an inverted repeat), purple for accessory sites that form direct repeats, and yellow for accessory sites that form inverted repeats. The recombinase dimers bound to the accessory sites are catalytically inactive, and these sites always differ from the crossover site in the length of their central spacers and/or the relative orientation of their half-sites. Sin and related resolvases require a DNA bending protein as well as additional recombinase subunits. The coding sequence for the resolvase protein is usually adjacent to its cognate res site. Figure adapted from reference (60) with permission. (B) Resolvase synaptosomes trap 3 supercoiling nodes. The resolvase and accessory proteins (if any) bound to each of the cognate res sites form a complex (the “synaptosome”) that traps 3 dsDNA-over-dsDNA crossings and juxtaposes the two site Is. Synaptosome formation activates the site I-bound resolvase subunits, which then introduce double-strand breaks. A 180° rotation of the bottom two subunits (as drawn) realigns the broken ends, which are then re-ligated. In a negatively supercoiled substrate a right-handed rotation is favored because it introduces a + supercoiling node (ΔLk = +1) that cancels one of the pre-existing (−) nodes trapped in the synaptosome and because it allows rewinding of each duplex by a half turn (ΔTw for each = +½). The remaining two crossings trapped by the synaptosome are no longer intramolecular and instead catenate the two daughter circles (which can be separated by a host type II topoisomerase). doi:10.1128/microbiolspec.MDNA3-0045-2014.f3 31)). (B) Activated mutant γδ resolvase with crossover site DNA in the covalent protein–DNA intermediate state. Note that each catalytic domain has undergone major conformational changes in the transition from dimer to tetramer (PDBid 2gm4; (48, 49)). (C) The same structure as in B, rotated by ~90° about a horizontal axis. (D) Activated Sin resolvase tetramer catalytic domain tetramer. Sulfate ions that mark the binding pockets for the scissile phosphate are shown as sticks (PDBid 3pkz) (50). doi:10.1128/microbiolspec.MDNA3-0045-2014.f4
STRUCTURAL BIOLOGY OF SERINE RESOLVASES
Serine resolvases have been the target of structural studies for three decades, and the combined results of these efforts provide an increasingly detailed three-dimensional view of the reaction pathway. A brief overview of the surprisingly convoluted history of using serine resolvase structures to understand function is presented here. The first structure, of the catalytic domain of WT γδ resolvase, reported in 1990, was as puzzling as it was informative (25). DNA-binding and footprinting studies had suggested that the WT protein forms a dimer in solution, and that dimer–dimer interactions form the synaptosome (reviewed in reference (26)). Although multiple protein–protein interfaces were seen in the crystal, additional biochemical data were needed to determine which mediated the solution dimer, which represented contacts formed between dimers within the synaptosome, and which were probably irrelevant in solution (27, 28).
The resolvase catalytic domain contains a small α/β fold containing the catalytic serine followed by a long helix (helix E) that is sometimes now considered a separate connector domain (Figure 4A). Although the overall fold has been noted to show similarities to that of 5′ → 3′ exonucleases and topoisomerases of the TOPRIM fold, the active site is unrelated to either of those (29, 30). In the solution dimer, the E helices of two subunits dock against each other and into a shallow groove on the partner’s surface. However, this dimer places the two active site serines much too far apart to attack the scissile phosphate groups in B-form DNA. Furthermore, because the serines lie on flexible solvent-exposed loops, they do not appear to be activated for nucleophilic attack. It was also unclear how two such dimers could dock together to form a recombination-competent tetramer, or, given the partially interdigitated nature of the interface, how subunit exchange could occur.
FIGURE 4.
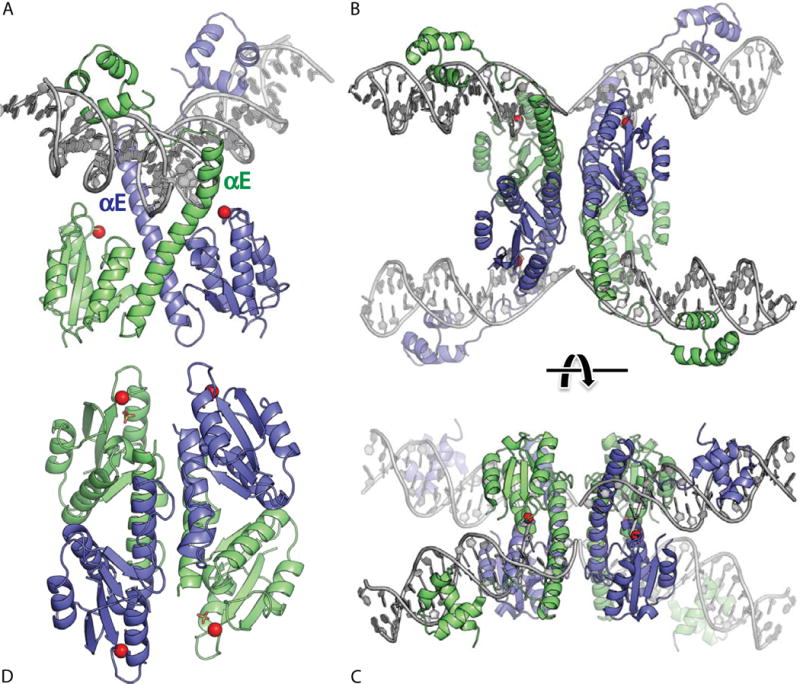
Serine resolvase structures. (A) Wild-type γδ resolvase bound to crossover site (or “site I”) DNA. The active site serine residues are marked with red spheres (PDBid 1gdt; (31)). (B) Activated mutant γδ resolvase with crossover site DNA in the covalent protein–DNA intermediate state. Note that each catalytic domain has undergone major conformational changes in the transition from dimer to tetramer (PDBid 2gm4; (48, 49)). (C) The same structure as in B, rotated by ∼90° about a horizontal axis. (D) Activated Sin resolvase tetramer catalytic domain tetramer. Sulfate ions that mark the binding pockets for the scissile phosphate are shown as sticks (PDBid 3pkz) (50). doi:10.1128/microbiolspec.MDNA3-0045-2014.f4
A similar catalytic domain dimer was seen in the structure of the WT γδ resolvase bound to crossover site DNA (Figure 4A) (31). That structure was very informative regarding resolvase–DNA contacts: it showed that while the most stringent aspects of protein–DNA recognition are, as expected, accomplished by the helix-turn-helix DNA-binding domain (32), additional contacts are made by an extension of helix E and the subsequent linker, which bind in the minor groove near the center of the site. The insertion of helix E into the central minor groove widens it, inducing it to bend away from the catalytic domains (a feature that recurs in the complexes with accessory sites described below). However, despite the DNA distortions seen in the resolvase–site I complex, the scissile phosphates were still not docked near the catalytic serines, and the mechanisms of catalysis and strand exchange remained puzzling.
Other lines of evidence also suggested that the conformation of the isolated WT dimer must be significantly different from that of the catalytically active species formed within the synaptosome. Constitutively active mutants of resolvases (and invertases) had been selected that, unlike the WT proteins, would recombine isolated crossover sites (and many more have been selected since that time) (33–41). Such mutants form tetramers rather than dimers in solution (36, 42–45). The positions of the activating mutations map to the dimer interface region: helix E, the turn leading into it, and the surfaces that it packs against. It had already been noticed when comparing multiple structures that the WT dimer is quite flexible, and nuclear magnetic resonance data showed that helix E is not integral to the folding of the catalytic domain (43, 46, 47). Thus, it seemed likely that activation might involve a rearrangement of helix E relative to the rest of the catalytic domain.
Despite the foreshadowing described above, the magnitude of the activating conformational change, finally revealed in 2005, was unexpected (Figures 4B, C and 5A) (48, 49). The breakthrough structure used an activated mutant of γδ resolvase that crystallized as a tetramer, with bound DNA in the double-strand break form and with each active site serine residue covalently linked to the 5′ end of a DNA segment. Within each subunit, the catalytic domain core had rotated and tilted with respect to helix E. Furthermore, the old dimer-mediating protein–protein contacts were completely rearranged. The mutations resulting in constitutive activation appeared to both destabilize the dimeric form and to stabilize the tetrameric form. In the center of the tetramer, holding the broken DNA ends together, was a hydrophobic but unusually flat interface. At last one could picture rotation of two subunits relative to the other two! However, the active site itself appeared to have undergone a post-catalytic conformational change: the displaced 3′OH groups were far (> 10 Å) from the phosphoserine linkages and not all of the residues known to be critical for catalysis pointed into the active site. As a result, it remained difficult to propose a structure-based model of the phosphotransfer reactions.
FIGURE 5.
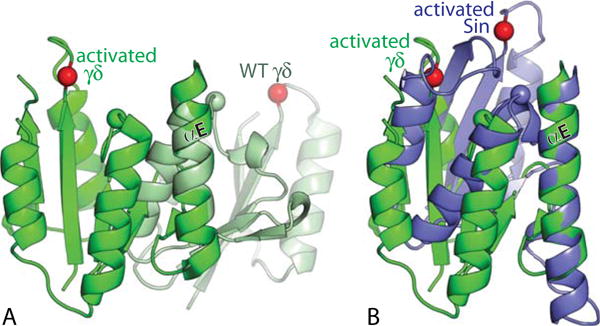
Intramolecular conformational changes upon activation. (A) Superposition, using the E-helices as guides, of one subunit from an activated, DNA-bound γδ resolvase tetramer (green) in a post-cleavage state with one from an inactive wild-type γδ dimer (light green) (PDBids 2gm4 and 2rsl (46, 49)). Red spheres mark the α carbons of the active site serines (S10 for γδ, S9 for Sin); green and blue spheres those of the probable general acid R71 (γδ)/R69 (Sin). (B) Similar superposition of the same activated γδ subunit as in (A), and one subunit from an activated Sin tetramer that appeared to be in the cleavage-ready state (PDBid 3pkz, (50)). doi:10.1128/microbiolspec.MDNA3-0045-2014.f5 49–51). doi:10.1128/microbiolspec.MDNA3-0045-2014.f6
In 2011, a high-resolution structure of an activated Sin resolvase catalytic domain provided a picture of the serine recombinase active site apparently fully assembled for catalysis (50). In this structure, the catalytic domain core is further tilted relative to Helix E (Figures 4D and 5B), and all of the catalytically important side chains assemble into a hydrogen-bonded network surrounding a sulfate ion that marks the scissile phosphate-binding pocket. The overall architecture of this tetramer also appears “ready for cleavage”: the relative orientation of the two halves differs by about 35° from that seen in the γδ tetramer, and that rotation brings pairs of active sites closer together. Duplex DNA taken from the inactive WT γδ dimer structure could be docked as a rigid body onto the activated Sin tetramer such that the scissile phosphate groups superimposed on the sulfate ions bound in the Sin active site. This docking exercise also implied that the bending of the DNA may be relatively constant throughout the reaction, and that the catalytic domains simply swing in to meet the scissile phosphates when necessary.
Although more than one structure was obtained for activated γδ resolvase tetramers they all adopted the same overall rotation angle. However, as described above, an activated Sin resolvase tetramer adopted a different angle. A third rotational state was later seen in the structure of an activated Gin invertase tetramer (Figure 6) (51), and most recently a different activated Sin mutant crystallized with three independent tetramers in the asymmetric unit: two similar to that seen previously for Sin, and one in a fourth rotational state (C.S. Trejo and P.A. Rice, unpublished data). These structures illustrate that serine recombinase tetramers can indeed adopt multiple rotational states.
FIGURE 6.
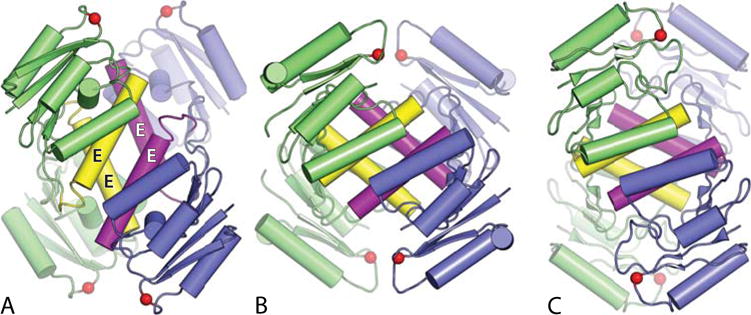
Structures of serine recombinase tetramers in three different rotational states. Colors are as in Figure 4, except that the E-helices are highlighted in yellow (green subunits) and magenta (blue subunits). (A) Activated γδ resolvase tetramer; (B) activated Sin resolvase tetramer; (C) activated Gin invertase tetramer. PDBids 2gm4, 3pkz, and 3uj3, respectively (49–51). doi:10.1128/microbiolspec.MDNA3-0045-2014.f6
MOLECULAR MODELS FOR THE SYNAPTOSOME
The history of modeling the synaptosomes within which resolution occurs is similarly long and convoluted. Early work focused on Tn3 and γδ resolvases, which are 81% identical and have similar res sites. The overall path of the DNA was defined by elegant in vitro studies of the Tn3 system showing that within the synaptosome, wrapping of the two res sites about one another traps three supercoiling nodes (see chapter by WM Stark). Early work also identified residues that make inter-dimer contacts in the crystal and are important in assembling the synaptosome (28). However, because the full Tn3/γδ synaptosome contains 12 copies of the same protein (Figure 3A), deciphering exactly which pairs of subunits those important contacts are between has been challenging (36, 52, 53). Modeling has been further complicated by the fact that the central spacer of the three dimer-binding sites of res varies by nearly a helical turn, and while biochemical data confirm that complexes with sites II and III are highly asymmetric (54–56), structural data have only been published for γδ resolvase bound to site I. Nevertheless, a number of increasingly sophisticated models for the Tn3/γδ synaptosome have been proposed over the years (19, 36, 53, 57–59).
Sin’s res site is shorter and its synaptosome proved simpler to model (Figure 3A) (60). The topology of the Sin synaptosome is the same as for Tn3 and γδ resolvases, but the res site contains only two Sin dimer-binding sites separated by a gap that is synergistically bound by a DNA bending protein (38). The activated γ δ resolvase–site I complex provides a good model for that of Sin because of the overall similarity of the proteins (~33% sequence identity and nearly identical folds) and the geometry of their site Is. In the natural Sin host Staphylococcus aureus, the DNA bending protein is most likely the highly conserved nucleoid-associated protein HU (originally named as Histone-like protein from strain U13). Structures are available for HU in complex with DNA, but its lack of sequence specificity complicates precise modeling (61). Fortunately, HU can be replaced with the structurally similar but sequence-specific Escherichia coli IHF (“Integration Host Factor”) protein if the WT site I–site II spacer DNA sequence is replaced with an IHF binding site in the correct register (62, 63). Hence for modeling the synaptosome, the only missing structure was that of the Sin–site II complex.
Sin’s site II is a direct rather than inverted repeat of the two half-sites. The structure of a WT Sin–site II complex showed how the dimeric protein can bind a direct repeat (Figure 7A). Although the catalytic domains form an approximately two-fold symmetric dimer similar to that of γδ resolvase, there is a break in one of the E-helices that allows major re-orientation of the DNA-binding domain (64). The extension of the other E helix binds in the minor groove, bending the DNA in the same way that γδ resolvase does. Nevertheless, when the γδ resolvase–site I complex is superimposed on the Sin– site II complex, the DNAs are almost mutually perpendicular. The observation that two Sin–site II complexes can pair even in the absence of the other synaptosome components could be explained by previously unsuspected protein–protein contacts between the DNA-binding domains seen in the crystal. The importance of these new contacts in site II–site II interactions was independently established by an elegant genetic screen (39, 64). The direct repeat arrangement of the binding sites perfectly orients the two pairs of DNA binding domains to make synergistic interactions, which explains why Sin–site I complexes do not form similar binding-domain-mediated tetramers.
FIGURE 7.
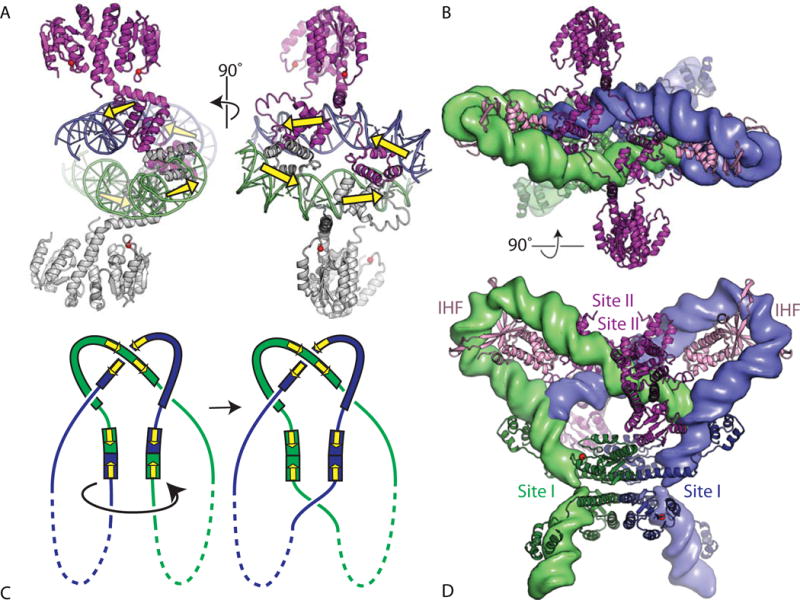
Modeling the Sin synaptosome. (A) Two orthogonal views of the structure of a tetramer of WT Sin resolvase bound to its cognate site II. In the left panel, the catalytic domains of the lower dimer are oriented in a similar way to those of wild-type γδ resolvase in Figure 4(A). The dimer–dimer contacts are mediated by the DNA-binding domains (PDBid 2r0q (64)). Yellow arrows show the orientations of the individual monomer-binding sites in the DNA. (B) Model for the synaptosome, created by rigid-body docking together of a symmetrized version of an activated γδ resolvase tetramer–DNA structure (blue and green proteins), two copies of an IHF–DNA complex structure (pink proteins), and the Sin–site II structure (purple proteins). The DNAs are shown as smoothed green and blue surfaces. The view is the same as for the right hand panel of (A). PDBids 1zr4, 1ihf, and 2r0q were used (48, 64, 84). (C) Cartoon similar to that in Figure 3B showing the expected synaptosome topology. Yellow arrows mark the orientations of the half-sites within each dimer-binding site. (D) Second view of the synaptosome model, rotated 90° about a horizontal axis. doi:10.1128/microbiolspec.MDNA3-0045-2014.f7 50)). A sulfate ion marks the scissile-phosphate binding pocket, and side chains important for catalysis are shown as sticks. Those residues whose mutation had the most deleterious effect on the rate of DNA cleavage by Tn3 resolvase are shown in magenta, shading to white for those whose mutation had more moderate effects (74). (B) Stereo view of one subunit from a site II-bound wild-type Sin dimer (PDBid 2r0q (64)). The same side chains are shown, similarly shaded from a dark color to white. doi:10.1128/microbiolspec.MDNA3-0045-2014.f9
Docking of the activated γδ-site I, IHF-DNA, and Sin-site II structures created a model of the Sin synaptosome that matches the known topology (Figure 7) (64). It also arranges the site I- and II-bound proteins such that a rotation of the site II-bound catalytic domains about helix E’s hinge point would lead to dimer– dimer contacts mediated by a patch of residues known to be important in stabilizing the synaptosome (and analogous to those that make inter-dimer contacts in Tn3 and γδ resolvases). The model was supported by careful analysis of the combined effects of mutations in this patch, mutations that interfere with DNA-binding domain-mediated interactions and activating mutations (39).
How similar are other serine resolvase synaptosomes to Sins? A general pattern among diverse res sites can be seen in Figure 3A: Site I, where the crossover occurs, is always an inverted repeat, and it is always followed by two additional “accessory” resolvase-binding and/or bending protein-binding sites. The additional resolvase binding sites vary in the relative orientation and spacing of their half-sites, but they always differ from site I. The only other resolvase with a Sin-like res site that has been studied is β recombinase, and it is likely to form a Sin-like synaptosome (14, 65, 66). However, under some conditions β can also catalyze inversions, implying that the requirements for catalytic activation may be less stringent than those for Sin (14).
In contrast to Sin and β, Tn3 resolvase appears to have found a different solution for constructing a three-noded synaptosome. A recent Tn3 resolvase–site III complex structure (S.P. Montaño and P.A. Rice, unpublished data) shows that its short central spacer allows only one E-helix to bind in the minor groove, giving the complex an overall asymmetry that is different yet again from the Sin–site II complex but that agrees well with previous footprinting data (54). It appears that the variations in the additional (noncrossover) resolvase-binding sites are tailored to induce particular geometries in the resulting protein–DNA complex. A Tn3 synaptosome model has been constructed using this new Tn3– site III structure, the activated γδ–site I structure, and SAXS-supported modeling of the Tn3–site II complex. The model also uses the inter-dimer contacts seen in crystals of Tn3 and γδ resolvases and new data that finally deconvolutes exactly which pairs of subunits interact (S.-J. Rowland, M.R. Boocock and W.M. Stark, unpublished data). Interestingly, while Sin and Tn3 use the same patch of protein to make those contacts, they differ in which subunits within the synaptosomes those contacts link. It appears that these systems have converged upon different ways to assemble three-noded synaptosomes because that topology is perfect for channeling the recombination reaction to produce only resolution products (rather than inversion or integration products).
ACTIVATION AND CATALYSIS
How does incorporation into a synaptosome activate the site-I bound proteins? It must at least transiently tip the conformational equilibrium from dimer to tetramer. By bringing the two site I-bound dimers into close proximity, the synaptosome can favor the tetramer simply by applying mass action. The local concentration of site-I-bound resolvase in the synaptosome can be roughly estimated as that of four subunits in a cube 130 Å on each side, which is 30 mM. In vitro, WT Sin bound to isolated crossover sites is normally inactive, but slight catalytic activity begins to be detectable if the concentration is raised to even 10 μM (45). In addition, the protein–protein contacts between crossover- and accessory-site-bound subunits may be activating as well as structurally stabilizing. The residues involved lie on the opposite side of the catalytic domain from helix E, and formation of these contacts may induce and/or stabilize rotation of the catalytic domain core relative to helix E, thus favoring the conformation found in the tetramer (Figure 8). This mechanism would explain why, even though the overall arrangement of accessory proteins is different in the Sin and Tn3 synaptosomes, the same patch of side chains is used to make these contacts. Furthermore, an overlapping patch is important in activating the serine invertase Hin, albeit through protein– DNA rather than protein–protein contacts (67).
FIGURE 8.
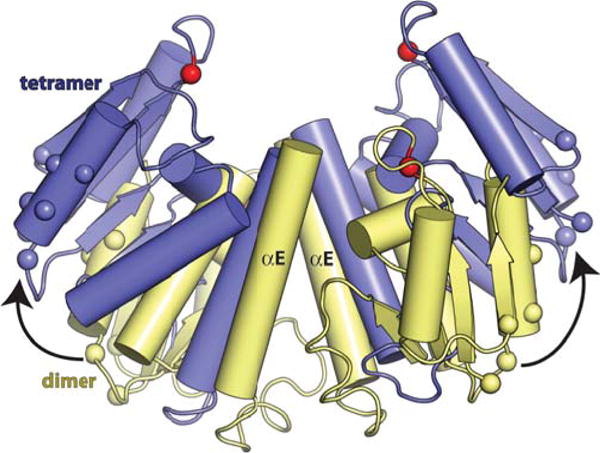
Interdimer contacts within the synaptosome may affect the dimer–tetramer equilibrium. A wild-type Sin dimer (yellow) is superimposed on two subunits from the activated Sin tetramer (blue). Red spheres mark the α carbons of the catalytic serines, and other spheres mark the positions of side chains whose mutation interferes with interdimer contacts in Sin and γδ resolvases and with catalytic domain–DNA contacts in the related Hin invertase (28, 39, 67). doi:10.1128/microbiolspec.MDNA3-0045-2014.f8
Once activated, how do serine recombinases catalyze DNA cleavage and re-ligation? The chemistry of phosphotransfer reactions has been extensively reviewed elsewhere (68, 69). Serine recombinases are unusual, although not unique, in that they do not require divalent cations such as Mg2+ but instead rely only on amino acid side chains to provide catalytic power (19, 70). Bio chemical studies, in conjunction with the evolving structural data, have identified a set of residues that are important for activity and proposed roles for them (42, 71–74). As shown in Figure 9, the “inner circle” surrounding the scissile phosphate is comprised of three arginines. Arginine is often found in phosphotransferase active sites, and may aid catalysis in several ways: it can localize the scissile phosphate through bidentate hydrogen bonds, it can stabilize additional partial negative charge on the transition state, and if properly oriented, it may preferentially stabilize the geometry of the transition state as well.
FIGURE 9.
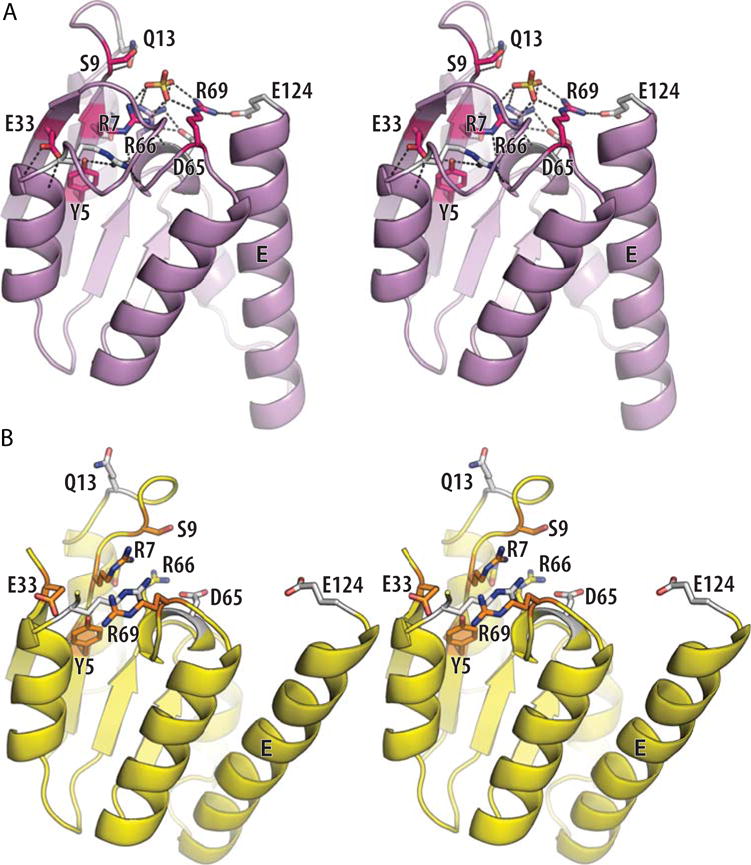
Details of the Sin active site. (A) Stereo view of one subunit from an activated Sin tetramer (PDBid 3pkz (50)). A sulfate ion marks the scissile-phosphate binding pocket, and side chains important for catalysis are shown as sticks. Those residues whose mutation had the most deleterious effect on the rate of DNA cleavage by Tn3 resolvase are shown in magenta, shading to white for those whose mutation had more moderate effects (74). (B) Stereo view of one subunit from a site II-bound wild-type Sin dimer (PDBid 2r0q (64)). The same side chains are shown, similarly shaded from a dark color to white. doi:10.1128/microbiolspec.MDNA3-0045-2014.f9
What is surprising about the serine recombinase active site is the lack of residues commonly associated with general acid–base catalysis such as histidine. The high intrinsic pKa of arginine makes it an unusual candidate for that role, but there is precedent (75). Furthermore, the clustering of several arginines in close proximity may alter the equilibrium between the positively charged and neutral forms, thus lowering their pKas. In the Sin active site, R7 is well-positioned to abstract a proton from the nucleophilic serine as it attacks, R66 to hold the scissile phosphate in place and stabilize the transition state, and R69 and R66 to donate a proton to the DNA 3′ oxygen as it leaves. Evidence that R69 is important in stabilizing the leaving group (during the cleavage reaction) and likely to be the general acid was supplied by experiments replacing the 3′O with an S, which is predicted to be a better leaving group in the absence of a general acid (76). Mutants of R69, but not of other arginines, were partially rescued by the 3′S (although it should be noted that R66 mutants were simply inactive with all substrates). Careful analysis of the kinetic effects of R to A versus R to K changes also showed that the residues in this positively charged cluster probably do collaborate to lower one another’s pKas (76). In this respect, the catalytic center should be considered as a cooperative network.
FUTURE QUESTIONS
Several decades of careful study have drawn an increasingly detailed picture of these unusual enzymes. Armed with buffets of designer variants and numerous structures, the field is poised to branch into new directions. From a basic science viewpoint, serine resolvases provide a rich system for investigating protein dynamics as well as the enzymology of phosphotransfer reactions. From a medical viewpoint, serine resolvases are under-appreciated functional components of many antibiotic resistance-bearing transposons and plasmids. Finally, from a biotech viewpoint, they can be useful genetic engineering tools.
The subunit rotation hypothesis, once rather puzzling at the molecular level, is now well-supported by many lines of evidence, from topological to structural. However, the detailed dynamics of this molecular swivel have not been examined. Does it move smoothly or are there significant energy minima along the way? Is there an intrinsic energy minimum at 180° that would cause pausing when re-ligation should occur, or is the only stop-the-merry-go-round signal the pairing of the 2-nucleotide overhangs? Although describing the protein conformational changes as simply dimer to tetramer is convenient, the superposition of structures shown in Figures 5B and 10 suggests that reality is more complicated. With the caveat that structures of different proteins (Sin vs. γδ) are used to compare different states, it appears that the ready-to-cleave tetramer conformation differs from the post-cleavage one. Figure 10 also highlights the flexibility of the inactive dimer (note the spread of blue and green spheres marking the position of its active site serine). Much, but not all, of this conformational variability reflects the motion of helix E. That helix and the region of protein it packs against are unusually rich in methionine residues, which are the most flexible of the large hydrophobic amino acids. Methionine-rich binding pockets are also used by the signal recognition particle and the chaperone DnaK to make favorable interactions with hydrophobic but variable substrates (77–79). While more structures, especially at high resolution, that trap additional rotational states would be very useful, even now the system seems ripe for computational exploration of its dynamics.
FIGURE 10.
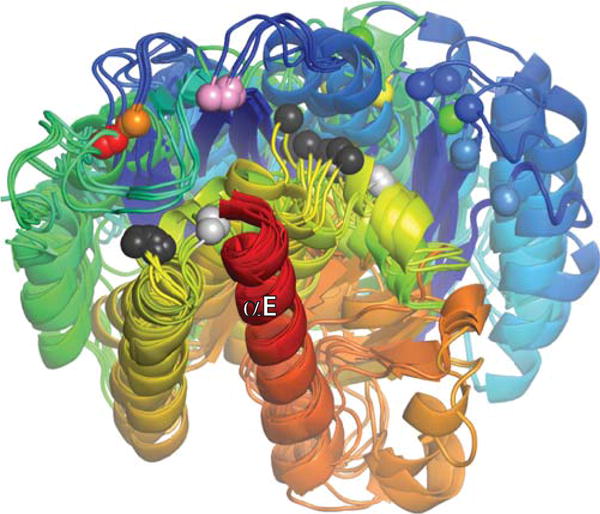
Superposition of all currently published serine resolvase catalytic domain structures, aligned using their E helices as guides. Each structure is shaded from blue (N-ter) to red (end of catalytic domain), and all unique copies from the asymmetric unit for each structure were used (two to four per structure). Colored spheres mark the positions of the active site serines. From right to left (roughly), they are: light blue, wild-type (WT) Sin from the Sin–site II structure (PDBid 2r0q); dark blue, WT γδ determined without DNA (PDBid 2rsl); green, WT γδ from the γδ–site I complex structure (PDBid 1gdt); yellow, activated γδ from a structure without DNA (PDBid 2gm5); orange and red, two different activated variants of γδ from structures with covalently linked crossover site DNA (PDBids 1zr4 and 2gm4, respectively), and pink, activated Sin tetramer (determined without DNA; PDBid 1pkz). Black and Gray spheres mark the Cα positions of the probable general acid R71 (γδ)/R69 (Sin). Those that cluster towards the left are in structures of activated mutants, whereas those that cluster towards the right are from WT structures. doi:10.1128/microbiolspec.MDNA3-0045-2014.f10
Although the residues that are critical for catalysis have been identified and recent structural and biochemical work has helped dissect their roles, several questions remain. Biochemical data link R69 with pro-tonating the leaving 3′ oxygen during the DNA cleavage reaction, but is there a general base that accepts a proton from S9 as it attacks? R7 is nicely poised to do so in the structure, but there is no direct experimental evidence to support its playing that role. It is reasonable to expect that the pKas of the active site arginines are shifted towards neutral, which could make them more efficacious as general acid–base catalysts, but their pKas have not been measured. Another question is, what protects the phosphoserine linkage from hydrolysis during the subunit rotation step? It may be that the active site is only fully assembled at the 0° and 180° points of rotation where it needs to be catalytically competent. The role effect of divalent and multivalent cations on the enzyme is unexplained in detail. While serine recombinases do not require Mg2+ to catalyze phosphotransfer reactions, the phosphoserine-linked intermediate tends to accumulate in its absence (80, 81). No structures have revealed binding pockets for Mg2+, and it can be replaced with Ca2+ or polyvalent cations such as spermidine (19, 70). Do these cations facilitate pairing of the 2-nucleotide overhangs before re-ligation through charge neutralization, perhaps stabilizing the catalysis-ready conformation, or do they affect some other stage of the strand exchange process? Finally, if the 2-nucleotide overhangs do not form Watson : Crick pairs after rotation, the complex simply rotates another 180° to realign the original partners, but how exactly is that base pairing checked (23, 82)?
The extensive structural and biochemical data regarding small serine recombinases (resolvases and invertases) have made them excellent targets for engineering. As discussed in more detail in the chapter by W.M. Stark, genomic rearrangements can be targeted to specific sequences by fusing the catalytic domain of an activated serine resolvase with the DNA binding domain of choice (7, 8). These simple, largely modular recombinases currently lack the directionality shown by large serine recombinases (83). Adding directionality to these designer-specificity recombinases will be an interesting protein engineering challenge for the future.
Acknowledgments
I thank Martin Boocock, Sally Rowland and Marshall Stark for comments on the manuscript, decades of informative discussion, and help with Figure 3A, and Caitlin S. Trejo and Nancy Craig for comments on the manuscript. Macromolecular structure figures were generated using The PyMOL Molecular Graphics System, Version 1.7 Schrödinger, LLC. (http://www.pymol.org/)
References
- 1.Grindley NDF, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 2.Rice PA, Mouw KW, Montaño SP, Boocock MR, Rowland S-J, Stark WM. Orchestrating serine resolvases. Biochem Soc Trans. 2010;38:384– 387. doi: 10.1042/BST0380384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boocock MR, Brown JL, Sherratt DJ. Structural and catalytic properties of specific complexes between Tn3 resolvase and the recombination site res. Biochem Soc Trans. 1986;14:214–216. doi: 10.1042/bst0140214a. [DOI] [PubMed] [Google Scholar]
- 4.Wasserman SA, Dungan JM, Cozzarelli NR. Discovery of a predicted DNA knot substantiates a model for site-specific recombination. Science. 1985;229:171–174. doi: 10.1126/science.2990045. [DOI] [PubMed] [Google Scholar]
- 5.Merickel SK, Johnson RC. Topological analysis of Hin-catalysed DNA recombination in vivo and in vitro. Mol Microbiol. 2004;51:1143–1154. doi: 10.1046/j.1365-2958.2003.03890.x. [DOI] [PubMed] [Google Scholar]
- 6.Garvie CW, Wolberger C. Recognition of specific DNA sequences. Mol Cell. 2001;8:937–946. doi: 10.1016/s1097-2765(01)00392-6. [DOI] [PubMed] [Google Scholar]
- 7.Akopian A, He J, Boocock MR, Stark WM. Chimeric recombinases with designed DNA sequence recognition. Proc Natl Acad Sci USA. 2003;100:8688–8691. doi: 10.1073/pnas.1533177100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mercer AC, Gaj T, Fuller RP, Barbas CF. Chimeric TALE recombinases with programmable DNA sequence specificity. Nucleic Acids Res. 2012;40:11163–11172. doi: 10.1093/nar/gks875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rutherford K, Van Duyne GD. The ins and outs of serine integrase site-specific recombination. Curr Opin Struct Biol. 2014;24:125–131. doi: 10.1016/j.sbi.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hickman AB, Chandler M, Dyda F. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev Biochem Mol Biol. 2010;45:50–69. doi: 10.3109/10409230903505596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesterlin C, Barre F-X, Cornet F. Genetic recombination and the cell cycle: what we have learned from chromosome dimers. Mol Microbiol. 2004;54:1151–1160. doi: 10.1111/j.1365-2958.2004.04356.x. [DOI] [PubMed] [Google Scholar]
- 12.Rojo F, Alonso JC. A novel site-specific recombinase encoded by the Streptococcus pyogenes plasmid pSM19035. J Mol Biol. 1994;238:159– 172. doi: 10.1006/jmbi.1994.1278. [DOI] [PubMed] [Google Scholar]
- 13.Ceglowski P, Boitsov A, Karamyan N, Chai S, Alonso JC. Characterization of the effectors required for stable inheritance of Streptococcus pyogenes pSM19035-derived plasmids in Bacillus subtilis. Mol Gen Genet. 1993;241:579–585. doi: 10.1007/BF00279900. [DOI] [PubMed] [Google Scholar]
- 14.Canosa I, López G, Rojo F, Boocock MR, Alonso JC. Synapsis and strand exchange in the resolution and DNA inversion reactions catalysed by the beta recombinase. Nucleic Acids Res. 2003;31:1038–1044. doi: 10.1093/nar/gkg166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulsen IT, Gillespie MT, Littlejohn TG, Hanvivatvong O, Rowland SJ, Dyke KG, Skurray RA. Characterisation of sin, a potential recombinase-encoding gene from Staphylococcus aureus. Gene. 1994;141:109– 114. doi: 10.1016/0378-1119(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 16.Shearer JES, Wireman J, Hostetler J, Forberger H, Borman J, Gill J, Sanchez S, Mankin A, Lamarre J, Lindsay JA, Bayles K, Nicholson A, O’Brien F, Jensen SO, Firth N, Skurray RA, Summers AO. Major families of multiresistant plasmids from geographically and epidemiologically diverse staphylococci. G3 Bethesda Md. 2011;1:581–591. doi: 10.1534/g3.111.000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stark WM, Boocock MR, Sherratt DJ. Site-specific recombination by Tn3 resolvase. Trends Genet. 1989;5:304–309. doi: 10.1016/0168-9525(89)90113-3. [DOI] [PubMed] [Google Scholar]
- 18.Watson MA, Boocock MR, Stark WM. Rate and selectively of synapsis of res recombination sites by Tn3 resolvase. J Mol Biol. 1996;257:317– 329. doi: 10.1006/jmbi.1996.0165. [DOI] [PubMed] [Google Scholar]
- 19.Stark WM, Sherratt DJ, Boocock MR. Site-specific recombination by Tn3 resolvase: topological changes in the forward and reverse reactions. Cell. 1989;58:779–790. doi: 10.1016/0092-8674(89)90111-6. [DOI] [PubMed] [Google Scholar]
- 20.Cozzarelli NR, Krasnow MA, Gerrard SP, White JH. A topological treatment of recombination and topoisomerases. Cold Spring Harb Symp Quant Biol. 1984;49:383–400. doi: 10.1101/sqb.1984.049.01.045. [DOI] [PubMed] [Google Scholar]
- 21.Sumners DW, Ernst C, Spengler SJ, Cozzarelli NR. Analysis of the mechanism of DNA recombination using tangles. Q Rev Biophys. 1995;28:253–313. doi: 10.1017/s0033583500003498. [DOI] [PubMed] [Google Scholar]
- 22.Vologodskii AV, Cozzarelli NR. Conformational and thermodynamic properties of supercoiled DNA. Annu Rev Biophys Biomol Struct. 1994;23:609–643. doi: 10.1146/annurev.bb.23.060194.003141. [DOI] [PubMed] [Google Scholar]
- 23.Stark WM, Grindley ND, Hatfull GF, Boocock MR. Resolvase-catalysed reactions between res sites differing in the central dinucleotide of subsite I. EMBO J. 1991;10:3541–3548. doi: 10.1002/j.1460-2075.1991.tb04918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark WM, Boocock MR. The linkage change of a knotting reaction catalysed by Tn3 resolvase. J Mol Biol. 1994;239:25–36. doi: 10.1006/jmbi.1994.1348. [DOI] [PubMed] [Google Scholar]
- 25.Sanderson MR, Freemont PS, Rice PA, Goldman A, Hatfull GF, Grindley ND, Steitz TA. The crystal structure of the catalytic domain of the site-specific recombination enzyme gamma delta resolvase at 2.7 A resolution. Cell. 1990;63:1323–1329. doi: 10.1016/0092-8674(90)90427-g. [DOI] [PubMed] [Google Scholar]
- 26.Grindley ND, Reed RR. Transpositional recombination in prokaryotes. Annu Rev Biochem. 1985;54:863–896. doi: 10.1146/annurev.bi.54.070185.004243. [DOI] [PubMed] [Google Scholar]
- 27.Hughes RE, Rice PA, Steitz TA, Grindley ND. Protein-protein interactions directing resolvase site-specific recombination: a structure– function analysis. EMBO J. 1993;12:1447–1458. doi: 10.1002/j.1460-2075.1993.tb05788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes RE, Hatfull GF, Rice P, Steitz TA, Grindley ND. Cooperativity mutants of the gamma delta resolvase identify an essential interdimer interaction. Cell. 1990;63:1331–1338. doi: 10.1016/0092-8674(90)90428-h. [DOI] [PubMed] [Google Scholar]
- 29.Yang W. Topoisomerases and site-specific recombinases: similarities in structure and mechanism. Crit Rev Biochem Mol Biol. 2010;45:520– 534. doi: 10.3109/10409238.2010.513375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Artymiuk PJ, Ceska TA, Suck D, Sayers JR. Prokaryotic 5′–3′ exonucleases share a common core structure with gamma-delta resolvase. Nucleic Acids Res. 1997;25:4224–4229. doi: 10.1093/nar/25.21.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang W, Steitz TA. Crystal structure of the site-specific recombinase gamma delta resolvase complexed with a 34 bp cleavage site. Cell. 1995;82:193–207. doi: 10.1016/0092-8674(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 32.Rimphanitchayakit V, Hatfull GF, Grindley ND. The 43 residue DNA binding domain of gamma delta resolvase binds adjacent major and minor grooves of DNA. Nucleic Acids Res. 1989;17:1035–1050. doi: 10.1093/nar/17.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klippel A, Cloppenborg K, Kahmann R. Isolation and characterization of unusual gin mutants. EMBO J. 1988;7:3983–3989. doi: 10.1002/j.1460-2075.1988.tb03286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haffter P, Bickle TA. Enhancer-independent mutants of the Cin recombinase have a relaxed topological specificity. EMBO J. 1988;7:3991–3996. doi: 10.1002/j.1460-2075.1988.tb03287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haykinson MJ, Johnson LM, Soong J, Johnson RC. The Hin dimer interface is critical for Fis-mediated activation of the catalytic steps of site-specific DNA inversion. Curr Biol. 1996;6:163–177. doi: 10.1016/s0960-9822(02)00449-9. [DOI] [PubMed] [Google Scholar]
- 36.Sarkis GJ, Murley LL, Leschziner AE, Boocock MR, Stark WM, Grindley ND. A model for the gamma delta resolvase synaptic complex. Mol Cell. 2001;8:623–631. doi: 10.1016/s1097-2765(01)00334-3. [DOI] [PubMed] [Google Scholar]
- 37.Burke ME, Arnold PH, He J, Wenwieser SVCT, Rowland S-J, Boocock MR, Stark WM. Activating mutations of Tn3 resolvase marking interfaces important in recombination catalysis and its regulation. Mol Microbiol. 2004;51:937–948. doi: 10.1046/j.1365-2958.2003.03831.x. [DOI] [PubMed] [Google Scholar]
- 38.Rowland SJ, Boocock MR, Stark WM. Regulation of Sin recombinase by accessory proteins. Mol Microbiol. 2005;56:371–382. doi: 10.1111/j.1365-2958.2004.04550.x. [DOI] [PubMed] [Google Scholar]
- 39.Rowland S-J, Boocock MR, McPherson AL, Mouw KW, Rice PA, Stark WM. Regulatory mutations in Sin recombinase support a structure-based model of the synaptosome. Mol Microbiol. 2009;74:282–298. doi: 10.1111/j.1365-2958.2009.06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heiss JK, Sanders ER, Johnson RC. Intrasubunit and inter-subunit interactions controlling assembly of active synaptic complexes during Hin-catalyzed DNA recombination. J Mol Biol. 2011;411:744–764. doi: 10.1016/j.jmb.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnold PH, Blake DG, Grindley ND, Boocock MR, Stark WM. Mutants of Tn3 resolvase which do not require accessory binding sites for recombination activity. EMBO J. 1999;18:1407–1414. doi: 10.1093/emboj/18.5.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olorunniji FJ, He J, Wenwieser SVCT, Boocock MR, Stark WM. Synapsis and catalysis by activated Tn3 resolvase mutants. Nucleic Acids Res. 2008;36:7181–7191. doi: 10.1093/nar/gkn885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nöllmann M, He J, Byron O, Stark WM. Solution structure of the Tn3 resolvase-crossover site synaptic complex. Mol Cell. 2004;16:127– 137. doi: 10.1016/j.molcel.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 44.Dhar G, McLean MM, Heiss JK, Johnson RC. The Hin recombinase assembles a tetrameric protein swivel that exchanges DNA strands. Nucleic Acids Res. 2009;37:4743–4756. doi: 10.1093/nar/gkp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mouw KW, Steiner AM, Ghirlando R, Li N-S, Rowland S-J, Boocock MR, Stark WM, Piccirilli JA, Rice PA. Sin resolvase catalytic activity and oligomerization state are tightly coupled. J Mol Biol. 2010;404:16–33. doi: 10.1016/j.jmb.2010.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice PA, Steitz TA. Refinement of gamma delta resolvase reveals a strikingly flexible molecule. Struct Lond Engl 1993. 1994;2:371–384. doi: 10.1016/s0969-2126(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 47.Pan B, Deng Z, Liu D, Ghosh S, Mullen GP. Secondary and tertiary structural changes in gamma delta resolvase: comparison of the wild-type enzyme, the I110R mutant, and the C-terminal DNA binding domain in solution. Protein Sci Publ Protein Soc. 1997;6:1237–1247. doi: 10.1002/pro.5560060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Kamtekar S, Xiong Y, Sarkis GJ, Grindley NDF, Steitz TA. Structure of a synaptic gammadelta resolvase tetramer covalently linked to two cleaved DNAs. Science. 2005;309:1210–1215. doi: 10.1126/science.1112064. [DOI] [PubMed] [Google Scholar]
- 49.Kamtekar S, Ho RS, Cocco MJ, Li W, Wenwieser SVCT, Boocock MR, Grindley NDF, Steitz TA. Implications of structures of synaptic tetramers of gamma delta resolvase for the mechanism of recombination. Proc Natl Acad Sci USA. 2006;103:10642–10647. doi: 10.1073/pnas.0604062103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keenholtz RA, Rowland S-J, Boocock MR, Stark WM, Rice PA. Structural basis for catalytic activation of a serine recombinase. Struct Lond Engl. 2011;19:799–809. doi: 10.1016/j.str.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritacco CJ, Kamtekar S, Wang J, Steitz TA. Crystal structure of an intermediate of rotating dimers within the synaptic tetramer of the G-segment invertase. Nucleic Acids Res. 2013;41:2673–2682. doi: 10.1093/nar/gks1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soultanas P, Oram M, Halford SE. Site-specific recombination at res sites containing DNA-binding sequences for both Tn21 and Tn3 resolvases. J Mol Biol. 1995;245:208–218. doi: 10.1006/jmbi.1994.0017. [DOI] [PubMed] [Google Scholar]
- 53.Murley LL, Grindley ND. Architecture of the gamma delta resolvase synaptosome: oriented heterodimers identity interactions essential for synapsis and recombination. Cell. 1998;95:553–562. doi: 10.1016/s0092-8674(00)81622-0. [DOI] [PubMed] [Google Scholar]
- 54.Mazzarelli JM, Ermácora MR, Fox RO, Grindley ND. Mapping interactions between the catalytic domain of resolvase and its DNA substrate using cysteine-coupled EDTA-iron. Biochemistry (Mosc) 1993;32:2979– 2986. doi: 10.1021/bi00063a008. [DOI] [PubMed] [Google Scholar]
- 55.Blake DG, Boocock MR, Sherratt DJ, Stark WM. Cooperative binding of Tn3 resolvase monomers to a functionally asymmetric binding site. Curr Biol CB. 1995;5:1036–1046. doi: 10.1016/s0960-9822(95)00208-9. [DOI] [PubMed] [Google Scholar]
- 56.Nöllmann M, Byron O, Stark WM. Behavior of Tn3 resolvase in solution and its interaction with res. Biophys J. 2005;89:1920–1931. doi: 10.1529/biophysj.104.058164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krasnow MA, Cozzarelli NR. Site-specific relaxation and recombination by the Tn3 resolvase: recognition of the DNA path between oriented res sites. Cell. 1983;32:1313–1324. doi: 10.1016/0092-8674(83)90312-4. [DOI] [PubMed] [Google Scholar]
- 58.Rice PA, Steitz TA. Model for a DNA-mediated synaptic complex suggested by crystal packing of gamma delta resolvase subunits. EMBO J. 1994;13:1514–1524. doi: 10.2210/pdb1gdr/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leschziner AE, Grindley NDF. The architecture of the gammadelta resolvase crossover site synaptic complex revealed by using constrained DNA substrates. Mol Cell. 2003;12:775–781. doi: 10.1016/s1097-2765(03)00351-4. [DOI] [PubMed] [Google Scholar]
- 60.Rowland S-J, Stark WM, Boocock MR. Sin recombinase from Staphylococcus aureus: synaptic complex architecture and transposon targeting. Mol Microbiol. 2002;44:607–619. doi: 10.1046/j.1365-2958.2002.02897.x. [DOI] [PubMed] [Google Scholar]
- 61.Swinger KK, Lemberg KM, Zhang Y, Rice PA. Flexible DNA bending in HU-DNA cocrystal structures. EMBO J. 2003;22:3749–3760. doi: 10.1093/emboj/cdg351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rowland S-J, Boocock MR, Stark WM. DNA bending in the Sin recombination synapse: functional replacement of HU by IHF. Mol Microbiol. 2006;59:1730–1743. doi: 10.1111/j.1365-2958.2006.05064.x. [DOI] [PubMed] [Google Scholar]
- 63.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 64.Mouw KW, Rowland S-J, Gajjar MM, Boocock MR, Stark WM, Rice PA. Architecture of a serine recombinase-DNA regulatory complex. Mol Cell. 2008;30:145–155. doi: 10.1016/j.molcel.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alonso JC, Weise F, Rojo F. The Bacillus subtilis histone-like protein Hbsu is required for DNA resolution and DNA inversion mediated by the beta recombinase of plasmid pSM19035. J Biol Chem. 1995;270:2938–2945. doi: 10.1074/jbc.270.7.2938. [DOI] [PubMed] [Google Scholar]
- 66.Rojo F, Alonso JC. The beta recombinase of plasmid pSM19035 binds to two adjacent sites, making different contacts at each of them. Nucleic Acids Res. 1995;23:3181–3188. doi: 10.1093/nar/23.16.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLean MM, Chang Y, Dhar G, Heiss JK, Johnson RC. Multiple interfaces between a serine recombinase and an enhancer control site-specific DNA inversion. eLife. 2013;2:e01211. doi: 10.7554/eLife.01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lassila JK, Zalatan JG, Herschlag D. Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu Rev Biochem. 2011;80:669–702. doi: 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cassano AG, Anderson VE, Harris ME. Understanding the transition states of phosphodiester bond cleavage: insights from heavy atom isotope effects. Biopolymers. 2004;73:110–129. doi: 10.1002/bip.10517. [DOI] [PubMed] [Google Scholar]
- 70.Castell SE, Jordan SL, Halford SE. Site-specific recombination and topoisomerization by Tn21 resolvase: role of metal ions. Nucleic Acids Res. 1986;14:7213–7226. doi: 10.1093/nar/14.18.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reed RR, Moser CD. Resolvase-mediated recombination intermediates contain a serine residue covalently linked to DNA. Cold Spring Harb Symp Quant Biol. 1984;49:245–249. doi: 10.1101/sqb.1984.049.01.028. [DOI] [PubMed] [Google Scholar]
- 72.Boocock MR, Zhu X, Grindley ND. Catalytic residues of gamma delta resolvase act in cis. EMBO J. 1995;14:5129–5140. doi: 10.1002/j.1460-2075.1995.tb00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leschziner AE, Boocock MR, Grindley ND. The tyrosine-6 hydroxyl of gamma delta resolvase is not required for the DNA cleavage and rejoining reactions. Mol Microbiol. 1995;15:865–870. doi: 10.1111/j.1365-2958.1995.tb02356.x. [DOI] [PubMed] [Google Scholar]
- 74.Olorunniji FJ, Stark WM. The catalytic residues of Tn3 resolvase. Nucleic Acids Res. 2009;37:7590–7602. doi: 10.1093/nar/gkp797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guillén Schlippe YV, Hedstrom L. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch Biochem Biophys. 2005;433:266–278. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 76.Keenholtz RA, Mouw KW, Boocock MR, Li N-S, Piccirilli JA, Rice PA. Arginine as a general acid catalyst in serine recombinase-mediated DNA cleavage. J Biol Chem. 2013;288:29206–29214. doi: 10.1074/jbc.M113.508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bernstein HD, Poritz MA, Strub K, Hoben PJ, Brenner S, Walter P. Model for signal sequence recognition from amino-acid sequence of 54K subunit of signal recognition particle. Nature. 1989;340:482–486. doi: 10.1038/340482a0. [DOI] [PubMed] [Google Scholar]
- 78.Keenan RJ, Freymann DM, Walter P, Stroud RM. Crystal structure of the signal sequence binding subunit of the signal recognition particle. Cell. 1998;94:181–191. doi: 10.1016/s0092-8674(00)81418-x. [DOI] [PubMed] [Google Scholar]
- 79.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reed RR, Grindley ND. Transposon-mediated site-specific recombination in vitro: DNA cleavage and protein–DNA linkage at the recombination site. Cell. 1981;25:721–728. doi: 10.1016/0092-8674(81)90179-3. [DOI] [PubMed] [Google Scholar]
- 81.Bai H, Sun M, Ghosh P, Hatfull GF, Grindley NDF, Marko JF. Single-molecule analysis reveals the molecular bearing mechanism of DNA strand exchange by a serine recombinase. Proc Natl Acad Sci USA. 2011;108:7419–7424. doi: 10.1073/pnas.1018436108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McIlwraith MJ, Boocock MR, Stark WM. Tn3 resolvase catalyses multiple recombination events without intermediate rejoining of DNA ends. J Mol Biol. 1997;266:108–121. doi: 10.1006/jmbi.1996.0765. [DOI] [PubMed] [Google Scholar]
- 83.Colloms SD, Merrick CA, Olorunniji FJ, Stark WM, Smith MCM, Osbourn A, Keasling JD, Rosser SJ. Rapid metabolic pathway assembly and modification using serine integrase site-specific recombination. Nucleic Acids Res. 2014;42:e23. doi: 10.1093/nar/gkt1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]