

Abstract
The contributions of coding mutations to tumorigenesis are relatively well known; however, little is known about somatic alterations in noncoding DNA. Here we describe GECCO (Genomic Enrichment Computational Clustering Operation) to analyze somatic noncoding alterations in 308 pancreatic ductal adenocarcinomas (PDAs) and identify commonly mutated regulatory regions. We find recurrent noncoding mutations are enriched in PDA pathways, including axon guidance and cell adhesion, and novel processes including transcription and homeobox genes. We identify mutations in protein binding sites correlating with differential expression of proximal genes and experimentally validate effects of mutations on expression. We developed an expression modulation score that quantifies the strength of gene regulation imposed by each class of regulatory elements, and find the strongest elements are most frequently mutated, suggesting a selective advantage. Our detailed single-cancer analysis of noncoding alterations identifies regulatory mutations as candidates for diagnostic and prognostic markers, and suggests novel mechanisms for tumor evolution.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is a highly lethal malignancy with a 5-year survival rate of 6%, due to therapy resistance and late stage at diagnosis1. A detailed understanding of the molecular alterations underlying PDA is required to uncover mechanisms of tumorigenesis and enable development of effective therapies. Exome sequencing efforts have revealed genes (KRAS, TP53, CDKN2A, SMAD4) and pathways (Wnt/Notch, transforming growth factor-β (TGF-β, axon guidance, cell adhesion) important for PDA progression2,3. However, the exome comprises less than 2% of the human genome. Whole-genome sequencing (WGS) analyses have uncovered an average somatic mutation rate of 2.64 mutations per megabase in PDA indicating that PDA tumors often carry thousands of mutations, the vast majority of which are located in noncoding regions and are completely uncharacterized.4
Relevance of noncoding mutations (NCMs) to cancer development was previously established with the discovery of highly recurrent mutations in the telomerase reverse transcriptase (TERT) promoter in sporadic and familial melanoma5,6. These mutations create binding motifs for ETS transcription factors and lead to increased TERT transcriptional activity5,7. Subsequent reports identified TERT promoter mutations in a wide-range of human tumors, including glioblastoma and hepatocellular carcinoma8. TERT promoter mutations are the most common genetic alterations in bladder cancer and correlate with recurrence and survival, demonstrating the potential of NCMs to act as clinical biomarkers9. NCMs have also been demonstrated to drive tumor progression from intergenic elements. Somatic mutations in a subset of T-cell acute lymphoblastic leukemia cases generate binding sites for the MYB transcription factor, creating a super-enhancer driving expression of the TAL1 oncogene10. Recent analyses have pooled WGS data from multiple cancer types and hundreds of patients, identifying recurrent mutations in regulatory elements of several genes, including TERT11–15. While multi-cancer studies can identify ubiquitous cancer variants, in-depth analysis of individual cancer subtypes is required for uncovering disease-specific alterations16.
To detect somatic NCMs in PDA, we developed a computational pipeline to analyze WGS data of 308 PDA tumors from the International Cancer Genome Consortium (ICGC)17. We used FunSeq218,19 to initiate prioritization of noncoding mutations, which revealed hundreds of thousands of noncoding somatic mutations with potential functional implications. To discriminate amongst this large number of NCMs, we developed GECCO (Genomic Enrichment Computational Clustering Operation) to identify candidate NCMs that drive differential gene expression. This approach reduced the number of putative gene-proximal regulatory regions by three orders of magnitude to a set of high confidence calls.
Using GECCO, we identify novel recurrent mutations and interrogate expression data from matched tumors to find variants associated with changes in mRNA levels. We find significant differential expression of 16 genes associated with NCMs. For two of these genes, PTPRN2 and SLC12A8 we uncover previously unidentified clinical relevance in PDA. Specifically, we find that PTPRN2 expression level is an independent prognostic variable for overall patient survival. Pathway analysis of the genes associated with recurrent NCMs identifies known and novel PDA pathways. Furthermore, we find enrichment for mutations in specific regulatory regions, suggesting that NCMs may be acted upon by selection during tumor formation. Our analysis provides a model for tumor evolution via the formation and selection for alterations in noncoding regulatory elements of specific genes as a means of control over specific biological pathways.
RESULTS
To analyze NCMs in PDA, we selected all 405 patients with WGS data from the ICGC Pancreatic Cancer Genome Project. We determined the total number of somatic single nucleotide variants (SNV) and small insertions or deletions (indels) for each patient, and retained those with mutation load no greater or less than 3 standard deviations from the mean (mean=7,937; range=1–440,471) to exclude the hyper-mutated tumors with unlocalized replication defects (Fig. 1a, Supplementary Fig. 1). In total, 2,248,158 SNVs/indels from 308 PDA patient samples were kept for analysis.
Figure 1. Identification of recurrent noncoding mutations in PDA.
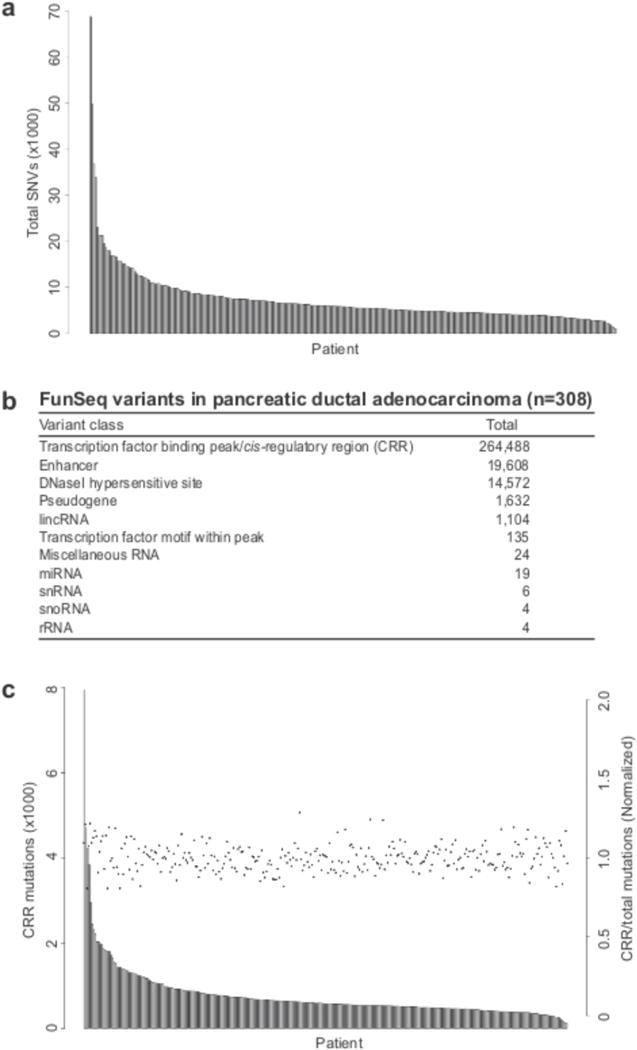
(a) The total number of single nucleotide variants (SNV) was plotted for each patient. (b) FunSeq2 was utilized to detect and characterize putatitve somatic noncoding mutations from 308 PDA whole genome sequences. Mutation counts for each functional category are displayed. (c) The number of cis-regulatory region (CRR) mutations (grey bars), and CRR/total SNV (black points) were plotted for each patient.
General features of GECCO
To discover the effect of noncoding mutations on PDA progression and patient outcome we developed the computational pipeline GECCO (Fig. 2). GECCO begins by selecting noncoding mutations falling within The Encyclopedia of DNA Elements20 (ENCODE)-defined transcription factor binding peaks – hereby referred to as cis-regulatory regions (CRRs) as not all proteins profiled are transcription factors and may be part of larger regulatory complexes – and then proceeds with downstream processing in two parallel modules. We define a “CRR class” to be all CRRs that are bound by the same DNA-binding protein (i.e. CTBP2, with 1781 CRRs across the genome) or proteins involved in DNA-binding complexes (i.e. SUZ12, with 1618 CRRs across the genome). The first module of GECCO associates NCMs with proximal genes and uses permutation testing to identify highly mutated clusters that correlate significantly with changes in gene expression. The second module calculates the mutation rate of each CRR to determine which specific CRR classes are more commonly mutated in PDA.
Figure 2. GECCO (Genomic Enrichment Computational Clustering Operation) flowchart.
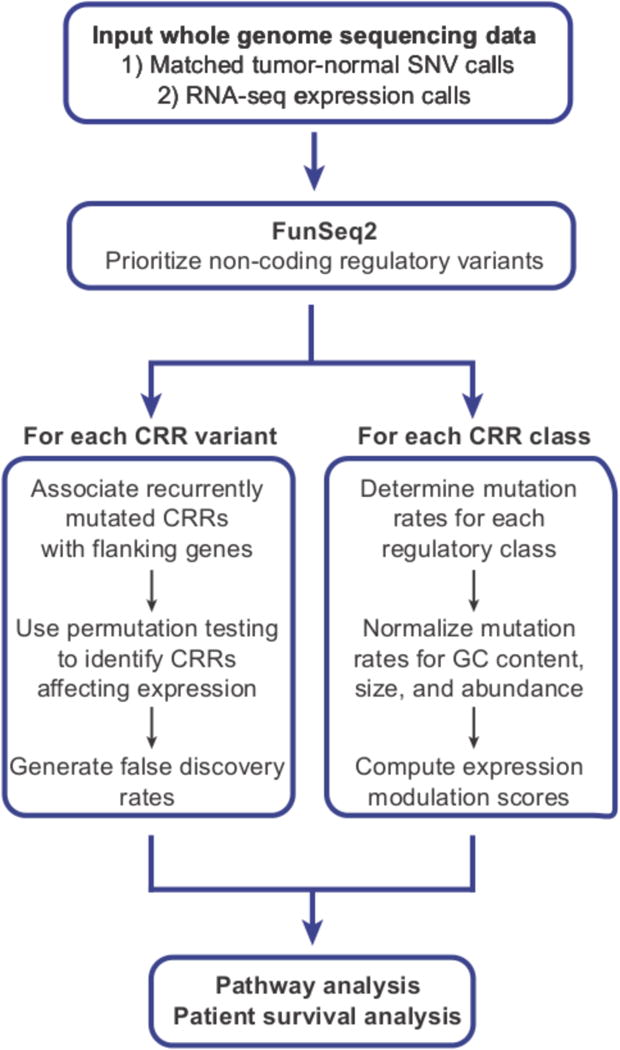
GECCO utilizes noncoding somatic mutation calls from tumor whole genome sequencing data to identify clusters of mutations within 2kb of genes, including those that correlate with changes in gene expression. GECCO also calculates the mutation rate of gene regulatory regions and determines the strength of each regulatory region in terms of the effect on gene expression (expression modulation score, EMS). These data can then be used for pathway analysis of genes proximal to noncoding clusters and genes downstream of specific regulatory regions. The gene lists can also be interrogated for patient survival analysis when coupled to outcome data for detection of clinically relevant interactions.
In the second module, GECCO computes an expression modulation score (EMS) using coupled gene expression data to determine the regulatory impact of each CRR class. The EMS can be used to generate a rank sorted list of CRRs based on the strength of their relative gene regulatory impact (such that the strongest activators and repressors fall at both ends of the list). Taken together, the results generated from GECCO provide information on the impact of NCMs on the expression level of individual genes and identifies potential driver transcription factors. Finally, GECCO merges the results of both modules to perform pathway and clinical survival analysis, allowing novel insights into PDA biology and patterns of somatic mutations in cancer.
Prioritization of non-coding mutations
We first identified NCMs in the exact same genomic position in multiple patients and removed common human variants (MAF > 5% in 1000 Genome Phase I) (Supplementary Table 1). This identified several variants reaching over 2% incidence (n ≥ 7 out of 308 patients) in the patient cohort (Supplementary Table 1). Among the 11 genes associated with these variants, 6 have been implicated in tumorigenesis, including WASF321, BNC222, ELMO123, GPR9824, PDE3B25 and SOX526. Interestingly, 10 of 11 of these mutations were found in introns. However, none of the exactly recurrent mutations disrupted, or created, transcription factor-binding motifs (as defined by the JASPAR transcription factor binding profile database27) or fell within known regulatory elements. This analysis is consistent with several pan-cancer analyses that found few exactly recurrent mutations outside of the well-characterized TERT promoter mutations11,12.
We extended this analysis by prioritizing NCMs by their association with functional annotations and clustering within regulatory elements. We used the FunSeq2 computational pipeline18,19 as a high-level filter to remove common variants and identify putative somatic regulatory mutations with functional impact. One important benefit of this approach is that it relies on functional information and thus drastically reduces any biases resulting from non-homogeneous mutation rates across the genome. This initial round of filtering identified 301,596 potential somatic drivers across all 308 patients (mean=1,988; range=203–17,902) (Fig. 1b). 264,488 of the somatic NCMs fell within ENCODE-defined transcription factor-binding peaks, with the majority of the remaining mutations within enhancers (19,608) or DNaseI hypersensitive sites (DHSs) (14,572) (Fig. 1b). We focused our analysis on the 264,488 NCMs within the ENCODE-defined CRRs. There was a direct correlation between CRR mutation rate and total SNVs (Fig. 1c). In contrast, no correlations between CRR mutation rate and coding mutations in KRAS, TP53, CDKN2A, SMAD4, and ARID1A were observed (Supplementary Fig. 3).
Analysis of cis-regulatory mutations
Starting with 264,488 candidate mutations, we used GECCO to focus our analysis on CRRs within 2kb of each gene (many of which overlap promoters), seeking to identify clusters of mutations in CRRs that directly impact gene expression (Fig. 3a). The requirement to be within 2kb of a gene excludes many distal enhancer regions but increases the likelihood that a given CRR topologically associates with, and therefore regulates, the expression of its proximal gene. The most frequently mutated CRR (17 patients, 5.52% of cohort) was in a TCF12-binding region proximal to LHX8 (LIM homeobox 8) (Fig. 3a). LHX8, a homeobox gene and regulator of craniofacial development, modulates the Hedgehog pathway, a known regulator of PDA pathogenesis28. We observed a cluster of mutations in a E2F1-binding region in proximity to BMP7 (bone morphogenetic protein 7). BMP7 is a TGF-β family member, with pleiotropic roles in development and cancer progression29. GECCO did not detect any recurrent variants in the TERT promoter, in concordance with a previous study that failed to detect TERT promoter mutations in 24 PDA samples8. To determine if the identified NCMs were within active promoters or enhancers in pancreatic cells, we interrogated H3K4me3 and H3K27ac regions from ENCODE in pancreatic carcinoma-derived PANC-1 cells. In PANC-1 cells, 37.6% of all transcription factor-binding peaks were found within active PANC-1-predicted promoters or enhancers. In contrast, 58.9% of recurrent NCMs (>5 patients) were found within at least one PANC-1-predicted active promoter or enhancer. The CRRs with recurrent NCMs did not differ significantly in size from those lacking recurrent NCMs. Therefore, recurrent NCMs are enriched in transcriptionally active regions of the genome in pancreatic cancer cells.
Figure 3. Clustered gene-proximal mutations and pathways in PDA.
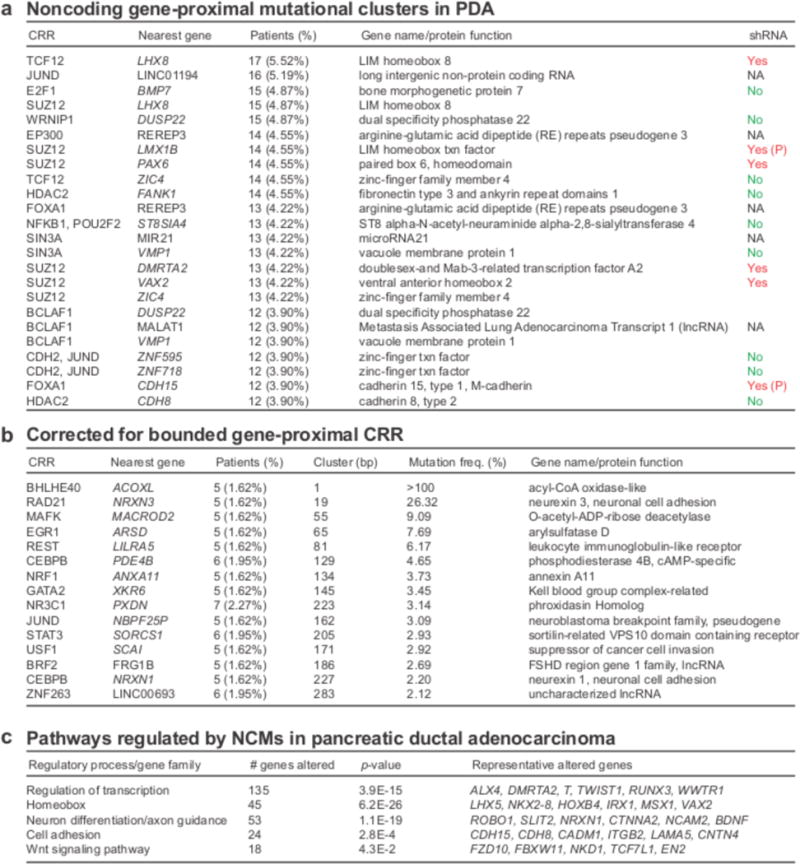
(a) The most common mutational clusters across the patient cohort as determined by GECCO, with associated genes; Yes = knockdown promoted cell death in shRNA cancer cell line screen. (P denotes PDA-specific); No = no evidence for effect on cell death in shRNA cancer cell line screen. (b) Most significant clusters when corrected for cluster size as determined by GECCO. (c) DAVID pathway analysis was used to identify regulatory processes and pathways from genes associated with recurrent NCMs.
We identified clusters of NCMs in regulatory regions of long intergenic non-protein coding RNAs (lncRNAs), including the oncogenic lncRNA Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1)30, and in microRNAs, including the oncogenic miR-2131 (Fig. 3a). To infer functional consequences of the most recurrently mutated gene-proximal CRRs, we used data from a published in vitro short hairpin RNA (shRNA) screen, which monitored survival in 102 cell lines, of which 13 were pancreas cancer-derived32. Knockdown of 6 (LHX8, LMX1B, PAX6, DMRTA2, VAX2, CDH15) of the top 15 genes was found to decrease cancer cell survival, providing potential functional relevance for these genes as cancer drivers (Fig. 3a). Knockdown of two genes, LMX1B and CDH15, showed selective killing of PDA cell lines amongst all cancers, suggesting tumor-specific vulnerabilities.
To control for variable CRR size, we calculated a mutational frequency for each cluster harboring at least 5 mutations, defined as the number of mutations across all patients divided by the number of nucleotides spanning the cluster (Fig. 3b). The highest scoring result was an exactly recurrent mutation in the same genomic position in 5 patients, flanking the acyl-CoA oxidase-like gene ACOXL, a known susceptibility locus for chronic lymphocytic leukemia33. This mutation was not found to be within a known transcription factor-binding site as defined by JASPAR. We also identified a cluster of 5 mutations within 19 nucleotides proximal to the neuronal cell adhesion gene NRXN3, a regulator of glioma cell proliferation and migration34.
While multi-cancer recurrent NCMs have been described11,12, we lack an understanding of their mutational patterns. For example, it is unknown if NCMs cluster near the same genes that show recurrent coding mutations for a given disease. Therefore, we looked for clusters of NCMs in association with known PDA genes, present in at least 5 patients (Supplementary Table 2). We did not detect any recurrent NCMs in CRRs within 2kb of KRAS, TP53, CDKN2A, SMAD4, ARID1A and MLL3, in addition to 24 of 26 other PDA genes identified from previous whole exome analyses (Supplementary Table 2)2,3. This result is consistent with defects in protein function, rather than alterations in expression, in the pathogenesis of these PDA genes.
Novel clinical outcomes from pathway analysis
Pathway analysis of recurrently mutated PDA genes has been used to identify signaling networks and biological processes underlying disease pathogenesis2,3. To detect patterns in NCM localization at the pathway level, we utilized The Database for Annotation, Visualization and Integrated Discovery (DAVID), a functional annotation enrichment algorithm for large-scale biological datasets35. Pathway analysis of genes near CRRs containing clusters of mutations (>5 patients) identified significant enrichment of several gene families and regulatory processes, including transcriptional regulation, homeobox genes, axon guidance, cell adhesion and Wnt signaling (Fig. 3c). The involvement of three of these pathways (axon guidance, cell adhesion, Wnt signaling) in PDA has been identified from previous exome sequencing studies2,3. Furthermore, several homeobox genes and transcription factors have been implicated in PDA pathogenesis, including PAX636, HOXB237, HOXB738 and RUNX339. Therefore, NCMs display preferential patterns of localization in the PDA genome and, although not found near canonical PDA genes, may act through modulation of canonical PDA pathways. In addition, we uncover a previously unrecognized localization of NCMs near transcriptional regulators and homeobox genes, suggesting a role for these factors in PDA.
The availability of matched gene expression data from a large number (n=96) of patient samples allowed association studies between specific clusters of mutations and changes in gene expression. For each of the 124,075 CRRs we determined differential gene expression between patients with mutations in a proximal CRR compared to patients without mutations. Using permutation testing we identified NCMs that significantly impacted expression of their proximal gene and calculated their false discovery rates (for details, see Online Methods). Many of the genes with the greatest number of mutations (Fig. 3a) did not reveal significant changes in gene expression. However, this analysis yielded 16 NCMs associated with significant changes in gene expression (≥3 patients, p<0.05, FDR<0.25) (Fig. 4a). Eight of the 16 NCMs were present in regions marked by H3K4me3 and H3K27ac in PANC-1 cells. None of the statistically significant mutations were associated with increases in gene expression. Three of the genes with statistically significant decreases in expression (KCNQ1, IKZF1, TUSC7) have been implicated as tumor suppressors40,41, while two (PTPRN2, SNRPN) are frequently hypermethylated42,43. Next, we looked for correlations between NCM-associated differential expression and clinical correlates in PDA. The small sample size precluded identification of specific NCMs associated with differences in patient outcome. Therefore, we looked for associations between expression of these 16 genes and patient outcome. Low mRNA expression of the phosphatase PTPRN2 and the ion transporter SLC12A8 were associated with decreased overall survival and decreased disease-free survival in a univariate analysis, respectively (Fig. 4b,c). Furthermore, a multivariate analysis revealed PTPRN2 as an independent prognostic variable for overall survival (Supplementary Table 3).
Figure 4. Recurrent gene-proximal mutations correlate with gene expression changes in PDA.
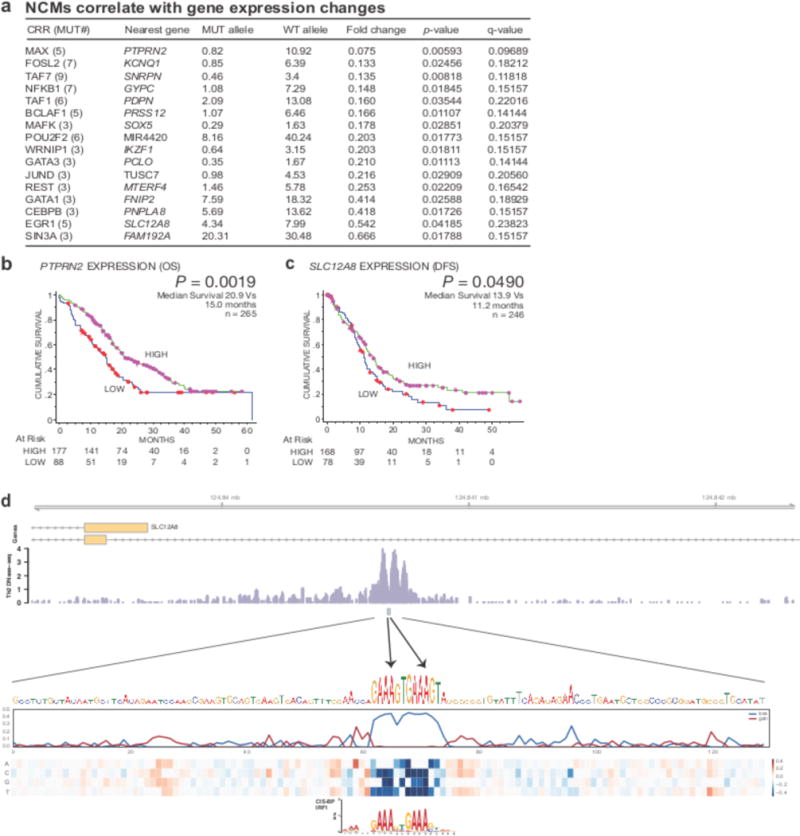
(a) GECCO used gene expression data from matched PDA patients to correlate NCMs with changes in gene expression “Mut allele” = mean expression of linked gene in patients with associated CRR mutations. “WT allele” = mean expression of linked gene in patients without associated CRR mutations. (b) Analysis of overall survival (OS) in PDA patients expressing high (upper 2/3) and low (lower 1/3) levels of PTPRN2. Purple dots represent patients with high expression of PTPRN2 “at risk” (alive). Red dots represent patients with low expression of PTPRN2 “at risk” (alive). (c) Analysis of disease-free survival (DFS) in PDA patients expressing high (upper 2/3) and low (lower 1/3) levels of SLC12A8. (d) Two A→C mutations in a regulatory site on chromosome 3 at positions 124,840,671 and 124,840,678 alter critical nucleotides in an IRF1 and/or PRDM1 binding site. The regulatory site lies in an intron of one isoform and promoter of an alternative isoform of SLC12A8. At the bottom, heat map displays predicted change in accessibility, considered here as DNase-seq signal in GM12865. The line plots above measure the maximum (gain) and minimum (loss) predicted change; the loss highlights nucleotides that significantly alter the overall signal upon mutation as both of these mutations do.
Mechanisms of NCM-modulated expression
To uncover mechanisms by which expression-correlated SNPs may influence transcription, we annotated mutations with their predicted influence on local DNase hypersensitivity using the software Basset44 (see Online Methods). The predicted influences of these 55 SNPs were significantly greater in magnitude after Bonferroni correction than a null model of sampling from the full set in 160 out of 164 examined cell types. For example, two different mutations in IRF1 and PRDM1 motifs altered critical positions that likely debilitate binding within an intron of SLC12A8 (Fig. 4d). Additional mutations modulate an NRF1 motif in the promoter of SNRPN and a GATA motif adjacent to a PU.1 binding site in an intron of LSAMP (Supplementary Fig. 4). Therefore, GECCO enriches for NCMs with predicted effects on DNase hypersensitivity and transcription factor binding.
While the Basset analysis identified NCMs predicted to affect DNase hypersensitivity, we sought to uncover NCMs directly modulating gene expression. To determine the functional relevance of specific NCMs, we performed luciferase reporter assays in non-transformed HEK-293 cells and the MiaPaCa2 and Suit2 PDA cell lines, comparing gene expression driven by wild type (WT) and mutant (MUT) sequences (Fig. 5). Among 11 regions tested, 7 (293) and 4 (MiaPaCa2, Suit2) mutations significantly altered luciferase expression. Importantly, NCMs associated with PTPRN2, PDPN, TUSC7, SNRNP and MTERF4 significantly decreased luciferase expression in one or multiple cell lines, consistent with decreased expression of these genes associated with NCMs in patient samples (Fig. 4a). Our validation rate was greater or comparable in terms of hit rate, and greater in terms of fold change, than other recent attempts to identify NCMs driving differential expression15,16, highlighting the power of GECCO to identify functionally significant NCMs from millions of candidate mutations.
Figure 5. - Noncoding mutations modulate luciferase gene expression.
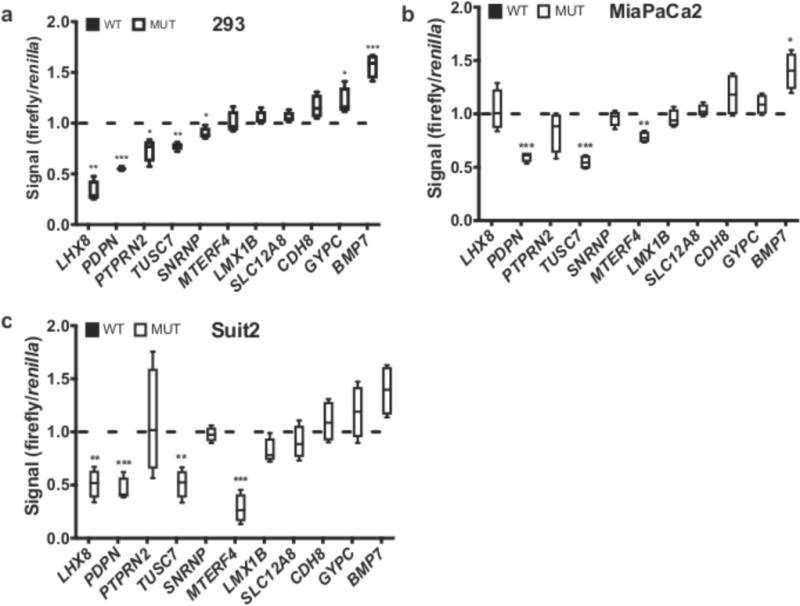
(a-c) Luciferase reporter assays of WT (black) and MUT sequences (white bars) are shown for selected NCMs associated with named genes. For each box-and-whisker plot, center line is the mean, box limits are min/max values, whiskers are s.d. Data from a representative experiment (n=3 replicates) with a total of n=4 independent transfected cultures for each cell line are shown. P values calculated by two-tailed unpaired t test. (*, p<0.05; **, p<0.01; ***, p<0.001)
Mutational and expression patterns of CRR classes
The second module of GECCO focuses on CRR classes, rather than individual genes, to identify mutational patterns and overall effects on gene expression of each CRR class (Figure 6). We computed the mutation rate for each CRR class correcting for element size and abundance in the genome. We found no significant effect of GC content on CRR class mutation rate. Noncoding mutations were specifically enriched in certain classes of gene-proximal CRRs (see Supplementary Note). Next, we sought to understand the molecular characteristics of each CRR class in terms of effect on gene expression. We calculated an expression modulation score (EMS) for each CRR class reflecting the impact of the presence of that CRR on the expression of the neighboring gene in relation to all other genes. This method compared, for each CRR class, mean expression of genes proximal to a CRR to those that are non-proximal. CRRs with strong predicted activating or repressing activity would be proximal to genes with expression levels substantially higher (for activators) or substantially lower (for repressors) than the basal genome expression level (Supplementary Table 4, see Online Methods). To determine if the strongest activators and repressors were enriched for those CRRs with the highest mutational frequencies, we considered any activator or repressor that was greater than 1 standard deviation from the mean EMS (12 activators, 9 repressors) (Fig. 6, green and orange bars). The mutational frequencies for each group (activators, repressors, all others with balanced expression) were then calculated and activators and repressors compared to the balanced group (p=0.02077 for activators vs. balanced; p=0.04982 for repressors vs. balanced). The CRR classes with the highest percentage of mutations across all PDA patients were enriched on either end of the spectrum (most repressive or most active), suggesting that recurrent NCMs are preferentially located in CRR classes with the strongest impact on gene expression. These highly active CRR classes have the largest effect on gene expression and may, therefore, confer a selective advantage to the cell. In addition, we noted that the 6 genes identified from the shRNA survival screen (Fig. 3a) were all associated with NCMs in highly repressive CRRs. In contrast, every gene that failed to score in the shRNA survival screen was associated with highly active CRRs (Fig. 3a).
Figure 6. Gene-proximal NCMs are enriched in specific classes of CRRs.

Percentage of CRRs with at least 2 mutations across the patient cohort, corrected for genome abundance and size, ordered from left to right by expression modulation score (EMS) (most repressive to most active). Dotted line represents mean mutation frequency across all CRRs.
Pathway dynamics between activating and repressing CRRs
Next, we investigated the patterns of noncoding SUZ12 mutations in our patient cohort, as SUZ12 had the highest repressive score and SUZ12 sites were frequently mutated (Supplementary Table 4, Fig. 6). We generated two distinct lists of SUZ12-associated genes. The first list contained those genes associated with recurrently mutated SUZ12 sites. The second list contained those genes associated with SUZ12 sites that never harbored recurrent NCMs. We then performed pathway analysis on each gene set to identify differences in biological functions (Fig. 7a). We found that genes without recurrent SUZ12 mutations were enriched in glycoproteins, intracellular signaling as well as the axon guidance/neuron differentiation pathway. In contrast, genes with recurrent SUZ12 mutations were more significantly enriched in homeobox genes, transcription factors, Wnt signaling, proto-oncogenes and the axon guidance/neuron differentiation pathway. Surprisingly, several categories, including glycoproteins, intracellular signaling and extracellular matrix, were completely absent within the mutant SUZ12 gene set. Therefore, there is specificity for the location of NCMs in PDA, not only for certain CRRs, but also for the corresponding cancer-associated genes and pathways.
Figure 7. Gene-proximal NCMs in repressors and activators cluster near distinct subsets of genes.

(a) Pathway analysis of genes associated with recurrently mutated repressive (SUZ12, CTBP2, SETDB1) sites (red bars), versus those never harboring NCMs in those CRRs (blue bars). (b) Pathway analysis of genes associated with recurrently mutated activator (KAT2A, BCLAF1, TAF7, WRNIP1) sites (red bars), versus those never harboring NCMs in those CRRs (blue bars). AG/ND, axon guidance/neuron differentiation.
To further characterize pathways downstream of commonly mutated repressive CRRs, we performed pathway analysis on genes with and without associated CTBP2 mutations (Fig. 7a). Genes without CTBP2 noncoding mutations showed a similar pattern of pathway regulation as SUZ12. These pathways were markedly enriched in the gene set associated with CTBP2 mutations, while alternative splicing and glycoproteins were completely absent. We extended this analysis to another repressive CRR with a high mutational frequency, SETDB1 (Fig. 6a). Genes associated with recurrent NCMs in SETDB1 binding sites were enriched in axon guidance/neuron differentiation, cell adhesion and disease mutation pathways. Therefore, mutations in highly repressive CRRs are enriched in PDA and selectively associated with genes regulating a core set of biological processes.
We performed a similar analysis for the commonly mutated activator CRRs, including KAT2A, BCLAF1, TAF7 and WRNIP1 (Fig. 7b) and again found specificity for the genes and pathways that are commonly mutated. For all CRRs, there were significant differences in the pathways regulated by genes with or without mutations in a given CRR. KAT2A, BCLAF1 and TAF7 shared a very similar pattern of pathway regulation, with significant increases in nucleosome assembly/organization, methylation and ubiquitin conjugation, all processes involved in chromatin dynamics. This suggests that genes associated with NCMs in transcriptional repressors regulate homeobox genes and PDA-associated pathways, while genes associated with NCMs in transcriptional activators may regulate transcriptional dynamics through modulation of chromatin states.
DISCUSSION
We developed a new computational method, GECCO, to systematically analyze the noncoding genome of PDA to uncover recurrent regulatory somatic mutations. We find patterns of NCMs associated with genes regulating canonical PDA pathways, but not associated with commonly mutated PDA genes. Therefore, NCMs may serve as a novel mechanism in cancer cells for regulating pathways critical for tumorigenesis. Furthermore, GECCO uncovers mutations correlated with changes in gene expression, including several known tumor suppressors and aberrantly methylated genes. GECCO produces a set of high confidence calls that enrich for predicted effects on DNase hypersensitivity and transcription factor binding, as well as functional effects on gene expression, as experimentally demonstrated by luciferase reporter assays. We find enrichment for NCMs in specific CRRs and distinct subsets of pathways associated with NCMs in highly repressive and transcriptionally active CRRs as identified by our EMS algorithm. To our knowledge, this is the first comprehensive analysis of noncoding alterations in PDA, providing novel insights into PDA pathogenesis and serving as a counterpart to the information gleaned from large-scale exome sequencing projects2,3.
Mutational analysis of patient tumors is increasingly informing treatment decisions, whereas complimentary techniques, including microarray, RNA sequencing, fluorescence in situ hybridization and immunohistochemistry are required to analyze changes in gene or protein expression of cancer drivers that lack coding mutations. As somatic mutations in DNA regulatory elements can alter gene expression of cancer drivers, targeted or whole genome sequencing may provide clinically useful information for these patients, both in terms of therapeutic decisions and clinical prognosis. Our analysis provides the first collection of NCMs that correlate with changes in gene expression in PDA. Furthermore, we uncover clinical outcome relationships for PTPRN2 and SLC12A8, neither of which has previously been implicated in PDA.
Functional validation of NCM-gene expression associations is a critical step in evaluating the robustness of an analysis pipeline. Our luciferase reporter assay experiments demonstrated that GECCO has a higher validation rate in cancer cell lines than any recent study of NCMs15,16. Furthermore, the validation rate in HEK293 cells, a standard cell line for luciferase assays, was 64%, concordant with the expected false discovery rate. Finally, GECCO accurately predicted the directionality of gene expression changes associated with NCMs. NCMs associated with PTPRN2, PDPN, TUSC7, SNRNP and MTERF4 significantly decreased luciferase expression in one or multiple cells lines, consistent with decreased gene expression of these genes associated with NCMs in patient samples. This is in contrast to a recent report where the directionality of gene expression changes in the luciferase assay was not consistent with the predicted response16. Therefore, GECCO represents a significant improvement in the ability to identify functionally relevant NCMs.
Pathway analysis of the gene lists generated by GECCO revealed several unexpected findings. Strikingly, we found that the most highly recurrent somatic NCMs were located near genes in known PDA-associated pathways, including axon guidance, cell adhesion and Wnt signaling, but not the most commonly mutated PDA genes. This suggests that NCMs may drive tumor progression through modulation of PDA-specific pathways, providing an alternative route for pathway activation and a novel mechanism of tumorigenesis. Furthermore, we provide evidence that NCMs in specific regulatory element classes are selected for during tumor evolution. These highly mutated regulatory element classes are predominantly those with the greatest impact on gene expression. Therefore, clusters of NCMs are enriched in gene-proximal regions with the greatest regulatory impact, again providing evidence for selection during tumorigenesis.
Pathway analysis of genes near NCMs within these highly mutated regulatory regions shows selectivity for PDA pathways. These pathways are not enriched when analyzing genes without associated clusters of NCMs, again arguing in favor of selection. Interestingly, many transcriptional regulators bind selectively to different regions of the genome in malignant versus non-neoplastc cells45. We propose that NCMs found within promoters of PDA pathway genes modify regulatory factor binding to alter gene transcription, thereby providing an additional mechanism to promote cancer.
ONLINE METHODS
1. Data Acquisition
All data used in this analysis were downloaded from the International Cancer Genome Consortium (IGCG) data portal (https://dcc.icgc.org/projects). At our last date of access (Feb 11, 2015), simple somatic mutations (SSM) for 405 pancreatic ductal adenocarcinoma samples were available from the Australian (PACA-AU) and Canadian (PACA-CA) groups. We download the clinical data, SSMs, and when available, sequence-based gene expression (EXP-S) data for all 405 patients.
2. Pre-processing
The whole genome sequencing (WGS) required to call SNVs across all 405 patients and the whole genome RNA-sequencing required to calculate gene expression were carried out by two distinct consortiums, one Canadian and one Australian. All SNV calls (SSMs) and gene expression calculations (EXP-S) by these two groups were consolidated by ICGC.
2.1. SNV calls from whole genome sequencing
For each of the 405 patients we extracted the chromosome, start location, end location, somatic allele, and mutated allele from the list of simple somatic mutations (file: ssm_open.tsv) and converted to bed format. Many of the SNVs were redundant within patients. For each patient, the list of SNVs were sorted by genomic coordinates and consolidated to contain only a single entry for each unique SNV. A subset of patients had extremely low numbers of SNVs (likely due to poor sequencing results) or high numbers of SNVs (likely due to hyper-mutated regions, unlocalized replication defects, or microsatellite instability). Across all 405 patients the number of unique SNVs ranged from 1 to 440,471 with a mean 7,937 and a standard deviation of 26,224. In order to remove outliers we eliminated all patients with less than 100 SNVs (92 patients in total) or an SNV count more than 3 standard deviations away from the mean (5 patients in total). This left 308 patients with a mean SNV count of 7,300 and ranging from 1,040 to 68,885.
2.2. Gene expression (FPKM) from whole genome RNA-sequencing
Of the 308 patients that passed the previous filtering step, 96 had expression data available from ICGC. For each of the 96 patients, we extracted the normalized read count (FPKM) and Ensembl gene id (file: exp_seq.tsv). While the vast majority of genes have expression data across all 96 patients, there were several thousand Ensembl genes that only contained expression data for a subset of patients. In order to streamline and simplify downstream analysis we kept only the 50,861 Ensembl genes that were shared by all 96 patients. In addition, there were three patients (DO33168, DO35098, DO35100) that had gene expression from either 2 or 3 independently sequenced samples. For these three patients, the gene expression for each gene was calculated by taking the mean across all samples.
3. Analyzing noncoding variants with GECCO
In order to identify potential noncoding cancer drivers, we first used FunSeq2 (v2.1.0) as a high level filter to prioritize our SNVs. The unique SNVs for each of the 308 patients were converted to bed format and analyzed by FunSeq2 using the command ./run.sh –inf bed –n to identify only noncoding variants. This analysis pipeline requires a suite of annotation data that is used to make calls and score noncoding variants. These were downloaded from (http://funseq2.gersteinlab.org/data/). One of these files, “ENCODE.annotation.gz” contains the full list of TFPs/CRRs used in our analysis along with their exact genomic coordinates.
3.1 Processing recurrently mutated cis-regulatory regions (CRRs)
FunSeq2 generates a number of output files including Recur. Summary, which contains a list of all noncoding elements, the genomic coordinates of these elements, the fraction of patients with a mutation in this element, and the full list of patient names along with the genomic locations of each mutation. While the ENCODE annotation data provides a number of different noncoding elements (enhancers, transcription factor binding sites (TFPs), DNase hypersensitivity, etc.) we chose to focus our analysis on TFPs – referred to in this manuscript as CRRs – as they were the most highly represented class of elements identified. CRR proximal genes were found by intersecting CRRs with genes that had been expanded by 2kb at their 5’ and 3’ ends.
3.2 Calculating CRR mutation rates
As described above, the full list of CRRs (121 distinct CRR classes in total) including their counts and genomic positions can be found in “ENCODE.annotation.gz.” GECCO makes two separate calculations across all 121 CRR classes using the CRR genomic information: (1) For a given CRR class, it calculates the fraction of distinct CRR sites that are mutated within the class and (2) the base level mutation rate for each CRR class (the number of mutations in all CRRs of a given class divided by the total number of base pairs of all CRRs in a given class). For an individual CRR, there are three ways in which GECCO calculates the mutational frequency: (1) by summing the number of mutations in a given CRR, (2) by calculating the fraction of bases in the CRR that are mutated (i.e. mutation counts normalized by read length), or (3) by calculating the fraction of bases in a CRR mutation cluster. Option (3) is computed by first determining the cluster size within a CRR, the number of bases required to span all mutations in a given CRR. For example, consider a 2kb CRR with 9 mutations. If the two most distantly separated of the 9 mutations are 100bps apart then the length of the mutation cluster is 100bp. The mutational frequency of the cluster is then computed by dividing the number of mutations in that cluster by the size of the cluster (9/100 = 9.0%). This approach weights exactly recurrent or proximal mutations more strongly than distant mutations.
4. Pathway analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID), a functional annotation enrichment algorithm for large-scale biological datasets was used for pathway analysis, with the following annotation categories: SP_PIR_KEYWORDS, GOTERM_BP_FAT, KEGG_PATHWAY, PANTHER_PATHWAY, SMART. A Bonferroni corrected p-value of 0.05 was used as a cutoff for enrichment significance.
5. Survival analysis
Median survival was estimated using the Kaplan-Meier method and the difference was tested using the log-rank Test. P values of less than 0.05 were considered statistically significant. Clinico-pathologic variables analyzed with a P value of less than 0.25 on log-rank test were entered into Cox Proportional Hazard multivariate analysis, and redundant variables were eliminated using a backward elimination method. Statistical analysis was performed using StatView 5.0 Software (Abacus Systems, Berkeley, CA, USA). Overall survival (OS) or disease-free survival (DFS) was used as the primary endpoint.
PTPRN2 Expression level > 4.98 defined as high
SLC12A8 Expression level > 7.03 defined as high
6. Computing differential expression
Differential expression was computed for each recurrently mutated CRR that was within 2kb of an Ensemble gene using permutation testing. For each CRR/gene pair, the 96 patients with mutation data were split into two groups – patients with mutations in the CRR and patients without mutations in the CRR. Using the expression data downloaded from ICGC for the gene of interest a t-test is performed to generate a single t-value, the observed t-value. The expression values for patients with mutations in CRRs and the expression values for patients without mutations are then permuted 100,000 times to generate 100,000 additional t-values, the permuted t-values. These t-values generally fit a Gaussian distribution to which the observed t-value is then compared to using a two-tailed test. The empirical p-value is computed as the fraction of times (x/100,000) that a “permuted t-value” falls further outside the Gaussian distribution than the “observed t-value”. Once p-values have been calculated for all recurrently mutated genes proximal to CRRs, GECCO estimate q-values (the false discovery rate) for each call. This is done using the “qvalue” package in R and measures the proportion of false positives incurred given the p-value distribution.
7. Luciferase Reporter Assay and Statistics
150 base pair sequences surrounding specific NCMs (wild type, WT or mutant, MUT) were synthesized (Integrated DNA Technologies) and cloned into pGL4.23 (Promega), containing a minimal promoter driving firefly luciferase. Five thousand cells per well (HEK-293, MiaPaCa2 or Suit2) were co-transfected in 96-well format with the specific WT or MUT vector and pRL-SV40P (Renilla luciferase, Addgene #27163) as a normalization control. Luciferase activity was measured 48 hours post-transfection with the Dual-Luciferase Reporter Assay System (Promega). Values reported are firefly luciferase divided by Renilla luciferase. Analytical statistics were generated in Prism 7.0 (GraphPad), and P values are from two-tailed unpaired t tests. All cell lines were obtained from ATCC and tested for mycoplasma contamination.
8. Computing Expression Modulation Scores (EMS)
Some CRRs bind transcription factors or transcription factor components with well-known expression modulation including SUZ12 and CTBP2, which act as transcriptional repressors, or BDP1 and BRF1, which act as transcriptional activators. However, many of the 121 CRRs used in this study have unexplored or unvalidated directions of expression modulation. We developed a method to infer the direction and effect of expression modulation for each CRR class by comparing the expression of genes proximal CRRs in a given CRR class to the mean expression of all other active genes in the genome.
Many genes are inactive in any given tissue and in a given RNA-seq experiment ~50% of genes show low to no expression. For all 96 patients with expression data, we found this also to be true with ~50% of genes showing 0 expression. When computing the expression modulation for each CRR class we ignored all genes that showed 0 expression in at least 90% of patients (86 patients or more). For a given CRR class and for each of the 96 patients we compute (1) the mean expression of all genes proximal to CRRs in that class and (2) the mean expression of all genes non-proximal to a CRR in that class. For a given CRR class we then compute the log of the ratio between (1) and (2) for each of the 96 patients and then take the mean of the log ratio for all 96 patients to get a single “expression modulation score” for each CRR class. The log of the ratio will be negative if the mean expression of genes proximal to a CRR class is lower than the genome average (repression) and will be positive if the mean expression of genes proximal to a CRR class is higher than the genome average (activation). This calculation is not meant to generate absolute numerical score for the repressive or activating activity of a CRR but is instead used to generate a rank-sorted list of CRR classes based on their expression modulation.
9. Basset Analysis
Basset is a recently introduced method based on convolutional neural networks to accurately predict DHSs from DNA sequence, thus enabling annotation of the influence of mutations on accessibility44. We trained the Basset deep convolutional neural network on DHSs from 164 cell types mapped by ENCODE and the Roadmap Epigenomics projects. From this, we predicted the influence of variants on the presence of DNase hypersensitivity in each cell type by computing the difference between predictions on sequences with each allele. Candidate high impact variants were further analyzed for interrupting known binding sites by converted Basset-learned first convolution layer filters to probabilistic position weight matrixes by counting nucleotide occurrences in the set of sequences that activate the filter to a value that is more than half of its maximum value. We identified the likely binding protein for the motifs by querying the CIS-BP database46 (accessed on June 12, 2015) using the TomTom v4.10.1 search tool47 and requiring an FDR q-value < 0.1.
Supplementary Material
Supplementary Figure 1 – Identification of recurrent noncoding mutations in PDA.
Distribution of SNV rates across the patient cohort.
Supplementary Figure 2 – Overlap of SNVs and common coding mutations in PDA.
Distribution of SNVs across the patient cohort, with common coding mutations (colored bars) in PDA genes.
Supplementary Figure 3 – Overlap of gene-proximal NCMs in CRRs and common coding mutations in PDA.
Distribution of CRR mutation rates across the patient cohort, with common coding mutations (colored bars) in PDA genes.
Supplementary Figure 4 – NCMs disrupt transcription factor binding motifs.
(a) A G→A mutation in a regulatory site on chromosome 15 at position 25,200,056 alters a critical nucleotide in an NRF1 binding site. The regulatory site lies in the promoter of SNRPN. At the bottom, the heat map displays the predicted change in binding, considered here as ChIP-seq signal for NRF1 in H1-hESCs. The line plots above measure the maximum (gain) and minimum (loss) predicted change; the loss highlights nucleotides that significantly alter the overall signal upon mutation as this mutation does. (b) A G->T mutation in a regulatory site on chromosome 3 at position 115,757,580 introduces a GATA factor binding site nearby an established PU.1 binding site. The heat map displays the predicted change in accessibility, considered here as DNase-seq signal in K562. In other cells, such as monocytes, the model predicts reduced accessibility, suggesting that GATA binding here may alter the combinatorial logic of the regulatory element in a complex fashion.
Supplementary Table 1 – Genome-wide exactly recurrent mutations in PDA.
The most common exactly recurrent mutations across the patient cohort. Sequence of mutant allele in parenthesis.
Supplementary Table 2 – Distribution of gene-proximal NCMs near known PDA genes.
Analysis of the association of NCM clusters as determined by GECCO with known PDA genes.
Supplementary Table 3 – PTPRN2 multivariate analysis.
Multivariate analysis of clinico-pathological variables and PTPRN2 expression in the patient cohort.
Supplementary Table 4 – CRR expression modulation scores.
Effect of CRR on activity of neighboring gene compared with all other genes in the genome (see Online Methods for analysis details). EM Score, expression modulation score.
Acknowledgments
The authors wish to thank the members of the Tuveson lab, C. Vakoc and A. Siepel for helpful discussions. DAT is a distinguished scholar of the Lustgarten Foundation and Director of the Lustgarten Foundation-designated laboratory of Pancreatic Cancer Research. DAT is also supported by the Cold Spring Harbor Laboratory Association, the Carcinoid Foundation, PCUK, and the David Rubinstein Center for Pancreatic Cancer Research at MSKCC. In addition, we are grateful for support from the following: the STARR foundation (I7-A718 for DAT), DOD (W81XWH-13-PRCRP-IA for DAT), Louis Morin Charitable Trust (MEF) and the NIH (5P30CA45508-26, 5P50CA101955-07, 1U10CA180944-01, 5U01CA168409-3, and 1R01CA190092-01 for DAT and R01HG006677 for MCS).
Footnotes
AUTHOR CONTRIBUTIONS
Wrote the manuscript: MEF, TG, MCS, DAT
Supervised the study: MCS, DAT
Performed FunSeq analysis and developed GECCO: TG
Performed pathway analysis: MEF
Contributed to data analysis: MEF, TG, SMG, AVB, EK, SS, LDS, SG, JDM
Performed patient outcome analysis: DC, PB
Performed Basset analysis: DRK
Performed germline sequence analysis: NW
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
DATA AVAILABILITY STATEMENT
All data used in this analysis were downloaded from the International Cancer Genome Consortium (IGCG) data portal (https://dcc.icgc.org/projects). At our last date of access (Feb 11, 2015), simple somatic mutations (SSM) for 405 pancreatic ductal adenocarcinoma samples were available from the Australian (PACA-AU) and Canadian (PACA-CA) groups. We download the clinical data, SSMs, and when available, sequence-based gene expression (EXP-S) data for all 405 patients.
All code can be requested by e-mailing MCS.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biankin AV, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waddell N, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang FW, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horn S, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 7.Bell RJ, et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015 doi: 10.1126/science.aab0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Killela PJ, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rachakonda PS, et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A. 2013;110:17426–17431. doi: 10.1073/pnas.1310522110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansour MR, et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 2014;46:1160–1165. doi: 10.1038/ng.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46:1258–1263. doi: 10.1038/ng.3141. [DOI] [PubMed] [Google Scholar]
- 13.Melton C, Reuter JA, Spacek DV, Snyder M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat Genet. 2015 doi: 10.1038/ng.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathelier A, et al. Cis-regulatory somatic mutations and gene-expression alteration in B-cell lymphomas. Genome biology. 2015;16:84. doi: 10.1186/s13059-015-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araya CL, et al. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat Genet. 2016;48:117–125. doi: 10.1038/ng.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujimoto A, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500–509. doi: 10.1038/ng.3547. [DOI] [PubMed] [Google Scholar]
- 17.International Cancer Genome, C. et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khurana E, et al. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science. 2013;342:1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu Y, et al. FunSeq2: A framework for prioritizing noncoding regulatory variants in cancer. Genome biology. 2014;15:480. doi: 10.1186/s13059-014-0480-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teng Y, Mei Y, Hawthorn L, Cowell JK. WASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene. 2014;33:203–211. doi: 10.1038/onc.2012.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winham SJ, et al. Genome-wide investigation of regional blood-based DNA methylation adjusted for complete blood counts implicates BNC2 in ovarian cancer. Genetic epidemiology. 2014;38:457–466. doi: 10.1002/gepi.21815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dulak AM, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–486. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman SK, et al. Gastric inhibitory polypeptide receptor (GIPR) is a promising target for imaging and therapy in neuroendocrine tumors. Surgery. 154:1206–1213. doi: 10.1016/j.surg.2013.04.052. discussion 1214 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uzawa K, et al. Targeting phosphodiesterase 3B enhances cisplatin sensitivity in human cancer cells. Cancer medicine. 2013;2:40–49. doi: 10.1002/cam4.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renjie W, Haiqian L. MiR-132, miR-15a and miR-16 synergistically inhibit pituitary tumor cell proliferation, invasion and migration by targeting Sox5. Cancer letters. 2015;356:568–578. doi: 10.1016/j.canlet.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic acids research. 2004;32:D91–94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flandin P, et al. Lhx6 and Lhx8 coordinately induce neuronal expression of Shh that controls the generation of interneuron progenitors. Neuron. 2011;70:939–950. doi: 10.1016/j.neuron.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boon MR, et al. Bone morphogenetic protein 7: a broad-spectrum growth factor with multiple target therapeutic potency. Cytokine & growth factor reviews. 2011;22:221–229. doi: 10.1016/j.cytogfr.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Gutschner T, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriyama T, et al. MicroRNA-21 modulates biological functions of pancreatic cancer cells including their proliferation, invasion, and chemoresistance. Molecular cancer therapeutics. 2009;8:1067–1074. doi: 10.1158/1535-7163.MCT-08-0592. [DOI] [PubMed] [Google Scholar]
- 32.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lan Q, et al. Genetic susceptibility for chronic lymphocytic leukemia among Chinese in Hong Kong. European journal of haematology. 2010;85:492–495. doi: 10.1111/j.1600-0609.2010.01518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun HT, Cheng SX, Tu Y, Li XH, Zhang S. FoxQ1 promotes glioma cells proliferation and migration by regulating NRXN3 expression. PLoS One. 2013;8:e55693. doi: 10.1371/journal.pone.0055693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 36.Mascarenhas JB, et al. AX6 is expressed in pancreatic cancer and actively participates in cancer progression through activation of the MET tyrosine kinase receptor gene. J Biol Chem. 2009;284:27524–27532. doi: 10.1074/jbc.M109.047209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segara D, et al. Expression of HOXB2, a retinoic acid signaling target in pancreatic cancer and pancreatic intraepithelial neoplasia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:3587–3596. doi: 10.1158/1078-0432.CCR-04-1813. [DOI] [PubMed] [Google Scholar]
- 38.Chile T, et al. HOXB7 mRNA is overexpressed in pancreatic ductal adenocarcinomas and its knockdown induces cell cycle arrest and apoptosis. BMC cancer. 2013;13:451. doi: 10.1186/1471-2407-13-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whittle MC, et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell. 2015;161:1345–1360. doi: 10.1016/j.cell.2015.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Than BL, et al. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene. 2014;33:3861–3868. doi: 10.1038/onc.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geimer Le Lay AS, et al. The tumor suppressor Ikaros shapes the repertoire of notch target genes in T cells. Science signaling. 2014;7:ra28. doi: 10.1126/scisignal.2004545. [DOI] [PubMed] [Google Scholar]
- 42.Anglim PP, et al. Identification of a panel of sensitive and specific DNA methylation markers for squamous cell lung cancer. Molecular cancer. 2008;7:62. doi: 10.1186/1476-4598-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benetatos L, et al. CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leukemia research. 2010;34:148–153. doi: 10.1016/j.leukres.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 44.Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome research. 2016;26:990–999. doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Squazzo SL, et al. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome research. 2006;16:890–900. doi: 10.1101/gr.5306606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weirauch MT, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome biology. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 – Identification of recurrent noncoding mutations in PDA.
Distribution of SNV rates across the patient cohort.
Supplementary Figure 2 – Overlap of SNVs and common coding mutations in PDA.
Distribution of SNVs across the patient cohort, with common coding mutations (colored bars) in PDA genes.
Supplementary Figure 3 – Overlap of gene-proximal NCMs in CRRs and common coding mutations in PDA.
Distribution of CRR mutation rates across the patient cohort, with common coding mutations (colored bars) in PDA genes.
Supplementary Figure 4 – NCMs disrupt transcription factor binding motifs.
(a) A G→A mutation in a regulatory site on chromosome 15 at position 25,200,056 alters a critical nucleotide in an NRF1 binding site. The regulatory site lies in the promoter of SNRPN. At the bottom, the heat map displays the predicted change in binding, considered here as ChIP-seq signal for NRF1 in H1-hESCs. The line plots above measure the maximum (gain) and minimum (loss) predicted change; the loss highlights nucleotides that significantly alter the overall signal upon mutation as this mutation does. (b) A G->T mutation in a regulatory site on chromosome 3 at position 115,757,580 introduces a GATA factor binding site nearby an established PU.1 binding site. The heat map displays the predicted change in accessibility, considered here as DNase-seq signal in K562. In other cells, such as monocytes, the model predicts reduced accessibility, suggesting that GATA binding here may alter the combinatorial logic of the regulatory element in a complex fashion.
Supplementary Table 1 – Genome-wide exactly recurrent mutations in PDA.
The most common exactly recurrent mutations across the patient cohort. Sequence of mutant allele in parenthesis.
Supplementary Table 2 – Distribution of gene-proximal NCMs near known PDA genes.
Analysis of the association of NCM clusters as determined by GECCO with known PDA genes.
Supplementary Table 3 – PTPRN2 multivariate analysis.
Multivariate analysis of clinico-pathological variables and PTPRN2 expression in the patient cohort.
Supplementary Table 4 – CRR expression modulation scores.
Effect of CRR on activity of neighboring gene compared with all other genes in the genome (see Online Methods for analysis details). EM Score, expression modulation score.