

Abstract
Mutations in GLE1 underlie Lethal Congenital Contracture syndrome and Lethal Arthrogryposis with Anterior Horn Cell Disease. Both Lethal Congenital Contracture syndrome and Lethal Arthrogryposis with Anterior Horn Cell Disease are characterized by reduced fetal movements, congenital contractures, and a severe form of motor neuron disease that results in fetal death or death in the perinatal period, respectively. We identified bi-allelic mutations in GLE1 in two unrelated individuals with motor delays, feeding difficulties and respiratory insufficiency who survived beyond the perinatal period. Each affected child had missense variants predicted to result in amino acid substitutions near the C-terminus of GLE1 that are predicted to disrupt protein-protein interaction or GLE1 protein targeting. We hypothesize that mutations that preserve function of the coiled-coil domain of GLE1 cause Lethal Arthrogryposis with Anterior Horn Cell Disease whereas mutations that abolish the function of the coiled-coil domain cause Lethal Congenital Contracture syndrome. The phenotype of Lethal Arthrogryposis with Anterior Horn Cell Disease is now expanded to include multiple individuals surviving into childhood suggesting that Lethal Arthrogryposis with Anterior Horn Cell Disease is a misnomer and should be re-named Arthrogryposis with Anterior Horn Cell Disease.
Keywords: phenotype expansion, GLE1, motor neuron disease, developmental delay, respiratory difficulties, arthrogryposis
INTRODUCTION
Exome sequencing (ES) is a powerful tool for dissecting the genetic basis of Mendelian conditions, including both the identification of novel disease genes and the diagnosis of monogenic diseases [Chong et al., 2015a]. Exome sequencing has been particularly useful for making a precise molecular diagnosis of phenotypically and genetically heterogeneous disorders. In turn, this has broadened the spectrum of phenotypes of many known Mendelian conditions and the number of conditions associated to single genes [Dyment et al., 2015; Thevenon et al., 2016].
Mutations in GLE1 have been reported to underlie two autosomal recessive conditions, Lethal Congenital Contracture Syndrome 1 (LCCS1; OMIM #253310) and Lethal Arthrogryposis with Anterior Horn Cell Disease (LAAHD; OMIM #611890), which result in death in the fetal and perinatal period, respectively [Nousiainen et al., 2008]. Lethal Congenital Contracture Syndrome 1 is characterized by a lack of fetal movements in the second trimester of pregnancy. Affected individuals typically present with intrauterine growth retardation (IUGR), fetal hydrops, micrognathia, pulmonary hypoplasia, and multiple joint contractures [Herva et al., 1985]. Individuals with LAAHD present with a similar albeit “milder” phenotype, with prenatal onset of diminished fetal mobility and contractures, and post-natal respiratory failure resulting in perinatal death [Vuopala and Herva, 1994].
Herein we report the delineation of two unrelated individuals with a phenotype similar to LAAHD, who survived beyond four years of life, and were either homozygous or compound heterozygous for missense variants in GLE1. These findings expand the phenotype of LAAHD and suggest that either some mutations in GLE1 are associated with better outcomes and / or differences in intervention early in life can improve outcomes.
CLINICAL FINDINGS
The study was approved by the Ethics Committee of the University Medical Center Hamburg-Eppendorf, Malta, and the Institutional Review Board of the University of Washington.
FAMILY A
Proband 1 (Figure 1; Supplementary Material, Table I) was the first-born male of healthy non-consanguineous parents of European and Maltese ancestry. Due to fetal bradycardia with initial uterine contractions he was delivered by emergency Caesarian at 38 weeks gestation with a birth weight of 2,480 g (−2.0 SD), a birth length of 46 cm (−2.3 SD) and an OFC of 35 cm (0 SD). Severe respiratory distress required resuscitation and admission to the neonatal intensive care unit where he was ventilated. Abnormal facial characteristics included a prominent forehead, down-slanting palpebral fissures, tent-shaped mouth, prominent frenulum, micrognatia and low-set ears (Figure 1A). Congenital contratures included bilateral camptodactyly of both hands, adducted thumbs, and bilateral clubfeet. His neurological exam at birth was notable for hypertonia of his upper and lower limbs with startling and jerky movements.
Figure 1. Photographs of individuals with LAAHD.
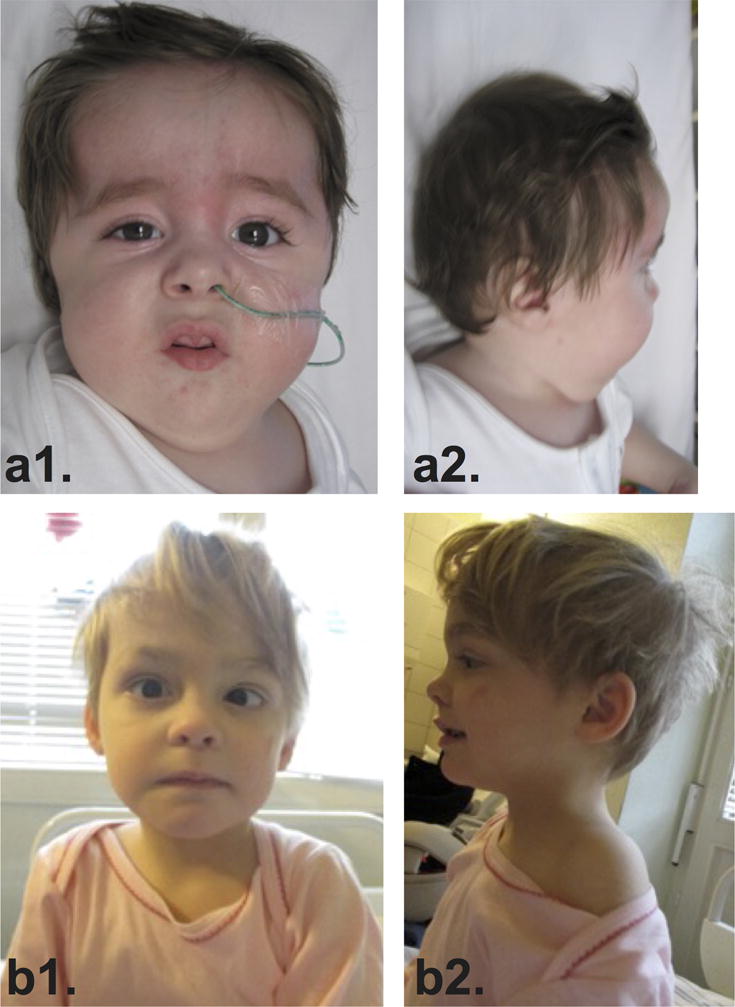
Images show facial features of proband 1 at 8 weeks (A-1) and 9 months (A-2, A-3) and proband 2 (B-1, B-2) at 3 years 5 months of age. Features include prominent forehead, down-slanting palpebral fissures, and micrognathia. See Supplementary Material, Table I for detailed clinical information on each affected individual.
At the age of 12 months, he had a flexion contracture of the left hip, decreased abduction of both shoulders and decreased extension of both elbows. He sat unsupported at 10 months and walked with a broad base gait at 3 years 8 months. Examination at 4 years 8 months of age revealed continued expressive speech delay due mainly to micrognathia but his receptive language was normal. His muscle bulk was markedly diminished. He could walk without assistance but occasionally used a K-walker.
FAMILY B
Proband 2 (Supplementary Material, Figure 1 and Table 1) was the first-born female of healthy non-consanguineous parents of European ancestry. A previous pregnancy was terminated at 20 weeks because of generalized non-immune hydrops fetalis, however no genetic analysis was performed and fetal DNA was not obtained. Proband 2 was born at 41 weeks gestational age via vacuum extraction due to fetal bradycardia. Her birth weight was 2,930 g (−1.4SD), birth length 48 cm (−1.8SD) and OFC 34.5 cm (−0.4SD).
She was evaluated at 12 months of age because of suspected developmental delay. She sat unsupported at age of 12 months, crawled at 15 months and stood at 15 months. At 2 years and 5 months, she could speak ten words, but not a full sentence. Examination at age of 3 years and 3 months (Figure 1B) revealed a restless girl with truncal hypotonia, dystonic movements of the upper limbs, involuntary facial grimacing and eye movements, and unmotivated yelling. She was unable to sit or walk unsupported, and protective reflexes were depressed. Supported gait was ataxic. Her weight, height and OFC were within the normal range.
Starting at the age of 14 months she developed frequent febrile infections that each resulted in loss of motor skills and extended recovery time. At the age 3 years 8 months, she had parainfluenza pneumonia which progressed to severe hypoxemic respiratory failure requiring ventilation. After prolonged weaning from the ventilator, she developed delirium and motor abnormalities with flailing of arms and legs, grimacing and writing movements, even while asleep. These involuntary movements improved over the next weeks but she had lost her gross motor skills including sitting and crawling. Repeated MRI of brain showed generalized parenchymal volume loss and a small brainstem. After her fourth bout of pneumonia she died of respiratory insufficiency at the age of four years.
GENETIC RESULTS
Exome sequencing and analysis were performed as described in Families A [Chong et al., 2015b] and B [Hempel et al., 2015]. A homozygous variant in GLE1 (GenBank: NM_001003722.1), c.2078C>T [p.(Ser693Phe)], was considered the best candidate given the gene’s known function and the similarity of phenotypes previously reported to be caused by mutations in GLE1. This variant is a founder mutation as it lies within a large 7.7 Mb run of homozygosity and the parents originate from the same island. In family B, bioinformatic filtering did not detect de novo variants. Filtering for rare candidate variants (alternate allele frequency <0.01, according to ExAC) under an autosomal recessive model of inheritance identified compound heterozygous alterations, c.1706G>A [p.(Arg569His)] and c.1750C>T [p.(Arg584Trp)] in only a single gene, GLE1.
The p.(Ser693Phe) variant in proband 1 has not been observed in over 76,000 population controls from multiple public databases. The variant p.(Arg569His) identified in proband 2, has been reported in individuals with a LCCS1/LAAHD-like phenotype [Nousiainen et al., 2008; Ellard et al., 2014] and has a global allele frequency of 0.0004 (ExAC). The second variant, p.(Arg584Trp), has a global allele frequency of 5×10−5. All three variants have high phred-scaled CADD (v1.3) scores (which incorporates conservation and other pathogenicity predictors such as PolyPhen-2 and SIFT), consistent with pathogenic recessive variants (Supplementary Material, Table I). Taken together, both the genetic and phenotypic data suggest that the GLE1 variants identified (Figure 2 and Supplementary Material, Table I) are causal for a phenotype overlapping LAAHD and LCCS1. Investigators studying the two affected individuals described here connected via GeneMatcher [Sobreira et al., 2015], a member of MatchMaker Exchange (MME) [Philippakis et al., 2015].
Figure 2. Genomic and protein structure of GLE1 and spectrum of pathogenic mutations in GLE1.
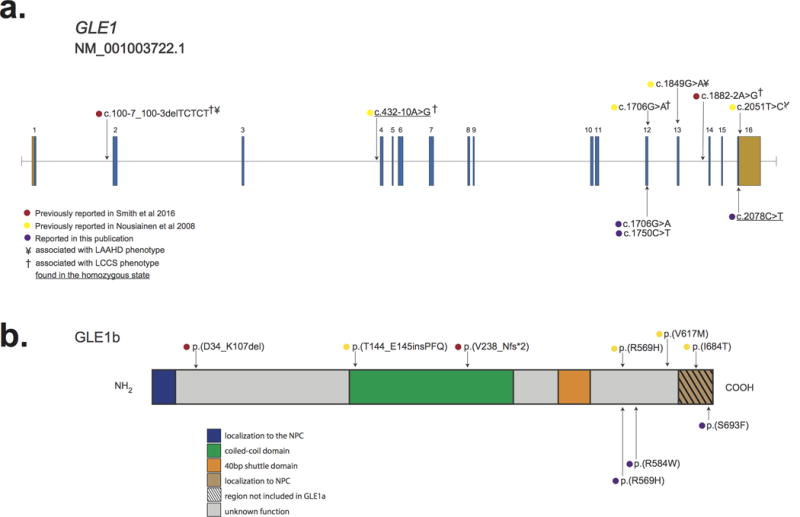
a.) GLE1 is composed of 16 exons, all which are protein-coding (blue) and two of which also contain non-coding sequence (orange). Lines with attached dots indicate the approximate locations of eleven different recessive variants that cause LCCS1 and LAAHD. The color of each dot reflects the report of each mutation. b.) Protein topology of GLE is comprised of six domains. Most mutations lie in the C-terminal portion of GLE1. No mutations in the N-terminal Nup155 binding domain or in the nucleocytoplasmic shuttling domain have been reported. The approximate positions of variants that cause LCCS1 and LAAHD are indicated by colored dots. Only p.(V238_Nfs*2) has been shown to result in complete lack of GLE1; all other variants have been shown or are expected to be expressed.
DISCUSSION
We identified two families without known Finnish ancestry who each had a child with a phenotype similar to LAAHD and LCCS1 and were either compound heterozygous or homozygous for missense variants in GLE1 predicted to be deleterious. In contrast to LAADH and LCCS1, both children survived the perinatal period and one is still alive (Supplementary Material, Table II). Indeed, no fetus with LCCS1 surviving beyond 35 weeks EGA or infant with LAADH surviving beyond eight weeks after birth has been reported until the recent publication of single exception [Smith et al., 2016]. Accordingly, the natural history of the children reported herein is considerably different from that of children with either LCCS1 or LAADH (Supplementary Material, Table II). This observation suggests that either the children reported herein have a condition distinct from both LCCS1 and LAADH, that mutations in GLE1 produce a single condition with a broad distribution of phenotypes, or that these children represent phenotypic expansion of LCCS1 or LAAHD.
Smith et al. [2016] recently reported a family with two affected brothers, one of whom died at two weeks of age while the other survived to 12 years, who were compound heterozygotes for GLE1 variants, c.100‐7_100‐3delTCTCT [p.(D34_K107del)] and c.1882‐2A>G [p.(V238_Nfs*2)]. Smith et al. [2016] proposed that LCCS1 and LAAHD constitute the same clinical entity with variable severity because the two disorders have “nearly indistinguishable phenotypes.” However, the observation that all individuals homozygous for p.T144_E145insPFQ died prenatally and exhibit no fetal movements during gestation (Supplementary Material, Table II) suggests that LCCS1 represents a distinct clinical entity as the prognosis is different than for individuals who have biallelic GLE1 mutations. This is similar to other human genes in which a specific or small number of mutations result in a distinct phenotype (e.g., FGFR3 and achondroplasia).
GLE1 is an essential mRNA export factor [Watkins et al., 1998] that also plays a role in both initiation and termination of protein translation in eukaryotes [Bolger et al., 2008; Folkmann et al., 2013]. GLE1 encodes at least two nearly identical isoforms, GLE1A and GLE1B. Both contain a N-terminal domain required for localization to the nuclear pore complex (NPC), a nucleocytoplasmic shuttling domain, and a coiled-coil domain that is necessary for oligomerization of GLE1—oligomerization is needed for NPC localization and mRNA shuttling [Kendirgi et al., 2003]. A C-terminal 43 amino acid segment unique to GLE1B (Figure 2b) is required for localization of GLE1 to the NPC [Kendirgi et al., 2003; 2005]. Nevertheless, this difference leads to distinct functions: GLE1B exports mRNAs from the nucleus to the cytoplasm via the NPC, while GLE1A localizes to the cytoplasm and is required for stress granule assembly and disassembly, thereby regulating protein translation [Aditi et al., 2015].
Each of the variants we identified affects a residue(s) located in the C terminus of GLE1 (Figure 2 and Supplementary Material, Table I), suggesting a possible relationship between affected domain(s) or affected isoform(s) and phenotype. However, other than proband A, who is homozygous for p.(S693F), a variant that lies in the domain unique to GLE1B, all individuals with GLE1 mutations reported to date have either one or two mutations predicted to affect both isoforms (Supplementary Material, Table 1). Therefore, we cannot readily distinguish any relationships between observed phenotype features, severity, and type or location of a variant.
We predict that biallelic mutations that truncate or otherwise result in loss of function of the coiled-coil domain of both GLE1 isoforms cause LCCS1. In contrast, individuals with mutations that preserve the function of the coiled-coil domain in both isoforms cause LAAHD. We also suggest that because the phenotype of LAAHD is now expanded to include multiple individuals surviving into childhood, LAAHD should be re-named Arthrogryposis with Anterior Horn Cell Disease (AAHD).
Supplementary Material
Acknowledgments
The authors are thankful to affected individuals and their family members for participation. University of Washington Center for Mendelian Genomics (UW-CMG) was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant U54HG006493 to Drs. Debbie Nickerson, Michael Bamshad, and Suzanne Leal. This work was also supported by a grant from the Eunice Kennedy Shriver National Institute of Child Health & Human Development (4R01HD048895-11 to M.J.B.). The authors thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. The authors would like to thank the University of Washington Center for Mendelian Genomics and all contributors to Geno2MP for use of data included in Geno2MP. The authors thank the NHLBI GO Exome Sequencing Project.
Footnotes
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Aditi, Folkmann AW, Wente SR. Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol Biol Cell. 2015;26:1476–1490. doi: 10.1091/mbc.E14-11-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Folkmann AW, Tran EJ, Wente SR. The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell. 2008;134:624–633. doi: 10.1016/j.cell.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, Mcmillin MJ, Wiszniewski W, Gambin T, Coban Akdemir ZH, Doheny K, Scott AF, Avramopoulos D, Chakravarti A, Hoover-Fong J, Mathews D, Witmer PD, Ling H, Hetrick K, Watkins L, Patterson KE, Reinier F, Blue E, Muzny D, Kircher M, Bilguvar K, López-Giráldez F, Sutton VR, Tabor HK, Leal SM, Gunel M, Mane S, Gibbs RA, Boerwinkle E, Hamosh A, Shendure J, Lupski JR, Lifton RP, Valle D, Nickerson DA, Centers for Mendelian Genomics. Bamshad MJ. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet. 2015a;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JX, Mcmillin MJ, Shively KM, Beck AE, Marvin CT, Armenteros JR, Buckingham KJ, Nkinsi NT, Boyle EA, Berry MN, Bocian M, Foulds N, Uzielli MLG, Haldeman-Englert C, Hennekam RCM, Kaplan P, Kline AD, Mercer CL, Nowaczyk MJM, Klein Wassink-Ruiter JS, McPherson EW, Moreno RA, Scheuerle AE, Shashi V, Stevens CA, Carey JC, Monteil A, Lory P, Tabor HK, Smith JD, Shendure J, Nickerson DA, University of Washington Center for Mendelian Genomics. Bamshad MJ. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet. 2015b;96:462–473. doi: 10.1016/j.ajhg.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyment DA, Tetreault M, Beaulieu CL, Hartley T, Ferreira P, Chardon JW, Marcadier J, Sawyer SL, Mosca SJ, Innes AM, Parboosingh JS, Bulman DE, Schwartzentruber J, Majewski J, Tarnopolsky M, Boycott KM, Consortium FC, Care4Rare C Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet. 2015;88:34–40. doi: 10.1111/cge.12464. [DOI] [PubMed] [Google Scholar]
- Ellard S, Kivuva E, Turnpenny P, Stals K, Johnson M, Xie W, Caswell R, Lango Allen H. An exome sequencing strategy to diagnose lethal autosomal recessive disorders. Eur J Hum Genet. 2014;23:401–404. doi: 10.1038/ejhg.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR. Gle1 Functions during mRNA Export in an Oligomeric Complex that Is Altered in Human Disease. Cell. 2013;155:582–593. doi: 10.1016/j.cell.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel M, Cremer K, Ockeloen CW, Lichtenbelt KD, Herkert JC, Denecke J, Haack TB, Zink AM, Becker J, Wohlleber E, Johannsen J, Alhaddad B, Pfundt R, Fuchs S, Wieczorek D, Strom TM, van Gassen KL, Kleefstra T, Kubisch C, Engels H, Lessel D. De Novo Mutations in CHAMP1 Cause Intellectual Disability with Severe Speech Impairment. Am J Hum Genet. 2015;97:493–500. doi: 10.1016/j.ajhg.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herva R, Leisti J, Kirkinen P, Seppanen U. A lethal autosomal recessive syndrome of multiple congenital contractures. Am J Med Genet. 1985;20:431–439. doi: 10.1002/ajmg.1320200303. [DOI] [PubMed] [Google Scholar]
- Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. The Journal of Cell Biology. 2003;160:1029–1040. doi: 10.1083/jcb.200211081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendirgi F, Rexer DJ, Alcázar-Román AR, Onishko HM, Wente SR. Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell. 2005;16:4304–4315. doi: 10.1091/mbc.E04-11-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousiainen HO, Kestilä M, Pakkasjärvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, Brunner HG, Buske OJ, Carey K, Doll C, Dumitriu S, Dyke SOM, Dunnen den JT, Firth HV, Gibbs RA, Girdea M, Gonzalez M, Haendel MA, Hamosh A, Holm IA, Huang L, Hurles ME, Hutton B, Krier JB, Misyura A, Mungall CJ, Paschall J, Paten B, Robinson PN, Schiettecatte F, Sobreira NL, Swaminathan GJ, Taschner PE, Terry SF, Washington NL, Zuchner S, Boycott KM, Rehm HL. The Matchmaker Exchange: A Platform for Rare Disease Gene Discovery. Hum Mutat. 2015;36:915–921. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Parboosingh JS, Boycott KM, Bonnemann CG, Mah JK, Care4Rare Canada Consortium. Lamont RE, Innes AM, Bernier FP. Expansion of the GLE1-associated arthrogryposis multiplex congenita clinical spectrum. Clin Genet. 2016 doi: 10.1111/cge.12876. [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, Chehadeh-Djebbar El S, St-Onge J, Steinmetz A, Huet F, Chouchane M, Darmency-Stamboul V, Callier P, Thauvin-Robinet C, Faivre L, Riviere JB. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. 2016;89:700–707. doi: 10.1111/cge.12732. [DOI] [PubMed] [Google Scholar]
- Vuopala K, Herva R. Lethal congenital contracture syndrome: further delineation and genetic aspects. J Med Genet. 1994;31:521–527. doi: 10.1136/jmg.31.7.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins JL, Murphy R, Emtage JL, Wente SR. The human homologue of Saccharomyces cerevisiae Gle1p is required for poly(A)+ RNA export. Proc Natl Acad Sci USA. 1998;95:6779–6784. doi: 10.1073/pnas.95.12.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.