

Abstract
Pediatric solid tumors arise from endodermal, ectodermal, or mesodermal lineages1. Although the overall survival of children with solid tumors is 75%, that of children with recurrent disease is below 30%2. To capture the complexity and diversity of pediatric solid tumors and establish new models of recurrent disease, we developed a protocol to produce orthotopic patient-derived xenografts (O-PDXs) at diagnosis, recurrence, and autopsy. Tumor specimens were received from 168 patients, and 67 O-PDXs were established for 12 types of cancer. The origins of the O-PDX tumors were reflected in their gene-expression profiles and epigenomes. Genomic profiling of the tumors, including detailed clonal analysis, was performed to determine whether the clonal population in the xenograft recapitulated the patient’s tumor. We identified several drug vulnerabilities and showed that the combination of a WEE1 inhibitor (AZD1775), irinotecan, and vincristine can lead to complete response in multiple rhabdomyosarcoma O-PDX tumors in vivo.
One barrier to identifying and validating biomarkers that predict sensitivity to molecularly targeted therapeutics is the lack of preclinical models that capture the diversity of pediatric solid tumors. For adult cancers, several important advances have been achieved in developing patient-derived organoids for colon, prostate, and pancreatic cancer3–5, and this has led to an international collaboration called the Human Cancer Model Initiative for developing cancer and normal organoids for the research community. There are also international efforts to develop patient-derived xenografts (PDXs) for adult leukemias and solid tumors, including the EuroPDX consortium, the Public Repository of Xenografts, and the NCI Patient-Derived Models Repository6–9. Pediatric solid tumors are rare, relative to adult cancers, and access to tissue is a barrier to developing pediatric organoids or PDX models of solid tumors10.
To obtain fresh tumor tissue from children with solid tumors, we developed a protocol (NCT01050296) called Molecular Analysis of Solid Tumors (MAST). Between 2010 and 2015, 225 patients consented to the protocol and we received 192 tumor specimens from 168 patients. Of the 192 specimens, 148 (77%) were injected into immunocompromised mice (Supplementary Table 1, Fig. 1a)11. Once the individual O-PDXs grow in the mice, they are expanded for banking and undergo molecular, cellular, and histologic characterization concurrently with the patient tumor.
Figure 1. Generation of pediatric solid tumor O-PDX models.
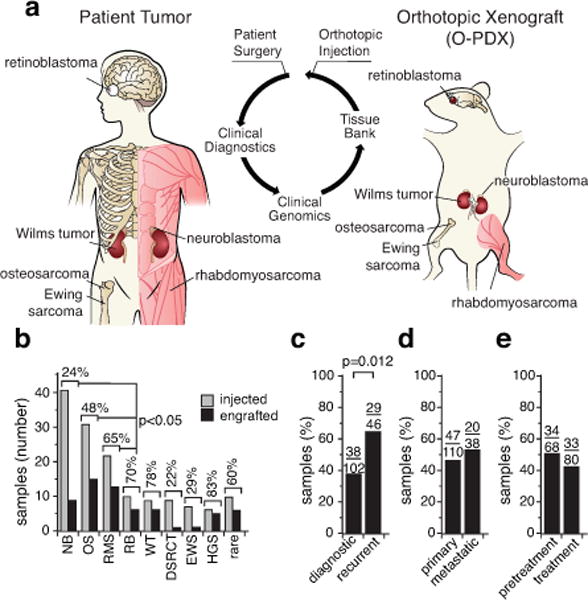
(a) Drawing of orthotopic implantation in immunocompromised mice. (b) Histogram of engraftment efficiency by tumor type. (c–e) Histograms of engraftment efficiency for diagnostic and recurrent samples (c), primary and metastatic samples (d), and pretreatment versus samples collected during treatment (e). The number of tumor specimens are indicated over each bar from Supplementary Table 1. Abbreviations: DSRCT, desmoplastic small round cell tumor; EWS, Ewing sarcoma; HGS, high-grade sarcoma; NB, neuroblastoma; OS, osteosarcoma; RB, retinoblastoma; RMS, rhabdomyosarcoma; WT, Wilms tumor.
In total, we injected 15 different types of pediatric solid tumors, including 41 neuroblastomas, 31 osteosarcomas, 20 rhabdomyosarcomas, 10 retinoblastomas, 9 Wilms tumors, 9 desmoplastic small round-cell tumors, 7 Ewing sarcomas, 6 high-grade sarcomas, and 5 adrenocortical carcinomas (Fig. 1b and Supplementary Table 1). Additionally, 10 tumor specimens representing 6 rare tumor types were injected. We have successfully established 67 O-PDXs from 12 different pediatric solid tumor types (Fig. 1b).
The overall engraftment efficiency was 45% (67/148) (Supplementary Table 1). The highest rates of engraftment were for high-grade sarcoma (83%), Wilms tumor (78%), retinoblastoma (70%), and rhabdomyosarcoma (65%) (Fig. 1b). Recurrent tumor samples were significantly more likely to engraft (63%) than were diagnostic samples (37%) (p=0.012; Fig. 1c). There was a similar engraftment rate for samples from metastatic (53%) and primary (43%) sites (Fig. 1d). The engraftment efficiency of samples obtained prior to chemotherapy was similar (50%) to that of samples obtained during chemotherapy (41%) (Fig. 1e).
We performed hematoxylin and eosin staining and immunohistochemistry on 51 O-PDX/patient tumor pairs (Extended Data Fig. 1a–d, Supplementary Table 2). 49 of the tumors were evaluable for immunostaining, and 98% (48/49) were concordant between the O-PDX and the patient tumor. We scored the proportion of proliferating cells as measured by Ki67 immunostaining and dying cells as measured by activated caspase 3 immunostaining. We also performed tumor type—specific protein immunohistochemical staining (Supplementary Table 2). Neuroblastomas were stained for synaptophysin (Extended Data Fig. 1e), rhabdomyosarcomas for myogenin and MyoD1, osteosarcomas for SATB2, Ewing sarcomas for FLI-1, high-grade sarcomas for vimentin, liposarcomas for S100, retinoblastomas for CRX, and rhabdoid tumors for INI1. In total, 1,173 slides were examined and evaluated microscopically. Generally, there was more proliferation (Ki67+) and less cell death (activated caspase 3+) in the O-PDXs than in the patient tumors. Each of the tumor type–specific immunohistochemical stains were concordant between the patient tumors and the O-PDXs, except for SJRHB010928_X1, which was discordant by histopathologic analysis.
Next, we surveyed 36 of the O-PDX tumors by transmission electron microscopy (TEM) to characterize subcellular features of each tumor type (Supplementary Table 2). Rhabdomyosarcomas had intracellular myofibrils; neuroblastomas had dense core vesicles; liposarcomas had large lipid droplets, and osteosarcomas had swollen endoplasmic reticulum and collagen deposits (Extended Data Fig. 1f–i). Overall, the O-PDX tumors retained the cellular and subcellular features of the patient tumors (Supplementary Table 2).
To determine whether the O-PDX tumors retain the somatic mutations in the patient tumors and whether they acquire additional mutations, we performed whole-genome sequencing (WGS) and whole-exome sequencing (WES) of the O-PDXs and the matched patient tumor and germline samples (51 O-PDX/tumor pairs). For some samples, mutations were detected in the O-PDX but not the patient tumor and vice versa (Extended Data Fig. 2 and Supplementary Table 3). These differences in detection of mutations may be due to patient tumor purity, clonal changes, or sequence coverage. To distinguish amongst these possibilities, we performed capture enrichment and Illumina sequencing for 32,113 single nucleotide variations (SNVs) identified across the sample cohort. The 2 mutations that were lost in the O-PDXs showed that both were due to shifts in the clonal composition. Among the 14 mutations that were gained in the O-PDXs, 6 were not surveyed due to copy number alterations (CNAs), 2 were due to low tumor purity, 1 was due to insufficient coverage in the patient tumor, and the remaining 5 were bona fide new SNVs acquired in the O-PDX. Together, these data suggest that the O-PDXs faithfully maintain many features of the genomic landscape of the patient tumor from which they were derived.
To determine whether the clonal composition of O-PDX tumors changes relative to the patient tumors, we performed clonal analysis using the deep-sequence reads for all the SNVs across 42 of the patient tumors and their matched O-PDXs (Supplementary Table 4). In total, 24% (10/42) of the O-PDXs faithfully preserved the clonal composition of the patient tumor (Group 1, Fig. 2a). Thirty-one percent (13/42) maintained the major clonal features of the patient tumor in the O-PDX but continued to clonally evolve in the mice (Group 2, Fig. 2b). Some O-PDX tumors (33%; 14/42) were derived from the major clone in the patient tumor but showed evidence of clonal loss (Group 3, Fig. 2c), and 12% (5/42) of the O-PDX tumors had clonal loss and were derived from a minor clone (<10% of patient tumor) (Group 4, Fig. 2d). Osteosarcomas had the best clonal preservation and neuroblastomas had the least (Fig. 2e).
Figure 2. Clonal preservation in O-PDX tumors.
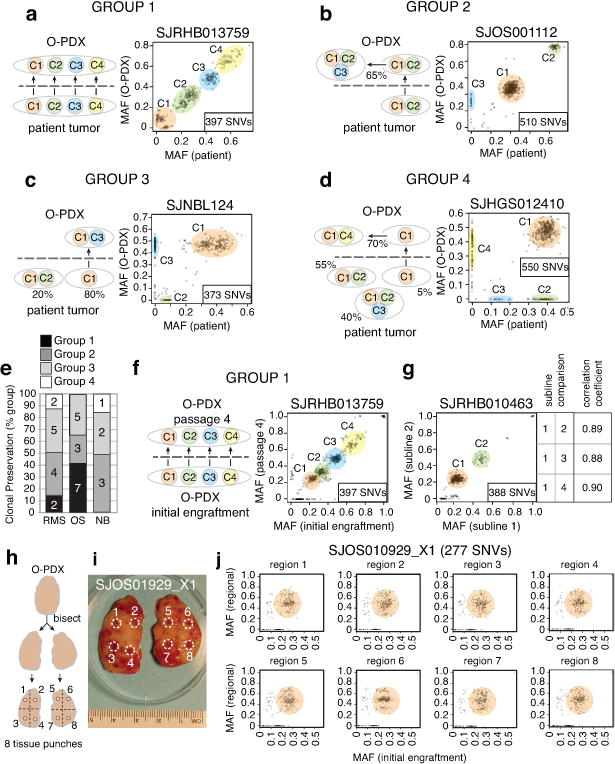
(a–d) Representative scatterplots of mutant allele frequency (MAFs) for the SNVs analyzed by capture-based Illumina sequencing. Individual clones are color-coded and labeled C1–C4. (a) Group 1 includes O-PDX tumors that preserved the clonal diversity and ratios of the patient tumors. (b) Group 2 includes O-PDX tumors that preserved the clonal diversity and ratios of the patient tumors and continued to evolve in the mice. (c) Group 3 includes O-PDX tumors that lost at least 1 clone in the patient tumor but were derived from a major clone. (d) Group 4 includes O-PDX tumors that were derived from a minor clone (<10%). (e) Stack histogram of the percentage of groups 1–4 clonal classification by tumor type. The number of samples in each group are indicated on the histogram. (f) Comparison of clonal composition at initial engraftment and after passage 4. (g) Comparison of multiple sublines from the same patient. Clonal composition across those sublines is indicated in the box. (h,i) Drawing and photograph of the sampling of 8 regions of the O-PDX tumors for analysis of regional clonal heterogeneity. (j) Clonal analysis for all 8 regions of the tumor shown in (i) showing clonal preservation across all 8 regions. Abbreviations: NB, neuroblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma; SNVs, single-nucleotide variations.
To determine if the clonal composition of O-PDX tumors was stable with passages in vivo, we performed clonal analysis on 18 early-passage (initial engraftment or passage 1) and late-passage (passages 4–6) O-PDX tumor pairs (Supplementary Table 4). Forty-four percent (8/18) of the O-PDX tumors had clonal preservation (Group 1) at late passage relative to early passage (Fig. 2f), and a similar number (7/18) maintained the major clonal features of the early-passage tumor but continued to evolve (Group 2).
We also analyzed the different O-PDX sublines and regional clonal heterogeneity in the tumor mass. The patient tumors were initially implanted in up to 10 immunocompromised mice, and each tumor that grew was designated as a separate subline. For this analysis, we selected 11 O-PDX tumors with multiple sublines (range, 2–5 sublines per tumor) and performed clonal analysis (Supplementary Table 4). All tumors were from initial engraftment or the first passage. Sixty-four percent (7/11) of the O-PDX models had clonal preservation across 2 sublines (Group 1, Fig. 2g), and the remaining 4 had features of the major clone but showed some clonal evolution (Group 2). To test the regional heterogeneity within tumors, we selected 6 O-PDX tumors and sampled 8 regions of the tumor using a biopsy punch (Fig. 2h,i). Clonal analysis of those samples showed clonal preservation (Group 1, Fig. 2j, and Supplementary Table 4).
To compare the gene-expression profiles of patient tumors to those of their matched O-PDXs, we performed RNA sequencing (RNA-seq) on all 102 samples that were analyzed by WGS and WES. We calculated the correlation coefficient from the Log2 transformed FPKM value for each pair and grouped the data by tumor type (Extended Data Fig. 3a and Supplementary Table 5). There was no difference in the O-PDX/patient tumor correlation coefficients for the RNA-seq data by tumor type (p = 0.62, Kruskal-Wallis test). However, there was a significant inverse correlation between the patient tumor purity calculated from the WGS data and the O-PDX/patient tumor RNA-seq correlation coefficients for neuroblastoma and rhabdomyosarcoma (Extended Data Fig. 3b–d). Patient tumors with low tumor purity contained more infiltrating normal cells than did those with higher tumor purity. Upon transplantation into immunocompromised mice, the normal cells were lost, which can lead to a lower correlation for the RNA-seq of the patient tumor and matched O-PDX sample (Extended Data Fig. 3).
To determine whether the developmental origins are preserved in the tumors, we identified the genes that were significantly upregulated in neuroblastoma, osteosarcoma and rhabdomyosarcoma (Supplementary Table 5). The most significantly enriched pathways in neuroblastoma were those for neuronal development and homeostasis, rhabdomyosarcomas were enriched for myogenesis and osteosarcomas were enriched for pathways in bone development and collagen production. To determine whether this association is reflected in the epigenetic landscape of the tumors, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for 9 histone marks (H3K27Ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K9-14Ac, H3K9me2, H3K9me3), RNA Pol II, CTCF, and BRD4. We used chromatin prepared from a representative embryonal rhabdomyosarcoma (ERMS, SJRHB000026_X1), an alveolar rhabdomyosarcoma (ARMS, SJRHB010463_X16), an osteosarcoma (SJOS001112_X1), and a neuroblastoma (SJNBL046_X). We performed ChIP-seq from biological replicates of each sample for a total of 96 ChIP-seq libraries. We performed chromatin Hidden Markov Modeling (chromo-HMM)12 using 16 states (Fig. 3a,b). States 1–6 had epigenetic marks consistent with enhancers and active promoters; state 7 contained epigenetic marks of bivalent (H3K27me3/H3K27Ac) poised promoters/enhancers; and states 8–10 included epigenetic marks of gene bodies. State 11 comprised polycomb-repressed genomic regions, and state 12 was an empty state lacking ChIP-seq peaks in our analysis. States 13 and 14 consisted of heterochromatin, and state 15 represented repressed chromatin (H3K27me3 and H3K9me3). The CTCF insulator protein–bound regions were enriched in state 16.
Figure 3. O-PDX tumors retain epigenetic and molecular signatures of their diverse cellular origins.
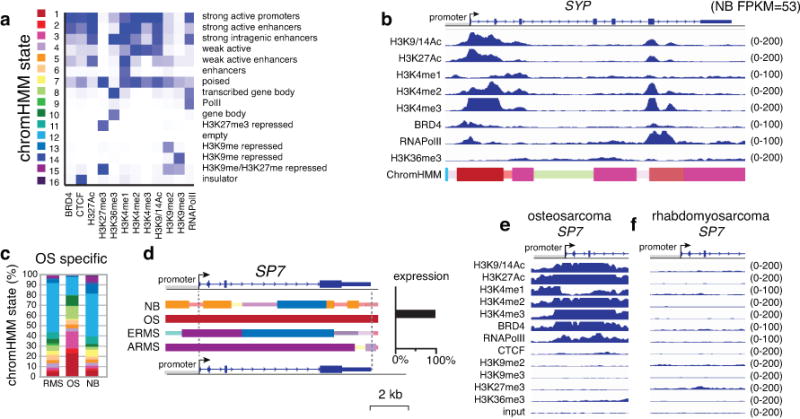
(a) Heatmap of the chromHMM states used in this study. (b) ChIP-seq data from the neuroblastoma O-PDX for the synaptophysin (SYP) gene (RNA-seq FPKM=53 in neuroblastoma but <1.0 for other tumor types). The chromHMM states for each region of the gene are indicated below the ChIP-seq peaks, and the scale for the ChIP-seq is indicated on the right of each track. The 2 states that are the highest proportion are full-height bars, and the remaining 9 states are half height. The intensity of each bar is proportional to the percentage of each state across all samples for that gene. For the bars that are half the height, the intensity is scaled, starting at 50% of maximum intensity. (c) Stack histogram of the percentage of each of the 16 chromHMM states for the genes expressed specifically in osteosarcoma across the 3 tumor types (OS, NB, RMS). (d) ChromHMM and expression of a gene (SP7) that is specifically expressed in osteosarcoma. (e,f) ChIP-seq peaks for all 12 antibodies for the SP7 promoter in osteosarcoma and rhabdomyosarcoma O-PDXs. Abbreviations: chromHMM, chromatin Hidden Markov Modeling; NB, neuroblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma.
Most lineage-specific genes that were expressed at high levels in a particular tumor type had tumor-specific enrichment in active chromo-HMM states (states 1–6) (Fig. 3c and Extended Data Fig. 4). The chromo-HMM state for those genes in the tumor types where they are not expressed were often classified as empty (chromo-HMM state 12) or H3K27me3-repressed (chromo-HMM states 7, 11, 15) (Fig. 3c and Extended Data Fig. 4). For example, the SP7 gene was expressed in osteosarcoma but not in neuroblastoma or rhabdomyosarcoma (Fig. 3d). The promoter of the SP7 gene has an HMM state of a strong active promoter (state 1) in osteosarcoma, but it is silent in rhabdomyosarcoma (Fig. 3d,e). Similar patterns were found for genes expressed specifically in rhabdomyosarcoma and neuroblastoma (Extended Data Fig. 4). Together, these data suggest that the lineage-specific developmental program is strongly preserved in the epigenome of pediatric solid tumors (Supplementary Table 5).
To determine whether our O-PDX tumors demonstrate differential drug sensitivity, we developed primary culture conditions for each tumor type for high throughput drug screening. We screened 16 rhabdomyosarcomas, 8 osteosarcomas, 4 neuroblastomas, 1 Ewing sarcoma, and 1 rhabdoid tumor. For comparison, we also included 21 pediatric solid tumor cell lines (Supplementary Table 6); 156 drugs were used in this analysis for a total of over 500,000 individual data points. Of the 1,960 plates screened, 49 (2.5%) had z-prime < 0 and were excluded from further analysis. Of the remaining plates, the average z-prime was 0.57, and 95% of all plates has z-prime between 0.27 and 0.82 (Extended Data Fig. 5a). Drugs with the same mechanism of action had similar activity across O-PDXs and cell lines (Extended Data Fig. 5b). Mutations in signal transduction pathways were predictive of tumor response in a subset of tumors (Extended data Fig. 5c). Validated data were entered into a central database (https://stjude.org/cstn-drug-sensitivity), and curve-fitting was performed to determine the EC50 of each drug for each tumor (Supplementary Table 6). Growth rate can influence drug sensitivity for some classes of drugs in high-throughput screening experiments13 so we corrected for differences in growth rate (Supplementary Table 6). The most commonly used broad-spectrum chemotherapeutics were active across several tumor types and histone deacetylase (HDAC) and proteasome inhibitors were particularly active across tumor types (Supplementary Table 6 and https://stjude.org/cstn-drug-sensitivity).
Among the drugs with tumor subtype–specific activity, the WEE1 inhibitor AZD1775 (MK-1775) was of particular interest for rhabdomyosarcoma because a phase I study of AZD1775 with irinotecan (IRN) was recently completed by the Children’s Oncology Group (COG trial: NCT02095132). This is important because IRN and vincristine (VCR) are used to treat recurrent rhabdomyosarcoma, and given the sensitivity of rhabdomyosarcoma O-PDXs to AZD1775, combining it with IRN and VCR may be justified for future clinical trials. Indeed, most of the rhabdomyosarcoma O-PDX tumors were sensitive to AZD1775, whether they were obtained at diagnosis or recurrence (Supplementary Table 6).
To establish a clinically relevant murine-equivalent dose for bortezomib (proteasome inhibitor), panobinostat (HDAC inhibitor), and AZD1775 (WEE1 inhibitor), we performed plasma and tumor pharmacokinetic studies of SJRHB000026_X1 tumor-bearing mice (Extended Data Fig. 6a–c and Supplementary Information). Preclinical phase I studies were performed on athymic nontumor-bearing mice to establish the tolerability of the drug combination at clinically relevant doses and schedules14,15. We used O-PDXs derived from 2 high-risk ERMS tumors that were resected at recurrence (SJRHB000026_X1 and SJRHB012_Y) and a high-risk ARMS tumor resected at recurrence (SJRHB013759_X1) (Supplementary Table 1). We introduced a luciferase reporter gene into the O-PDXs, orthotopically injected the tumor cells into immunocompromised mice and several weeks later, the mice were enrolled in a preclinical phase II trial as described previously14,15. Mice were screened weekly using bioluminescence, and 4–6 courses (21 days per course) of therapy were administered (Extended Data Fig. 6d). Mice with a tumor burden greater than 20% of their body weight were removed from the study and scored as progressive disease (PD). Stable disease (SD), partial responses (PR), and complete responses (CR) were scored as done previously14,15. All of the mice treated with panobinostat and bortezomib had PD for all 3 O-PDX tumors tested (Fig. 4a,b, Extended Data Fig. 6e and Supplementary Table 7). The mice treated with AZD1775+VCR+IRN had a better response than those treated with VCR+IRN alone (Extended Data Fig. 6f–h and Supplementary Table 7).
Figure 4. Preclinical phase III using O-PDX models.
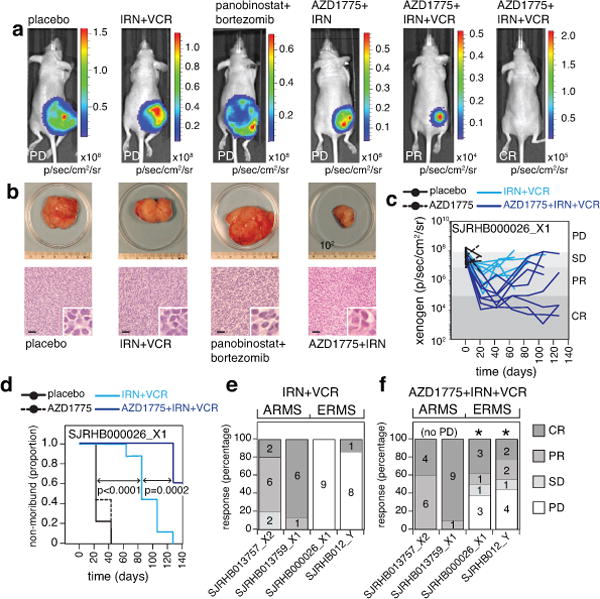
(a) Xenogen images showing luciferase bioluminescence for progressive disease (PD), partial response (PR), and complete response (CR). (b) Photographs of tumors for each treatment group corresponding to the images above in (a) and micrographs of corresponding hematoxylin and eosin–stained sections. (c) Line graph of tumor response for 1 of the 4 RMS O-PDX models used in the preclinical phase III study. (d) Survival curve for the mice shown in (c) for each of the 4 treatment groups. (e,f) Stack bar plots of the percentage response for each of the 4 O-PDX models in the preclinical phase III study for standard of care (IRN+VCR) (e) and the addition of AZD1775 (f). The (*) indicates those models that had a significant difference in tumor progression for the AZD1775+IRN+VCR relative to IRN+VCR. The source data are in Supplementary Tables 7,8. Abbreviations: IRN, irinotecan; VCR, vincristine; ARMS, alveolar rhabdomyosarcoma; ERMS, embryonal rhabdomyosarcoma. Scale bars: b, 50 μm.
Based on the activity of AZD1775 combined with VCR and IRN in multiple O-PDX rhabdomyosarcoma models, we performed a double-blind, randomized, placebo-controlled preclinical phase III study, as described previously14,15. We used the same O-PDX models as above (SJRHB000026_X1, SJRHB012_Y, SJRHB013759_X1) and added another ARMS O-PDX (SJRHB013757_X2) (Supplementary Table 1). We enrolled 140 mice and randomized them to 4 treatment groups (placebo, AZD1775, VCR+IRN, and AZD1775+VCR+IRN; Supplementary Table 8). Each of the O-PDX tumors had an improved outcome with the triple-drug combination relative to the standard of care (VCR+IRN), AZD1775 alone, or placebo (Fig. 4c–f). Individual O-PDX tumors showed marked differences in response to VCR+IRN and AZD1775+VCR+IRN, highlighting the importance of using multiple O-PDX models for preclinical studies (Figure 4e,f and Supplementary Table 8). In addition, we performed clonal analysis on 17 O-PDX tumors from SJRHB000026_X1 mice with PD in 3 different treatment groups (AZD1775, VCR+IRN, and AZD1775+VCR+IRN). Fifty-nine percent (10/17) of those tumors showed clonal selection for the minor clone (Supplementary Table 4), which was similar to results from previously published data on patient tumors before and after treatment16.
Many of the O-PDXs that we developed are from patients with recurrent disease, which is important because there were previously few models of recurrent pediatric solid tumors. The O-PDXs retained the molecular and cellular features of the patient tumor and the epigenetic landscape of their developmental origins. The clonal composition of the patient tumors was compared to the O-PDX tumors at early and late passages, from different sublines and distinct locations within the tumor mass. Some O-PDX tumors retained the clonal composition of the patient tumor even after several passages in mice but others underwent clonal changes. For some tumors, different sublines captured different clones from the patient tumor suggesting that separately propagating individual sublines may be important for capturing the clonal heterogeneity in patient tumors. The drug screening identified chemotherapeutic agents that had broad activity as well as drugs that were more selective for particular cancer types. This approach led to the discovery that the combination of a WEE1 inhibitor with IRN and VCR can improve response across 4 different O-PDXs of high-risk rhabdomyosarcoma, including recurrent disease, relative to IRN and VCR alone. We demonstrated clonal selection in the treated tumors, as shown previously in patients16 and it will be important to directly relate the clonal selection in O-PDX models to patients in future studies. This is proof of principle for using O-PDX tumors for basic and translational research on pediatric solid tumors and it provides a useful platform for understanding clonal selection in recurrent disease. All of our O-PDX models and associated data are freely shared through the Childhood Solid Tumor Network (CSTN; www.stjude.org/CSTN/) with no obligation to collaborate.
METHODS
Animals
Athymic nude immunodeficient mice were purchased from Jackson Laboratories (strain code 007850). NSG mice were purchased from Jackson Laboratories (strain code 005557). C57BL/6 scid mice were purchased from Jackson Laboratories (strain code 001913). This study was carried out in strict accordance with the recommendations in the Guide to Care and Use of Laboratory Animals of the National Institute of Health. The protocol was approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital. All efforts were made to minimize suffering. All mice were housed in accordance with approved IACUC protocols. Animals were housed on a 12–12 light cycle (light on 6am off 6pm) and provided food and water ad libitum.
Patient Consent and MAST Protocol
Excess, deidentified tumor material was collected from solid tumor patients at St. Jude Children’s Research Hospital in agreement with local institutional ethical regulations and institutional review board approval. Patient consent for tissue acquisition was obtained under the guidelines of the MAST protocol.
Primary Patient Tissue Processing
Primary tumor tissue was processed for implantation within 2 hours of surgical resection in the majority of cases. Primary tumor was enzymatically dissociated into a single cell suspension and injected into an anatomically correct location for the disease type when possible. If the initial tumor sample was too small for dissociation, tumor tissue was implanted in the flank location. Initial implantation was primarily done into recipient NSG female mice with the exception of retinoblastoma which was implanted into C57BL/6 scid mice. After engraftment and sufficient tumor growth, the tumor was harvested and passaged into athymic nude mice using the same dissociation and implantation techniques.
Orthotopic Injections
(Bone Marrow Injections) To minimize distress and movement during the procedure, isoflurane gas anesthetic is used. The mouse is placed in the supine position on a nose cone prior to injection. The skin of the knee joint is prepped with alternating iodine scrubs and 70% isopropyl alcohol wipes. The prepped leg is flexed at the knee joint and secured to the work surface. The femur is palpated until the femoral condyles become visible. A 25-gauge needle on a 50 μL Hamilton glass syringe (Hamilton Cat# 80920) is held at a 45-degree angle to the mouse, and the needle tip is inserted into the femur via the femoral intercondylar notch, while retracting the patella and patella tendon medially to avoid ligament damage. The needle is advanced down to the femoral head, which is approximately 5–10 mm depending on the size of the mouse, and 10 μL of cell suspension is slowly injected into the femur. (Intramuscular injections) The mouse is restrained gently but firmly by the scruff method. The rear foot nearest to the investigator is secured beneath the little finger and lower thumb. The area to be injected is swabbed with 70% ethanol. The needle is inserted, bevel up, into the caudal thigh at a 45 degree angle and 50–100 μL of cell suspension is slowly injected into the muscle while avoiding injury to the sciatic nerve. (Intravitreal injections)C57BL/6 scid mice are given general anesthesia via isoflurane inhalation continuously at 1–3% concentration with an oxygen flow rate of 2 Liter/minute. The mouse is placed under the microscope where the eye is proptosed and a small incision using the tip of an 18 gauge needle is created between the sclera and the cornea. Using a 5 μL glass Hamilton syringe (Hamilton Company, 7633-01, 65RN), cells are injected through a 33 gauge small hub needle into the vitreous of the eye at 5 μL per injection. (Para-adrenal injections) All ultrasound procedures are performed using the VEVO 2100 high frequency ultrasound equipped with a MS-550S transducer running at 40 MHz. Recipient immunocompromised mice are given general anesthesia via isoflurane inhalation continuously at 1–3% concentration with an oxygen flow rate of 2 Liter/minute. Anesthetized recipients are placed laterally on the imaging bed with left flank facing upward. In order to provide a channel for delivery of the implant, a 22 gauge catheter (BD Worldwide, Cat# 381423) is gently inserted through the skin and back muscle into the para-adrenal region and the hub is removed. A chilled 50 μL Hamilton glass syringe (Hamilton Cat# 80920) fitted with a 27 gauge 1.5 inch needle is loaded with 10 μl of suspension and guided steretoactically through the catheter and positioned between the kidney and adrenal gland using ultrasound. The cell suspension is then injected into the region and the needle is left in place for 30 seconds to permit the matrigel component to set. The needle is then slowly removed, followed by gentle removal of the catheter. The mouse is placed in a clean cage on a warmer to recover from anesthesia. (Flank implantation) For some tumor with sufficient material, we implanted tissue into the flank of immunocompromised mice as a backup to the orthotopic injection. Recipient immunocompromised mice are given general anesthesia via isoflurane inhalation continuously at 1–3% concentration with an oxygen flow rate of 2 Liter/minute. The mouse is placed ventral side down with a nose-cone to provide continuous anesthesia. The area from mid-spine to the tail base is cleaned with 70% ethanol. A small horizontal 5mm incision in the flank area is made using sterile small surgical scissors. The tip of the sterile scissors is inserted into the incision, directly over the flank, and the scissors are opened to introduce a pocket in the subcutaneous space. One individual piece of tumor tissue is inserted into the pocket using sterile forceps. One drop of 100X penicillin/streptomycin solution is inserted into the opening over the tissue piece. The incision is closed with Vetbond tissue adhesive (3M Cat# 1469SB). The overlying skin is held together for 3–5 seconds with forceps to allow adequate time for drying.
Enzymatic Tumor Dissociation
(Neuroblastoma) Tumor was minced with sterile scalpels and rinsed in phosphate buffered saline without calcium or magnesium (PBS-minus solution). Tumor suspension was transferred to a 50 mL conical tube and filled with PBS-minus solution. Dissociation was done by adding 600 μl of trypsin (10 mg/ml, Sigma Cat#T9935) and tube placed in 37 degree water bath for 10 minutes. Dissociation was stopped by adding 60μL of Soybean Trypsin Inhibitor (10 mg/ml, Sigma Cat#T6522). Deoxyribonuclease I (2 mg/ml, Sigma Cat#D4513) and magnesium chloride (1 M) were added in equal amounts of 60 μL increments until tumor fragments easily settle at the bottom of the tube. Tumor suspension is filtered with a 40 micron cell strainer and then centrifuged at 500g (g=RCF) for 5 minutes. Supernatant was discarded and 10 ml of red blood cell lysis solution (5 Prime Cat#2301310) added and allowed to incubate at room temperature for 10 minutes. Solution of phosphate buffered saline without calcium or magnesium (PBS-minus, Lonza Cat#17-516F)/10% Fetal Bovine Serum (FBS, Biowest Cat#SO1520) was added to fill 50 ml conical tube and cell suspension centrifuged at 500g (g=RCF) for 5 minutes. Supernatant was discarded and cell pellet was resuspended in PBS-minus/10%FBS for counting. Cells were then resuspended in Matrigel™ basement membrane matrix (BD Biosicences cat. 354234) at a concentration of 2 × 105 cells per 10 μL and placed on ice for injection. (Soft tissue sarcoma) Tumor was placed through a tumor press and then rinsed with Dulbecco’s modified Eagle’s medium (DMEM) (Lonza catalog no. 12- 604F). The tumor suspension was transferred to a 50 ml conical tube and filled with DMEM. Dissociation was done by adding 600 μL of trypsin (10 mg/ml; Sigma catalog no. T9935) and 50 mg of type II collagenase (275 U/mg; Worthington Biochemical catalog no. 4177), and then the tube was placed in a 37C water bath for 1 hour. Dissociation was stopped by adding 600 μL of soybean trypsin inhibitor (10 mg/ml; Sigma catalog no. T6522). Deoxyribonuclease I (2 mg/ml; Sigma catalog no. D4513) and magnesium chloride (1 M) were added in equal amounts in 60 μL increments until tumor fragments easily settled at the bottom of the tube. The tumor suspension was filtered with a 40 micron cell strainer and centrifuged at 500g (g=RCF) for 5 min. The supernatant was discarded, and 10 ml of red blood cell lysis solution (5 PRIME catalog no. 2301310) was added and allowed to incubate at room temperature for 10 min. A solution of PBS without calcium or magnesium (PBS-minus; Lonza catalog no. 17-516F)/10% fetal bovine serum (FBS) (Biowest catalog no. SO1520) was added to fill a 50 ml conical tube, and the cell suspension was centrifuged at 500g for 5 min. The supernatant was discarded, and the cell pellet was resuspended in PBS-minus/10% FBS for counting. Cells were then resuspended in matrigel (BD Worldwide catalog no. 354234) at a concentration of 1 × 106 per 100 μL and placed on ice for injection. (Osteosarcoma) The tumor was placed through a tumor press and then rinsed with PBS without calcium or magnesium (PBS-minus; Lonza catalog no. 17-516F). The tumor suspension was transferred to 100 ml screw cap glass bottle and filled with PBS-minus to the 100 ml mark. Dissociation was done by adding 600 μL of trypsin (10 mg/ml; Sigma catalog no. T9935) and 200 mg of type II collagenase (275 U/mg; Worthington Biochemical catalog no. 4177), and placed in a warm 37 degree water bath for 90 min, agitating with a magnetic bead at ~200 rpm aiming to have all tissue circulating and lifted off the bottom of the glass bottle. Dissociation was stopped by adding 600 μL of soybean trypsin inhibitor (10 mg/ml; Sigma catalog no. T6522). Deoxyribonuclease I (2 mg/ml; Sigma catalog no. D4513) and magnesium chloride (1 M) were added in equal amounts in 60 μL increments until tumor fragments easily settled at the bottom of the tube. The tumor suspension was filtered with a 70 micron cell strainer and centrifuged at 500 g (g=RCF) for 5 min. The supernatant was discarded, and 10 ml of red blood cell lysis solution (5 PRIME catalog no. 2301310) was added and allowed to incubate at room temperature for 10 min. A solution of PBS-minus/10% fetal bovine serum (FBS) (Biowest catalog no. SO1520) was added to fill a 50 ml conical tube, and the cell suspension was centrifuged at 500 g for 5 min. The supernatant was discarded, and the cell pellet was resuspended in PBS-minus/10% FBS for counting. Cells were then resuspended in Matrigel™ basement membrane matrix (BD Biosicences cat. 354234) at a concentration of 1 × 106 cells per 10 μL and placed on ice for injection. (Retinoblastoma) Tumor was minced with sterile scalpels and rinsed in RPMI (Lonza catalog no 12-167F). Tumor suspension was transferred to a 50 mL conical tube and filled with RPMI. Dissociation was done by adding 600 μL of trypsin (10 mg/ml, Sigma Cat#T9935) and tube placed in 37 degree water bath for 10 minutes. Dissociation was stopped by adding 600 μL of Soybean Trypsin Inhibitor (10 mg/ml, Sigma Cat#T6522). Deoxyribonuclease I (2 mg/ml, Sigma Cat#D4513) and magnesium chloride (1 M) were added in equal amounts of 60 μL increments until tumor fragments easily settle at the bottom of the tube. Tumor suspension is filtered with a 40 micron cell strainer and then centrifuged at 500g (G=RCF) for 5 minutes. Supernatant was discarded and 10ml of red blood cell lysis solution (5 Prime Cat#2301310) added and allowed to incubate at room temperature for 10 minutes. Solution of phosphate buffered saline without calcium or magnesium (PBS-minus, Lonza Cat#17-516F)/10% Fetal Bovine Serum (FBS, Biowest Cat#SO1520) was added to fill 50 ml conical tube and cell suspension centrifuged at 500g (G=RCF) for 5 minutes. Supernatant was discarded and cell pellet was resuspended in PBS-minus/10%FBS for counting. Cells were then resuspended in RPMI at a concentration of 1 × 105 cells per 5 μL for injection.
Tumor cryopreservation
After dissociation, tumor cells that were not utilized for passaging or high throughput screening were cryopreserved for banking and later usage. Cells were counted and resuspended in chilled FBS/10% DMSO at a concentration of 6 × 106 cells per 1 ml per tube for all tumor types with the exception of retinoblastoma at a concentration of 1 × 106 cells per 1 ml. Cryo tubes were placed in styrofoam containers and frozen at −80° C for 3 days and then transferred to liquid nitrogen for long term storage.
Histopathologic scoring
Histopathologic features of the patient H&E-stained slides and the O-PDX derived slides were compared for histologic similarity. These patient-xenograft pairs were placed into similarity categories based on evaluation of the following attributes: overall tumor cellularity, growth pattern, cytomorphology including degree of pleomorphism, and mitotic activity. The following classification system was used:
Same: No difference noted between patient and xenograft-derived samples.
Similar: Changes in growth pattern and overall cellularity allowed with no difference in pleomorphism or mitotic activity.
Morphologic Shift: Significant change such that the two lesions are morphologically dissimilar with significant difference in cellularity, cytomorphologic features, degree of pleomorphism, growth pattern, and mitotic activity.
Immunohistochemical Staining and Scoring
Patient samples and the matched xenograft tissues were assessed for Caspase 3 (total), Caspase 3 (cleaved), and Ki-67. In addition, a number of diagnostically delineating markers were performed based on the histopathologic diagnosis and included: Fli-1 (Ewing sarcoma), Vimentin (high-grade sarcoma not otherwise specified), S100-protein (liposarcoma), INI-1 (malignant rhabdoid tumor), CRX (retinoblastoma), synaptophysin (neuroblastoma), SATB2 (osteosarcoma), and myogenin and MyoD1 (rhabdomyosarcoma). In total, excluding controls, 446 immunohistochemical stained slides were reviewed and scored. 51 xenograft samples were reviewed by H&E staining, as were all pertinent slides from the primary patient specimens (~676 H&E stained slides). Total slides examined microscopically: 1,173. Formalin-fixed paraffin embedded tissue (4 μm in thickness) was utilized for staining with appropriate positive and negative controls evaluated. The antibodies are provided in Supplementary Table 2. Patient sample-xenograft pairs were evaluated for overall immunohistochemical concordance. Cases were considered discordant if any of the selected diagnostic markers (Fli-1, Vimentin, S100-protein, INI-1, SATB2, and CRX) were discrepant (positive vs. negative in the tumor cells). Only the expected localization of staining was considered evidence of positivity (or loss of staining as is the case with INI-1). As Myogenin and MyoD1 are known to display different degrees of positivity in rhabdomyosarcoma subtypes, these stains were scored to the nearest 5% of nuclear positivity. A case was considered discordant if there was >25% discrepancy between the estimated percentages for either Myogenin or MyoD1 between the patient and xenograft samples. Histology images were obtained using Aperio® ImageScope (Leica Biosystems).
Electron microscopy
Tumor samples were fixed with 2.5% glutaraldehyde, 2% parqaformadehyde in 0.1 M sodium cacodylate buffer pH 7.4 and post fixed in 2% osmium tetroxide in 0.1 M cacodylate buffer with 0.3% potassium ferrocyanide for 1.5 hours. After rinsing in same buffer the tissue was dehydrated through a series of graded ethanol to propylene oxide, infiltrated and embedded in epoxy resin and polymerized at 70° C overnight. Semithin sections (0.5 micron) were stained with toluidine blue for light microscope examination. Ultrathin sections (80 nm) were cut and imaged using an FEI Tecnai 200Kv FEG Electron Microscope with an ATM XR41 Digital Camera.
Whole Genome Sequencing
Whole genome sequencing and library construction was performed as described previously17–19 with the following modifications; 250–500 ng of genomic DNA was input for library construction using Illumina compatible adapters, and 4–6 cycles of amplification was performed with Kapa HiFi Hotstart ReadyMix (KAPA Biosystems). Identification of SNV, CNVs, SVs and Indels was performed as described previously17–19.
Whole Exome Sequencing
Whole exome sequencing was conducted using the SeqCap EZ HGSC VCRome (Roche) according to manufactures instructions.
RNA Preparation and RNA-Seq
RNA was isolated from individual TRIzol (Life Technologies) preparations via a phenol-chloroform extraction. Samples were first homogenized at 17,000 rpm for 30 seconds with a tissue homogenizer (Polytron, PT10-35GT). A 1:4 volume of chloroform (Sigma) was then added to each sample and incubated at room temperature for 3 minutes followed by centrifugation at 12,000 rcf at 4°C for 15 minutes. The aqueous layer was then transferred to a siliconized eppendorf tube followed by the addition of 2.0 μL glycogen (Roche) and 500 μL isopropanol (Fisher). Samples were incubated at room temperature for 10 minutes followed by centrifugation at 12,000 rcf at 4°C for 15 minutes. Samples were then washed twice with ice-cold 80% EtOH (Fisher) to remove salts, resuspended in DEPC H20, and the concentration was determined with a Nanodrop (Thermoscientific). RNA was extracted from freshly isolated retinas using RNeasy Plus Mini kit (Qiagen #74134). Libraries were prepared from approximately 500 ng total RNA with the TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer’s directions (Illumina). Paired-end 100-cycle sequencing was performed on HiSeq 2000 or HiSeq 2500 sequencers, per the manufacturer’s directions (Illumina). Gene based FPKM quantification in RNA-Seq was performed as described previously1. Spearman correlation was calculated for each primary tumor-O-PDX pair.
Clonal analysis
Targeted enrichment was performed using the Seqcap EZchoice Kit (Roche) according to vendor instructions for the Kapa workflow with 500 ng of genomic DNA as the starting input for library construction. Clonal analysis was performed as described previously1. To test for clonal changes within different regions of a single tumor, a subset of orthotopic xenograft tumors were harvested for segmental clonal analysis. The excised tumor was cut in half with the cut edge facing upwards. Each tumor half was visually segmented into 4 quadrants and a biopsy punch needle was used to sample the tissue in each quadrant producing 8 samples from a single tumor. Clonal analysis was performed for each of the 8 quadrant samples as described above. Samples were divided into 4 groups based on the tumor clonal evolution between primary and O-PDX tumors: Group 1 O-PDX tumors retained the clonal structure of the primary tumor. Group 2 O-PDX tumors acquired significant amount of new mutations compared to the matching primary tumor. A minor clone in the primary tumor was lost in O-PDX tumors in Group 3 samples. In Group 4 samples, O-PDX tumors were derived from a minor clone in the primary tumor.
ChIP-Seq
SJRHB000026_X1, SJRHB010463_X16, SJOS001112_X1 and SJNBL046_X were submitted to Active Motif® for ChIP-sequencing for 7 histone marks (H3K27Ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K9-14Ac, H3K9me3), RNA Pol II, CTCF, and BRD4. We first employ BWA (version 0.5.9-r26-dev, default parameter) to align the ChIP-Seq reads to human genome hg19(GRCh37-lite). Picard(version 1.65(1160)) then have been used for marking duplicated reads. Then only non-duplicated reads with have been kept by samtools (parameter “-q 1 -F 1024” version 0.1.18 (r982:295)). We followed the ENCODE criterion to quality control (QC) the data that non-duplicated version of SPP (version 1.11) have been used to draw cross-correlation and calculated relative strand correlation value (RSC) under support of R (version 2.14.0) with packages caTools(version 1.17) and bitops(version 1.0–6) and estimated the fragment size. We required > 10M unique mapped reads for point-source factor (H3K4me2/3, H3K9/14Ac, H3K27Ac, CTCF, RNAPolII, BRD4) and RSC > 1. We required 20M unique mapped reads for broad markers (H3K9me3, H3K27me3, H3K36me3). We required 10M unique mapped reads for INPUTs and RSC < 1. We noticed H3K4me1 is point-source factor in some stages while broad in other stages so we QC H3K4me1 as broad markers. Then upon manually inspection, the cross-correlation plot generated by SPP, the best fragment size estimated (the smallest fragment size estimated by SPP in all our cases) were used to extend each reads and generate bigwig file to view on IGV (version 2.3.40). All profiles were manually inspected for clear peaks and good signal to noise separation.
Chromatin Hidden Markov Modeling
Non-duplicated aligned reads were extend by fragment size define above and ChromHMM (version 1.10, with “-colfields 0,1,2,5 –center” for BinarizeBed) was used for chromatin state modeling. To choose the state number we first modeled all mouse development stages together from 7 states to 33 states and selected the model with 16 states upon manual inspection. For better visualization of the dynamics of HMM state across stages, we normalized color intensity by the max total percentage of a state covering a gene and flanking region. To determine the best region representing a gene, we first filter annotated isoforms by TSS within 2kb of any H3K4me3/H3K4me2/H3K27Ac/H3K9-14Ac peaks at any development stage and then we selected the highest expressed isoform at any development stage estimated by cuffdiff or the longest isoform if no expression level estimated by cuffdiff. At last, we reduced the interval for a HMM state to half bar and the intensity to half of the normalized intensity if it didn’t ranked top 2 HMM state for a gene. As HMM states could be assigned by multiple genes, the max total percentage across genes has been used for normalization. Percentage of HMM states were calculated for individual genes across 4 tumors with ChIP-seq profiles. Tumor type specific up-regulated genes were selected based on the following criteria: differentially expressed among tumor types (FDR < 0.05), highly expressed in the specific tumor type (FPKM > 8), and highly overexpressed compared to other tumors (at least 4-fold higher). Tumor type specific repressed genes were selected as: differentially expressed among tumor types (FDR < 0.05), highly expressed in at least 1other tumor type (FPKM > 8), and highly repressed compared to other tumors (at least 4-fold lower). Tumor type specific epigenetically regulated genes were selected as tumor specific up-regulated or repressed genes with at least one state with a fraction change >= 0.25 between the specific tumor and other tumors.
Statistical analysis
The p values were adjusted for multiple comparisons using the Benjamini & Hochberg method20. For comparing engraftment efficiency, we used the MedCalc software (https://www.medcalc.org/calc/comparison_of_proportions.php) for comparison of proportions using the N-1 Chi-squared test as recommended by Cambell and Richardson21,22. For comparison of time to engraftment, we used the comparison of means on MedCalc. Pre-study consultation for the preclinical phase III study was done with the Department of Biostatistics at St. Jude Children’s Research Hospital. The data include a total of 140 mice treated in 4 different treatment groups from 4 O-PDX models. The goal of the study was to assess and compare the tumor response and tumor progression free survival among treatment groups. Mice were randomized into each treatment group with a randomization code provided by the Department of Biostatistics. The survival curves of time to tumor progression were generated by Kaplan-Meier method. The log-rank tests were used to compare survival curves in each sub-group. Tumor response was defined by bioluminescence at the end of therapy as shown in the tables below. If a mouse was taken off study at any point in time after enrollment due to tumor size greater than 20% of mouse weight, they were automatically assigned a response of progressive disease. Signals less than 105 photons/sec/cm2/sr were classified as a complete response (CR). Animals with signal between 105–106 were classified as partial response (PR). Stable disease (SD) was the same as enrollment signal 106–108 photons/sec/cm2/sr and progressive disease (PD) was greater than 108 photons/sec/cm2/sr. The observed response data were recorded in Supplementary Tables 7 and 8. The results showed that the triple combination AZD1755 + Irinotecan + VCR had the highest complete response (CR) rate 40% (4/10) for SJRHB013757_X2, 90% (9/10) for SJRHB013759_X1, 37.5% (3/8) for SJRHB000026_X1, 22% (2/9) for SJRHB012_Y, 65% (13/20) for both ARMS O-PDXs and 29% (5/17) for both ERMS O-PDXs combined. Time to tumor progression was defined as time interval between date of enrollment and date of tumor size > 20% of mouse weight or xenogen signal > 108 at the end of study. The survival probabilities at day 25, day 66 and day 126 for treatment groups were summarized in following tables. Again, two triple combination AZD1755 + Irino + VCR had the highest progression-free survival probabilities with survival probabilities at the end of study (day 126) 100%, 100%, 62.5%, 55.6%, 100% and 59% for SJRHB013757_X2, SJRHB013759_X1, SJRHB000026_X1, SJRHB012_Y, alveolar combined and embryonal combined, respectively.
The Kaplan-Meier survival curves were given in Figures with log-rank test p-values (all log-rank test p-values were 0 for the comparisons among 4 treatment groups for all studies).
Cell screening
(Plating Density and DMSO-Sensitivity) Cells from each tumor model were plated on a flat-bottomed, white 384-well plate (Corning Cat#8804BC), at 6–8 different cell densities ranging from 125 cells/well to 15000 cells/well. Twenty-four hours after plating, cells were treated with varying DMSO concentrations (0%, 0.083%, 0.197%, and 0.443% DMSO), resulting in twelve wells per cell density at a given DMSO concentration. Seventy-two hours following the addition of DMSO, CellTiter-Glo® (‘CTG’, Promega Cat#G7573) reagent was added at 25 μl/well and the plate was read using a PerkinElmer EnVision plate reader. The relative light unit (RLU) signal was processed to determine which plating density yielded optimal signal to noise separation while maintaining logarithmic growth at 72-hr post-plating and minimal DMSO sensitivity. (Positive Control Compound Selection) Cells from each tumor model were plated in a 384-well plate at optimal density as determined above. Twenty-four hours post-plating, cells were drugged with a compound plate containing six positive control compound candidates (doxorubicin HCl, staurosporine, etoposide, SN-38, bortezomib, and cycloheximide) arrayed at a high single-point concentration and in a 1:3 serial dilution series (16 points per series). At 72-hr post-drugging, the plate was read using the CTG protocol described above. Positive controls were selected based on the compound’s ability to achieve complete cell killing (RLUs near background levels) with a well-defined, sigmoidal dose-response curve shape. We selected staurosporine (primary) and bortezomib (secondary) to be the positive controls for all drug sensitivity experiments. (Assay Validation) For each tumor model, we assessed the variability of the assay in 384-well plates by running the CTG assay using three independent plates with alternating max/min/mid control signals on at least two different days, and then examined Z-prime and other relevant statistics. The maximum signal corresponded to cells treated with DMSO only; the minimum signal had cells treated with ca. 30 uM staurosporine; and the mid signal used the cell line specific EC50 value for staurosporine determined during the positive control selection described above. In general, we observed no systematic location-dependent effect across wells, the three signal levels were well separated, and z-prime values were above 0.4. (Cell Culture Experiments in Dose Response) Cell lines were prepared via standard cell culture techniques. Xenograft cells were obtained from freshly dissociated tumors. Cell lines and O-PDX cells were counted and plated in 384 well plates (Corning Cat#8804BC) and plating densities and plating conditions are shown in Supplementary Table 6. 24 hours following plating, each cell line or O-PDX sample was drugged with both a compound plate(s) and a positive control plate using a Biomek FX (Beckman Coulter) liquid handler equipped with a pin tool. Compounds were obtained as dry powder and dissolved in DMSO to a target concentration of either 10mM or 2mM (for compounds with reported high cellular potency or solubility limitations). The pin tool transferred ca. 65 nL compound stock, resulting in ca. 370-fold compound dilution. At 72-hr post-drugging, cells were lysed with the addition of 25 μL/well CTG reagent as described above. The compound plate contained compounds dissolved in DMSO arrayed in a 1:3 fold dilution series in columns 1–20 with 32 compounds per plate. The positive control plate was empty from columns 1–20; in columns 21–24, it contained single-point and 1:3 serial dilutions of the primary and secondary positive controls (staurosporine and bortezomib, respectively) dissolved in DMSO to give ca. 10 mM top stock concentration, and also wells containing DMSO alone (negative control). There were three technical replicates per biological replicate. The purity of each compound used in these studies was verified to be >95% using UV UV/TWC and evaporative light scattering detection (ELSD) spectroscopy, and the concentration of each solution was quantified using nitrogen chemo-luminescence where possible.
Cell screening analysis
(Raw data processing – log2 RLU dose-response fits) Raw luminescence RLU (relative light unit) values for each compound at each concentration were log2 transformed, normalized to obtain % activity using the following equation: 100* [(mean(negctrl) – compound)/(mean(negctrl) – mean(posctrl))]; and then pooled from replicate experiments prior to fitting. Here, negctrl and posctrl refer to the negative and positive controls on each plate. Outliers for controls were detected using the robust statistic (below the [first quartile – 1.5×IQR] or above the [third quartile + 1.5×IQR], where IQR = interquartile range), and removed prior to calculating mean values. (QC metrics) The z-prime statistic was calculated using the following formula: 1 − ((3×sd(negctrl)) + (3×sd(posctrl)))/abs(mean(negctrl) – mean(posctrl)). Outliers among the negative and positive controls were removed as described above prior to the calculation of the mean and standard deviation statistics. Of the 1960 plates screened in total for this project, 49 (2.5%) had z-prime < 0 and were excluded from further analysis. Of the remaining plates, the average z-prime was 0.57, and 95% of all plates has z-prime between 0.27 and 0.82. (Fitting log2 RLU dose-response curves) Dose-response curves were fit using the drc [REF: Ritz et al. Bioassay Analysis using R, Journal of Statistical Software, 2005, 12, 5] package in R [REF: R Core Team (2012). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org/]. Both a three parameter (with y0, the response without drug, set to zero) and a four parameter model (y0 allowed to vary) were fit using the sigmoidal function LL2.4. The hill slope was constrained to be between −10 and 0, and ec50 was constrained to be between 10−10 and 10−4 (which equated to the drug concentration range tested in these experiments). For the 3 parameter model, yFin, the maximum response of the dose-response curve, was constrained to be between zero and the maximum of the median activities calculated at each concentration over all pooled measurements. For the 4 parameter model, y0 and yFin were both constrained to be between the minimum and the maximum of the median activities calculated at each concentration over all pooled measurements. Values from the highest concentration tested for each compound were weighted at 10% to reduce curve fitting artifacts. The model with the lowest corrected Akaike information criterion (AICc) was selected as the best fit model. Area under the curve (auc) was calculated from the fitted curve using the trapezoid rule. In the event of a failure to fit a sigmoidal dose response curve, the smooth.spline option in R was used to fit a curve that could be used to determine area under the curve. (Growth rate determination) Growth rates for each tumor cell model in this study were determined by quantifying RLU values at days 1–4 following cell plating at densities ranging from 39 to 15,000 cells/well. RLU values were log2 transformed to fit a linear model with y-intercept constrained to zero. The resulting slope was the change in log2 RLU signal per day. A value of 1 meant one doubling of RLU signal per day. (Dose-response analysis based on observed growth rate) We fit a three parameter model using the sigmoidal function LL2.4 in the drc package by regressing against the ‘observed growth rate’ as a function of concentration. The observed growth rate was calculated by estimating x0 from xctrl (given the growth rate of a specific tumor model as determined above, and the number of days of growth – 4 days in these experiments), and then determining the growth rate required to produce x(c), the cell count in the presence of drug at concentration c at the end of the experiment (4 days). We assumed cell count was proportional to RLU, and thus performed all calculations using RLU. For these regressions, the value of y0 was set to the growth rate for untreated cells and the following constraints were employed: 10 ≤ hill slope ≤ 0; minimum of the median observed growth rate in the presence of drug calculated at each concentration over all pooled measurements ≤ yFin ≤ growth rate for untreated cells; 10−10 ≤ ec50 ≤ 10−4. As before, observed growth rates from the highest concentration tested for each compound were weighted at 10% to reduce curve fitting artifacts. Two useful metrics were calculated from the dose-response curves based on observed growth rates. ED0 is the effective dose required to achieve zero growth, or cytostatic behavior. For curves that never reach zero observed growth rate, this value was set to the highest concentration tested. For curves from tumor models with a negative growth rate, this value was set to the lowest concentration tested. The second metric, AUC0, is the area under the curve where the observed growth rate dose-response curve is below zero.
Pharmacokinetics
(AZD1775) The total plasma and tumor homogenate PK of AZD1775 in female tumor-bearing athymic nude mice (Jax Laboratories, aged 8–16 weeks) was assessed after a single oral gavage dose of 120 mg/kg. AZD1775 (AbMole, M2143, purity >98%, MolWt 500.60) was suspended in 0.5% methylcellulose (type 400 cPs, Sigma) at a nominal concentration of 12 mg/mL for a 10 mL/kg gavage volume. Mice were sacrificed using an IACUC-approved method at 30 min, 1, 2, 4, and 8 hr post-dose, with 3 mice per time point. Whole blood was collected with sodium heparin via cardiac puncture, immediately centrifuged to plasma, and stored on dry ice for remainder of study. Mice were then perfused with PBS via the aorta, the RMS orthotopic xenografts excised from the hind limb, rinsed with PBS, and placed on dry ice. At the end of the in vivo procedures, all samples were transferred from dry ice and placed at −80 °C until analysis. Total plasma and tumor homogenate AZD1775 concentrations were assessed using a sensitive and specific liquid chromatography, tandem mass spectrometry assay. First, tumor samples were macerated, diluted with a 5:1 volume of Ringer Solution (Frey Scientific), and homogenized with a bead-based technique7 on a FastPrep-24 system (MP Biomedicals, Santa Ana, CA). Steel lysing matrix beads (MP Biomedicals, Metal Bead Lysing Matrix, 3 mg per mg of tumor) were added to the microcentrifuge tubes containing samples. The samples were then subjected to three 60 M/S vibratory cycles of 1 min each on the FastPrep-24 system. To prevent over-heating due to friction, samples were placed on wet ice for 5 min between each cycle. The homogenates were then stored at −80 °C until analysis. AZD1775 (AbMole, M2143, purity >98%) stock solutions were prepared in methanol and used to spike matrix calibrators and quality controls. Plasma and tumor homogenate samples, 25 μL each, were protein precipitated with 100 μL of 5 ng/mL crizotinib (ApexBio, A8802, Batch 1, purity >99%) in methanol as an internal standard. A 2 μL aliquot of the extracted supernatant was injected onto a Shimadzu LC-20ADXR high performance liquid chromatography system via a LEAP CTC PAL autosampler. The LC separation was performed using a Phenomenex Synergi Hydro-RP 80Å LC column (4.0 μm, 30 mm × 2.0 mm) maintained at 60 °C with gradient elution at a flow rate of 0.35 mL/min. The binary mobile phase consisted of 0.1% formic acid in ultra-pure water in reservoir A and 0.1% formic acid in methanol in reservoir B. The initial mobile phase consisted of 5% B with a linear increase to 70% B in 1.5 minutes. The column was then rinsed for 2.5 minutes at 100% B and then equilibrated at the initial conditions for 2 minutes for a total run time of 6 minutes. Under these conditions, the analyte and IS eluted at 0.98 and 0.93 minutes, respectively. Analyte and IS were detected with tandem mass spectrometry using a SCIEX API 5500 Q-TRAP in the positive ESI mode with monitoring of the following mass transitions: AZD1775 501.20 −> 442.20, crizotinib 450.10 −> 260.20. The method qualification and bioanalytical runs all passed our acceptance criteria for non-GLP assay performance. A quadratic model (1/X2 weighting) fit the calibrators across the 0.5 to 100 ng/mL range, with a correlation coefficient (R) of ≥0.9982. The lower limit of quantitation (LLOQ), defined as a peak area signal-to-noise ratio of 5 or greater verses a matrix blank with IS, was 0.5 ng/mL. The intra-run precision and accuracy was < 6.28% CV and 88.2% to 107%, respectively. The resultant AZD1775 concentration-time (Ct) data were grouped by matrix and time point, and summary statistics (arithmetic mean, standard deviation, %CV, minimum, median, maximum) generated using R software8. The AZD1775 arithmetic mean Ct data for each matrix was then subjected to noncompartmental pharmacokinetic analysis (NCA) using Phoenix WinNonlin 6.4 (Certara USA, Inc., Princeton, NJ). The extravascular model (Model 202) was applied, and area under the Ct curve (AUC) values were estimated using the “linear up log down” method. The terminal phase was defined as the three time points at the end of the Ct profile, and the elimination rate constant (Ke) was estimated using an unweighted log-linear regression of the terminal phase. The terminal elimination half-life (T1/2) was estimated as 0.693/Ke, and the AUC from time 0 to infinity (AUCinf) was estimated as the AUC to the last time point (AUClast) + predicted Clast/Ke. Other NCA parameters estimated included observed maximum concentration (Cmax), time of Cmax (Tmax), concentration at the last observed time point (Clast), time of Clast (Tlast), apparent clearance (CL/F = Dose/AUCinf), and apparent terminal volume of distribution (Vz/F). The average concentration over a dosing interval (Cavg) was estimated as AUCinf/dosing interval in hours. The apparent partition coefficient of AZD1775 from the plasma to tumor (Kp,tumor) was estimated as the ratio of the AUCinf, tumor to AUCinf plasma when available. To estimate a clinically relevant mouse dosage, the resultant mouse plasma AUCinf and Cavg was compared with the reported human plasma PK values at the putative single agent AZD1775 maximum tolerated dose or Phase 2 recommended dose23. All inferences were made under the assumption of time-independent, linear and dose-proportional PK in mice and humans. (Bortezomib) The total plasma and tumor PK of bortezomib in tumor-bearing female athymic nude mice (Jax Laboratories, aged 8–16 weeks) was assessed after a single intraperitoneal (IP) injection of 1 mg/kg. Bortezomib (LC Labs, Lot BBZ-106, purity >99%, MolWt 384.24) was dissolved in DMSO and further diluted with 0.9% sodium chloride for injection, USP (NS, Baxter) for a final nominal concentration of 0.1 mg/mL in 2% DMSO/98% NS and a 10 mL/kg injection volume. The IP route was found to be a convenient method, given the intermittent twice weekly dosing required in efficacy studies. Mice were sacrificed using an IACUC-approved method at 10 min, 1, 3, 24, and 72 hr post-dose, with 3 mice per time point. Whole blood was collected with sodium heparin via cardiac puncture, immediately centrifuged to plasma, and stored on dry ice for remainder of study. Mice were then perfused with PBS via the aorta, the RMS orthotopic xenograft tumors excised, rinsed with PBS, and placed on dry ice. At the end of the in vivo procedures, all samples were transferred from dry ice and placed at -80 °C until analysis. Total plasma and tumor homogenate bortezomib concentrations were assessed using a sensitive and specific liquid chromatography, tandem mass spectrometry assay. First, tissue samples were macerated, diluted with a 5:1 volume of ultrapure water, and homogenized with a bead-based technique24 on a FastPrep-24 system (MP Biomedicals, Santa Ana, CA). Ceramic lysing matrix beads (MP Biomedicals, Ceramic Bead Lysing Matrix, 3 mg per mg of tissue) were added to the microcentrifuge tubes containing tumor samples. Samples were then subjected to three 60 M/S vibratory cycles of 1 min each on the FastPrep-24 system. To prevent over-heating due to friction, samples were placed on wet ice for 5 min between each cycle. The homogenates were then stored at −80 °C until analysis. Bortezomib (LC Labs, BBZ-106, purity >99%) stock solutions were prepared in 80% methanol/20% ultrapure water with 0.1% formic acid and used to spike matrix calibrators and quality controls. Plasma and tissue homogenate samples, 25 μL each, were treated with 10 μL of internal standard (bortezomib-d8, Toronto Research Chemicals, 3-GBH-170-2, purity 98%) 50 ng/mL spiking solution and then protein precipitated with 100 μL of acetonitrile with 0.1% formic acid, vortexed vigorously for 1 minute and centrifuged at 4 °C and 13000g for 5 minutes. A 3 μL aliquot of the extracted supernatant was injected onto a Shimadzu LC-20ADXR high performance liquid chromatography system via a Shimadzu SIL-20ACXR autosampler. The LC separation was performed using a Phenomenex Luna C8 80Å LC column (4.0 μm, 30 mm × 2.0 mm) maintained at ambient temperature with gradient elution at a flow rate of 0.25 mL/min. The binary mobile phase consisted of 0.1% formic acid in ultra-pure water in reservoir A and 0.1% formic acid in acetonitrile in reservoir B. The initial mobile phase consisted of 30% B with a linear increase to 100% B in 2 minutes. The column was then rinsed for 2.5 minutes at 100% B and then equilibrated at the initial conditions for 2 minutes for a total run time of 6.5 minutes. Under these conditions, the analyte and IS eluted at 2.56 and 2.53 minutes, respectively. Analyte and IS were detected with tandem mass spectrometry using a SCIEX API 4000 in the positive ESI mode with monitoring of the following mass transitions: bortezomib 367.4 −> 226.2, bortezomib-d8 374.8 −> 234.0. The experimental bioanalytical runs were all found to be acceptable for the purpose of a singlicate non-GLP, preclinical PK assessment. A linear model (1/X2 weighting) fit the calibrators across the 1 to 100 ng/mL range, with a correlation coefficient (R) of ≥0.99. Above the calibration range quality control samples were diluted with adequate precision and accuracy. The lower limit of quantitation (LLOQ), defined as a peak area signal-to-noise ratio of 5 or greater verses a matrix blank with IS, was 1 ng/mL for plasma and 6 ng/mL for tissues due to the dilution factor. The intra-run precision and accuracy was < 5.56% CV and 87.1% to 107%, respectively across the matrices. The resultant bortezomib concentration-time (Ct) data were grouped by matrix and time point, and manual imputation of data below the lower limit of quantitation (BLOQ) was as follows: IF at any time point ≥ 2/3rds of the Ct results were above the LLOQ, the BLOQ data were replaced with a value of ½ LLOQ, ELSE the entire time point’s data were treated as missing. Then, using Phoenix WinNonlin 6.4 (Certara USA, Inc., Princeton, NJ), Ct data summary statistics (arithmetic mean, standard deviation, %CV, minimum, median, maximum) were generated, and the bortezomib arithmetic mean Ct data for each matrix was subjected to noncompartmental pharmacokinetic analysis (NCA). The extravascular model (Model 202) was applied, and area under the Ct curve (AUC) values were estimated using the “linear up log down” trapezoidal rule. The terminal phase was defined as the two to three time points at the end of the Ct profile, and the elimination rate constant (Ke) was estimated using an unweighted log-linear regression of the terminal phase. The terminal elimination half-life (T1/2) was estimated as 0.693/Ke, and the AUC from time 0 to infinity (AUCinf) was estimated as the AUC to the last time point (AUClast) + predicted Clast/Ke. Other NCA parameters estimated included observed maximum concentration (Cmax), time of Cmax (Tmax), concentration at the last observed time point (Clast), time of Clast (Tlast), apparent clearance (CL/F = Dose/AUCinf), and apparent terminal volume of distribution (Vz/F). The average concentration over a dosing interval (Cavg) was estimated as AUCinf/dosing interval in hours. The apparent partition coefficient of bortezomib from the plasma to the tissue of interest (Kp,tissue) was estimated as the ratio of the AUCinf, tissue to AUCinf plasma when available. To estimate a clinically relevant mouse dosage, the resultant mouse plasma AUCinf and Cavg was compared with the reported human plasma PK values at the putative single agent bortezomib maximum tolerated dose at 1.3 mg/m2 IV on Day 11 of therapy25. All inferences were made under the assumption of time-independent, linear and dose-proportional PK in mice and humans. (Panobinostat) The total plasma and tissue PK of panobinostat in female tumor-bearing athymic nude mice (Jax Laboratories, aged 8–16 weeks) was assessed after a single intraperitoneal (IP) injection of 20 mg/kg. Panobinostat (LC Labs, Lot PNB-101, purity >99%, MolWt 349.43) was suspended as the free base form in dextrose 5% for injection, USP (D5W, Baxter) at a nominal concentration of 2 mg/mL for a 10 mL/kg injection volume. The IP route was chosen given the low oral bioavailability reported in rodents,26 and as it appeared to be the preferred route in reviewed mouse studies27,28. Mice were sacrificed using an IACUC-approved method at 15 min, 40 min, 2.25, 8, and 24 hr post-dose, with 3 mice per time point. Whole blood was collected with sodium heparin via cardiac puncture, immediately centrifuged to plasma, and stored on dry ice for remainder of study. Mice were then perfused with PBS via the aorta, the RMS orthotopic xenografts and tissues excised, rinsed with PBS, and placed on dry ice. At the end of the in vivo procedures, all samples were transferred from dry ice and placed at −80 °C until analysis.Total plasma and tissue homogenate panobinostat concentrations were assessed using a sensitive and specific liquid chromatography, tandem mass spectrometry assay. First, tissue samples were macerated, diluted with a 5:1 volume (for tumor and brain) or a 3.1–7.0:1 volume (for varying masses retina and vitreous) of ultrapure water, and homogenized with a bead-based technique7 on a FastPrep-24 system (MP Biomedicals, Santa Ana, CA). Ceramic lysing matrix beads (MP Biomedicals, Lysing Matrix D, 10 mg per mg of tissue) were added to the microcentrifuge tubes containing samples. The samples were then subjected to three 60 M/S vibratory cycles of 1 min each on the FastPrep-24 system. To prevent over-heating due to friction, samples were placed on wet ice for 5 min between each cycle. The homogenates were then stored at −80 °C until analysis. Panobinostat (LC Labs, Lot PNB-101, purity >99%) stock solutions were prepared in methanol and used to spike matrix calibrators and quality controls. Plasma and tissue homogenate samples, 25 μL each, were protein precipitated with 100 μL of 240 ng/mL panobinostat-d8 hydrochloride salt (Toronto Research Chemicals, Inc., P180502, Lot 5-KSS-175-5, purity 96%) in methanol as an internal standard. A 2 μL aliquot of the extracted supernatant was injected onto a Shimadzu LC-20ADXR high performance liquid chromatography system via a LEAP CTC PAL autosampler. The LC separation was performed using a Waters XBridge BEH C18 LC column (2.5 μm, 75 mm × 2.1 mm) maintained at 60 °C with gradient elution at a flow rate of 0.35 mL/min. The binary mobile phase consisted of ultra-pure water - 100 mM ammonium formate, pH=3.0 – methanol (850:50:100 v/v) in reservoir A and methanol – acetonitrile – 100 mM ammonium formate, pH=3.0 (475:475:50 v/v) in reservoir B. The initial mobile phase consisted of 42.5% B and was maintained for 1.6 minutes. The column was then rinsed for 1.4 minutes at 100% B and then equilibrated at the initial conditions for 2 minutes for a total run time of 5 minutes. Under these conditions, the analyte and IS eluted at 0.85 and 0.83 minutes, respectively.
Analyte and IS were detected with tandem mass spectrometry using a SCIEX API 5500 Q-TRAP in the positive ESI mode with monitoring of the following mass transitions: panobinostat 350.18 −> 158.18, panobinostat-d8 357.91 −> 147.19. The experimental bioanalytical runs were all found to be acceptable for the purpose of a singlicate non-GLP, preclinical PK assessment. A linear model (1/X2 weighting) fit the calibrators across the 5 to 1000 ng/mL range, with a correlation coefficient (R) of ≥0.99. The lower limit of quantitation (LLOQ), defined as a peak area signal-to-noise ratio of 5 or greater verses a matrix blank with IS, was 5 ng/mL for plasma and 30 ng/mL for tissues due to the dilution factor. The intra-run precision and accuracy was < 0.312% CV and 99.4% to 113%, respectively for plasma, brain, retina, and vitreous. Tumor matrix demonstrated less-favorable precision and accuracy, with values of < 1.79% CV and 108% to 119%, respectively. The resultant panobinostat concentration-time (Ct) data were grouped by matrix and time point, and manual imputation of data below the lower limit of quantitation (BLOQ) was as follows: IF at any time point ≥ 2/3rds of the Ct results were above the LLOQ, the BLOQ data were replaced with a value of ½ LLOQ, ELSE the entire time point’s data were treated as missing. Then, using Phoenix WinNonlin 6.4 (Certara USA, Inc., Princeton, NJ), Ct data summary statistics (arithmetic mean, standard deviation, %CV, minimum, median, maximum) were generated, and the panobinostat arithmetic mean Ct data for each matrix was subjected to noncompartmental pharmacokinetic analysis (NCA). The extravascular model (Model 202) was applied, and area under the Ct curve (AUC) values were estimated using the linear trapezoidal method and the sparse sampling option. The terminal phase was defined as the three time points at the end of the Ct profile, and the elimination rate constant (Ke) was estimated using an unweighted log-linear regression of the terminal phase. The terminal elimination half-life (T1/2) was estimated as 0.693/Ke, and the AUC from time 0 to infinity (AUCinf) was estimated as the AUC to the last time point (AUClast) + Clast/Ke. Other NCA parameters estimated included observed maximum concentration (Cmax), time of Cmax (Tmax), concentration at the last observed time point (Clast), time of Clast (Tlast), apparent clearance (CL/F = Dose/AUCinf), and apparent terminal volume of distribution (Vz/F). The average concentration over a dosing interval (Cavg) was estimated as AUCinf/dosing interval in hours. The apparent partition coefficient of panobinostat from the plasma to the tissue of interest (Kp,tissue) was estimated as the ratio of the AUCinf, tissue to AUCinf plasma when available. To estimate a clinically relevant mouse dosage, the resultant mouse plasma AUCinf and Cavg was compared with the reported human plasma PK values at the putative single agent panobinostat maximum tolerated dose or the FDA-approved dose. All inferences were made under the assumption of time-independent, linear and dose-proportional PK in mice and humans.
Preclinical testing
(Preclinical Phase I) Non-tumor bearing athymic nude mice were used for tolerability testing of the following combinations: VCR + IRN, AZD1775 + VCR + IRN, and panobinostat + bortezomib. Mice were given 4 cycles of chemotherapy with 21 days per cycle. The chemotherapeutic drug combinations and schedule used were designed to mimic potential human clinical trials. Each treatment group contained 3 mice. Vincristine was dosed by intraperitoneal injection once a week on days 1, 8, and 15. Irinotecan was dosed by intraperitoneal injection once daily on days 1–5 and 8–12. AZD1775 was dosed by oral gavage twice daily on days 1–5. Panobinostat was dosed by intraperitoneal injection once daily on days 1, 3, 5, 8, 10 and 12. Bortezomib was dosed by intraperitoneal injection once daily on days 1, 4, 8 and 11. Mouse weight and standard complete blood counts were monitored for each treatment group. The health of the animals was monitored daily throughout therapy. (Preclinical Phase II) RMS orthotopic xenografts were created by injecting luciferase labeled cells from SJRHB000026_X1 (ERMS), SJRHB012_Y (ERMS), and SJRHB013759_X1 (ARMS) into recipient CD-1 nude mice using the intramuscular injection technique previously described. Mice were screened weekly by Xenogen and the bioluminescence was measured. Mice were enrolled in the study after achieving a target bioluminescence signal of 106 –107 photons/sec/cm2 or greater for 2 weeks or a palpable tumor, and chemotherapy was started the following Monday. The following preclinical phase II trials were performed (Table S10):
SJRHB000026_X1 (ERMS)
Mice were randomized to 6 treatment groups: AZD1775 + VCR + IRN, AZD1775 + IRN, VCR + IRN, panobinostat + bortezomib, IRN alone, and placebo. The dose of AZD1775 was 60 mg/kg oral gavage twice daily for days 1–5. IRN was 1.25 mg/kg intraperitoneal once daily on days 1–5 and 8–12. Vincristine was administered at 0.38 mg/kg intraperitoneal once daily on days 1,3,5,8,10 and 12. Bortezomib was administered at 1 mg/kg intraperitoneal once daily on days 1,4,8 and 11. Mice received 4 courses of chemotherapy (3 weeks per course) and bioluminescence was monitored weekly and at the end of therapy. Disease response was classified according to bioluminescence signal. Mice with a signal of 105 photons/sec/cm2 or less (similar to background) were classified as complete response, 105–106 photons/sec/cm2 as partial response, 107 up to 108 photons/sec/cm2 (similar to enrollment signal) as stable disease, and greater than 108 photons/sec/cm2 as progressive disease. Mice with tumor burden at any time greater than 20% of body weight were also classified as progressive disease. Mice were monitored daily while receiving chemotherapy.
SJRHB012_Y (ERMS)
AZD1775 groups and panobinostat + bortezomib groups were tested in 2 separate phase II preclinical trials. In the first trial, mice were randomized to 4 treatment groups: AZD1775 + VCR + IRN (low dose protracted schedule), AZD1775 + VCR + IRN (standard 5 day schedule), VCR + IRN (low dose protracted schedule), and placebo. The second trial randomly enrolled mice into 2 treatment groups, panobinostat + bortezomib, and placebo. The dose of AZD1775 was 60 mg/kg oral gavage twice daily for days 1–5. IRN was 1.25 mg/kg intraperitoneal once daily on days 1–5 and 8–12. Another regimen of IRN was also used in these mice at 3.125 mg/kg intraperitoneal once daily on days 1–5. These represent the two most commonly used schedules in children in the clinic.Vincristine was administered at 0.38 mg/kg intraperitoneal once daily on days 1,3,5,8,10 and 12. Bortezomib was administered at 1 mg/kg intraperitoneal once daily on days 1,4,8 and 11. Mice received 6 courses of chemotherapy and bioluminescence was monitored weekly in the first trial and with each course (every 3 weeks) in the second trial. Disease response was classified according to bioluminescence signal. Mice with a signal of 105 photons/sec/cm2 or less (similar to background) were classified as complete response, 105–106 photons/sec/cm2 as partial response, 107 up to 108 photons/sec/cm2 (similar to enrollment signal) as stable disease, and greater than 108 photons/sec/cm2 as progressive disease. Mice with tumor burden at any time greater than 20% of body weight were also classified as progressive disease. Mice were monitored daily while receiving chemotherapy.
SJRHB013759_X1 (ARMS)
AZD1775 groups and panobinostat + bortezomib groups were tested in 2 separate phase II preclinical trials. In the first trial, mice were randomized to 3 treatment groups: AZD1775 + VCR + IRN (low dose protracted schedule), VCR + IRN (low dose protracted schedule), and placebo. The second trial randomly enrolled mice into 2 treatment groups, panobinostat + bortezomib, and placebo. The dose of AZD1775 was 60 mg/kg oral gavage twice daily for days 1–5. IRN was 1.25 mg/kg intraperitoneal once daily on days 1–5 and 8–12. Vincristine was administered at 0.38 mg/kg intraperitoneal once daily on days 1,3,5,8,10 and 12. Bortezomib was administered at 1 mg/kg intraperitoneal once daily on days 1,4,8 and 11. Mice received 6 courses of chemotherapy and bioluminescence was monitored weekly in the first trial and with each course (every 3 weeks) in the second trial. Disease response was classified according to bioluminescence signal. Mice with a signal of 105 photons/sec/cm2 or less (similar to background) were classified as complete response, 105–106 photons/sec/cm2 as partial response, 107 up to 108 photons/sec/cm2 (similar to enrollment signal) as stable disease, and greater than 108 photons/sec/cm2 as progressive disease. Mice with tumor burden at any time greater than 20% of body weight were also classified as progressive disease. Mice were monitored daily while receiving chemotherapy.
(Preclinical Phase III) We performed a randomized, double-blind placebo controlled preclinical phase III trial using 140 mice from 4 different O-PDX lines. RMS orthotopic xenografts were created by injecting luciferase labeled cells from SJRHB000026_X1 (ERMS), SJRHB012_Y (ERMS), SJRHB013757_X2 (ARMS) and SJRHB013759_X1 (ARMS) into recipient athymic nude mice using the intramuscular injection technique previously described. Mice were screened weekly by Xenogen and the bioluminescence was measured. Mice were enrolled in the study after achieving a target bioluminescence signal of 106 –107 photons/sec/cm2 or greater for 2 weeks or a palpable tumor, and chemotherapy was started the following Monday. Individuals giving the chemotherapy as well as those doing the bioluminescence imaging were blinded to the drugs being administered as well as the specific O-PDX tumor line. The following drugs and doses were used in the preclinical phase III trial based off AUC-guided dosing obtained from pharmacokinetic studies matched to human doses that are clinically relevant in patients. The dose of AZD1775 was 48 mg/kg oral gavage twice daily for days 1–5. IRN was 1.25 mg/kg intraperitoneal once daily on days 1–5 and 8–12. Vincristine was administered at 0.38 mg/kg intraperitoneal once daily on days 1,3,5,8,10 and 12. Mice received 6 courses of chemotherapy (3 weeks per course) and bioluminescence was monitored on week 3 of each course and at the end of therapy. Disease response was classified according to bioluminescence signal as above. AZD1775 was reconstituted in 0.5% methylcellulose, IRN and VCR in sterile normal saline, panobinostat in 5% dextrose with sterile water (D5W) and bortezomib in 2% DMSO/98% sterile normal saline.
Xenogen imaging and quantification
Mice were given intraperitoneal injections of Firefly D-Luciferin (Caliper Life Sciences 3 mg/mouse). Bioluminescent images were taken five minutes later using the IVIS® 200 imaging system. Anesthesia was administered throughout image acquisition (isoflurane 1.5% in O2 delivered at 2 liters/min). The Living Image 4.3 software (Caliper Life Sciences) was used to generate a standard region of interest (ROI) encompassing the largest tumor at maximal bioluminescence signal. The identical ROI was used to determine the average radiance (photons/s/cm2/sr) for all xenografts.
Data availability
All sequence data is deposited in the EMBL-EBI (EGA accession number: EGAS00001002528).
Extended Data
Extended Data Figure 1. O-PDX models retain the cellular features of the corresponding patient tumors.
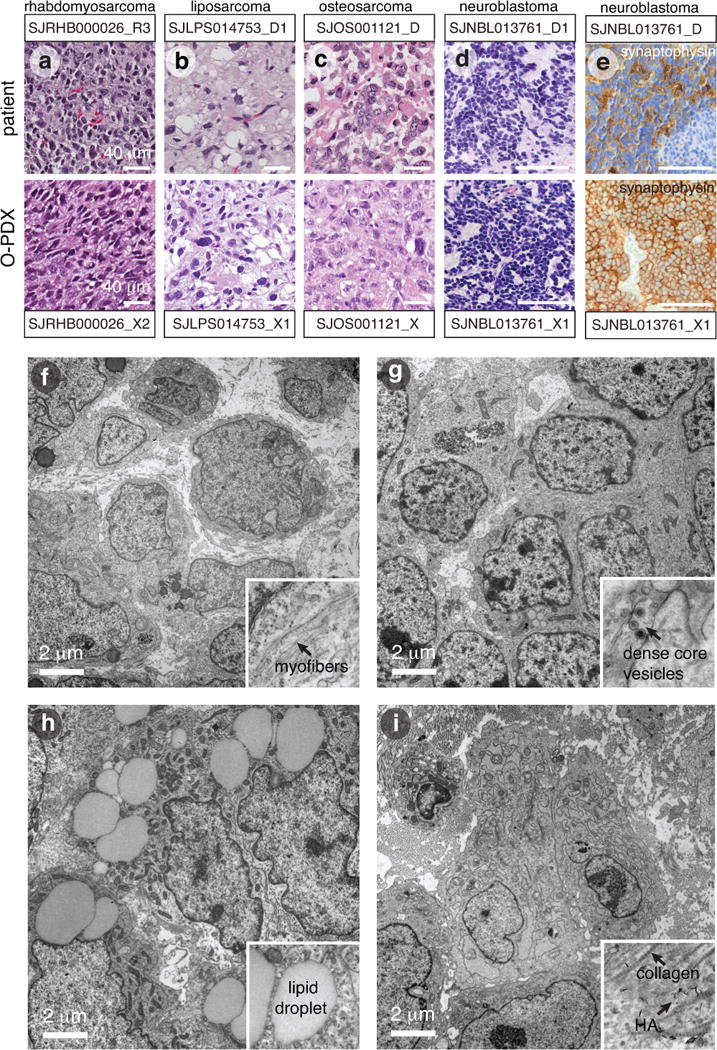
(a–d) Micrographs of hematoxylin and eosin–stained patient tumors and corresponding O-PDX tumors for a representative rhabdomyosarcoma (a), liposarcoma (b), osteosarcoma (c), and neuroblastoma (d). (e) Immunohistochemical staining for synaptophysin (brown) for SJNBL013761_D and the matched O-PDX. (f–i) Transmission electron micrographs of a representative rhabdomyosarcoma (f), neuroblastoma (g), liposarcoma (h), and osteosarcoma (i). Inset for each micrograph is a higher magnification view of a key cellular hallmark of each tumor. Abbreviations: HA, hydroxyapatite. Scale bars a–e, 40 μm; f–i, 2 μm.
Extended Data Figure 2. O-PDX tumors retain the genomic features of the patient tumors.
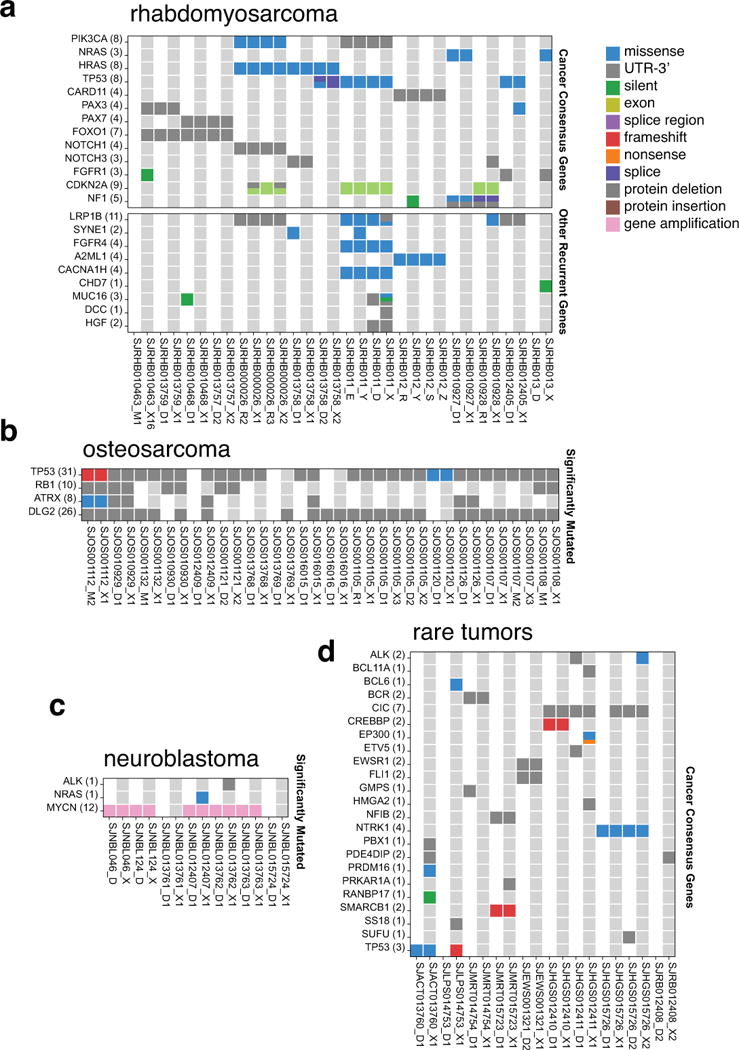
(a–d) Heat maps of the 51 tumors that had sufficient material for whole-genome sequencing and whole-exome sequencing analyses. Individual O-PDX/patient tumor pairs are indicated by alternating gray and white columns. Heat maps are grouped based on the disease: rhabdomyosarcoma (a), osteosarcoma (b), neuroblastoma (c), and rare tumors (d).
Extended Data Figure 3. Molecular and cellular clonal heterogeneity.
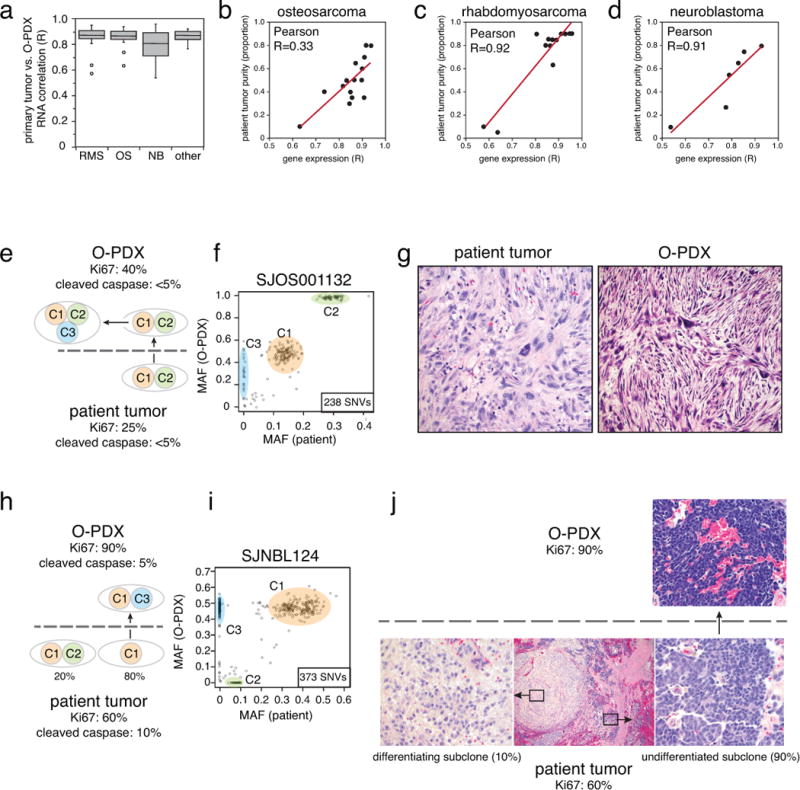
(a) Boxplot of the correlation coefficient (R) between each O-PDX and the corresponding patient tumor. (b–d) Scatterplots for the gene-expression correlation coefficients for the patient tumor compared to the O-PDX relative to the patient tumor purity determined from whole genome sequencing. The Pearson correlation between those 2 variables is indicated for each tumor type. The line of best fit for the data is shown (red). (e,f) Diagram and scatterplot of the clonal changes in SJOS001132 between the patient tumor and the O-PDX. The proportion of proliferating cells as measured by Ki67 immunostaining and dying cells as measured by cleaved caspase 3 immunostaining. The patient tumor had a major clone with two clusters of SNVs (C1 and C2) that then continued to evolve and diverge in the O-PDX into two distinct clones. One clone contains the original SNVs found in the patient tumor (C1 and C2) and the other clone has an additional cluster of SNVs (C3). The total number of SNVs analyzed in this sample was 238. g) Hematoxylin and eosin staining of the patient tumor and the O-PDX showing an expansion of cells with more aggressive pleiomorphic cellular features. h,i) Diagram and scatterplot of the clonal changes in SJNBL124 between the patient tumor and the O-PDX. The proportion of proliferating cells as measured by Ki67 immunostaining and dying cells as measured by cleaved caspase 3 immunostaining. The patient tumor had a major clone (80% of the tumor) with one clusters of SNVs (C1) and a minor clone (20% of the tumor) with two clusters of SNVs (C1 and C2). In the O-PDX, the minor clone was lost and the tumor continue to evolve and acquire an additional cluster of SNVs (C3). The total number of SNVs analyzed in this sample was 373. j) In the hematoxylin and eosin stained patient tumor, there was a major clone (90% of the tumor) with proliferating small round cells and a minor clone (10% of the tumor) that had features of differentiated neuroblastoma cells. The cells with features of differentiated cells were lost in the O-PDX.
Extended Data Figure 4. Epigenetic landscape reflects cellular origins.
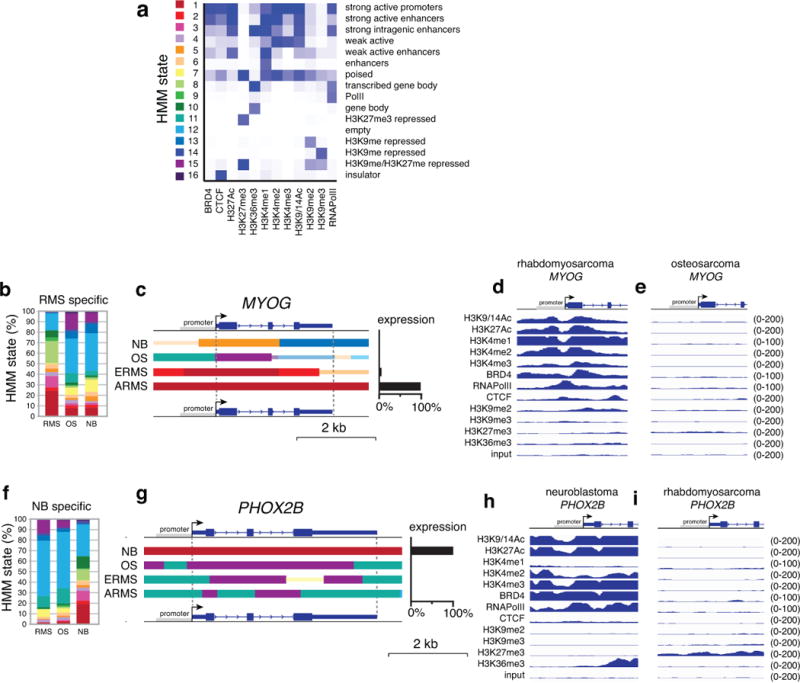
(a) Heatmap of the HMM states used in this study. (b) Stack histogram of the percentage of each of the 16 HMM states for the genes expressed specifically in rhabdomyosarcoma across the 3 tumor types (OS, NB, RMS). (c) Representative HMM and expression of a gene (MYOG) that is specifically expressed in rhabdomyosarcoma. (d,e) Corresponding ChIP-seq peaks for all 12 antibodies and the input sample for the MYOG promoter in the rhabdomyosarcoma and osteosarcoma O-PDXs. (O) Stack histogram of the percentage of each of the 16 HMM states for the genes expressed specifically in neuroblastoma across the 3 tumor types (OS, NB, RMS). (f) Representative HMM and expression of a gene (PHOX2B) that is specifically expressed in neuroblastoma. (g,h) Corresponding ChIP-seq peaks for all 12 antibodies and the input sample for the PHOX2B promoter in the neuroblastoma and rhabdomyosarcoma O-PDXs. Abbreviations: HMM, Hidden Markov Modeling; NB, neuroblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma.
Extended Data Figure 5. Drug screening quality control.
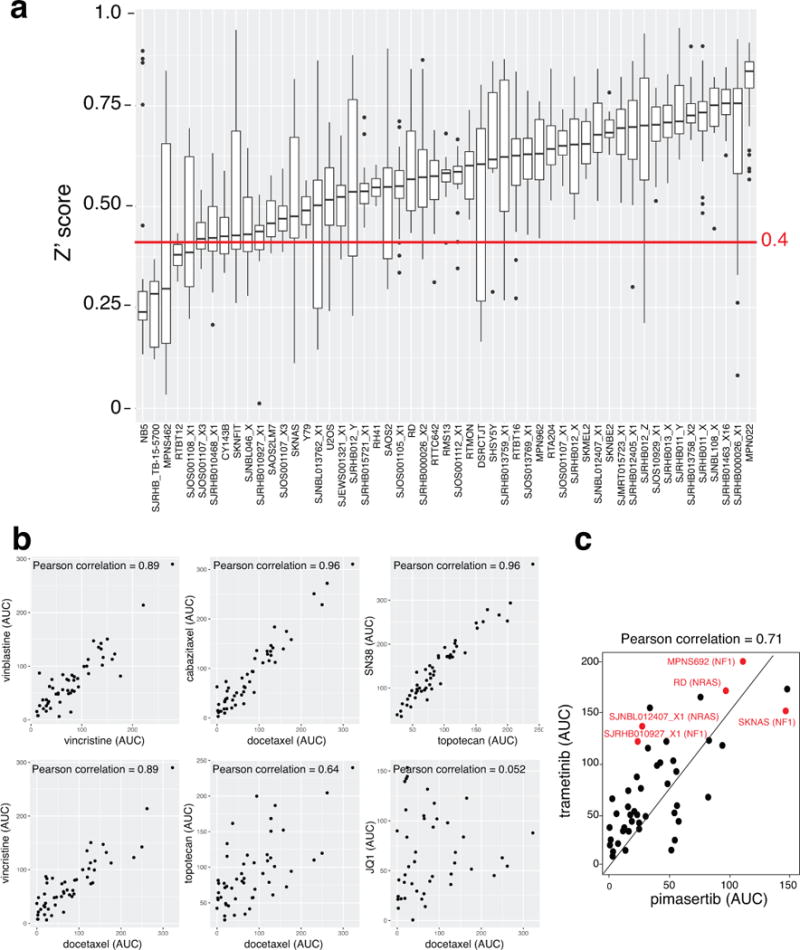
a) Box plot for the Z-prime score for each of the 1,911 plates screened in this study that passed our quality control metrics. Most cell models (O-PDXs and cell lines) had a z-prime score above 0.4 (red line). The average z-prime was 0.57 and 95% of plates had z-prime between 0.27 and 0.82. b) Scatterplot of area under the curve (AUC) for the dose response curves for pairs of drugs with similar and dissimilar mechanisms. Larger AUC values represent greater drug potency (killing). The Pearson correlation is shown for each drug pair. c) Scatterplot of AUC for the dose response curve for two MEK inhibitors (trametinib and selumetinib). The cell models highlighted in red have mutations in the RAS/NF1/MEK pathway.
Extended Data Figure 6. Preclincial pharmacokinetics and in vivo preclinical phase II.
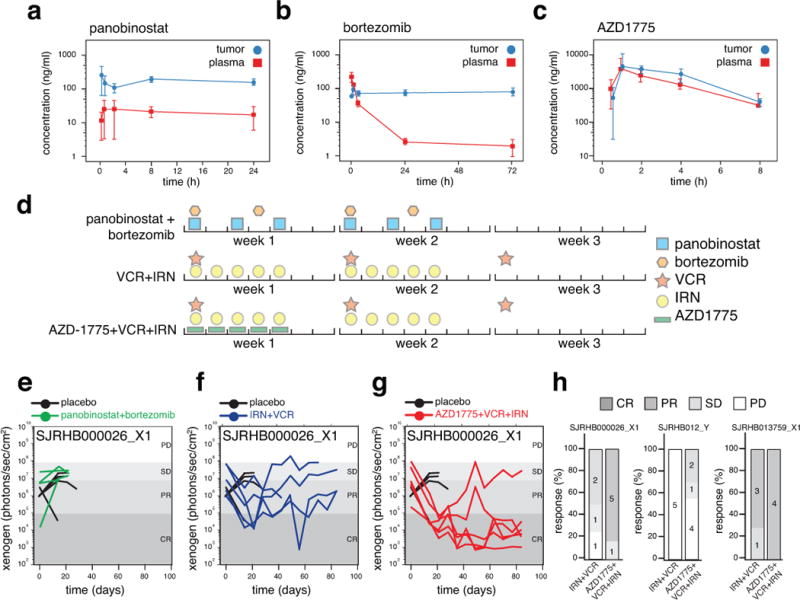
(a–c) Concentration-time curves for panobinostat, bortezomib and AZD1775 in mice bearing O-PDX tumors. Each data point is the mean and standard deviation of triplicate animals. The pharmacokinetics for IRN and VCR had been determined previously. (d) Outline of the schedule used for the combination of panobinostat with bortezomib, VCR+IRN as the standard of care and AZD1775+VCR+IRN. The schedules were selected to match that used in patients and the dose was selected to provide the same plasma exposure based on the pharmacokinetics in (a–c). (e–g) Line graphs of tumor response for individual SJRHB000026_X1 mice on the preclinical phase II study for placebo-treated mice and each of the 3 treatment groups. Each line indicates an individual mouse with tumor. (h) Stack bar plots of the response percentages for 3 different RMS O-PDX models with standard of care for recurrent RMS (IRN+VCR) and the AZD1775+VCR+IRN treatment regimen. The numbers of mice in each response category are indicated on the plot.
Supplementary Material
Acknowledgments
We thank Angela McArthur for editing the manuscript. This work was supported by Cancer Center Support (CA21765), grants to M.A.D from the NIH (EY014867, EY018599, and CA168875), and American Lebanese Syrian Associated Charities (ALSAC). M.A.D. was also supported by HHMI, Alex Lemonade Stand, Tully Family and Peterson Foundations. E.S. was supported by the St. Baldrick’s Foundation and the National Pediatric Cancer Foundation (NPCF).
Footnotes
AUTHOR CONTRIBUTIONS E.S. and M.A.D. designed the study and wrote the paper. S.F. wrote and supervised the protocol. X.C. and B.X. performed all computational analyses. C.B., B.G, V.H., M.O., K.B., J.D. assisted with preclinical animal studies and cell screening. J.E. assisted with molecular analyses. M.C. and A.B. provided pathology review. B.F. performed pharmacokinetic analysis. J.W. performed biostatistics. N.T. and A.S. analyzed drug sensitivity data. A.K. manages the CSTN and assisted with drug screening. X.Z. developed the protein paint visualization. E.R.M., R.K.W., J.D., J.Z., A.P. and M.A.D. contributed to the Pediatric Cancer Genome Project.
AUTHOR INFORMATION The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to those sections appear only in the online paper.
References
- 1.Chen X, Pappo A, Dyer MA. Pediatric solid tumor genomics and developmental pliancy. Oncogene. 2015;34:5207–5215. doi: 10.1038/onc.2014.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith MA, Altekruse SF, Adamson PC, Reaman GH, Seibel NL. Declining childhood and adolescent cancer mortality. Cancer. 2014;120:2497–2506. doi: 10.1002/cncr.28748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van de Wetering M, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boj SF, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao D, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Townsend EC, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016;30:183. doi: 10.1016/j.ccell.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Bruna A, et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell. 2016;167:260–274 e222. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klco JM, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25:379–392. doi: 10.1016/j.ccr.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao H, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 10.Zarzosa P, et al. Patient-derived xenografts for childhood solid tumors: a valuable tool to test new drugs and personalize treatments. Clin Transl Oncol. 2017;19:44–50. doi: 10.1007/s12094-016-1557-2. [DOI] [PubMed] [Google Scholar]
- 11.Stewart E, et al. The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev Biol. 2016;411:287–293. doi: 10.1016/j.ydbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hafner M, Niepel M, Chung C, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nature Methods. 2016;13:521–527. doi: 10.1038/nmeth.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart E, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9:829–841. doi: 10.1016/j.celrep.2014.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langenau DM, Sweet-Cordero A, Wechsler-Reya RJ, Dyer MA. Preclinical Models Provide Scientific Justification and Translational Relevance for Moving Novel Therapeutics into Clinical Trials for Pediatric Cancer. Cancer Res. 2015;75:5176–5186. doi: 10.1158/0008-5472.CAN-15-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013;24:710–724. doi: 10.1016/j.ccr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinto EM, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. 2015;6:6302. doi: 10.1038/ncomms7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirode F, et al. Genomic Landscape of Ewing Sarcoma Defines an Aggressive Subtype with Co-Association of STAG2 and TP53 Mutations. Cancer Discov. 2014;4:1342–1353. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 21.Campbell I. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat Med. 2007;26:3661–3675. doi: 10.1002/sim.2832. [DOI] [PubMed] [Google Scholar]
- 22.Richardson JT. The analysis of 2 × 2 contingency tables–yet again. Stat Med. 2011;30:890. doi: 10.1002/sim.4116. author reply 891–892. [DOI] [PubMed] [Google Scholar]
- 23.Do K, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33:3409–3415. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang X, et al. Evaluation of homogenization techniques for the preparation of mouse tissue samples to support drug discovery. Bioanalysis. 2011;3:1923–1933. doi: 10.4155/bio.11.181. [DOI] [PubMed] [Google Scholar]
- 25.Reece DE, et al. Pharmacokinetic and pharmacodynamic study of two doses of bortezomib in patients with relapsed multiple myeloma. Cancer Chemother Pharmacol. 2011;67:57–67. doi: 10.1007/s00280-010-1283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009;280:233–241. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 27.Xie C, et al. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS One. 2013;8:e79106. doi: 10.1371/journal.pone.0079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ocio EM, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95:794–803. doi: 10.3324/haematol.2009.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data is deposited in the EMBL-EBI (EGA accession number: EGAS00001002528).