

Abstract
Many viruses trigger innate and adaptive immune responses and must circumvent the negative consequences to successfully establish infection in their hosts. Human Cytomegalovirus (HCMV) is no exception, and devotes a significant portion of its coding capacity to genes involved in immune evasion. Activation of the NFκB signalling pathway by viral binding and entry results in induction of antiviral and pro-inflammatory genes that have significant negative effects on HCMV infection. However, NFκB signalling stimulates transcription from the Major Immediate Early Promoter (MIEP) and pro-inflammatory signalling is crucial for cellular differentiation and viral reactivation from latency. Accordingly, HCMV encodes proteins that act to both stimulate and inhibit the NFκB signalling pathway. In this Review we will highlight the complex interactions between HCMV and NFκB, discussing the known agonists and antagonists encoded by the virus and suggest why manipulation of the pathway may be critical for both lytic and latent infections.
Keywords: NFκB signalling pathway, Human cytomegalovirus, HCMV lifecycle
Viruses and the NFκB Signalling Pathway
The innate immune response to virus infection results in activation of the NFκB transcription factors, which regulate a vast array of antiviral and pro-inflammatory effector functions. Viruses often trigger the NFκB signalling pathway either through activation of Pattern Recognition Receptors (PRRs) or in response to membrane fusion events. In order to successfully establish an infection viruses encode genes to subvert or utilize this ubiquitous signalling pathway to their own advantage [1]. Some viruses, such as Human Immunodeficiency Virus (HIV) and Herpes Simplex Virus (HSV) utilize NFκB signalling to stimulate viral gene expression [2,3]. Oncogenic gamma-herpesviruses like Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) and Epstein Barr Virus (EBV) encode proteins that activate NFκB signalling in order to utilize pro-survival signals during latency [4,5]. More commonly, viruses inhibit the NFκB signalling pathway using a diverse array of strategies [1,6]. Many RNA and DNA viruses target the PRRs and their adaptors either via downregulation or blocking their activities [7–10]. Others target downstream components of the signalling pathway [11–14] or the NFκB subunits themselves [15–18]. While strategies for manipulation of the NFκB signalling pathway using viral proteins are diverse, new approaches, most recently using viral non-coding RNAs [19–23], are regularly being discovered.
NFκB signalling is a paradigm for the principles of signal transduction and transcriptional activation. Transcriptional regulation is mediated by the NFκB subunits (the transcriptional activators p65/RelA, RelB and c-Rel and the DNA binding proteins p105/p50 and p100/p52), which are abundant, potent, broadly expressed and modulate numerous important cellular functions allowing the cell to respond and adapt to environmental changes. Activation of the NFκB subunits requires phosphorylation- induced ubiquitination and proteasomal degradation of the inhibitor of NFκB proteins (most commonly IκBα, IκBβ and IκBε) that retain the NFκB subunits in the cytosol. For example, phosphorylation on the Ser32 and Ser36 residues results in degradation of IκBα via the 26S proteasome and releases the NFκB subunits to transit to the nucleus, homo- and heterodimerize and bind specific κB binding sites in the promoters of regulated genes. Canonical NFκB signalling is initiated by ligand binding to upstream cell surface receptors (including IL1β, TNFα and TLR receptors), which transduce these extracellular signals via activation of both kinases and ubiquitin ligases. Multiple upstream signalling pathways converge at the IκB kinase (IKK) complex composed of the catalytic subunits IKKα and IKKβ and the structural component IKKγ (or NEMO). Linear ubiquitination of NEMO assembles the IKK complex and activation is the result of phosphorylation of IKKα or IKKβ on serine residues in their activation loops either by upstream kinases or through trans-autophosphorylation. The activated IKK complex plays a critical role by phosphorylating the IκBs and thus activation of this complex is a highly regulated step in the NFκB signalling cascade [24]. In contrast, the non-canonical NFκB signalling pathway is induced by lymphotoxin B, B Cell Activating Factor (BAFF) or CD40 ligand and results in phosphorylation of IKKα dimers by the NFκB Inducing Kinase (NIK). Stimulation of the non-canonical NFκB signalling pathway results in the release of RelB and p52 heterodimers [25]. Termination of the NFκB response is complex and occurs in part through a negative feedback loop resulting in NFκB-dependent expression of the IκB proteins. Newly synthesized IκB relocalizes the NFκB subunits from the DNA to the cytosol thus resulting in a self-limiting inflammatory response.
Human Cytomegalovirus Modulation of the NFκB Signalling Pathway
Herpesviruses have co-evolved with their hosts over millions of years in order to succeed in establishing a life-long infection in the face of constant immune surveillance. In order to persist for the lifetime of the host, herpesviruses have evolved myriad strategies to utilize and evade the host innate and adaptive immune responses. Human cytomegalovirus (HCMV/HHV-5) is a member of the beta-herpesvirus family with high prevalence in the human population; in the United States 50–90% of adults are seropositive and seropositivity is closer to 100% in developing countries [26]. While HCMV infection is generally subclinical in healthy individuals, serious disease can arise when the host immune system is compromised and viral reactivation occurs. HCMV replicates in numerous cell types including macrophages, dendritic cells, fibroblasts, epithelial and endothelial cells as well as smooth muscle cells, neuronal cells, hepatocytes and trophoblasts. In these cell types, HCMV undergoes a lytic replication cycle involving viral binding and entry of the capsid into the cytoplasm releasing tegument proteins that act to immediately disarm intrinsic cellular immune responses. After injection of the viral DNA into the nucleus, cellular transcriptional trans activators act to stimulate transcription from the Major Immediate Early Promoter (MIEP), which results in the transcription of multiple Immediate Early (IE) genes including the major isoforms IE protein 72 (IE72/IE1) and IE86/ IE2. Expression of IE1 and IE2 is critical for the efficient launch of the lytic replication cycle [27,28]. The MIEP enhancer region is highly complex, containing an array of positive and negative cis-acting elements including binding sites for numerous cellular transcription factors such as CREB/ATF, AP-1, Elk-1, SRF and NFκB [29]. These Cis-acting elements work both cooperatively and independently to initiate RNA polymerase II transcription from the MIEP thus ensuring activation of the promoter by a variety of cellular signalling pathways regardless of the differentiation and activation state of the cell. IE proteins help to stimulate expression of Early (E) phase proteins, many of which are involved in DNA replication. E proteins also help to stimulate Late (L) gene expression, whose products are involved in virion assembly and release. HCMV replicates poorly in less differentiated cell types such as CD14+ monocytes and CD34+ Hematopoietic Progenitor Cells (HPCs). In these cells most viral genes are not expressed and the viral genome is maintained in the absence of progeny virus production. The limited viral proteins and non-coding RNAs expressed during latency play important roles in suppressing viral gene expression and regulating intracellular signalling pathways [30]. To uncover how HCMV successfully evades host innate and adaptive immunity in such a diverse array of cell types and during fundamentally disparate lifecycles an understanding of the role of both viral proteins and non-coding RNAs in manipulating cellular signal transduction pathways is required.
The role of NFκB signalling in the HCMV lifecycle is exceedingly complex and evidence suggests that the virus activates both canonical and non-canonical signalling pathways. In turn, HCMV encodes both agonists and antagonists of NFκB signalling in order to aid in viral replication and dissemination, establishment of latency and reactivation. Early work examining regulation of the MIEP identified multiple 18 nucleotide repeats within the MIEP enhancer region containing consensus NFκB binding sites [31–33]. It was postulated that induction of the NFκB signalling pathway at early times after infection could enhance expression from the MIEP and thus help initiate the lytic cascade of gene expression [32,34,35]. It was shown that TNFα, a potent inducer of the NFκB signalling pathway, enhances expression from the MIEP via increased binding of p50 and p65 to the 18 nucleotide repeat [36]. In fact, later work demonstrated that activation of the NFκB signalling cascade is initiated by viral binding [35,37] mediated by gB and gH interacting with their cognate receptors in human fibroblasts [38,39] and monocytes [40] at least in part via interactions with TLR2 [41,42]. The signalling initiated by viral binding results in depletion of preformed cytosolic stores of p50 and p65. Subsequently, de novo synthesis of p50 and p65 occurs through a positive feedback signalling [34] and transactivation by IE proteins [37] involving regulation of the SP1 transcription factor [43]. In addition, Casein Kinase II (CKII) packaged in the virion has been proposed to rapidly phosphorylate IkBα following viral entry, allowing for an additional means of releasing the NFκB subunits which may be necessary for infection of diverse cell types [44]. Interestingly, studies of NFκB activation in primary Monocyte-Derived Macrophages (MDMs) determined that although canonical p50/p65 heterodimers are present at the MIEP very early after viral infection [40,45], complexes composed of p52 and Bcl-3 are found at the MIEP at 5 days post-infection, suggesting context dependent changes in NFκB signalling in different cellular environments [45]. Similar stimuli are known to activate distinct NFκB complexes in cell-type dependent manners [46,47], but how and why the non-canonical NFκB signalling pathway is activated in MDMs remains unclear. p52/Bcl-3 heterodimers are not as efficient at stimulating expression from MIEP reporter constructs [45]; therefore one possibility is that non-canonical NFκB signalling may act to limit MIEP expression in MDMs.
This early work clearly indicated that viral binding and entry induces activation of NFκB signalling and results in expression from the MIEP. However, the MIEP contains numerous binding sites for additional cellular transcriptional activators and repressors and thus the relative importance of NFκB in the overall stimulation of the MIEP and ultimately virus replication was unclear. Additionally, activation of NFκB signalling results in induction of numerous cellular genes, including cell adhesion molecules, complement and acute phase proteins as well as pro-inflammatory cytokines and chemokines which can have antiviral effects on HCMV replication. Thus, the contribution of the NFκB signalling pathway to full viral replication has been studied extensively in vitro - with conflicting results. Growth curves of AD169 and Toledo HCMV strains in human fibroblasts overexpressing a Dominant Negative (DN) mutant of IκBα, suggested that blocking NFκB signalling in fibroblasts was neutral to viral replication [48]. Additionally, when an NFκB site-mutated HCMV MIEP replaces its MCMV counterpart in the MCMV genome the resulting virus replicates with Wild Type (WT) kinetics in fibroblasts [48]. In contrast, using pharmacological inhibition of the NFκB pathway, as well as the IκBα DN mutant, it was suggested that blocking NFκB signalling resulted in a modest increase in AD169 replication, and prevented exogenous TNFα and IFNγ from negatively affecting virus replication [49]. In addition, this study utilized a constitutively active mutant of IKKβ and showed that constitutive activation of canonical NFκB signalling inhibited viral replication through the production of IFNβ. In order to directly test the requirement of NFκB signalling in regulation of the MIEP during viral infection, Gustems et al. [50] constructed an HCMV AD169 mutant containing point mutations in all 4 NFκB binding sites within the MIEP and showed no deleterious effects on IE expression or viral replication in human fibroblasts. This work indicated that in the context of lytic AD169 infection of fibroblasts, transactivation of the MIEP can be accomplished through the additional transcription factor binding sites found within the enhancer region [29]. In fact, our work and that of others (unpublished observations, [51,52]) suggest that AD169 does not trigger or modulate the NFκB signalling pathway in the same manner as clinical strains of HCMV and may account for the relative resistance of AD169 replication to inhibition of the NFκB signalling pathway.
In contrast to the studies described above, work by several groups [53–59], using both AD169 and clinical strains of HCMV and various NFκB inhibitors as well as DN IκBα, IKKα and IKKβ constructs demonstrate that IE and subsequent gene expression as well as viral yields are reduced when NFκB signalling is blocked in fibroblasts and endothelial cells. Intriguingly, expression of the IκBα DN protein had the greatest deleterious effect on MIEP transactivation compared to DN IKKα and IKKβ constructs [59]. These observations suggest that there are multiple signalling pathways activated by HCMV infection that converge at the phosphorylation of IκBα, some of which do not include activation of the IKK complex, such as direct phosphorylation of IκBα by tegument-associated CKII [44]. These studies also indicated that IKKα plays a more important role in MIEP transaction than IKKβ [59] and hints at the involvement of the non-canonical NFκB signalling pathway in fibroblasts as has been observed in MDMs [45]. Interestingly, when the later phase of NFκB signalling that occurs as a result of IE1 transactivation of the p50 and p65 promoters [37] was blocked by addition of pharmacological inhibitors, viral replication was still impaired [58], suggesting an essential role for sustained NFκB signalling during HCMV infection. The apparently contradictory observations about the importance of NFκB signalling during viral infection could be at least partially resolved by studies which examined the role of NFκB signalling in replicating and growth arrested cells [55]. Using DN IKKβ constructs and viruses lacking the NFκB target sequences within the MIEP the authors demonstrate that virus replication is only restricted in growth arrested, and not proliferating fibroblasts and endothelial cells. These data suggest that the differentiation and activation state of the infected cell plays a significant role in NFκB-mediated MIEP transactivation and lytic replication. Further experimentation to address the contradictory requirement of NFκB signalling to the HCMV lifecycle is required to resolve this essential question. Finally, the role of NFκB signalling in regulating gene expression at other stages of the HCMV lifecycle has not been thoroughly investigated. US3 contains NFκB binding sites [60,61] that may contribute to the requirement of NFκB at later times in the infection cycle and additional κB binding sites exist within the HCMV genome [55].
Whether NFκB signalling and transactivation of the MIEP is essential to virus replication both in vitro and in vivo remains an ongoing question, but microarray data indicates that expression of NFκB-inducible genes is more robust when viral gene expression is inhibited [62], suggesting that some viral gene products act to dampen the NFκB response. It was first reported that different lab-adapted and clinical strains of HCMV could block signalling through the canonical NFκB pathway initiated by IL1β or TNFα at or above the point of convergence of the NFκB signalling pathways [63,64]. IκBα phosphorylation and degradation was abrogated and expression of several pro- inflammatory cytokines was prevented in infected fibroblasts and endothelial cells treated with IL1β or TNFα after 72 h of infection [63,64]. Similarly, phosphorylation and degradation of IκBα was not detected at 5 days post-infection in MDMs [45]. In fact, IκBα transcript [40] and protein levels [45] are increased during infection of MDMs, suggesting that canonical NFκB signalling is also actively blocked in this cell type at later times of infection [65]. The antagonism of NFκB signalling requires expression of both early [64] and late gene products [63,64]. Interestingly, when infected cells are treated with IL1β a near-complete block in IkBα degradation is observed, while treatment of infected cells with TNFα resulted in residual IkBα phosphorylation and degradation, suggesting that HCMV antagonism of the NFκB signalling pathway is dependent upon which upstream signalling pathway triggers IκBα phosphorylation [63]. Using AD169 mutants the ability to block TNFα-mediated NFκB signalling could be genetically separated from blocking IL1β-mediated signalling [64]. To date, the viral gene product(s) necessary for this late block in NFκB signalling have not been identified, but several gene products have been implicated in interfering with the NFκB signalling pathway.
HCMV-Encoded Antagonists of the NFκB Signalling Pathways
Viral proteins involved in blocking NFκB signalling
Figure 1 Illustrates the HCMV proteins and non-coding RNAs that interfere with the NFκB signalling pathway.
Figure 1.

HCMV-encoded antagonists of the NFκB signalling pathway. NFκB signalling can be induced by activation of a variety of cell surface receptors as well as HCMV binding and entry. Upstream signalling cascades culminate at the activation of the IKK complex. Several HCMV proteins and miRNAs (shown in red) block activation of the IKK complex or downstream binding of the NFκB transcription factors to their cognate sequences.
The tegument protein pp65 was the first HCMV protein shown to interfere with NFκB signalling [66]. Using DNA arrays, it was demonstrated that pp65-deficient viruses induced anti-viral and pro-inflammatory genes to a greater extent than WT virus and exogenous expression of pp65 could block type I IFN signalling. pp65-deficient viruses induce NFκB subunit binding to a greater extent than WT, but have no effect on IRF3 binding, suggesting that pp65 interferes specifically with the NFκB signalling pathway.
The immediate early protein IE86 also blocks NFκB signalling in infected cells [67–69]. IE86 attenuates the production of IFNβ during HCMV infection either by preventing NFκB subunit binding to the IFN promoter [68] or by blocking interactions between the subunits and other transcriptional activators [70]. In addition, expression of IE86 blocks NFκB-dependent gene expression in response to external stimuli, such as Sendai virus and TNFα treatment indicating that IE86 alone is sufficient to block NFκB signalling [67]. These studies examined the effects of IE86 in isolation or at early times post-infection, well before the late block to NFκB signalling observed in studies by Jarvis et al. [63] and Montag et al. [64]. Thus HCMV likely encodes multiple gene products from different kinetic classes that block NFκB signalling. It remains an intriguing question as to why HCMV encodes an inhibitor of canonical NFκB signalling that is expressed with IE kinetics when the MIEP is transactivated by NFκB subunit binding. Perhaps this is a mechanism of negative feedback utilized by the virus to prevent over-activation of NFκB signalling and pro-inflammatory cytokine production, given the functional redundancy of transcription factor binding to the MIEP.
HCMV cmv-IL-10 (UL111a) is a functional homolog of cellular IL-10, itself a potent inhibitor of pro-inflammatory responses. Like cellular IL-10, recombinant cmv-IL-10 treatment of THP-1 cells can block NFκB signalling at or above the level of IκBα degradation, although the exact mechanism for the inhibition has not been further elucidated [71].
The tegument protein UL26 has most recently been demonstrated to possess NFκB inhibiting functions [52]. Expression of UL26 can block TNFα and Sendai-virus-induced IKK activation, IκBα degradation and IL6 production, suggesting that it functions at or above the point of convergence of multiple NFκB signalling pathways and may contribute to the late block in NFκB signalling observed in HCMV-infected cells [63,64]. An UL26-mutant virus induces canonical NFκB signalling with similar kinetics to WT infection, suggesting tegument-associated UL26 does not block early induction of the pathway. Interestingly, the UL26 mutant virus induces higher expression of the RelB NFκB subunit, especially at later time of infection, suggesting that UL26 may play a role in suppressing non-canonical NFκB signalling.
HCMV Non-coding RNAs Involved in Blocking NFκB Signalling
Along with viral proteins, HCMV also expresses non-coding RNAs that interfere with different aspects of NFκB signalling. MicroRNAs (miRNAs) are small, ~22 nucleotide RNAs that act to post-transcriptionally regulate gene expression. miRNAs normally interact with short regions of complementarity in the 3′ UTR of targeted transcripts which results in recruitment of cellular protein complexes that ultimately lead to translations repression and/or mRNA degradation [72]. Thus, by targeting regions of complementarity in genes involved in the NFκB signalling pathway, HCMV miRNAs could participate in the late block to NFκB signalling observed in HCMV infected cells [63,64]. In fact, most HCMV miRNAs are expressed with early kinetics, accumulate throughout the course of lytic infection [73,74] and are abundant at the late stages of infection. Additionally, several HCMV miRNAs are expressed during latency in CD34+ HPCs [75] and could act to modulate NFκB signalling when most viral proteins are no longer expressed. HCMV miR-US5-1 and miR-UL112-3p have recently been demonstrated to block NFκB signalling induced by IL1β and TNFα at late times post-infection [20]. Both miRNAs target IKKα and IKKβ, limit the phosphorylation and degradation of IκBα and attenuate the downstream expression of the pro-inflammatory cytokines RANTES, IL6 and TNFα in fibroblasts, endothelial cells and THP-1 cells. Infection of cells with an HCMV TB40/E mutant lacking expression of miR-US5-1 and miR-UL112-3p results in higher levels of IKKα and IKKβ proteins compared to WT-infected cells, allows for partial IκBα degradation following exogenous IL1β or TNFα treatment and increased secretion of pro-inflammatory cytokines compared to WT infected cells. By replacing the miRNA sequences with shRNAs targeting IKKα and IKKβ, the expression and secretion of pro- inflammatory cytokines could be reduced to WT levels, indicating that the mutant phenotype was due to the loss of IKK complex targeting [20]. In addition, miR-UL112- 3p also targets the TLR2 receptor, thereby blocking TLR2-induced IRAK1 activation and subsequent expression of pro-inflammatory cytokines [21]. Given that TLR2 signalling results in activation of the IKK complex, it is likely that at least some of the observed effects of miR-UL112-3p on pro-inflammatory cytokine expression is also due to its effects on IKKα and IKKβ expression [20]. miR-US5-1 and miR-UL112-3p also work in concert with a third HCMV miRNA, miR-US5-2, to interfere with the endocytic recycling compartment and severely attenuate the secretion of pro-inflammatory cytokines [76]. Additionally, miR-UL112-3p may target IL-32, an inducer of NFκB signalling [77]. Finally, HCMV miR-UL148D targets RANTES [78] and ACVR1B of the activin signalling axis which promotes increased IL6 secretion upon activin stimulation [75]. These studies underscore how HCMV miRNAs can interfere with NFκB signalling at numerous steps to limit the deleterious effects of pro-inflammatory cytokine production.
HCMV-encoded agonists of NFκB signalling pathways
Paradoxically, while encoding numerous proteins and non-coding RNAs that block NFκB signalling in fibroblasts, endothelial cells and monocytes, HCMV also encodes several agonists of NFκB signalling. It has long been postulated that certain NFκB- responsive genes and the effects of activation of the NFκB signalling pathway could also be beneficial to viral replication and spread, especially in vivo [79]. Pro-inflammatory cytokines and chemokines recruit cells to the site of lytic infection that can be used for dissemination and seeding new viral infections [80]. Additionally, anti-apoptotic genes induced by NFκB signalling may help to prolong the life of the cell for efficient virus production [81]. Finally, an intriguing possibility is that HCMV encodes proteins that help to enhance NFκB signalling specifically in latently infected cells in order to augment transactivation of the MIEP to promote reactivation of the virus from latency. Figure 2 highlights the proteins that act to stimulate signalling through the NFκB pathway.
Figure 2.
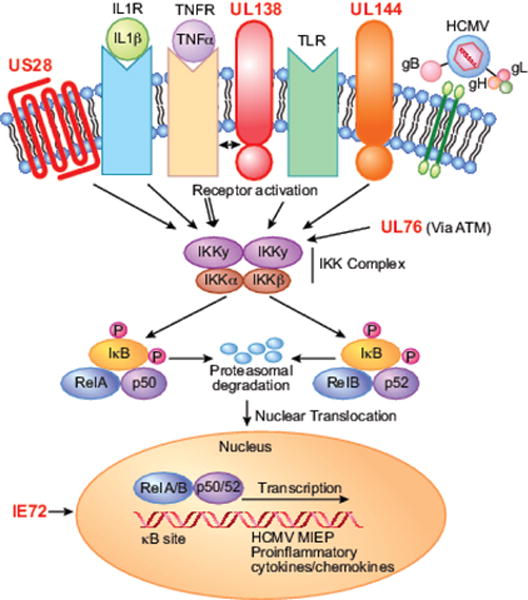
HCMV-encoded agonists of the NFκB signaling pathway. HCMV encodes three cell surface proteins (US28, UL138 and UL144, shown in red) that can activate or enhance NFκB signaling. In addition, HCMV UL76 and IE1 can activate NFκB signaling through unknown mechanisms.
In contrast to the NFκB-inhibiting functions of IE2, IE1 transactivates numerous cellular and viral genes utilizing the NFκB signalling pathway. Although many of its ascribed functions are due to positive feedback on the MIEP, IE1 alone induces NFκB signalling in several cell types [32]. IE1 transactivates the p65 promoter [37,38], IL6 promoter [82], TNFα promoter [83], and the IL8 promoter [84] through the NFκB signalling pathway. Interestingly, it was determined that IE1 selectively induces RelB/p50 subunits rather than the canonical p65/p50 complexes in smooth muscle cells and fibroblasts [85].
UL144 is a transmembrane protein with properties similar to the TNF Receptor (TNFR) family that potently activates the NFκB signalling pathway and expression of the chemokine CCL22 in a TRAF6- and TRIM23-dependent manner [86,87]. In light of the ability of IE86 to block NFκB subunit binding, Poole et al. [88] determined that UL144- mediated activation of CCL22 was insensitive to IE86 expression during infection suggesting that the ability of IE86 to block NFκB subunit binding is promoter- and context-dependent.
UL76, a putative endonuclease, induces the NFκB signalling pathway through activation of ATM and the DNA damage response. Activation of ATM ultimately results in the phosphorylation of NEMO leading to p65 translocation to the IL8 promoter, increased IL8 expression and enhancement of HCMV replication [89]. IL8 is an important chemokine for neutrophil attraction, which the authors postulate may be important for viral replication and dissemination [90,91].
US28 is a 7-transmembrane chemokine receptor that activates multiple cellular signalling pathways in ligand-dependent and -independent manners that is expressed during latency in CD34+ HPCs. US28 constitutively activates NFκB signalling utilizing Gq/11 protein-dependent pathways [92]. US28 has been postulated to play a role inactivation of the MIEP through its NFκB signalling activity [93] and activation of the NFκB signalling pathway by US28 has been linked to increased COX2 expression and angiogenesis in endothelial cells [94].
UL138 was described in two reports to enhance TNFR1 expression on the cell surface [95,96]. UL138 physically interacts with TNFR1, prolonging its half-life and signalling capacity [96]. Interestingly, in comparing a UL138 mutant virus to AD169 strains lacking the ULb’ region, additional TNF-regulating factors were postulated [96]. It is possible that during latent infection of CD34+ HPCs, UL138 acts to enhance the TNF- responsiveness of infected cells. Given the importance of TNF signalling to HCMV reactivation [65,97], and the role of NFκB signalling in enhancing MIEP expression [32,33,36,37,59], it is intriguing to postulate that the virus modulates NFκB signalling to regulate reactivation from latency.
Perspectives
While HCMV has evolved to utilize the NFκB signalling pathway to launch its lytic replication cycle it has also had to evolve to control the antiviral responses thus induced. Evidence suggests that NFκB signalling that is tightly controlled by the virus at early times post-infection is beneficial to viral replication. However, the virus has evolved mechanisms to block any strong NFκB signalling induced by extrinsic signals that could be detrimental to viral replication [20,52,63,64]. Moreover, HCMV modulates both canonical and non-canonical NFκB signalling. At early times activation of the canonical pathway predominates [37,38], but evidence of both activation [45,85,87] and suppression [52] of the non-canonical signalling pathway at later times post-infection has also been demonstrated. Activation of the non-canonical NFκB pathways by exogenous stimuli results in IFNβ production [98] suggesting extrinsic activation of non-canonical signalling, like extrinsic activation of canonical signalling [20,52,63,64] can be detrimental to virus replication. The intricate modulation of these different arms of the NFκB pathways may allow HCMV to enhance the pro-viral effects, while limiting the antiviral effects of NFκB signalling.
On the surface, the apparently contradictory roles of NFκB signalling during HCMV infection are confusing, but likely underlie the complexity of the HCMV replication cycle in the host. During lytic infection, NFκB signalling is used to enhance MIEP expression and viral replication, prolong the life of the infected cell while aiding in viral dissemination by recruiting additional cell types to the site of infection. During HCMV infection of monocytes, NFκB signalling helps to initiate a differentiation program resulting in a unique macrophage phenotype [99,100]. Additionally, NFκB-mediated up-regulation of ICAM-1 and ICAM-3 is essential for monocyte motility and firm adhesion to endothelial cells [101], a function key to the ability of monocytes to disseminate and seed new viral infections. Interestingly, HCMV-infected MDMs do not basally express high levels of NFκB-dependent cytokines, but can potentiate cytokine expression induced by lipopolysaccharide [102], suggesting that infected MDMs are poised to reactivate virus upon pro-inflammatory cytokine expression. Allogeneic T cell stimulation produces high levels of IL-6, TNFα and IFNγ and results in HCMV reactivation in monocytes from the peripheral blood [97]. Neutralization of TNFα or IFNγ prevents HCMV reactivation, suggesting that a highly inflammatory environment is critical for viral reactivation [65]. Thus, the virus must maintain a careful balancing act to manipulate the outcomes of NFκB activation for its own benefit depending on the cell type infected.
The role of NFκB signalling in latent HCMV infection of CD34+ cells has not been investigated. Whether viral binding and entry stimulates NFκB signalling in CD34+ HPCs as it does in other cell types is an intriguing question. NFκB signalling pathway components are transcriptionally up regulated in HPCs protected from FAS-mediated apoptosis [103], suggesting that HCMV-induced NFκB signalling may help protect and prolong the life of infected HPCs [104]. Non-canonical NFκB signalling, which is induced by HCMV infection [45,85,87], has been implicated in CD34+ HPC differentiation towards the myeloid lineage [105]. In addition, TNFα-mediated activation of NFκB signalling in HPCs prevents erythropoiesis [106,107], which is markedly suppressed during HCMV infection of HPCs [108]. NFκB signalling is also critical for CD34+ -derived myeloid DC differentiation and function [109], which may highlight a critical link between NFκB signalling, myeloid differentiation and viral reactivation. UL138 and US28, two viral gene products essential for latency in CD34+ HPCs [110,111], stimulate the NFκB signalling pathway and thus may play a role in both transactivation of the MIEP and cellular differentiation in order to promote reactivation. HCMV miRNAs are also expressed during latency, and at least some HCMV miRNAs act to block NFκB signalling [20,21]. One possibility is that viral proteins help to poise the latently infected cell for reactivation, but viral miRNAs act as fine-tuners of the NFκB response, blocking any low-level signals that would result in sub-optimal differentiation and viral reactivation. The mechanistic details of how HCMV limits the antiviral effects while enhancing the pro-viral facets of NFκB signalling remain a mystery. What is clear is that both viral proteins and non-coding RNAs participate in altering the intracellular signalling pathways in HCMV-infected cells in order to successfully establish life-long infections in vivo.
Acknowledgments
We wish to thank Patrizia Caposio and Jessica Smith for insightful comments during the preparation of this manuscript and are grateful to Andrew Townsend for technical assistance.
Funding Information
NIH grants AI21064 to Jay A. Nelson. The funders had no role in study design, data collection and interpretation or decision to submit work for publication.
References
- 1.Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene. 2006;25:6844–6867. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel A, Hanson J, McLean TI, Olgiate J, Hilton M, et al. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virol. 1998;247:212–22. doi: 10.1006/viro.1998.9243. [DOI] [PubMed] [Google Scholar]
- 3.Varin A, Manna SK, Quivy V, Decrion AZ, Van Lint C, et al. Exogenous Nef proteinactivates NF-kappa B, AP-1, and c-Jun N-terminal kinase and stimulates HIVtranscription in promonocytic cells. Role in AIDS pathogenesis. J Biol Chem. 2003;278:2219–2227. doi: 10.1074/jbc.M209622200. [DOI] [PubMed] [Google Scholar]
- 4.Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004;199:993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Malinin NL, Wallach D, Gilmore TD, Kieff E, Mosialos G, et al. Epstein-Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-kappaB through a pathway that includes the NF-kappaB-inducing kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc Natl Acad Sci USA. 1998;95:10106–10111. doi: 10.1073/pnas.95.17.10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulhern O, Harrington B, Bowie AG. Modulation of innate immune signaling pathways by viral proteins. Adv Exp Med Biol. 2009;666:49–63. doi: 10.1007/978-1-4419-1601-3_4. [DOI] [PubMed] [Google Scholar]
- 7.Mesman AW, Zijlstra Willems EM, Kaptein TM, de Swart RL, Davis ME, et al. Measles virus suppresses RIG-I-like receptor activation in dendritic cells via DC-SIGN-mediated inhibition of PP1 phosphatases. Cell Host Microbe. 2014;16:31–42. doi: 10.1016/j.chom.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bussey KA, Reimer E, Todt H, Denker B, Gallo A, et al. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the Toll-like receptor-induced proinflammatory cytokine response. J Virol. 2014;88:9245–9259. doi: 10.1128/JVI.00841-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, et al. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J Virol. 2010;84:10802–10811. doi: 10.1128/JVI.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D, Fang L, Wei D, Zhang H, Luo R, et al. Hepatitis A Virus 3C Protease Cleaves NEMO To Impair Induction of Beta Interferon. J Virol. 2014;88:10252–10258. doi: 10.1128/JVI.00869-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao S, Song L, Li J, Zhang Z, Peng H, et al. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol. 2012;14:1849–1866. doi: 10.1111/cmi.12005. [DOI] [PubMed] [Google Scholar]
- 13.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, et al. Poxvirus targeting of E3 ligase beta-TrCP by molecular mimicry: a mechanism to inhibit NF-kappaB activation and promote immune evasion and virulence. PLoS Pathog. 2013;9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morelli M, Dennis AF, Patton JT. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-kappaB activation by using molecular mimicry to target beta-TrCP. MBio. 2015;6 doi: 10.1128/mBio.02490-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Wang K, Wang S, Zheng C. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J Virol. 2013;87:12935–12948. doi: 10.1128/JVI.01952-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Burles K, Couturier B, Randall CM, Shisler J, et al. Ectromelia virus encodes a BTB/kelch protein, EVM150, that inhibits NF-kappaB signaling. J Virol. 2014;88:4853–4865. doi: 10.1128/JVI.02923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ni L, Wang S, Wang K, Lin R, Zheng C, et al. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol. 2013;87:9788–9801. doi: 10.1128/JVI.01440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sloan E, Henriquez R, Kinchington PR, Slobedman B, Abendroth A. Varicella- zoster virus inhibition of the NF-kappaB pathway during infection of human dendritic cells: role for open reading frame 61 as a modulator of NF-kappaB activity. J Virol. 2012;86:1193–1202. doi: 10.1128/JVI.06400-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramalingam D, Kieffer KP, Uldrick TS, Yarchoan R, Ziegelbauer JM, et al. Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/ interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol. 2012;86:11663–11674. doi: 10.1128/JVI.01147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meaghan HH, Lauren MH, Jennifer M, Jay AN. Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-kappaB-Activating Factors through Direct Downregulation of IKKalpha and IKKbeta. MBio. 2017;8 doi: 10.1128/mBio.00109-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landais I, Pelton C, Streblow D, DeFilippis V, Weeney MS, et al. Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFkappaB Signaling Pathway. PLoS Pathog. 2015;11:e1004881. doi: 10.1371/journal.ppat.1004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei X, Bai Z, Ye F, Xie J, Kim CG, et al. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skalsky RL, Kang D, Linnstaedt SD, Cullen BR. Evolutionary conservation of primate lymphocryptovirus microRNA targets. J Virol. 88:1617–1635. doi: 10.1128/JVI.02071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol1. 2009:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cildir G, Low KC, Tergaonkar V. Noncanonical NF-kappaB Signaling in Health and Disease. Trends Mol Med. 22:414–429. doi: 10.1016/j.molmed.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Mussi PMM, Yamamoto AY, Moura BRM, Isaac ML, Oliveira PF, et al. Birth prevalence and natural history of congenital cytomegalovirus infection in a highly seroimmune population. Clin Infect Dis. 2009;49:522–528. doi: 10.1086/600882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchini A, Liu H, Zhu H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol. 2001;75:1870–1878. doi: 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mocarski ES, Kemble GW, Lyle JM, Greaves RF. A deletion mutant in the human cytomegalovirus gene encoding IE1 (491aa) is replication defective due to a failure in autoregulation. Proc Natl Acad Sci USA. 1996;93:11321–11326. doi: 10.1073/pnas.93.21.11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stinski MF, Isomura H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med Microbiol Immunol. 2008;197:223–223. doi: 10.1007/s00430-007-0069-7. [DOI] [PubMed] [Google Scholar]
- 30.Goodrum F, Caviness K, Zagallo P. Human cytomegalovirus persistence. Cell Microbiol. 2012;14:644–655. doi: 10.1111/j.1462-5822.2012.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stinski MF, Roehr TJ. Activation of the major immediate early gene of human cytomegalovirus by cis-acting elements in the promoter-regulatory sequence and by virus-specific trans-acting components. J Virol. 1985;55:431–441. doi: 10.1128/jvi.55.2.431-441.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sambucetti LC, Cherrington JM, Wilkinson GW, Mocarski ES. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. Embo J. 1989;8:4251–4258. doi: 10.1002/j.1460-2075.1989.tb08610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cherrington JM, Mocarski ES. Human cytomegalovirus ie1 transactivates the alpha promoter-enhancer via an 18-base-pair repeat element. J Virol. 1989;63:1435–1440. doi: 10.1128/jvi.63.3.1435-1440.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowalik TF, Wing B, Haskill JS, Azizkhan JC, Baldwin AS, Jr, et al. Multiple mechanisms are implicated in the regulation of NF-kappa B activity during human cytomegalovirus infection. Proc Natl Acad Sci USA. 1993;90:1107–1111. doi: 10.1073/pnas.90.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boldogh I, Fons MP, Albrecht T. Increased levels of sequence-specific DNA-binding proteins in human cytomegalovirus-infected cells. Biochem Biophys Res Commun. 1993;197:1505–1510. doi: 10.1006/bbrc.1993.2647. [DOI] [PubMed] [Google Scholar]
- 36.Prosch S, Staak SK, Liebenthal C, Stamminger T. Stimulation of the human cytomegalovirus IE enhancer/promoter in HL-60 cells by TNFalpha is mediated via induction of NF-kappaB. Virol. 1995;208:197–206. doi: 10.1006/viro.1995.1143. [DOI] [PubMed] [Google Scholar]
- 37.Yurochko AD, Kowalik TF, Huong SM, Huang ES. Human cytomegalovirus upregulates NF-kappa B activity by transactivating the NF-kappa B p105/p50 and p65 promoters. J Virol. 1995;69:5391–5400. doi: 10.1128/jvi.69.9.5391-5400.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yurochko AD, Wang ESH, Rasmussen L, Keay S, Pereira L, et al. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J Virol. 1997;71:5051–5059. doi: 10.1128/jvi.71.7.5051-5059.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlquist JF, Edelman L, Bennion DW, Anderson JL. Cytomegalovirus induction of interleukin-6 in lung fibroblasts occurs independently of active infection and involves a G protein and the transcription factor, NF-kappaB. J Infect Dis. 1999;179:1094–1100. doi: 10.1086/314734. [DOI] [PubMed] [Google Scholar]
- 40.Yurochko AD, Huang ES. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J Immunol. 1999;162:4806–4816. [PubMed] [Google Scholar]
- 41.Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77:4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol. 2006;177:7094–7102. doi: 10.4049/jimmunol.177.10.7094. [DOI] [PubMed] [Google Scholar]
- 43.Yurochko AD, Mayo MW, Poma EE, Baldwin AS, Jr, Huang ES. Induction of the transcription factor Sp1 during human cytomegalovirus infection mediates upregulation of the p65 and p105/p50 NF-kappaB promoters. J Virol. 1997;71:4638–4648. doi: 10.1128/jvi.71.6.4638-4648.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nogalski MT, Podduturi JP, DeMeritt IB, Milford LE, Yurochko AD. The human cytomegalovirus virion possesses an activated casein kinase II that allows for the rapid phosphorylation of the inhibitor of NF-kappaB, IkappaBalpha. J Virol. 2007;81:5305–5314. doi: 10.1128/JVI.02382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan KA, Coaquette A, Davrinche C, Herbein G. Bcl-3-regulated transcription from major immediate-early promoter of human cytomegalovirus in monocyte-derived macrophages. J Immunol. 2009;182:7784–7794. doi: 10.4049/jimmunol.0803800. [DOI] [PubMed] [Google Scholar]
- 46.Lernbecher T, Muller U, Wirth T. Distinct NF-kappa B/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365:767–770. doi: 10.1038/365767a0. [DOI] [PubMed] [Google Scholar]
- 47.Beg AA, Baldwin AS., Jr Activation of multiple NF-kappa B/Rel DNA-binding complexes by tumor necrosis factor. Oncogene. 1994;9:1487–1492. [PubMed] [Google Scholar]
- 48.Benedict CA, Angulo A, Patterson G, Ha S, Huang H. Neutrality of the canonical NF-kappaB-dependent pathway for human and murine cytomegalovirus transcription and replication in vitro. J Virol. 2004;78:741–750. doi: 10.1128/JVI.78.2.741-750.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eickhoff JE, Cotton M. NF-kappaB activation can mediate inhibition of human cytomegalovirus replication. J Gen Virol. 2005;86:285–295. doi: 10.1099/vir.0.80458-0. [DOI] [PubMed] [Google Scholar]
- 50.Gustems M, Borst E, Benedict CA, Perez C, Messerle M. Regulation of the transcription and replication cycle of human cytomegalovirus is insensitive to genetic elimination of the cognate NF-kappaB binding sites in the enhancer. J Virol. 2006;80:9899–9904. doi: 10.1128/JVI.00640-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho CM, Donovan-Banfield IZ, Tan L, Zhang T, Gray NS. Inhibition of IKKalpha by BAY61-3606 Reveals IKKalpha-Dependent Histone H3 Phosphorylation in Human Cytomegalovirus Infected Cells. PLoS One. 2016;11:e0150339. doi: 10.1371/journal.pone.0150339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathers C, Schafer X, Martinez-Sobrido L, Munger J. The human cytomegalovirus UL26 protein antagonizes NF-kappaB activation. J Virol. 2014;88:14289–14300. doi: 10.1128/JVI.02552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prosch S, Wuttke R, Kruger DH, Volk HD. NF-kappaB–a potential therapeutic target for inhibition of human cytomegalovirus (re)activation? Biol Chem. 2002;383:1601–1609. doi: 10.1515/BC.2002.181. [DOI] [PubMed] [Google Scholar]
- 54.Prosch S, Priemer C, Hoflich C, Liebenthaf C, Babel N. Proteasome inhibitors: a novel tool to suppress human cytomegalovirus replication and virus-induced immune modulation. Antivir Ther. 2003;8:555–567. [PubMed] [Google Scholar]
- 55.Caposio P, Luganini A, Hahn G, Landolfo S, Gribaudo G. Activation of the virus- induced IKK/NF-kappaB signalling axis is critical for the replication of human cytomegalovirus in quiescent cells. Cell Microbiol. 2007;9:2040–2054. doi: 10.1111/j.1462-5822.2007.00936.x. [DOI] [PubMed] [Google Scholar]
- 56.Caposio P, Musso T, Luganini A, Inoue H, Gariglio M. Targeting the NF- kappaB pathway through pharmacological inhibition of IKK2 prevents human cytomegalovirus replication and virus-induced inflammatory response in infected endothelial cells. Antiviral Res. 2007;73:175–184. doi: 10.1016/j.antiviral.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Caposio P, Dreano M, Garotta G, Gribaudo G, Landolfo S. Human cytomegalovirus stimulates cellular IKK2 activity and requires the enzyme for productive replication. J Virol. 2004;78:3190–3195. doi: 10.1128/JVI.78.6.3190-3195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeMeritt IB, Podduturi JP, Tilley AM, Nogalski MT, Yurochko AD. Prolonged activation of NF-kappaB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virol. 2006;346:15–31. doi: 10.1016/j.virol.2005.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DeMeritt IB, Milford LE, Yurochko AD. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol. 2004;78:4498–4507. doi: 10.1128/JVI.78.9.4498-4507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thrower AR, Bullock GC, Bissell JE, Stinski MF. Regulation of a human cytomegalovirus immediate-early gene (US3) by a silencer-enhancer combination. J Virol. 1996;70:91–100. doi: 10.1128/jvi.70.1.91-100.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan YJ, Tseng WP, Hayward GS. Two distinct upstream regulatory domains containing multicopy cellular transcription factor binding sites provide basal repression and inducible enhancer characteristics to the immediate-early IES (US3) promoter from human cytomegalovirus. J Virol. 1996;70:5312–5328. doi: 10.1128/jvi.70.8.5312-5328.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Browne EP, Wing B, Coleman D, Shenk T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J Virol. 2001;75:12319–12330. doi: 10.1128/JVI.75.24.12319-12330.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jarvis MA, Borton JA, Keech AM, Wong J, Britt WJ. Human cytomegalovirus attenuates interleukin-1beta and tumor necrosis factor alpha proinflammatory signaling by inhibition of NF-kappaB activation. J Virol. 2006;80:5588–5598. doi: 10.1128/JVI.00060-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montag C, Wagner J, Gruska I, Hagemeier C. Human cytomegalovirus blocks tumor necrosis factor alpha- and interleukin-1beta-mediated NF-kappaB signaling. J Virol. 2006;80:11686–11698. doi: 10.1128/JVI.01168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soderberg-Naucler C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest. 1997;100:3154–3163. doi: 10.1172/JCI119871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Browne EP, Shenk T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci USA. 2003;100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taylor RT, Bresnahan WA. Human cytomegalovirus IE86 attenuates virus- and tumor necrosis factor alpha-induced NFkappaB-dependent gene expression. J Virol. 2006;80:10763–10771. doi: 10.1128/JVI.01195-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taylor RT, Bresnahan WA. Human cytomegalovirus immediate-early 2 protein IE86 blocks virus-induced chemokine expression. J Virol. 2006;80:920–928. doi: 10.1128/JVI.80.2.920-928.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taylor RT, Bresnahan WA. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced beta interferon production. J Virol. 2005;79:3873–3877. doi: 10.1128/JVI.79.6.3873-3877.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gealy C, Humphreys C, Dickinson V, Stinski M, Caswell R. An activation-defective mutant of the human cytomegalovirus IE2p86 protein inhibits NF-kappaB-mediated stimulation of the human interleukin-6 promoter. J Gen Virol. 2007;88:2435–2440. doi: 10.1099/vir.0.82925-0. [DOI] [PubMed] [Google Scholar]
- 71.Nachtwey J, Spencer JV. HCMV IL-10 suppresses cytokine expression in monocytes through inhibition of nuclear factor-kappaB. Viral Immunol. 2008;21:477–482. doi: 10.1089/vim.2008.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 73.Grey F, Antoniewicz A, Allen E, Saugstad J, McShea A. Identification and characterization of human cytomegalovirus-encoded microRNAs. J Virol. 2005;79:12095–12099. doi: 10.1128/JVI.79.18.12095-12099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stark TJ, Arnold JD, Spector DH, Yeo GW. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J Virol. 2012;86:226–235. doi: 10.1128/JVI.05903-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lau B, Poole E, Krishna B, Sellart I, Wills MR. The Expression of Human Cytomegalovirus MicroRNA MiR-UL148D during Latent Infection in Primary Myeloid Cells Inhibits Activin A-triggered Secretion of IL-6. Sci Rep. 2016;6:31205. doi: 10.1038/srep31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe. 2014;15:363–373. doi: 10.1016/j.chom.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang Y, Qi Y, Ma Y, He R, Ji R. The expression of interleukin-32 is activated by human cytomegalovirus infection and down regulated by hcmv-miR-UL112-1. Virol J. 2013;10:51. doi: 10.1186/1743-422X-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim Y, Lee S, Kim S, Kim D, Ahn JH. Human cytomegalovirus clinical strain-specific microRNA miR-UL148D targets the human chemokine RANTES during infection. PLoS Pathog. 2012;8:e1002577. doi: 10.1371/journal.ppat.1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu H, Cong JP, Yu D, Bresnahan WA, Shenk TE. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci U S A. 2002;99:3932–3937. doi: 10.1073/pnas.052713799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grundy JE, Lawson KM, MacCormac LP, Fletcher JM, Yong KL. Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J Infect Dis. 1998;177:1465–1474. doi: 10.1086/515300. [DOI] [PubMed] [Google Scholar]
- 81.Eickhoff J, Hanke M, Stein-Gerlach M, Kiang TP, Herzberger K. RICK activates a NF-kappaB-dependent anti-human cytomegalovirus response. J Biol Chem. 2004;279:9642–9652. doi: 10.1074/jbc.M312893200. [DOI] [PubMed] [Google Scholar]
- 82.Geist LJ, Dai LY. Cytomegalovirus modulates interleukin-6 gene expression. Transplantation. 1996;62:653–658. doi: 10.1097/00007890-199609150-00020. [DOI] [PubMed] [Google Scholar]
- 83.Geist LJ, Hopkins HA, Dai LY, He B, Monick MM. Cytomegalovirus modulates transcription factors necessary for the activation of the tumor necrosis factor-alpha promoter. Am J Respir Cell Mol Biol. 1997;16:31–37. doi: 10.1165/ajrcmb.16.1.8998076. [DOI] [PubMed] [Google Scholar]
- 84.Murayama T, Mukaida N, Sadanari H, Yamaguchi N, Khabar KS. The immediate early gene 1 product of human cytomegalovirus is sufficient for up-regulation of interleukin-8 gene expression. Biochem Biophys Res Commun. 2000;279:298–304. doi: 10.1006/bbrc.2000.3923. [DOI] [PubMed] [Google Scholar]
- 85.Jiang HY, Petrovas C, Sonenshein GE. RelB-p50 NF-kappa B complexes are selectively induced by cytomegalovirus immediate-early protein 1: differential regulation of Bcl-x(L) promoter activity by NF-kappa B family members. J Virol. 2002;76:5737–5747. doi: 10.1128/JVI.76.11.5737-5747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Poole E, Groves I, MacDonald A, Pang Y, Alcami A. Identification of TRIM23 as a cofactor involved in the regulation of NF-kappaB by human cytomegalovirus. J Virol. 2009;83:3581–3590. doi: 10.1128/JVI.02072-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Poole E, King CA, Sinclair JH, Alcami A. The UL144 gene product of human cytomegalovirus activates NFkappaB via a TRAF6-dependent mechanism. EMBO J. 2006;25:4390–4399. doi: 10.1038/sj.emboj.7601287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Poole E, Atkins E, Nakayama T, Yoshie O, Groves I. NF-kappaB-mediated activation of the chemokine CCL22 by the product of the human cytomegalovirus gene UL144 escapes regulation by viral IE86. J Virol. 2008;82:4250–4256. doi: 10.1128/JVI.02156-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Costa H, Nascimento R, Sinclair J, Parkhouse RM. Human cytomegalovirus gene UL76 induces IL-8 expression through activation of the DNA damage response. PLoS Pathog. 2013;9:e1003609. doi: 10.1371/journal.ppat.1003609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Murayama T, Kuno K, Jisaki F, Obuchi M, Sakamuro D, et al. Enhancement human cytomegalovirus replication in a human lung fibroblast cell line by interleukin-8. J Virol. 1994;68:7582–7585. doi: 10.1128/jvi.68.11.7582-7585.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Craigen JL, Yong KL, Jordan NJ, MacCormac LP, Westwick J, et al. Human cytomegalovirus infection up-regulates interleukin-8 gene expression and stimulates neutrophil transendothelial migration. Immunol. 1997;92:138–145. doi: 10.1046/j.1365-2567.1997.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Casarosa P, Bakker RA, Verzijl D, Navis M, Timmerman H, et al. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J Biol Chem. 2001;276:1133–1137. doi: 10.1074/jbc.M008965200. [DOI] [PubMed] [Google Scholar]
- 93.Boomker JM, The TH, de Leij LF, Harmsen MC. The human cytomegalovirus-encoded receptor US28 increases the activity of the major immediate-early promoter/enhancer. Virus Res. 2006;118:196–200. doi: 10.1016/j.virusres.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 94.Maussang D, Langemeijer E, Fitzsimons CP, Stigter van WM, Dijkman R, et al. The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res. 2009;69:2861–2869. doi: 10.1158/0008-5472.CAN-08-2487. [DOI] [PubMed] [Google Scholar]
- 95.Montag C, Wagner JA, Gruska B, Vetter L, Wiebusch, et al. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor alpha (TNF-alpha) signaling by upregulating TNF-alpha receptor 1 cell surface expression. J Virol. 2011;85:11409–11421. doi: 10.1128/JVI.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Le VT, Trilling M, Hengel H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb’-encoded modulation of TNF-alpha signaling. J Virol. 2011;85:13260–13270. doi: 10.1128/JVI.06005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Soderberg Naucler C, Fish KN, Nelson JA. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. 1997;91:119–126. doi: 10.1016/s0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 98.Benedict CA, Banks TA, Senderowicz L, Ko M, Britt WJ, et al. Lymphotoxins and cytomegalovirus cooperatively induce interferon-beta, establishing host-virus detente. Immunity. 2001;15:617–626. doi: 10.1016/s1074-7613(01)00222-9. [DOI] [PubMed] [Google Scholar]
- 99.Chan G, Bivins Smith ER, Smith MS, Yurochko AD. NF-kappaB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced atypical M1/M2 polarization of monocytes. Virus Res. 2009;144:329–333. doi: 10.1016/j.virusres.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chan G, Bivins Smith ER, Smith MS, Yurochko AD. Transcriptome analysis of NF-kappaB- and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J Virol. 2008;82:1040–1046. doi: 10.1128/JVI.00864-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smith MS, Bivins Smith ER, Tilley AM, Bentz GL, Chan G, et al. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol. 2007;81:7683–7694. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith MS, Bentz GL, Alexander JS, Yurochko AD. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol. 2004;78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mizrahi K, Kagan S, Stein J, Yaniv I, Zipori D, et al. Resistance of hematopoietic progenitors to Fas-mediated apoptosis is actively sustained by NFkappaB with a characteristic transcriptional signature. Stem Cells Dev. 2014;23:676–686. doi: 10.1089/scd.2013.0270. [DOI] [PubMed] [Google Scholar]
- 104.Pyatt DW, Stillman WS, Yang Y, Gross S, Zheng JH, et al. An essential role for NF-kappaB in human CD34(+) bone marrow cell survival. Blood. 1999;93:3302–3308. [PubMed] [Google Scholar]
- 105.De Molfetta GA, Luciola ZD, Alexandre PR, Santos ARD, Silva WA, Jr, et al. Role of NF⊠B2 on the early myeloid differentiation of CD34+ hematopoietic stem/progenitor cells. Differentiation. 2010;80:195–203. doi: 10.1016/j.diff.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 106.La Ferla K, Reimann C, Jelkmann W, HellwigBurgel T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-kappaB. FASEB J. 2002;16:1811–1813. doi: 10.1096/fj.02-0168fje. [DOI] [PubMed] [Google Scholar]
- 107.Imagawa S, Nakano Y, Obara N, Suzuki N, Doi T, et al. A GATA-specific inhibitor (K-7174) rescues anemia induced by IL-1beta, TNF-alpha, or L-NMMA. FASEB J. 2003;17:1742–1744. doi: 10.1096/fj.02-1134fje. [DOI] [PubMed] [Google Scholar]
- 108.Rakusan TA, Juneja HS, Fleischmann WR., Jr Inhibition of hemopoietic colony formation by human cytomegalovirus in vitro. J Infect Dis. 1989;159:127–130. doi: 10.1093/infdis/159.1.127. [DOI] [PubMed] [Google Scholar]
- 109.Vande L, vanden BLA, vander KSW, Janssen HL, Coffer PJ, et al. A nonredundant role for canonical NF-kappaB in human myeloid dendritic cell development and function. J Immunol. 2010;185:7252–7261. doi: 10.4049/jimmunol.1000672. [DOI] [PubMed] [Google Scholar]
- 110.Petrucelli A, Rak M, Grainger L, Goodrum F. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol. 2009;83:5615–5629. doi: 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Humby MS, O’Connor CM. Human Cytomegalovirus US28 Is Important for Latent Infection of Hematopoietic Progenitor Cells. J Virol. 2015;90:2959–2970. doi: 10.1128/JVI.02507-15. [DOI] [PMC free article] [PubMed] [Google Scholar]