

Perovskite quantum dot coupling tuned by A-site cation halide treatment yields a record quantum dot solar cell efficiency.
Abstract
We developed lead halide perovskite quantum dot (QD) films with tuned surface chemistry based on A-site cation halide salt (AX) treatments. QD perovskites offer colloidal synthesis and processing using industrially friendly solvents, which decouples grain growth from film deposition, and at present produce larger open-circuit voltages (VOC’s) than thin-film perovskites. CsPbI3 QDs, with a tunable bandgap between 1.75 and 2.13 eV, are an ideal top cell candidate for all-perovskite multijunction solar cells because of their demonstrated small VOC deficit. We show that charge carrier mobility within perovskite QD films is dictated by the chemical conditions at the QD-QD junctions. The AX treatments provide a method for tuning the coupling between perovskite QDs, which is exploited for improved charge transport for fabricating high-quality QD films and devices. The AX treatments presented here double the film mobility, enabling increased photocurrent, and lead to a record certified QD solar cell efficiency of 13.43%.
INTRODUCTION
Colloidal quantum dot (QD) materials offer unique properties over those afforded by conventional thin films for optoelectronic and other applications. While it is well known that colloidal QD systems offer immense tunability in the material bandgap, energetic position of the electronic states, and surface chemistry, more unique features are being discovered (1–4). For photovoltaics (PVs), colloidal QDs exhibit efficient multiple exciton generation, which led to the first PV and photoelectrochemical cells with external quantum efficiency (EQE) exceeding unity within the solar spectrum (5, 6). Additionally, it has recently been shown that colloidal CsPbI3 QD materials stabilize the cubic perovskite crystal phase (4, 7), whereas thin-film CsPbI3 materials relax to an orthorhombic phase at ambient temperature (8).
CsPbI3 with cubic symmetry exhibits the lowest bandgap (Eg = 1.73 eV) (9) among the all-inorganic Pb-halide perovskite materials for PV. However, at room temperature, the orthorhombic phase with Pnma space group symmetry (Eg = 2.82 eV) is thermodynamically preferred (10). To overcome this phase instability, the addition of bromide (CsPbI3−xBrx) has been shown to reduce the phase transition temperature from over 300° to ~110°C (10); however, for PV applications, this reduction in temperature is not low enough, and alloying comes at the expense of increasing the bandgap to ~1.9 eV for CsPbI2Br (11, 12). In an alternative approach, we previously demonstrated that, by leveraging the surface energy of QDs, the cubic phase can be stabilized at room temperature and below, leading to a PV device with efficiency >10% (4). Although we observed a high open-circuit voltage (VOC) (~85% of the maximum voltage from the Shockley-Queisser analysis for the given bandgap), the short-circuit current density (JSC) was limited by transport. This is a common trade-off in QD solar cells (QDSCs) whereby increasing light absorption with thick absorber layers reduces charge extraction efficiency in transport-limited QD films (13). Here, we show, for the first time, an AX posttreatment [where A = formamidinium (FA+), methylammonium (MA+), or cesium (Cs+) and X = I− or Br−] that tunes and greatly improves the electronic coupling between QDs, which enhances carrier mobility. We characterize the structural, optical, and electrical properties of the resultant CsPbI3 QD films and determine that the fabrication process enables the films to retain nanocrystalline character to preserve quantum confinement and that the AX salt species coats the QDs in the array rather than alloying or inducing grain growth in the films (these AX-posttreated CsPbI3 QD films are thus subsequently referred to as AX-coated).
RESULTS
PV device architecture and certified efficiency
Metal halide salts, such as CdCl2 and PbI2 (13–16), and molecular halides (17) have been used to improve QD coupling, passivate surfaces, tune device energetics, and improve stability of the more mature Pb-chalcogenide QDSC technology (14, 18). Banking on some potential similarities in the surface chemistry between CsPbI3 and PbSe or PbS, we explore a variety of AX salts to improve electronic coupling in CsPbI3 QDs. Figure 1 details one of the better performing devices made in our laboratory using formamidinium iodide (FAI) as the AX treatment. A schematic of the device architecture used (Fig. 1A) is shown mapped to a scanning electron microscopy (SEM) cross-sectional image (Fig. 1B) with false coloring applied to delineate the various layers used in the stack. Figure 1 (C to E) shows measurements performed by the National Renewable Energy Laboratory (NREL) PV Performance Characterization Team including a current density–voltage (J-V) scan (Fig. 1C) for determining standard PV performance characteristics such as VOC, JSC, and the maximum power point. The device was held at a voltage of 0.95 V, and a measurement of the output current versus time was recorded and is shown in Fig. 1D. Because of the hysteretic nature of perovskite solar cells (19, 20), this stabilized current output at a set voltage is used to determine the certified AM1.5G efficiency after the spectral mismatch (calculated, in part, from the EQE shown in Fig. 1E), and device active area were determined. The stabilized current at 0.95 V matches well to the reverse J-V scan shown in Fig. 1C, and the results indicate an overall efficiency of 13.43%, which is currently the highest efficiency colloidal QDSC reported to date (21).
Fig. 1. High-efficiency FAI-coated CsPbI3 QDSCs.
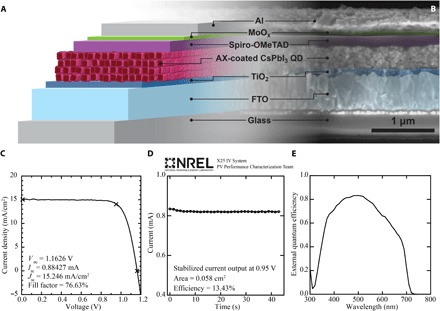
(A) Schematic cross section of a solar cell. FTO, fluorine-doped tin oxide. (B) SEM image of a device cross section. (C to E) NREL-certified (C) J-V characteristics from forward bias to reverse bias, (D) stabilized current at a constant voltage of 0.95 V, and (E) EQE.
Characterization of AX-coated CsPbI3 QDSCs and films
A variety of AX salts were investigated as potential treatments for modifying the QD array. To fabricate the CsPbI3 QD films for this work, CsPbI3 QDs were synthesized as described previously (4, 7) and deposited in a layer-by-layer fashion by spin coating from octane. To partially remove the native ligands and allow for further layers to be deposited without redispersing the existing layers, we immersed the film into a saturated lead(II) nitrate [Pb(NO3)2] solution in methyl acetate (MeOAc) after each QD layer (4). After building up a sufficiently thick CsPbI3 QD film (three to four deposition cycles, leading to 200- to 400-nm-thick films), the film was immersed in a saturated AX salt solution in ethyl acetate (EtOAc) for ~10 s. Figure 2A shows a flow diagram depicting this process for film assembly.
Fig. 2. Effect of AX salts on CsPbI3 QD films and PV performance.
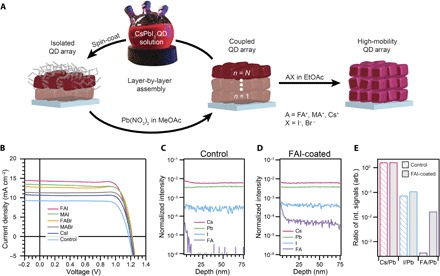
(A) Schematic of the film deposition process and AX salt posttreatment. (B) J-V characteristics of CsPbI3 QD devices treated with FAI (pink), methylammonium iodide (MAI) (green), formamidinium bromide (FABr) (yellow), methylammonium bromide (MABr) (gray), cesium iodide (CsI) (dark blue), and neat EtOAc control (blue). (C and D) ToF-SIMS depth profile of CsPbI3 QD films (C) without and (D) with an FAI posttreatment. Intensity is normalized to total counts at each data point. (E) Ratios of Cs/Pb, I/Pb, and FA/Pb calculated from the integrated ToF-SIMS signal intensities throughout the film. int., integrated; arb., arbitrary.
A series of solar cells with varied composition of the AX salt posttreatment, as well as a control device where only neat EtOAc is used, were fabricated and investigated to determine the role of the various A and X components as well as the structural makeup of the resulting films. Figure 2B compares J-V scans for devices with the various AX salt treatments. In comparison to the control, all the AX salt posttreatments markedly increase the performance of the CsPbI3 QDSCs, demonstrating the generality and effectiveness of this surface treatment scheme. Among the different AX salt posttreatments explored in this series, the highest power conversion efficiency (PCE) of 13.4% was achieved from the FAI treatment with a JSC of 14.37 mA/cm2, which was remarkably better than the neat EtOAc control with a PCE and JSC of 8.5% and 9.22 mA/cm2, respectively. As seen in Fig. 2B, the posttreatments have little impact on the VOC or fill factor (FF), and the PCE improvement is almost entirely attributed to the increased JSC (see Table 1). This suggests that the different AX salt treatments affect the QD coupling to varying degrees and may be used to tune the electronic properties of the QD films.
Table 1. Effect of AX salts on PV parameters extracted from J-V scans.
| AX salt posttreatment | VOC (V) | JSC (mA cm−2) | FF | PCE (%) |
| FAI (EtOAc) | 1.20 | 14.37 | 0.78 | 13.4 |
| FABr (EtOAc) | 1.22 | 12.70 | 0.81 | 12.6 |
| MAI (EtOAc) | 1.20 | 13.39 | 0.79 | 12.6 |
| MABr (EtOAc) | 1.21 | 11.27 | 0.82 | 11.2 |
| CsI (EtOAc) | 1.20 | 10.64 | 0.81 | 10.3 |
| Neat EtOAc | 1.17 | 9.22 | 0.78 | 8.5 |
By comparing the champion device treated with FAI to those treated with neat EtOAc (fig. S1), we measure a ~60% improvement in JSC, improving the PCE obtained from a J-V scan from 8.5 to 13.8% and the stabilized power output (SPO) from 6.1 to 13.5%. This photocurrent increase occurs because of a broadband improvement in the overall EQE rather than a change in the EQE onset, which would indicate a change in the bandgap (fig. S1). Moreover, the absorbance spectra of the films using neat EtOAc and FAI treatments show similar onsets at ~700 nm, consistent with the EQE spectra, but also show similar overall light absorption (fig. S2), ruling out the possibility that the improvement seen in JSC arises from improved light absorption. We find a similar broadband improvement in EQE for all of the investigated AX salt posttreatments (fig. S3), suggesting that this AX posttreatment scheme is a general way to improve the charge carrier collection efficiency in the CsPbI3 QD films. We investigated the case of FAI in detail; however, other AX salt posttreatments after optimization could yield even higher performance. In addition to an improvement in the PCE, we also observe a noticeable reduction in hysteresis between the forward (that is, JSC to VOC) and reverse (that is, VOC to JSC) J-V scans, as well as better agreement between the SPO and reverse J-V scan after the FAI posttreatment (fig. S1). Histograms of the PCE, VOC, JSC, and FF of 78 devices fabricated with this FAI posttreatment are provided in fig. S4 to demonstrate the reproducibility of these high-efficiency (>12%), high-voltage (>1.15 V) QDSCs.
We confirm the presence of the FAI salt in the film after the posttreatment via time-of-flight secondary ion mass spectrometry (ToF-SIMS) depth profiling, as seen in Fig. 2 (C to E) [see Materials and Methods for description of mass/charge ratio (m/z) values assigned to ions used to track each component]. Comparing the FAI-coated film (Fig. 2D) to the control film (Fig. 2C), the Cs, Pb, and I signals remained approximately the same, whereas there is a significant increase in the FA signal intensity. To directly compare the Cs, I, and FA, we calculate ratios of the signal intensities integrated over the thickness of the film (Fig. 2E). The FAI-coated film has a significant increase in the FA/Pb ratio (as expected, the signal at m/z = 45 is at the instrument response limit before treatment) and a slight increase in the I/Pb ratio, whereas the Cs/Pb ratio is unchanged. Furthermore, the ToF-SIMS trace of the FA component indicates a higher concentration of FA+ at the sample surface, which is corroborated by x-ray photoemission spectroscopy (XPS) (fig. S5), confirming the presence of FA+ on the surface. The slight increase in the I/Pb ratio is confirmed for the sample surface by XPS as well. We also identify the presence of FA+ in the film by transmission mode Fourier transform infrared (FTIR) spectroscopy (fig. S6) from the emergence of a peak at 1712 cm−1 following FAI treatment, which is characteristic of the C═N stretch in FA+.
It is clear that FA+ is present in the treated films; therefore, four scenarios regarding the interaction between FAI and the CsPbI3 QD film are hypothesized: (i) The FAI interactions could be limited to the QD surface, either binding to the QD or infilling the void space between QDs; (ii) a partial cation exchange could result in an alloyed CsxFA1−xPbI3 QD core; (iii) a perovskite FAPbI3 matrix or shell could form around the CsPbI3 QDs if excess of Pb2+ and I- are present; and (iv) the FAI salt could be inducing grain growth in the CsPbI3 QD film, similar to the observed Ostwald ripening of MAPbI3 thin films treated with MABr (22). In scenario (iv), there would no longer be individual QDs present in the treated films, and the films would resemble bulk CsPbI3 or alloyed CsxFA1−xPbI3 films.
Because the EQE onset (figs. S1 and S3) and absorption (fig. S2) are effectively unchanged after FAI treatment, drastic changes in the chemical composition of the film are unlikely, as we would expect a greater red shift in the case of a CsxFA1−xPbI3 alloying and an absorption onset at ~840 nm in the case of a FAPbI3 matrix (6). X-ray diffraction (XRD) patterns before and after the FAI treatment (fig. S7) are absent of any detectable peak shifts or new peaks, which would indicate alloying or formation of a new perovskite phase, respectively. Therefore, scenarios (ii) and (iii) are ruled out. Additionally, we find a decrease in the C–H modes associated with the as-synthesized oleate and oleylammonium ligands near 3000 cm−1 in the FTIR, suggesting that additional native ligands have been removed following the FAI treatment. Removal of native ligands reduces interparticle spacing and increases QD coupling [scenario (i)], which is well known to induce a bathochromic shift in QD samples (23).
To assess whether the increased charge carrier collection efficiency is the result of CsPbI3 QD grain growth induced by the FAI treatment [scenario (iv)], we characterized the photoluminescence (PL) from the films of three different sizes of QDs, which are readily synthesized by varying the reaction temperature (with lower reaction temperatures producing smaller QDs with increasing bandgap) (4, 7). After performing the FAI treatment, there is still a significant blue shift in the spectral position of the PL peak with decreasing QD size (Fig. 3A) compared to bulk CsPbI3, which verifies that most of the quantum confinement is retained following the FAI treatment. High-resolution atomic force microscopy (AFM) (Fig. 3, B and C) and SEM (fig. S8) images of the QD films before and after FAI treatment readily resolve individual QDs in the films and confirm that the QD film morphology is maintained after FAI treatment. Note that the grains shown in the AFM images in Fig. 3 (B and C) appear larger than those observed in SEM images (fig. S8) because of the tip-sample convolution. Thus, we conclude that the CsPbI3 QD structure remains intact, despite the addition of FA to the film. Given that the QD films perform well with <30-nm grains is encouraging because it departs from the growing notion that large grains are absolutely essential for high-performance perovskite devices. The QD films described here decouple the grain growth from the film formation, which could be highly beneficial for rapid manufacturing.
Fig. 3. Confinement in FAI-coated films.
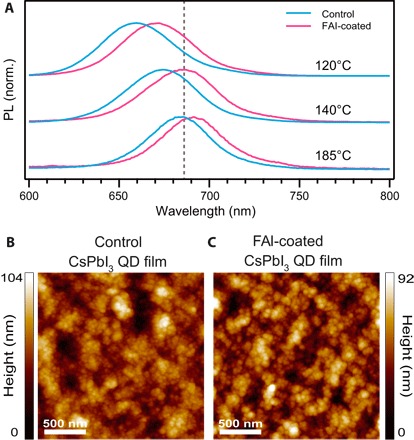
(A) Normalized (norm.) PL of CsPbI3 QD films fabricated with three different sizes (as indicated by the injection temperature during synthesis) of QDs with (pink) and without (blue) FAI treatment. (B and C) AFM images of CsPbI3 QD films (B) without and (C) with an FAI posttreatment.
The combined evidence of absorption, EQE, PL, ToF-SIMS, FTIR, XRD, XPS, and multimodal microscopy are consistent with the addition of the FAI to the CsPbI3 QD film without significant changes in the composition of the QD core. We therefore refer to the treated CsPbI3 QD films as AX-coated films. Given the fast anion exchange reported in the CsPbX3 QD systems and some limited reports on the A-site cation exchange, it is notable that the AX treatment is limited to QD surface interactions but is able to penetrate through the depth of the QD film. A more detailed study of the interaction of the FAI treatment on the CsPbI3 QD surface is forthcoming.
Because it is clear that quantum confinement is preserved in the FAI-coated films, we hypothesize that the slight red shift seen in the absorbance and PL following FAI treatment arises from the improved electronic coupling between individual QDs. Increasing the wave function overlap between neighboring QDs relaxes confinement in the QDs and improves charge carrier transport, which explains the increased photocurrent following AX coating. We therefore probed the mobility of FAI-coated CsPbI3 QD films using time-resolved terahertz spectroscopy (TRTS), which is a contactless pump-probe optical method capable of characterizing both conductivity and carrier dynamics in semiconductor films. In addition to the CsPbI3 QD films, we also probed conventional MAPbI3 thin films as well as PbSe and PbS QD films to compare CsPbI3 QDs with more heavily studied materials (24–27). The PbS QD sample was composed of 3-nm QDs used commonly in literature for high-efficiency QDSCs, whereas larger 6-nm PbSe QDs were chosen for their higher reported mobility (25).
Enhanced mobility in FAI-coated CsPbI3 QDs
The TRTS measurements were performed under identical conditions for a 3-nm PbS QD film, a 6-nm PbSe QD film, a CsPbI3 QD film, an FAI-coated CsPbI3 QD film, and a conventional, nonquantized MAPbI3 film. For thin films, conductivity is directly proportional to the change in terahertz transmission (28). Figure 4A shows the time-resolved change in terahertz transmission (ΔE/E) for each of the films probed. Mobility and lifetime values were extracted from the traces in Fig. 4B, as described in Materials and Methods and summarized in Fig. 4 (C and D, respectively). We find that the mobility for the control CsPbI3 QD film is 0.23 cm2 V−1 s−1, already significantly higher than that of the lead chalcogenide QD films (mobility of PbS and PbSe QD films were measured to be 0.042 and 0.090 cm2 V−1 s−1, respectively). When the CsPbI3 QD films are treated with FAI, the mobility improves from 0.23 to 0.50 cm2 V−1 s−1. The mobility reaches a factor of 5 lower than the MAPbI3 thin film (2.3 cm2 V−1 s−1 in this study, similar to other literature reports) (27). We also compare the carrier lifetimes that are extracted from the terahertz measurements. The lifetimes of the control CsPbI3 and FAI-coated CsPbI3 QD films were found to be 1.8 and 1.6 ns, respectively (neglecting the early sub–50-ps dynamics), largely similar to the 2.2-ns lifetime of the MAPbI3 film. The dynamics for the PbS and PbSe QD films are quite distinct. At the earliest times, the response decays rapidly; however, this is likely not recombination of carriers but rather a reduction in carrier mobility as carriers find lower-energy states with lower carrier mobility. After this initial decay, the mobility is reduced by another order of magnitude, and we find the mobility to be ~0.003 cm2 V−1 s−1, which falls in-line with other widely reported measurements of the carrier mobility in these QD films (24–26).
Fig. 4. Improved mobility with FAI coating.
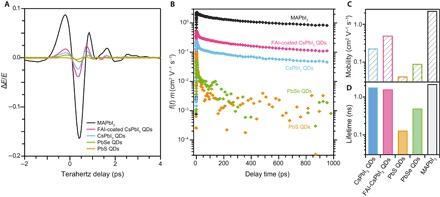
(A) Time-domain terahertz pulses through each of the films described in the text recorded at a pump delay time of ~1ps. (B) Time-resolved terahertz photoconductivity measurements of the films of CsPbI3 control sample (blue) compared to the FAI-coated CsPbI3 QD films (pink) along with a traditional MAPbI3 (black) thin film and films of 6-nm PbSe QDs (orange) and 3-nm PbS QDs (green). (C and D) Comparison of the extracted (C) mobility and (D) terahertz lifetimes for each of the films.
Note that the carrier dynamics extracted with terahertz spectroscopy for both CsPbI3 QD films are more similar to MAPbI3 than PbS and PbSe carrier dynamics. This finding strongly suggests that the carrier transport mechanism is quite distinct in perovskite QD films compared to lead chalcogenide QD films. More work is under way exploring observed differences to understand their microscopic origins. For PV applications, mobility-lifetime product is of particular interest because of its relation to charge carrier diffusion length. The mobility-lifetime product was larger for the FAI-coated CsPbI3 QD film than the untreated CsPbI3 QD film (fig. S9) and justifies the enhanced charge extraction. Time-resolved PL (tr-PL) measurements (fig. S10) show an increase in PL lifetime for FAI-coated films on a longer time scale than what is measured using terahertz spectroscopy, whereas the decay dynamics within the terahertz window are similar.
DISCUSSION
Although QDs have the advantages of widely tunable bandgap and energetic position and, in the case of CsPbI3, offer the added benefit of phase stability, QDSCs have traditionally suffered from both low VOC relative to their bandgap and poor carrier collection efficiency. We demonstrate that both of these disadvantages of QDSCs can be, in large part, overcome by the coupling of CsPbI3 QD films. The AX treatment strategy developed in this work provides a general method for tuning the electronic properties of the CsPbI3 QD films. More specifically, the FAI coating yields a doubling of the already-high mobility of CsPbI3 QD films and results in a certified record PCE of 13.43%, positioning QDSCs above the best reported PCE for dye-sensitized solar cells, organic PVs, and CZTSSe PV technologies. Additionally, the high VOC, exceeding 80% of the Shockley-Queisser limit, and tunability of the bandgap make these devices well suited for use in high-efficiency tandem solar cell architectures.
MATERIALS AND METHODS
Materials
All chemicals were purchased from Sigma-Aldrich and used as received unless otherwise specified. Cesium carbonate (Cs2CO3; 99.9%), PbI2 (99.9985%; Alfa Aesar), oleic acid (OA; technical grade, 90%), oleylamine (OAm; technical grade, 70%), 1-octadecene (ODE; technical grade, 90%), hexane (reagent grade, ≥95%), octane (anhydrous, ≥99%), MeOAc (anhydrous, 99.5%), Pb(NO3)2 (99.999%), EtOAc (anhydrous, 99.8%), CsI (99.999%), FAI (Dyesol), FABr (Dyesol), MAI (Dyesol), MABr (Dyesol), ethanol (EtOH; 200 proof, ≥99.5%), titanium ethoxide (≥97%), hydrochloric acid (HCl; 37% in water), 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD; ≥99.5%; Lumtec), chlorobenzene (anhydrous, 99.8%), 4-tert-butylpyridine (4-TBP; 96%), bis(trifluoromethane)sulfonimide lithium salt (Li-TFSI), and acetonitrile (anhydrous, 99.8%).
QD synthesis and purification
CsPbI3 QDs were synthesized (7) and purified (4) using previously reported methods with slight modifications. Cs oleate in ODE solution (0.125 M) was synthesized by first degassing 0.407 g of Cs2CO3, 1.25 ml of OA, and 20 ml of ODE in a 100-ml three-neck flask under vacuum at 120°C for 30 min under stirring. After purging the flask with N2, the flask was heated to 150°C until the reaction was complete and yielded a clear solution. The Cs-oleate in ODE solution was cooled and stored in N2 until needed for a QD synthesis. To synthesize CsPbI3 QDs, 0.5 g of PbI2 and 25 ml of ODE were degassed in a 100-ml three-neck flask under vacuum at 120°C for 30 min under stirring. Preheated OA and OAm (130°C, 2.5 ml each) were added to the reaction flask and degassed briefly until the PbI2 had completely dissolved. The reaction flask was purged with N2 and then heated to the desired reaction temperature (typically 185°C) to achieve the desired nanocrystal size. Once the reaction temperature was reached, 2 ml of preheated Cs-oleate in ODE (130°C, 0.125 M) was injected into the reaction flask. The reaction was quenched in an ice bath after 5 to 10 s. To isolate the QDs, 35 ml of MeOAc was added to 15 ml of the QD reaction liquor and then centrifuged at 7500 rpm for 5 min. The supernatant was discarded, and the QD pellet was redispersed in ~5 ml of hexane. A minimal amount of MeOAc (~5 to 8 ml) was added to the QDs until the dispersion appeared cloudy, and then it was immediately centrifuged at 7500 rpm for 5 min. The resultant QD pellet was dispersed in 15 ml of hexane and then stored in the dark at 4°C for at least 24 hours to precipitate excess Cs-oleate and Pb-oleate. Before use, these solid precipitates were removed from the QD solution via centrifugation at 7500 rpm for 5 min.
Film fabrication
Coupled CsPbI3 QD films were fabricated using previously reported methods (4). Saturated Pb(NO3)2 in MeOAc solution and AX salt (where AX = FAI, FABr, MAI, MABr, or CsI) in EtOAc solution were prepared by sonicating 20 mg of Pb(NO3)2 (or AX salt) and 20 ml of MeOAc (or EtOAc) for 10 min. Because all the salts were only slightly soluble in their corresponding solvent, excess salt was removed via centrifugation at 3500 rpm for 5 min. Each layer of CsPbI3 QDs was spin-cast from a concentrated QD solution in octane (~75 mg/ml) at spin speeds of 1000 rpm for 20 s and 2000 rpm for 5 s. The film was then briefly dipped (~1 s) into the Pb(NO3)2 in MeOAc solution, rinsed in a solution of neat MeOAc, and then immediately dried in a stream of dry air. This process of spin-coating QDs and dipping in Pb(NO3)2 in MeOAc solution was repeated three to five times to achieve a total film thickness of 100 to 500 nm. Once the desired film thickness was achieved, the films were posttreated by soaking the film in the AX salt in EtOAc solution for 10 s before rinsing in MeOAc and then immediately drying under a stream of dry air. All film fabrications were carried out under dry ambient conditions (relative humidity, ~16 to 20%).
Film characterization
Ultraviolet-visible (UV-vis) absorption spectra were recorded using a Shimadzu UV-3600 UV-vis–near-infrared spectrophotometer. Absorptance was calculated from the reflectance and transmittance spectra acquired with an integrating sphere. Steady-state PL was measured using a Horiba Jobin Yvon FluoroMAX-4 spectrophotometer. Powder XRD data were recorded using a Bruker D8 Discover x-ray diffractometer with a Hi-Star 2D area detector using Cu Kα radiation (1.54 Å). FTIR spectra were acquired using a Nicolet 6700 FTIR spectrometer on QD films on Si substrates.
Time-of-flight secondary ion mass spectrometry
SIMS is a powerful analytical technique for determining elemental and isotopic distributions in solids, as well as the structure and composition of organic materials (29–31). An ION-TOF ToF-SIMS 5 ToF-SIMS spectrometer was used to depth-profile the perovskite QD films. Analysis was completed using a three-lens 30-kV BiMn primary ion gun, and the Bi3+ primary ion beam (operated in bunched mode; 20 ns pulse width, analysis current 0.7 pA) was scanned over a 25 × 25 micron area. Depth profiling was accomplished with a 3-kV oxygen ion sputter beam (10.8 nA of sputter current) raster of 150 × 150 micron area. All spectra during profiling were collected at or below a primary ion dose density of 1 × 1012 ions/cm2 to remain under the static SIMS limit. The data were plotted with the intensity for each signal at each data point normalized to the total ion counts measured at the data point, which diminishes artifacts from a changing ion yield in different layers as well as artifacts due to minor fluctuations in the primary ion beam current. We assigned the m/z signals at 45, 127, 206, and 266 to FA+, I+, 206Pb+, and Cs2+, respectively, and simply referred to these signals as the FA, I, Pb, and Cs in the manuscript, respectively. The Cs2+ and 206Pb+ signals were tracked because they were the most intense species for Cs+ and Pb2+, respectively, which did not lead to detector saturation. In contrast, Cs+, Pb2+, and other abundant lead isotopes saturated the detector under the measurement conditions used.
Atomic force microscopy
QD films were prepared on FTO-coated glass as described above. Samples were mounted using double-sided adhesive strips to metal disks exposing the QD layer. Imaging was performed with multimode scanning probe microscope equipped with a NanoScope IV controller (Bruker). Height images were obtained in soft-tapping mode using etched silicon probes (TESP, Bruker) with an autotuned resonance frequency range of 250 to 300 kHz at a scan rate of 2 Hz. Images were analyzed with NanoScope Analysis v1.2 software.
PV device fabrication
A ~50-nm TiO2 layer was deposited via a sol-gel method onto prepatterned FTO on glass substrates (Thin Film Devices Inc.). Sol-gel TiO2 was prepared by mixing 5 ml of EtOH, two drops of HCl, 125 μl of deionized water, and 375 μl of titanium ethoxide, resulting in a clear solution. The headspace of the vial was filled with nitrogen, and the solution was stirred for 48 hours and then kept in the freezer until use. The sol-gel was spin-cast at 3000 rpm for 20 s and annealed at 115° and 450°C for 30 min each. The CsPbI3 QD photoactive layer was deposited using the procedure described above, resulting in a total thickness of 100 to 400 nm. The spiro-OMeTAD hole transporting material was spin-cast at 4000 rpm for 30 s from a solution with a nominal concentration of 72.3 mg of spiro-OMeTAD, 1 ml of chlorobenzene, 28.8 μl of 4-TBP, and 17.5 μl of Li-TFSI stock solution (520 mg/ml in acetonitrile). All the spin-coating processes were performed under ambient conditions. MoOx was deposited at a rate of 0.2 to 1.0 Å/s at a base pressure lower than 2 × 10−7 torr for a total thickness of 15 nm. Al electrodes were evaporated at a rate ranging from 0.5 to 2 Å/s for a total thickness of 200 nm. A 70-nm layer of MgF2 was evaporated on the glass side as an antireflective coating for the best-performing, certified PV device.
Device characterization
Devices were tested in a N2-filled glovebox with a Newport Oriel Sol3A solar simulator with a xenon lamp source. Light intensity was calibrated with a KG5 filtered Si reference diode to minimize the spectral mismatch and to set the intensity of the lamp to 100 mW/cm2 AM1.5 conditions. Devices were illuminated through a metal aperture (0.058 cm2). SPO was measured by holding the device at a constant voltage corresponding to the voltage at the maximum power point of the J-V scan. EQE measurements were taken using a Newport Oriel IQE200.
Certification
Devices certified by the NREL PV Performance Characterization Team were loaded into an air-free holder in a N2-filled glovebox. The device active area was illuminated through a metal aperture with an area of 0.058 cm2, which was measured by the PV Performance Characterization Team. As an accredited PV testing facility, the PV Performance Characterization Team received and tested the packaged solar cell in accordance with their independent testing procedures.
Terahertz spectroscopy
The films for terahertz measurements were deposited directly onto 1 × 2–cm quartz substrates following similar spin-coating procedures described above. PbS and PbSe QD samples were synthesized and fabricated as previously published using PbI2 ligand exchange treatment (13, 32). MAPbI3 films were prepared, as described by Ahn et al. (33), by spin coating from a N,N′-dimethylformamide/dimethyl sulfoxide solution containing stoichiometric MAI/PbI2. The change in terahertz transmission was recorded as a function of delay from an optical pump pulse (wavelength, 516 nm) with a fluence of 5 × 1015 photons/cm2 per pulse. For thin films, the mobility and carrier lifetimes are directly proportional to the change in terahertz transmission through the following relationship: (μe ⋅ fe(τ) + μh ⋅ fh(τ)) = [ΔE(τ)/E] ⋅ [(1 + ns)cε0/(e ⋅ Jabs)] (28). Here, ΔE/E is the measured terahertz response (see Fig. 4A) at a pump delay of τ, ns is the refractive index of the substrate (ns = 2.19 here), c is the speed of light, ε0 is the permittivity of free space, e is the charge of an electron, and Jabs is the absorbed photon fluence and is related to the film optical density by Jabs = (1 − R) ⋅ (1 − 10−OD) ⋅ Jin, where R is the reflection coefficient, and Jin is the measured photon fluence at the sample position. At τ = 0, both the normalized electron and hole carrier decay are 1 (fe(0) = fh(0) = 1), and the signal in Fig. 4B is the sum of the electron and hole mobilities: (μe + μh) = μs.
Supplementary Material
Acknowledgments
We thank T. Moriarty and D. Levi for certified device measurements, A. Hicks for assistance with graphics, B. To for SEM imaging, G. Pach for helping with time-resolved PL measurements, and J. Berry for insightful discussions. Funding: We acknowledge support from the Center for Advanced Solar Photophysics, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences. E.M.S. and L.Y.L. were supported by a NASA Space Technology Research Fellowship. J.A.C. was supported by the DOE/Energy Efficiency and Renewable Energy (EERE) Postdoctoral Research Award under the EERE/Solar Energy Technologies Office (SETO) administered by the Oak Ridge Institute for Science and Education for the DOE under DOE contract no. DE-SC00014664. P.S. was supported by the hybrid perovskite solar cell program of the National Center for Photovoltaics funded by the DOE, Office of EERE, and SETO. DOE funding was provided to NREL through contract no. DE-AC36-08G028308. Author contributions: E.M.S. and A.R.M. synthesized the QDs and fabricated devices. J.M.L. has verified all data and claims responsibility for each figure. All authors contributed to QD film or device characterization, analysis, figures, and edits to the manuscript. Competing interests: E.M.S., J.M.L., and A.R.M. are authors on a patent application related to this work (U.S. application no. 15/609,401; PCT application no. PCT/US17/35156; filed 31 May 2017). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/eaao4204/DC1
Supplementary Materials and Methods
fig. S1. Performance of FAI-coated and control devices.
fig. S2. Light absorption following FAI posttreatment.
fig. S3. Comparison of EQE with AX posttreatment.
fig. S4. Reproducibility of FAI-coated CsPbI3 QD device performance.
fig. S5. XPS spectroscopy.
fig. S6. FTIR spectra of CsPbI3 QDs.
fig. S7. Crystal structure of CsPbI3 QDs.
fig. S8. CsPbI3 QD film morphology.
fig. S9. Comparison of terahertz μs × τ product.
fig. S10. PL lifetime of CsPbI3.
Reference (34)
REFERENCES AND NOTES
- 1.Kovalenko M. V., Manna L., Cabot A., Hens Z., Talapin D. V., Kagan C. R., Klimov V. I., Rogach A. L., Reiss P., Milliron D. J., Guyot-Sionnnest P., Konstantatos G., Parak W. J., Hyeon T., Korgel B. A., Murray C. B., Heiss W., Prospects of nanoscience with nanocrystals. ACS Nano 9, 1012–1057 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Lan X., Masala S., Sargent E. H., Charge-extraction strategies for colloidal quantum dot photovoltaics. Nat. Mater. 13, 233–240 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Shirasaki Y., Supran G. J., Bawendi M. G., Bulović V., Emergence of colloidal quantum-dot light-emitting technologies. Nat. Photonics 7, 13–23 (2012). [Google Scholar]
- 4.Swarnkar A., Marshall A. R., Sanehira E. M., Chernomordik B. D., Moore D. T., Christians J. A., Chakrabarti T., Luther J. M., Quantum dot–induced phase stabilization of α-CsPbI3 perovskite for high-efficiency photovoltaics. Science 354, 92–95 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Semonin O. E., Luther J. M., Choi S., Chen H.-Y., Gao J., Nozik A. J., Beard M. C., Peak external photocurrent quantum efficiency exceeding 100% via MEG in a quantum dot solar cell. Science 334, 1530–1533 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Yan Y., Crisp R. W., Gu J., Chernomordik B. D., Pach G. F., Marshall A. R., Turner J. A., Beard M. C., Multiple exciton generation for photoelectrochemical hydrogen evolution reactions with quantum yields exceeding 100%. Nat. Energy 2, 17052 (2017). [Google Scholar]
- 7.Protesescu L., Yakunin S., Bodnarchuk M. I., Krieg F., Caputo R., Hendon C. H., Yang R. X., Walsh A., Kovalenko M. V., Nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I): Novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 15, 3692–3696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eperon G. E., Paternò G. M., Sutton R. J., Zampetti A., Haghighirad A. A., Cacialli F., Snaith H. J., Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A 3, 19688–19695 (2015). [Google Scholar]
- 9.Eperon G. E., Stranks S. D., Menelaou C., Johnston M. B., Herz L. M., Snaith H. J., Formamidinium lead trihalide: A broadly tunable perovskite for efficient planar heterojunction solar cells. Energy Environ. Sci. 7, 982–988 (2014). [Google Scholar]
- 10.Sharma S., Weiden N., Weiss A., Phase diagrams of quasibinary systems of the type: ABX3—A′BX3; ABX3—AB′X3, and ABX3—ABX′3; X = halogen. Zeitschrift Phys. Chemie 175, 63–80 (1992). [Google Scholar]
- 11.Sutton R. J., Eperon G. E., Miranda L., Parrott E. S., Kamino B. A., Patel J. B., Hörantner M. T., Johnston M. B., Haghighirad A. A., Moore D. T., Snaith H. J., Bandgap-tunable cesium lead halide perovskites with high thermal stability for efficient solar cells. Adv. Energy Mater. 6, 1502458 (2016). [Google Scholar]
- 12.Beal R. E., Slotcavage D. J., Leijtens T., Bowring A. R., Belisle R. A., Nguyen W. H., Burkhard G. F., Hoke E. T., McGehee M. D., Cesium lead halide perovskites with improved stability for tandem solar cells. J. Phys. Chem. Lett. 7, 746–751 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Crisp R. W., Kroupa D. M., Marshall A. R., Miller E. M., Zhang J., Beard M. C., Luther J. M., Metal halide solid-state surface treatment for high efficiency PbS and PbSe QD solar cells. Sci. Rep. 5, 9945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall A. R., Young M. R., Nozik A. J., Beard M. C., Luther J. M., Exploration of metal chloride uptake for improved performance characteristics of PbSe quantum dot solar cells. J. Phys. Chem. Lett. 6, 2892–2899 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Tang J., Kemp K. W., Hoogland S., Jeong K. S., Liu H., Levina L., Furukawa M., Wang X., Debnath R., Cha D., Chou K. W., Fischer A., Amassian A., Asbury J. B., Sargent E. H., Colloidal-quantum-dot photovoltaics using atomic-ligand passivation. Nat. Mater. 10, 765–771 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Ip A. H., Thon S. M., Hoogland S., Voznyy O., Zhitomirsky D., Debnath R., Levina L., Rollny L. R., Carey G. H., Fischer A., Kemp K. W., Kramer I. J., Ning Z., Labelle A. J., Chou K. W., Amassian A., Sargent E. H., Hybrid passivated colloidal quantum dot solids. Nat. Nanotechnol. 7, 577–582 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Lan X., Voznyy O., Kiani A., García de Arquer F. P., Abbas A. S., Kim G.-H., Liu M., Yang Z., Walters G., Xu J., Yuan M., Ning Z., Fan F., Kanjanaboos P., Kramer I., Zhitomirsky D., Lee P., Perelgut A., Hoogland S., Sargent E. H., Passivation using molecular halides increases quantum dot solar cell performance. Adv. Mater. 28, 299–304 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Stavrinadis A., Pradhan S., Papagiorgis P., Itskos G., Konstantatos G., Suppressing deep traps in PbS colloidal quantum dots via facile iodide substitutional doping for solar cells with efficiency >10%. ACS Energy Lett. 2, 739–744 (2017). [Google Scholar]
- 19.Christians J. A., Manser J. S., Kamat P. V., Best practices in perovskite solar cell efficiency measurements. Avoiding the error of making bad cells look good. J. Phys. Chem. Lett. 6, 852–857 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Snaith H. J., Abate A., Ball J. M., Eperon G. E., Leijtens T., Noel N. K., Stranks S. D., Wang J. T.-W., Wojciechowski K., Zhang W., Anomalous hysteresis in perovskite solar cells. J. Phys. Chem. Lett. 5, 1511–1515 (2014). [DOI] [PubMed] [Google Scholar]
- 21.National Renewable Energy Laboratory, Research Cell Record Efficiency Chart (NREL).
- 22.Yang M., Zhang T., Schulz P., Li Z., Li G., Kim D. H., Guo N., Berry J. J., Zhu K., Zhao Y., Facile fabrication of large-grain CH3NH3PbI3−xBrx films for high-efficiency solar cells via CH3NH3Br-selective Ostwald ripening. Nat. Commun. 7, 12305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talapin D. V., Murray C. B., PbSe nanocrystal solids for n- and p-channel thin film field-effect transistors. Science 310, 86–89 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Zhang J., Tolentino J., Smith E. R., Zhang J., Beard M. C., Nozik A. J., Law M., Johnson J. C., Carrier transport in PbS and PbSe QD films measured by photoluminescence quenching. J. Phys. Chem. C 118, 16228–16235 (2014). [Google Scholar]
- 25.Liu Y., Gibbs M., Puthussery J., Gaik S., Ihly R., Hillhouse H. W., Law M., Dependence of carrier mobility on nanocrystal size and ligand length in PbSe nanocrystal solids. Nano Lett. 10, 1960–1969 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Balazs D. M., Dirin D. N., Fang H.-H., Protesescu L., ten Brink G. H., Kooi B. J., Kovalenko M. V., Loi M. A., Counterion-mediated ligand exchange for PbS colloidal quantum dot superlattices. ACS Nano 9, 11951–11959 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herz L. M., Charge-carrier mobilities in metal halide perovskites: Fundamental mechanisms and limits. ACS Energy Lett. 2, 1539–1548 (2017). [Google Scholar]
- 28.Bergren M. R., Simonds B. J., Yan B., Yue G., Ahrenkiel R., Furtak T. E., Collins R. T., Taylor P. C., Beard M. C., Electron transfer in hydrogenated nanocrystalline silicon observed by time-resolved terahertz spectroscopy. Phys. Rev. B 87, 081301 (2013). [Google Scholar]
- 29.F. A. Stevie, Secondary Ion Mass Spectrometry: Applications for Depth Profiling and Surface Characterization (Momentum Press, 2015). [Google Scholar]
- 30.De Souza R. A., Martin M., Probing diffusion kinetics with secondary ion mass spectrometry. MRS Bull. 34, 907–914 (2009). [Google Scholar]
- 31.R. G. Wilson, F. A. Stevie, C. W. Magee, Secondary Ion Mass Spectrometry: A Practical Handbook for Depth Profiling and Bulk Impurity Analysis (Wiley-Interscience, 1989). [Google Scholar]
- 32.Chernomordik B. D., Marshall A. R., Pach G. F., Luther J. M., Beard M. C., Quantum dot solar cell fabrication protocols. Chem. Mater. 29, 189–198 (2017). [Google Scholar]
- 33.Ahn N., Son D.-Y., Jang I.-H., Kang S. M., Choi M., Park N.-G., Highly reproducible perovskite solar cells with average efficiency of 18.3% and best efficiency of 19.7% fabricated via Lewis base adduct of lead(II) iodide. J. Am. Chem. Soc. 137, 8696–8699 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Hoye R. L. Z., Schulz P., Schelhas L. T., Holder A. M., Stone K. H., Perkins J. D., Vigil-Fowler D., Siol S., Scanlon D. O., Zakutayev A., Walsh A., Smith I. C., Melot B. C., Kurchin R. C., Wang Y., Shi J., Marques F. C., Berry J. J., Tumas W., Lany S., Stevanović V., Toney M. F., Buonassisi T., Perovskite-inspired photovoltaic materials: Toward best practices in materials characterization and calculations. Chem. Mater. 29, 1964–1988 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/eaao4204/DC1
Supplementary Materials and Methods
fig. S1. Performance of FAI-coated and control devices.
fig. S2. Light absorption following FAI posttreatment.
fig. S3. Comparison of EQE with AX posttreatment.
fig. S4. Reproducibility of FAI-coated CsPbI3 QD device performance.
fig. S5. XPS spectroscopy.
fig. S6. FTIR spectra of CsPbI3 QDs.
fig. S7. Crystal structure of CsPbI3 QDs.
fig. S8. CsPbI3 QD film morphology.
fig. S9. Comparison of terahertz μs × τ product.
fig. S10. PL lifetime of CsPbI3.
Reference (34)