

Abstract
Multiple sclerosis (MS) is a neurodegenerative disease that is estimated to affect over 2.3 million people worldwide. The exact cause for this disease is unknown but involves immune system attack and destruction of the myelin protein surrounding the neurons in the central nervous system. One promising class of compounds that selectively prevent the activation of immune cells involved in the pathway leading to myelin destruction are Bifunctional Peptide Inhibitors (BPIs). Treatment with BPIs reduces neurodegenerative symptoms in Experimental Autoimmune Encephalomyelitis (EAE), a mouse model of MS. In this work, as an effort to further improve the bioactivity of BPIs, BPI peptides were conjugated to the N- and C-termini of the fragment crystallizable (Fc) region of the human IgG1 antibody. Initially, the two peptides were conjugated to IgG1 Fc using recombinant DNA technology. However, expression in yeast resulted in low yields and one of the peptides being heavily proteolyzed. To circumvent this problem, the poorly expressed peptide was instead produced by solid phase peptide synthesis and conjugated enzymatically using a sortase-mediated ligation. The sortase-mediated method showed near-complete conjugation yield as observed by SDS-PAGE and mass spectrometry in small-scale reactions. This method was scaled up to obtain sufficient quantities for testing the BPI-Fc fusion in mice induced with EAE. Compared to the PBS-treated control, mice treated with the BPI-Fc fusion showed significantly reduced disease symptoms, did not experience weight loss, and showed reduced demyelination. These results demonstrate that the BPI peptides were highly active at suppressing EAE when conjugated to the large Fc scaffold in this manner.
TOC image
INTRODUCTION
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system.1 The disease affects approximately 2.3 million people worldwide.2 Those affected experience a wide variety of neurological disabilities including impairments in mobility3, cognition4, and psychological health5. These neurological disabilities result from the loss of myelin, the lipoprotein sheath coating the axons of the central nervous system. Demyelination can be observed in the brain scans of MS patients as brain lesions.6 The etiology of MS is still unknown but suspected reasons include genetic, environmental, geographical, viral, and lifestyle factors.7–10 The disease is extremely complex and heterogeneous, with involvement by both the humoral and cellular immune responses.11–13 The disease involves the activation of autoreactive T-cells against myelin proteins that infiltrate the brain to damage the myelin sheath of the neuronal axons.14–16 The myelin proteins that are recognized by the autoreactive T-cells include proteolipid protein (PLP), myelin oligodendrocyte protein (MOG), and myelin basic protein (MBP). Currently, there is no cure for MS, and the currently available treatments such as beta interferons, glatiramer acetate, fingolimod, teriflunomide, dimethyl fumarate, and monoclonal antibodies are geared toward reducing symptom severity and frequency of attack.17.18, 19 Some of the current treatments suppress general immune responses, which can increase pathogenic infections in treated patients. Therefore, there is a need to develop MS treatments that selectively suppress autoreactive T-cells against the myelin proteins.
Bifunctional Peptide Inhibitors (BPIs) are a promising new class of peptide conjugates that are designed to selectively inhibit the maturation of T-cells specific for myelin protein.20 BPIs are composed of a myelin-specific antigenic peptide tethered to a signal-2-blocking peptide derived from lymphocyte function-associated antigen-1 (LFA-1), a protein found on T-cells that binds to intercellular adhesion molecule-1 (ICAM-1) (Table 1). For example, a myelin antigenic peptide (e.g., PLP139–15121–26 or MOG38–5025) linked to a LABL (CD11a237–246) peptide,21–23, 25 derived from the I-domain of LFA-1, through a short linker is a BPI. It is hypothesized that BPIs suppress autoreactive T-cells by blocking the formation of the immunological synapse (IS) at the interface of a T-cell and antigen presenting cell (APC) because the mechanism of activating T-cells is initiated by the formation of the IS (Figure 1).20, 27, 28 The IS is formed by at least two signals in which the first signal (signal-1) is generated via the interactions between the complex of antigen-major histocompatibility complex class II (Ag-MHC-II) and a T-cell receptor (TCR). The second signal (signal-2) can be generated by ICAM-1/LFA-1 interactions. Initially, signal-2 is formed in the center of the interface between an APC and a T-cell while signal-1 is formed at the periphery of the interface to form a bullseye-like arrangement. Then, the signal-1 molecules translocate to cluster at the center while the signal-2 molecules migrate to peripheral region of the bullseye to form an IS. The IS formation initiates the activation of a naïve T-cell into a proinflammatory T-cell (Figure 1). This proinflammatory T-cell promotes antigen-specific immune system attack on myelin, causing its inflammation and breakdown.15 BPI molecules are hypothesized to bind simultaneously to MHC-II and ICAM-1 on the surface of an APC and inhibit the formation of the IS. As a result, BPIs alter the commitment of naïve T-cells from an inflammatory phenotype to regulatory or suppressor phenotypes, and this suppresses autoimmune diseases in an antigen-specific manner. While antigenic peptides and signal-2 blocker peptides have been shown to have some ability to reduce T-cell activation on their own, Kobayashi et al. have demonstrated that linking the two types of peptides to form BPIs results in significantly lower clinical scores in an EAE model.21 However, one potential problem with BPIs is the short in vivo half-lives of a few hours (2–3 hours) as measured in rat plasm; therefore, there is a need to investigate methods to lengthen the in vivo half-lives of these types of molecules.22 Table 1. Peptide sequences that were used in the preparation of BPI Fc fusions. The active regions of the sequences are shown in bold. ICAM-1 Binding Peptide CD11a237–246 (LABL) is a signal-2-blocking peptide derived from LFA-1 that binds ICAM-1. For recombinant expression as a Fc fusion, an additional N-terminal and two C-terminal glycine residues were added to this peptide. Proteolipid Protein139–151 and Myelin Oligodendrocyte Glycoprotein38-50 are both myelin-derived antigenic peptides. For use as nucleophilic ligands in the sortase-mediated ligation, the PLP peptide contains three additional N-terminal glycine residues and the MOG peptide contains two additional N-terminal glycine residues. On the C-terminus, the PLP peptide contains an additional glycine residue, and the MOG peptide contains an additional glycine residue followed by two additional arginine residues. The sortase A substrate tag is recognized by sortase A and can be utilized as a peptide tag for C-terminal protein ligations. This tag contains three additional N-terminal alanine residues and two additional C-terminal glycine residues.
Table 1.
BPI-Fc Fusion Peptides
| Peptide Name | Representation | Sequence |
|---|---|---|
| ICAM-1 Binding Peptide CD11a237–246 (LABL) |
|
GITDGEATDSGGG |
| Proteolipid Protein139–151 (PLP) |
|
GGGHSLGKWLGHPDKFG |
| Myelin Oligodendrocyte Glycoprotein38–50 (MOG) |
|
GGGWYRSPFSRVVHLGGR |
| Sortase A Substrate Tag (ST) |
|
AAALPETGGG |
Figure 1.
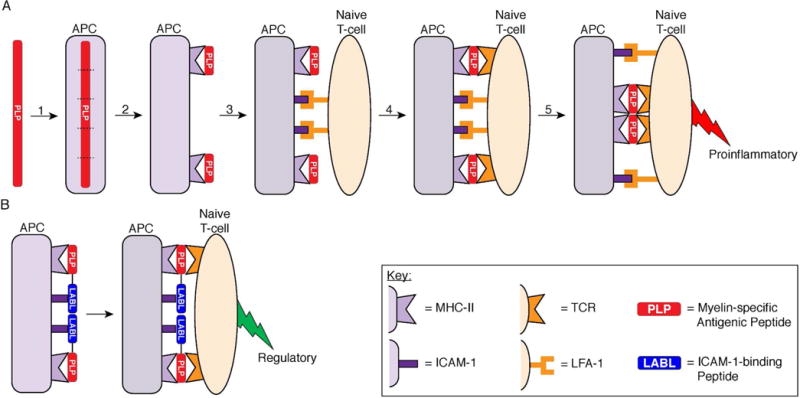
Two-signal model of T-cell activation highlighting the role of the ICAM-1–LFA-1 receptor pairing for both adhesion and costimulation and the alteration of T-cell activation by a BPI. (A) Mechanism of proinflammatory T-cell activation against myelin protein. (1) Myelin-specific antigen is internalized by an APC and broken into fragments. (2) The antigen fragments are presented on the APC cell surface by MHC-II. (3) A naïve CD4+ T-cell binds to the APC through the adhesion receptor pairings of ICAM-1 on the APC and LFA-1 on the T-cell. (4) If the naïve T-cell contains TCRs capable of recognizing the particular antigen presented by MHC-II, then the TCR binds the Ag-MHC-II complex to initiate signal-1 on the periphery. The ICAM-1–LFA-1 receptor pairings also function as costimulatory molecules to constitute signal-2 in a central cluster. (5) The two signals reorganize so that signal-1 pairings localize in the central cluster and the signal-2 pairings localize to the periphery of the signal-1 cluster to form the immunological synapse, which directs the maturation of a naïve T-cell into a proinflammatory T-cell against the myelin antigen. (B) Intervention of myelin-specific T-cell activation by a BPI. A BPI molecule consists of two peptides, a myelin-specific antigenic peptide (PLP) and a signal-2 blocking peptide (LABL), conjugated together. The antigenic peptide of the BPI allows formation of Ag–MHC-II pairing with the TCR to form signal-1 but inhibits the ICAM-1–LFA-1 receptor pairing to prevent formation of signal-2. Because the BPI peptides are conjugated together, the BPI is hypothesized to inhibit formation of the IS and alter the maturation of the naïve T-cell into a regulatory T-cell.
In this work, a BPI-Fc fusion was prepared in which the two BPI peptides, a myelin-specific antigenic peptide and a signal-2-blocking peptide, were conjugated to the N- and C-termini of the fragment crystallizable (Fc) region of a human IgG1 antibody. Initial difficulties in recombinant production of these Fc fusion proteins led us to utilize a sortase mediated ligation to attach antigenic peptides and form BPI-Fc fusion proteins. BPI-Fc fusion proteins were designed with the goal of suppressing experimental autoimmune encephalomyelitis (EAE) in mice. Using the Fc as a platform may increase the half-life of the BPI-Fc due to recycling by the FcRn receptor as well as reduced renal clearance because of increased molecular weight (4 kDa for a traditional BPI compared to 57 kDa for a BPI-Fc).29 Additionally, the Fc is a homodimer which allows the attachment of two of each of the BPI peptides to the Fc scaffold, potentially increasing avidity in binding interactions of the BPI peptides to their respective receptors. Furthermore, fusing to the Fc allows for increased solubility of insoluble peptides. This work was a proof-of-concept project to determine whether a BPI-Fc fusion could be prepared and whether the BPI peptides could still function to protect against EAE when they are separated by a 223 amino acid Fc protein.
RESULTS AND DISCUSSION
Construction of a BPI-Fc fusion
The fusion of small, rapidly cleared peptides and proteins to the Fc region of IgG antibodies is an effective way to improve their half-lives, and because of this Fc fusion proteins have become a prominent class of biopharmaceutics.30, 31 The initial attempt to prepare a BPI-Fc fusion used recombinant DNA technology to fuse both the antigenic and signal-2 blocking peptides to the human IgG1 Fc. A DNA construct was prepared consisting of the PLP and LABL peptides fused to the N- and C-termini of the Fc, respectively (+H3N-HSLGKWLGHPDKFGGG-Fc-AAAGGGITDGEATDSG, Supporting Information); however, recombinant expression of this construct in glycosylation-deficient yeast resulted in severe proteolysis of the PLP region as detected by intact protein mass spectrometry. Three attempts were performed to express this dual fusion construct. The first attempt was performed using a 1 L spinner flask and the conventional 3-day induction time (Supporting Information Figure S1). Mass spectrometry showed that unproteolyzed PLP-Fc-LABL dual fusion protein was present; however, it was not the major peak. Several other peaks of smaller molecular weight were present, all of which corresponded to proteolysis within the PLP region. Based on the ratio of peak heights in the mass spectrum, only 33% of the PLP-Fc-LABL protein was unproteolyzed. In a second attempt, the PLP-Fc-LABL dual fusion construct was expressed in a fermentor. Expression in a fermentor allows for more precise control over air flow, agitation rate, pH, dissolved oxygen concentration, and carbon source feed rate. Having more precise control over these expression conditions may help to reduce proteolysis. Expression in a fermentor did result in unproteolyzed PLP-Fc-LABL being the major peak in the mass spectrum (Supporting Information Figure S2). However, proteolysis was still severe. In fact, there were additional proteolyzed species in the fermentor expression compared to the spinner flask expression. Similar to the spinner flask expression trial, all of the observed proteolysis occurred within the PLP region. Based on the mass spectrometry peak heights, expression of the PLP-Fc-LABL protein in a fermentor resulted in only 31% of the protein being unproteolyzed.
The PLP-Fc-LABL DNA construct was cloned in frame of the Saccharomyces cerevisiae alpha factor prepro sequence to allow for secretion of the target protein into the media. During protein expression, proteases may also be secreted and can cleave the secreted PLP-Fc-LABL protein. In order to reduce the level of proteases present in the media, a third expression was attempted. In this attempt, the cells were grown to density in a 1 L spinner flask, pelleted by centrifugation under sterile conditions, and transferred to 1 L of fresh media immediately before methanol-induced protein expression. Transferring the cells to fresh media before induction resulted in fewer proteolysis products. Based on the peak heights in the mass spectrum, 46% of unproteolyzed protein was obtained (Supporting Information Figure S3). Although the third attempt produced the most uncut protein (46%), proteolysis was unfortunately still severe. Since IgG1 Fc is a dimer linked by inter-chain disulfide bonds, the presence of only 46% uncut monomer in the reduced protein detected by mass spectrometry translates to only approximately 21% of the disulfide-bonded dimer having two full PLP peptides. It was decided that even with the reduced proteolysis, having only 21% full-length dimer would potentially compromise future experiments.
Since changing the expression conditions did not improve the proteolysis observed within the PLP region sufficiently, expression of a different antigenic peptide fused to IgG1 Fc was attempted to determine if the proteolysis problem was specific to the PLP peptide sequence. The MOG peptide was fused to the N-terminus of the Fc (+H3N-GWYRSPFSRVVHLGGG-Fc) and expressed as a single Fc fusion. However, expression of this fusion showed that proteolysis was even more severe within the MOG region than observed in the PLP fusion (Supporting Information Figure S4). Based on the mass spectrometry peak heights, only 5% of unproteolyzed MOG-Fc was obtained. For both the PLP-Fc-LABL and MOG-Fc fusions, proteolysis was observed within the antigenic peptide sequence. Fortunately, the LABL peptide sequence showed no proteolysis in all three expression attempts of PLP-Fc-LABL. Given these results, an alternative method to prepare the BPI-Fc fusion was pursued.
IgG1 Fc fusions are most commonly prepared by recombinant protein expression.32, 33 However, the applicability of recombinant expression for the production of peptides and proteins is limited by proteolysis and low expression yields. To circumvent these limitations, the fusion partners can be prepared separately and conjugated in-vitro using a variety of bioconjugation techniques.34, 35 Such bioconjugation techniques can target natural conjugation sites on proteins such as the thiol of cysteine residues, the primary amine of lysine residues, and particular sugar residues of attached glycans. Additionally, non-native sites can be added to the protein such as unnatural amino acid residues, sugar residues, and peptide tags. Peptide tags are attractive, because the fusion partners can be conjugated site-specifically, and the conjugation occurs on the peptide tag instead of the native protein. A popular method for protein conjugation that requires a peptide tag substrate is sortase-mediated ligation.36 Over the past decade, sortase A from Staphylococcus aureus has served as an important conjugation tool to attach a variety of materials to proteins in a site-specific manner.36–41 In addition to site-specificity, this enzymatic conjugation method is very attractive for proteins as it is performed under mild conditions and requires only that one species contain the LPXTG recognition motif, where X is any amino acid, while the other species contains at least a single N-terminal glycine (Table 1).42 The mechanism of this reaction is as follows: sortase A recognizes the LPXTG motif, cleaving between the threonine and glycine residues using an active site cysteine. The glycine is released, and a thioester intermediate is formed between the C-terminal threonine of the sortase-tagged substrate protein and the active site cysteine of sortase A. Next, a ligand containing an N-terminal glycine acts as a nucleophile, attacking the thioester to displace sortase A and form an amide bond between the protein and ligand (Figure 2).36, 40
Figure 2.
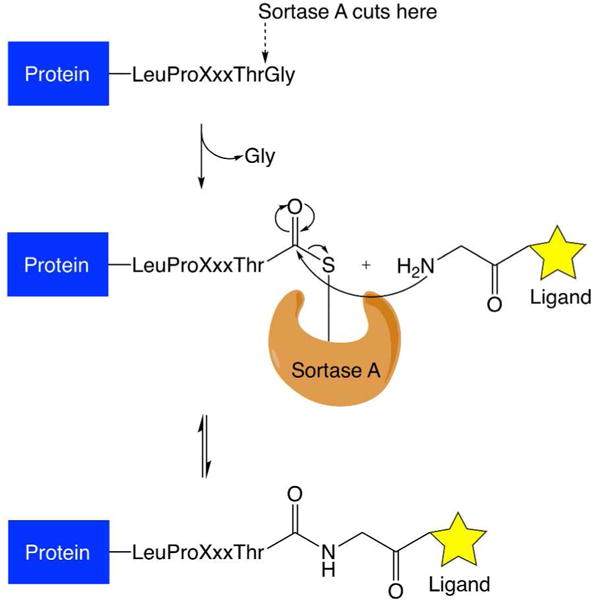
Mechanism of sortase-mediated ligations. The ligation requires a substrate to bear a LPXTG peptide tag. This peptide tag is recognized by sortase A. The enzyme cleaves between threonine and glycine residues to form an acylenzyme intermediate. The acyl-enzyme intermediate is then susceptible to nucleophilic attack by a ligand bearing an N-terminal glycine residue, leading to the formation of an amide bond between the substrate and the ligand.
A new Fc fusion protein was prepared that consisted of the LABL sequence fused to the N-terminus of the Fc, and a sortase A recognition tag (AAALPETGGG = ST) fused to the C-terminus. This construct was named LABL-Fc-ST (+H3N-GITDGEATDSGGG-Fc-AAALPETGGG). This approach avoids expression of the problematic antigenic peptides and instead allows conjugation of antigenic peptides after expression using a C-terminal sortase-mediated ligation (Figure 3). The LABL sequence was capped with an N-terminal glycine so that the positively charged N-terminus was not directly part of the LABL sequence. Also, a two glycine spacer was added in between the LABL and Fc to reduce the likelihood of the Fc sterically hindering the binding activity of the LABL peptide. Similarly, a three alanine spacer was added in between the Fc and ST recognition tag to reduce the likelihood of the Fc sterically hindering the sortase-mediated ligation. LABL-Fc-ST was expressed in yeast and showed no proteolysis (Supporting Information Figure S5). Furthermore, the 1 L spinner flask expression yield of LABL-Fc-ST was 5x higher than PLP-Fc-LABL (15 mg vs. 3 mg, respectively).
Figure 3.
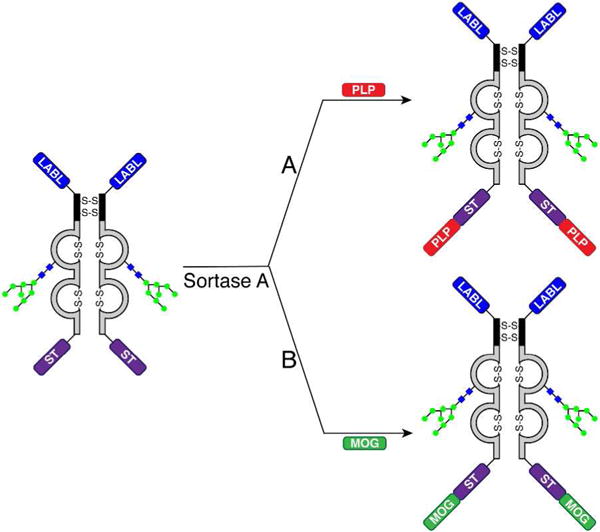
Sortase-mediated addition of two different antigenic peptides to the C-terminus of LABL-Fc-ST. An IgG1 Fc fusion containing an N-terminal LABL peptide and a C-terminal sortase A recognition tag was reacted in a sortase-dependent manner with the (A) PLP and (B) MOG peptides to produce LABL-Fc-ST-PLP and LABL-Fc-ST-MOG, respectively.
Preparation of Antigenic Peptides
The PLP peptide was purchased at ~60% purity and then RP-HPLC purified in our laboratory. The peptide was constructed with three glycine residues N-terminal to PLP since at least one N-terminal glycine residue was required for this peptide to be a nucleophile in a sortase-mediated ligation. The PLP peptide was designed with three sequential N-terminal glycine residues because a previous study comparing the number of sequential glycine residues on ligation yield showed that three N-terminal glycine residues gave a higher yield than two or one but more than three didn’t result in a much higher yield.43 An additional C-terminal glycine was also added to the PLP peptide so that the negative charge of the carboxy terminus of the polypeptide backbone was spaced away from the PLP peptide.
The MOG peptide was chemically synthesized with two N-terminal glycine residues instead of three because the native MOG38–50 sequence begins with glycine. However, the MOG peptide is very hydrophobic and doesn’t dissolve well in the aqueous buffer required for the sortase-mediated ligation. Previously, active BPIs have been produced with linkers and peptides attached to the C-terminus of MOG, and therefore we explored increasing the solubility of MOG by C-terminal modification.25 In order to increase solubility under aqueous conditions, two arginine residues were added to the C-terminus. Additionally, a one glycine spacer was added in between MOG and the two arginine residues to reduce the likelihood the positive charges of the arginine side changes interfering with binding of MOG38–50 to the MHC-II and TCR.
Sortase-mediated Ligations
On sub-milligram scales, the two antigenic peptides, PLP and MOG, were enzymatically ligated to the C-terminus of LABL-Fc-ST in a sortase-mediated manner to produce the ligation products LABL-Fc-ST-PLP and LABL-Fc-ST-MOG, respectively. These reactions were performed on small-scale to optimize the ligation yields, and because there was no downstream application, they were not purified prior to characterization. Based on the peak heights in the mass spectra, the ligation yields were both 90% (Figure 4B–C). The addition of the antigenic peptides to LABL-Fc-ST was also observed by SDS-PAGE, which showed an upward gel shift for LABL-Fc-ST-PLP and LABL-Fc-ST-MOG compared to LABL-Fc-ST (Figure 4D). The method to prepare LABL-Fc-ST-PLP using a sortase-mediated ligation was scaled up to produce enough material to conduct a mouse study to determine whether the BPI-Fc was active in mice induced with EAE. A drawback of the sortase-mediated ligation is that the product still contains the LPXTG recognition tag, so the product is also a sortase substrate. In order to drive the sortase-mediated ligation to near completion, a large excess of peptide was required (≥1 mM). In order to prepare enough LABL-Fc-ST-PLP for a mouse study, 52 mg of PLP peptide was prepared. Also, the expression of LABL-Fc-ST protein was scaled up from a 1 L spinner flask to a 5 L fermentor to produce enough protein for the mouse study. Fermentor expression of LABL-Fc-ST provided 105 mg of protein, and scaling up production of LABL-Fc-ST did not result in any proteolysis (Supporting Information Figure S5 vs. Figure 4A).
Figure 4.
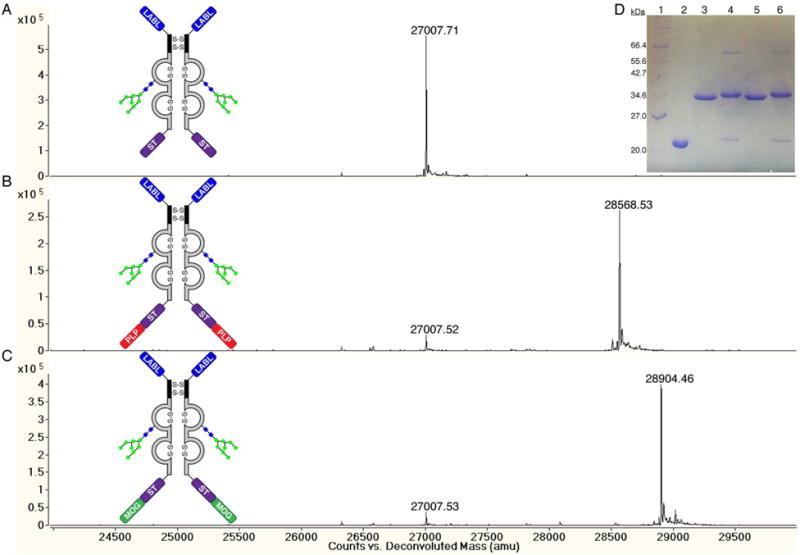
Characterization of LABL-Fc-ST and small-scale sortase-mediated ligations of LABL-Fc-ST with the PLP and MOG peptides. Prior to characterization, the proteins were deglycosylated with PNGase F and reduced to their monomeric states using dithiothreitol (DTT). (A) Mass spectrum of recombinant expression of LABL-Fc-ST. LABL-Fc-ST MW (Da): Expected = 27010.41and Observed = 27007.71. (B) Mass spectrum of small-scale sortase-mediated ligation of LABL-Fc-ST + PLP peptide. LABL-Fc-ST-PLP MW (Da): Expected = 28571.19 and Observed = 28568.53. (C) Mass spectrum of small-scale sortase-mediated ligation reactions of LABL-Fc-ST + MOG peptide. LABL-Fc-ST-MOG MW (Da): Expected = 28908.62 and Observed = 28904.46. (D) SDS-PAGE of LABL-Fc-ST and sortase-mediated ligations of LABL-Fc-ST + PLP and MOG peptides. Lanes: 1 = 2–212 kDa MW marker, 2 = SrtAΔ1–59, 3&5 = LABL-Fc-ST, 4 = sortase-mediated ligation of LABL-Fc-ST + PLP peptide, and 6 = sortase-mediated ligation of LABL-Fc-ST + MOG peptide.
In order to conserve peptide, almost a four-fold higher concentration of LABL-Fc-ST was used (10 μM for small scale vs. 37 μM for large scale). Based on the mass spectrometry peak height ratio of LABL-Fc-ST-PLP/(LABL-Fc-ST + LABL-Fc-ST-PLP), the large scale the ligation yield was 87% (Figure 5A). This ligation yield was slightly lower than what was observed for the small-scale reaction, but it was still nearly complete. The near-completeness of the ligation can also be observed by SDS-PAGE (Figure 5B) where the LABL-Fc-ST-PLP migrated slower (higher) than LABL-Fc-ST. However, SDS-PAGE also showed side products of higher molecular weight than the expected product. These products could have been caused by the N-terminal glycine that was added to LABL-Fc-ST, which may have caused the N-terminus of LABL-Fc-ST to act as a nucleophile in the sortase-mediated ligation to form LABL-Fc-ST–LABL-Fc-ST dimers. Also, another possibility is that lysine residues have been shown to act as nucleophiles in the sortase-mediated ligation because their primary amine side chains can compete with the GGG-peptide for thioester attack.44 The LABL-Fc-ST sequence contains 19 lysine residues in each chain, and SrtAΔ1–59 also contains 19 lysine residues. Additionally, the mass spectrum from the large-scale ligation showed that approximately 14% of the product had a loss of a terminal glycine. Because the terminal glycines are not part of the active PLP and MOG peptides, the loss of this glycine was not seen as a problem that would interfere with the testing of this fusion protein. After the reaction, the ligation product was purified by Protein G affinity chromatography. The TIC overlay of LABL-Fc-ST-PLP before and after purification showed that the SrtAΔ1–59 and unreacted PLP peptide were no longer present in the purified sample (Supporting Information Figure S6).
Figure 5.
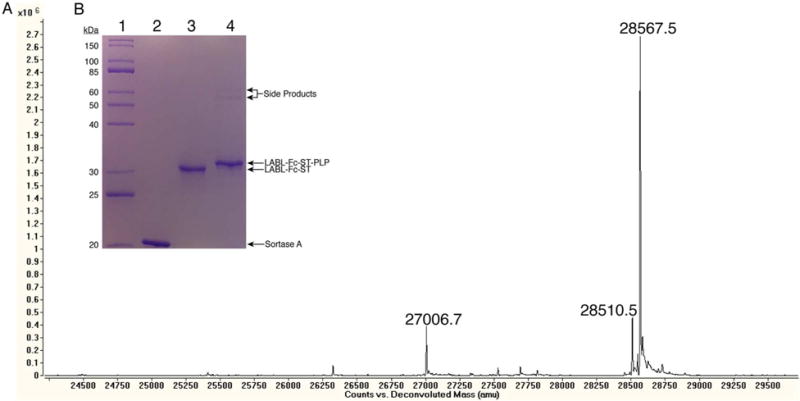
Characterization of the large-scale sortase-mediated ligation of LABL-Fc-ST + PLP peptide. Prior to characterization, the protein was deglycosylated with PNGase F and reduced to its monomeric state using DTT. (A) Mass spectrum of LABL-Fc-ST + PLP peptide ligation reaction. LABL-Fc-ST MW (Da): Expected = 27010.4 and Observed = 27006.7, LABL-Fc-ST-PLP MW (Da): Expected = 28571.2 and Observed = 28567.5, and LABL-Fc-ST-PLP (-Glycine) MW (Da): Expected = 28514.1 and Observed = 28510.5 (B) SDS-PAGE of large-scale LABL-Fc-ST + PLP peptide ligation. Lanes: 1) 10–200 broad range protein marker, 2) SrtAΔ1–59, 3) LABL-Fc-ST, and 4) LABL-Fc-ST-PLP.
Evaluation of the BPI-Fc in EAE Mice
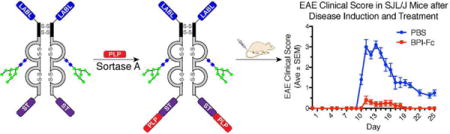
Ten 5–7 week old SJL/J mice were induced with EAE by immunizing them with an emulsion of PLP139–151 peptide in CFA as well as pertussis toxin. To test whether the LABL-Fc-ST-PLP fusion conjugate called BPI-Fc was able to suppress the effects of EAE, on days 4 and 7 post-immunization, five mice were intravenously injected with 25 nmol of the BPI-Fc and five with a PBS control. The study required approximately 15 mg of BPI-Fc. In order to inject two doses of 25 nmol/dose into five mice, the protein had to be concentrated to 168 μM (9.6 mg/mL) to allow for injection volumes of 150 μL per dose. During the course of the study, the mice were weighed and their physical symptoms associated with the disease were scored (Figure 6). The BPI-Fc treated mice showed significantly milder symptoms compared to the PBS control (Figure 6A). At the height of symptoms, the PBS control mice experienced complete paralysis of one or two hind limbs (average score = 3), whereas the BPI-Fc treated mice experienced, at most, tail weakness (average score = 0.4). Furthermore, at no point during the study did the BPI-Fc treated mice weigh less than their respective weights at day 0 (Figure 6B). In fact, the overall trend in the % change in body weight showed that the BPI-Fc treated mice were gaining weight throughout the study. On the other hand, the PBS control mice lost an average of 16% of body weight at the height of symptoms (day 12). These results show that the BPI-Fc protected the mice against the physical disablements and weight loss caused by EAE.
Figure 6.
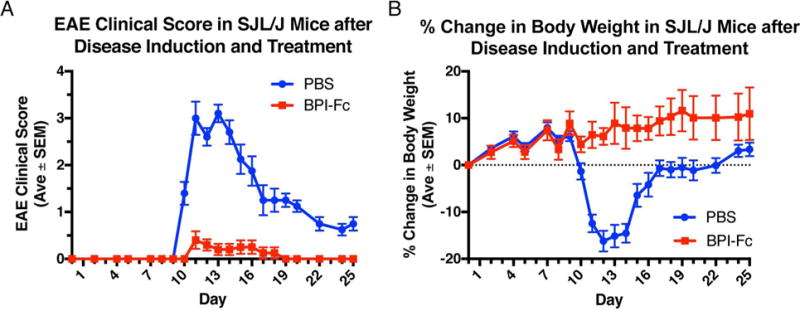
Comparison of EAE mice treated with BPI-Fc (LABL-Fc-ST-PLP) vs. PBS control (n = 5). (A) EAE clinical score in SJL/J mice after disease induction and treatment. Mice were scored during the course of the study from 0–5 based on the physical symptoms observed. (B) % change in body weight in SJL/J mice after disease induction and treatment. Each mouse was weighed during the course of the study and the weight at each of the measured days compared against the mouse’s weight at day 0.
On day 14, one mouse from both the BPI-Fc treated and PBS control groups were sacrificed along with a normal mouse that was neither induced with EAE nor treated. A brain slice from each mouse was sent to IHC World for Luxol Fast Blue (LFB) staining (Figure 7). The amount of staining is representative of the amount of myelin present in the brain slice, which is representative of the severity of the disease. A one-way ANOVA showed that there was a statistical difference between the three tissue groups (p = 0.0004). A Tukey multiple comparisons test showed no statistical difference in the amount of myelin staining between the normal control mouse that was neither induced with the EAE disease nor treated and the BPI-Fc-treated mouse (p = 0.2288). On the other hand, there was a statistical difference in the amount of myelin staining between the normal control mouse and the PBS control mouse (p = 0.0004). These results show that the BPI-Fc protected the mice against the demyelination caused by EAE.
Figure 7.
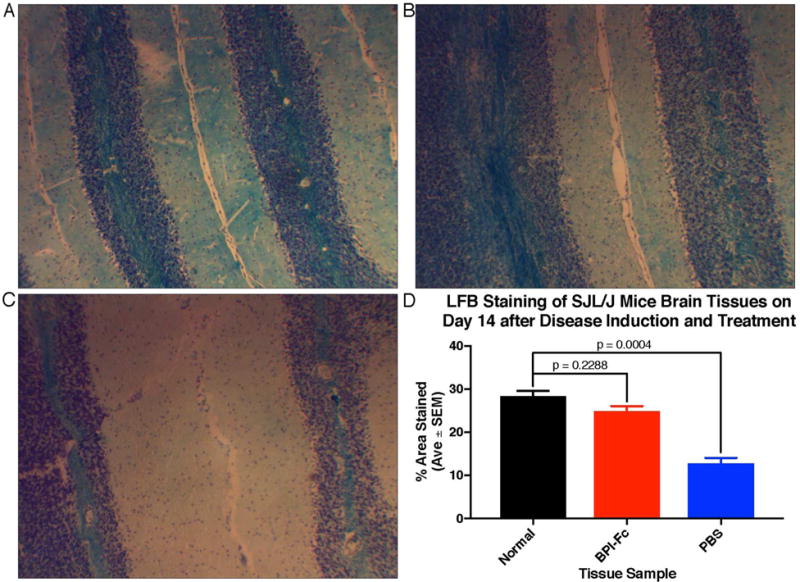
Luxol fast blue staining of mice brain slices on day 14 after EAE induction. (A) Normal: mouse neither induced with EAE nor treated. (B) BPI-Fc: mouse induced with EAE and treated with BPI-Fc. (C) PBS: mouse induced with EAE and treated with PBS. (D) Quantification of the area stained by LFB in each tissue. The % Area Stained by LFB for each tissue was measured in triplicate, and the results between the three tissue samples were compared for statistical significance using a one-way ANOVA and Tukey multiple comparisons test.
CONCLUSIONS
MS is an autoimmune disease that causes demyelination of the neurons in the central nervous system. There is currently no cure for MS, and current therapies globally suppress the immune system. BPIs are a promising novel approach to treating autoimmune diseases like MS and can potentially be designed to specifically prevent the activation of immune cells against a particular antigen. They are composed of an antigenic peptide fused to a signal-2 blocking peptide. In this work, we set out to test if the BPI peptides could be prepared as Fc fusions and if the BPI peptides could still work when fused to an IgG1 Fc scaffold. The initial strategy was to fuse both peptides (antigenic peptide PLP139–151 and signal-2 blocking peptide LABL) to the Fc using recombinant DNA technology. However, this strategy didn’t work because expression in yeast resulted in proteolysis of the PLP peptide. Three different expression conditions were tested, but severe proteolysis of the PLP peptide was observed in all three expressions. Furthermore, a different antigenic peptide (MOG38–50) was tested instead of PLP. However, proteolysis of the MOG region was even more severe than PLP.
Alternatively, a sortase-ligation strategy was developed to conjugate the antigenic peptides to a human IgG1 Fc fused to LABL. When IgG1 Fc fused to LABL and a sortase tag was expressed in glycosylation-deficient yeast, there was no proteolysis and good yield of the protein. The sortase ligation worked well to attach both the PLP and MOG antigenic peptides to the LABL-Fc-ST fusion protein in high yields (90% for both ligations). The advantage of this approach is that it avoids the proteolysis and expression problems that seem to be associated with the antigenic peptides that we have experienced. It also allows a single fusion protein (LABL-Fc-ST) to be expressed in quantity and then ligated to multiple antigenic peptides, which may be helpful to combat antigen spreading.45 Additionally, this method may be used to screen peptides for improved BPI-Fcs. The ligation strategy to produce LABL-Fc-ST-PLP was scaled up to produce enough material (19 mg) to conduct a mouse study. Scaling up reduced the ligation yield some, but not much (90% small scale vs. 87% large scale). The mouse study was conducted in EAE-induced mice, and it was found that the BPI-Fc worked very well and significantly suppressed EAE in a mouse model. BPI-Fc fusions are a promising line of research and will be explored further in the future.
EXPERIMENTAL PROCEDURES
Materials
DNA primers were ordered from Eurofins (Huntsville, AL). IgG1 cDNA (MGC: 12853) was previously purchased from the American Tissue Culture Collection (Manassas, VA).46 pGAPzαA and pPICzαA vectors were purchased from Invitrogen (USA). All restriction enzymes, T4 Polynucleotide Kinase (PNK), T4 DNA Ligase, and protein markers were purchased from New England Biolabs (Ipswich, MA). All final DNA constructs were confirmed by DNA sequencing. Zeocin was purchased from InvivoGen (San Diego, CA). Tryptone and yeast extract were both purchased from Becton, Dickinson and Company (Sparks, MD). Yeast nitrogen base was purchased from Sunrise Science Products (San Diego, CA). Fmoc-PAL-PEG-PS resin and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) were obtained from Applied Biosystems (Foster City, CA) and Chem-Impex International, Inc. (Wood Dale, IL), respectively. Protected amino acids were purchased from Peptides International (Louisville, KY), Bachem (Torrance, CA), and Sigma-Aldrich (St. Louis, MO). Triisopropylsilane (TIPS) and N,N-diisopropylethylamine (DIEA) were also purchased from Sigma-Aldrich. Detoxi-Gel Endotoxin Removing Gel was purchased from Thermo Scientific (Waltham, MA). All other chemicals were obtained from commercial sources without further purification. Protein G resin was prepared in house as previously described.47, 48 All materials and reagents, except for the protein markers, used for SDS-PAGE were purchased from Bio-Rad Laboratories (Hercules, CA), and the experimental procedure used for SDS-PAGE is described in the Supporting Information. All protein mass spectra, except those pertaining to PLP-Fc-LABL (spinner flask with media change), MOG-Fc, and LABL-Fc-ST (spinner flask) proteins were acquired using an Agilent 6520 electrospray ionization time-of-flight liquid chromatography mass spectrometer (LC-MS) using a method previously described.48 Mass spectra for PLP-Fc-LABL (spinner flask with media change), MOG-Fc, and LABL-Fc-ST (spinner flask) proteins were acquired using a Waters Synapt G2 quadrupole time-of-flight LC-MS using a method previously described.49 Peptide-N-Glycosidase F (PNGase F) was prepared in house using a method previously described.50
Cloning of pPICzαA-LABL-Fc-ST
A construct consisting of a human IgG1 Fc cDNA (T225➔K447) subcloned into pGAPzαA with a silent mutation to introduce a SacI restriction site near the 5′ end of the Fc coding region (termed pGAPzαA-Fc) was produced previously in our laboratory and used as a starting point for producing the peptide-Fc fusion proteins described herein. The sortase A recognition tag (ST), a peptide, was attached C-terminal to the Fc polypeptide sequence. This was accomplished by inserting DNA between the NotI site, which occurs after the last human IgG1 Fc encoded amino acid, and the XbaI site. Primers containing the ST DNA sequence (5′-ggccgccttgccagaaactggaggtggataat-3′ and 5′-ctagattatccacctccagtttctggcaaggc-3′) were designed so that when they anneal, they create the proper NotI and XbaI sticky ends. The 5′-termini of the primers were phosphorylated by PNK. pGAPzαA-Fc was digested with NotI-HF and XbaI, gel purified, and the ST DNA sequence was ligated to the 3′ side of the Fc DNA using T4 DNA Ligase to create pGAPzαA-Fc-ST. The LABL DNA sequence was cloned on the 5′ side of the Fc DNA in pGAPzαA-Fc-ST. Primers containing the LABL DNA sequence (5′-tcgagaaaagaggaattactgatggagaagctactgattctggaggtggaacatgcccaccgtgcccagcacctgagct-3′ and 5′-caggtgctgggcacggtgggcatgttccacctccagaatcagtagcttctccatcagtaattcctcttttc-3′) were designed so that when they anneal, they create the proper NotI and XbaI sticky ends. The 5′-termini of the primers were phosphorylated by PNK. pGAPzαA-Fc-ST was digested with XhoI and SacI, gel purified, and the digested LABL DNA sequence was ligated into pGAPzαA-Fc-ST on the 5′ side of the Fc DNA using T4 DNA Ligase to create pGAPzαA-LABL-Fc-ST. To allow for inducible expression under the strong AOX promoter, the LABL-Fc-ST was cut out of pGAPzαA and ligated into pPICzαA. To perform this transfer, both pGAPzαA-LABL-Fc-ST DNA and pPICzαA were digested with XhoI and XbaI and gel purified. LABL-Fc-ST and pPICzαA digested products were ligated together using T4 DNA Ligase to create pPICzαA-LABL-Fc-ST. The plasmid was linearized using PmeI and transformed into a strain of P. pastoris with the OCH1 gene deleted as previously described.46
Fermentor Expression and Purification of LABL-Fc-ST
A colony of P. pastoris OCH1 KO transformed with pPICzαA-LABL-Fc-ST was inoculated in a culture tube containing 2 mL of YPD media and 100 μg/mL Zeocin. The culture was grown in a shaker/incubator at 25°C. After 3 days, the 2 mL culture was transferred to a baffled shake flask containing 250 mL of YPD media, 100 μg/mL Zeocin, and 1 drop of antifoam. The culture was grown in a shaker/incubator at 25°C. After 3 days, the 250 mL culture was transferred to a New Brunswick BioFlo 415 fermentor (Hauppauge, NY) containing 5 L of sterilized Buffered Glycerol Complex Medium and PTM1 salts at pH 6.0. The pH was maintained by the fermentor-controlled addition of concentrated NH4OH, which also served as the nitrogen source. Agitation and gas flow were set under cascade control, which maintained an agitation range of 200–1000 rpm and a gas flow range of 1–20 L/min. The cells were grown on a carbon source feed of 50% glycerol in water for two days. After which, the carbon source was changed to 100% methanol to allow for induction of target protein. After 3 days of induction, the culture was harvested by centrifugation at 6,693 × g for 20 min using a Beckman JLA 10.5 rotor in a Beckman Avanti J-series centrifuge (USA). The supernatant was collected and filtered using a Buon Vino Mini Jet Wine Filter with 0.5 μm filter pads (Cambridge, Ontario). The filtered supernatant was then concentrated to 1 L using a Sartorius 30,000 MWCO tangential flow concentrator (USA). The concentrated supernatant was purified using Protein G affinity chromatography. First, 10 mL of Protein G resin was packed in a 1.5×20 cm diameter Bio-Rad Econo-column (Hercules, CA) and equilibrated with 250 mL of 20 mM Sodium Phosphate buffer pH 6.0. Then the concentrated supernatant was loaded onto the column and washed with 500 mL of 20 mM Sodium Phosphate buffer and 0.5 M NaCl at pH 6.0. The captured protein was eluted using 0.1 M Glycine-Cl buffer, pH 2.7. The column was re-equilibrated with 250 mL Sodium Phosphate buffer pH 6.0. This process was repeated until no more protein eluted from the column. The collected protein was dialyzed against 4 L of TBS, pH 7.5 using Spectra/Por 3500 MWCO dialysis tubing (USA) at 4°C with two buffer changes. 105 mg of purified protein was obtained as determined by measuring the optical density of the solution at 280 nm (MW = 54006.8 Da, ε280 = 71570 M−1cm−1).51
Preparation of the PLP peptide (GGGHSLGKWLGHPDKFG)
The PLP peptide used in this work was purchased as 60% pure from Shanghai Mocell Biotech Co. (Shanghai, China) and RP-HPLC purified. The peptide was purified on a Thermo Separation Products HPLC (preparative run) using a C18 Vydac column 218TP1022 with a linear gradient of water/acetonitrile (6 mL/min, detection 220 nm, eluent A is 5% acetonitrile and 0.1% TFA in water and eluent B is acetonitrile, 14–90% B in 40 min). The fractions were analyzed by HPLC on a 5 μm C18 Econosphere column (4.6 × 250 mm) and lyophilized. The purity was determined by HPLC analysis using a Shimadzu UFLC and UV detector at 220 nm. The mass was determined using a Matrix-assisted laser desorption/ionization (MALDI) mass spectrometer (Supporting Information Figure S7). [M+H]+ mass (Da): Expected = 1749.9 and Observed = 1750.0.
Solid-Phase Peptide Synthesis of the MOG peptide (GGGWYRSPFSRVVHLGRR)
The MOG peptide was synthesized manually using Fmoc-PAL-PEG-PS resin (0.18 meq/g) and employing standard Fmoc solid-phase peptide synthesis strategy. Couplings were performed using 4-fold molar excess of each Fmoc L-amino acid, 4-fold molar excess of HATU and 8-fold molar excess of DIEA. The peptide was then removed from the resin by treatment with TFA/TIPS/H2O (95/2.5/2.5 by volume) for 2 hours under nitrogen at room temperature. The resin beads were filtered; the filtrate was precipitated in cold diethyl ether, centrifuged, washed twice by diethyl ether, and dissolved in water. The crude peptide was purified by RP-HPLC using the same conditions that were used for purification of the PLP peptide. The mass was determined by MALDI mass spectrometry (Supporting Information Figure S8). [M+H]+ mass (Da): Expected = 2087.1 and Observed = 2087.2.
Expression and Purification of Sortase A
A construct of sortase A (SrtAΔ1–59) cloned with a C-terminal His6 tag into pET23 (a gift from Richard DiMarchi, Indiana University) was transformed into Escherichia coli Rosetta 2 strain. SrtAΔ1–59 was expressed and purified according to a similar procedure previously described (Supporting Information Figure S9).39 242 mg/L of purified enzyme was obtained as determined by measuring the optical density of the solution at 280 nm (MW = 17719.0 Da, ε280 = 14440 M−1cm−1).51
Small-scale Sortase-mediated Ligations of LABL-Fc-ST + PLP and MOG peptides
The PLP and MOG peptides were separately ligated to the C-terminus of LABL-Fc-ST using sortase-mediated ligations (Figure 3) on a sub-milligram scale. In each ligation, 10 μM LABL-Fc-ST protein was combined with 6 mM CaCl2, 1 mM GGG-peptide (either PLP or MOG), and 5 μM sortase in TBS, pH 7.5 in a 150 μL reaction.42 The ligations were incubated at 35°C for 24 hours. After which, they were quenched by the addition of 25 mM EDTA. The ligations were then observed by mass spectrometry and SDS-PAGE, which showed a 90% ligation yield for both peptides.
Large-scale Sortase-mediated Ligation of LABL-Fc-ST + PLP peptide
On a 60 mg protein scale (the total of 6 pooled reactions), the PLP peptide was ligated to the C-terminus of LABL-Fc-ST protein using a sortase-mediated ligation. In total, the ligation contained: 37 μM LABL-Fc-ST protein (60 mg), 6 mM CaCl2, 1 mM PLP peptide (52 mg), and 7 μM SrtAΔ1–59 (3.7 mg). The pH was adjusted to pH 7.5 using 1 M Tris base. The ligation was allowed to proceed 24 hours at 35°C and then stored at −20°C until purification.
Purification of Large-scale LABL-Fc-ST-PLP
The reaction mixture was adjusted to pH 7.0 using 1 N HCl. The Fc-containing protein was separated from SrtAΔ1–59 and unreacted PLP peptide using protein G affinity chromatography. The column was first equilibrated using TBS, pH 7.0. Then the reaction mixture was loaded onto the Protein G column, and the captured Fc protein was washed with 500 mL of 50 mM Tris and 500 mM NaCl, pH 7.0. Then the Fc-containing protein (unreacted LABL-Fc-ST and product LABL-Fc-ST-PLP) was eluted using 0.1 M glycine-HCl buffer pH 2.7 and immediately dialyzed against TBS, pH 7.5. The protein was then passed through a Detoxi-Gel endotoxin removal column (Thermo Scientific) to remove endotoxin contaminants that may have been introduced during the expression and/or purification steps.52 The flow through from the endotoxin removal column was concentrated using an EMD Millipore 10 kDa MWCO centrifugal concentrator (USA). Concentration was stopped when the volume of the protein solution was 2 mL because precipitation was observed. The concentrated protein was dialyzed against 4 L of 1× PBS pH 7.4 with one buffer change at 4°C overnight and then filtered through an EMD Millipore 0.2 μm syringe filter. The concentration of the end product was determined to be 168 μM (9.6 mg/mL, 19 mg total) by optical density of the solution at 280 nm (MW = 57128.3 Da, ε280 = 82570 M−1cm−1).51
Evaluation of BPI-Fc (LABL-Fc-ST-PLP) in EAE mice
The LABL-Fc-ST-PLP fusion protein was tested in mice induced with EAE using a similar protocol that was used by Manikwar et al. to test the I-domain protein, a 197 amino acid region of the αL subunit of LFA-1, conjugated to PLP139–151 peptide.53,54 Mice were housed and maintained in the University of Kansas Animal Care Unit and the protocol for this animal work had been approved by the Institutional Animal Care and Use Committee (IACUC). Ten 5–7 week old Female SJL/J mice (Charles River Labs) were immunized with PLP139–151 peptide in CFA on day 0. The PLP/CFA emulsion was injected above the shoulders and flanks of the mice (4 doses at 50 μL/dose) by subcutaneous injection. Additionally, on both days 0 and 2, the mice were administered 200 ng of pertussis toxin by intraperitoneal injection. On both days 4 and 7, five mice were administered 150 μL of 168 μM LABL-Fc-ST-PLP protein (2 doses at 25 nmol/dose) and five mice were administered 150 μL of PBS by intravenous injection. During the 25-day study, the mice were scored based on the severity of the physical symptoms associated with EAE. Scores ranged from 0 to 5 as follows: 0 = no symptoms, 1 = tail weakness, 2 = incomplete paralysis of one or two hind limbs, 3 = complete paralysis of one or two hind limbs, 4 = complete paralysis of one or two hind limbs plus paralysis one or two fore limbs, and 5 = moribund or dead. Additionally, the mice were weighed throughout the study to observe their change in weight relative to their weight on day 0. On day 14, one mouse from each group was sacrificed and 5 μm slices of each mouse’s brain tissue were prepared as previously described.24 The brain sections were sent to IHC World (Woodstock, MD) for staining with LFB and cresyl violet as previously described.55 The stained tissues were imaged by IHC World using a EPI Fluorescence Trinocular Microscope (Model# FM320-9M). IHC World also quantified the amount of LFB staining in each brain tissue using ImageJ software. Statistical significance between the stained tissue samples was determined using a one-way ANOVA (α=0.05) followed by a Tukey multiple comparisons test.
Supplementary Material
Acknowledgments
We thank the National Institute of Health for funding this work (NIH R01 GM090080 and R56AI063002). We also thank Ishan Shah at the University of Kansas for performing the mass spectrometry operation for the PLP-Fc-LABL (spinner flask and fermentor expressed), LABL-Fc-ST (fermentor expressed), LABL-Fc-ST-PLP (small and large scale), and LABL-Fc-ST-MOG (small scale) proteins, and the Analytical Proteomics Laboratory at the University of Kansas for performing the mass spectrometry operation for the PLP-Fc-LABL (spinner flask with media change), MOG-Fc, LABL-Fc-ST (spinner flask) proteins and PLP and MOG peptides. Furthermore, we thank Allison Kukuch for cloning the pGAP-Fc construct and introducing a silent mutation near the 5′ end of the Fc DNA, the DiMarchi Laboratory at Indiana University for providing us with the pET23-SrtAΔ1–59 expression plasmid, and the Lott Laboratory at Massey University for providing us with an E. coli strain that overexpresses PNGase F.
ABBREVIATIONS
- MS
Multiple Sclerosis
- BBB
Blood-Brain Barrier
- PLP
Proteolipid Protein
- MBP
Myelin Basic Protein
- MOG
Myelin Oligodendrocyte Glycoprotein
- BPI
Bifunctional Peptide Inhibitor
- EAE
Experimental Autoimmune Encephalomyelitis
- MHC-II
Major Histocompatibility Complex class II
- ICAM-1
Intracellular Adhesion Molecule-1
- LFA-1
Leukocyte Function-associated Antigen-1 (CD11a)
- IS
Immunological Synapse
- Fc
Fragment crystallizable
- ST
Sortase Substrate Tag
- DTT
Dithiothreitol
- SDS-PAGE
Sodium Dodecyl Sulfate – Polyacrylamide Gel Electrophoresis
- LFB
Luxol Fast Blue
- PNGase F
Peptide:N-Glycosidase F
- SrtAΔ1–59
Sortase AΔ1–59
- TBS
Tris Buffered Saline
- PBS
Phosphate Buffered Saline
- MALDI
Matrix-assisted laser desorption/ionization
Footnotes
SUPPORTING INFORMATION
Cloning procedures for pPICzαA-PLP-Fc-LABL and pPICzαA-MOG-Fc. Expression procedures for PLP-Fc-LABL (spinner flask, fermentor, and spinner flask with media change), MOG-Fc, and LABL-Fc-ST (spinner flask). Characterization of PLP-Fc-LABL (spinner flask, fermentor, and spinner flask with media change), MOG-Fc, LABL-Fc-ST (spinner flask), LABL-Fc-ST-PLP purification, PLP peptide, MOG peptide, and Sortase A.
References
- 1.Compston A, Coles A. Multiple sclerosis. The Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Browne P, Chandraratna D, Angood C, Tremlett H, Baker C, Taylor BV, Thompson AJ. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014;83:1022–1024. doi: 10.1212/WNL.0000000000000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sosnoff JJ, Socie MJ, Boes MK, Sandroff BM, Pula JH, Suh Y, Weikert M, Balantrapu S, Morrison S, Motl RW. Mobility, balance and falls in persons with multiple sclerosis. PLoS One. 2011;6:e28021. doi: 10.1371/journal.pone.0028021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiaravalloti ND, DeLuca J. Cognitive impairment in multiple sclerosis. The Lancet Neurology. 2008;7:1139–1151. doi: 10.1016/S1474-4422(08)70259-X. [DOI] [PubMed] [Google Scholar]
- 5.Bakshi R, Shaikh Z, Miletich R, Czarnecki D, Dmochowski J, Henschel K, Janardhan V, Dubey N, Kinkel P. Fatigue in multiple sclerosis and its relationship to depression and neurologic disability. Multiple Sclerosis. 2000;6:181–185. doi: 10.1177/135245850000600308. [DOI] [PubMed] [Google Scholar]
- 6.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassman H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Annals of neurology. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 7.Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. The Lancet Neurology. 2014;13:700–709. doi: 10.1016/S1474-4422(14)70041-9. [DOI] [PubMed] [Google Scholar]
- 8.Ascherio A, Munger KL. Seminars in neurology. Thieme Medical Publishers; 2016. pp. 103–114. [Google Scholar]
- 9.Ramagopalan SV, Dobson R, Meier UC, Giovannoni G. Multiple sclerosis: risk factors, prodromes, and potential causal pathways. The Lancet Neurology. 2010;9:727–739. doi: 10.1016/S1474-4422(10)70094-6. [DOI] [PubMed] [Google Scholar]
- 10.Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmunity reviews. 2010;9:A387–A394. doi: 10.1016/j.autrev.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 11.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nature immunology. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 12.Disanto G, Morahan J, Barnett M, Giovannoni G, Ramagopalan S. The evidence for a role of B cells in multiple sclerosis. Neurology. 2012;78:823–832. doi: 10.1212/WNL.0b013e318249f6f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krumbholz M, Meinl E. B cells in MS and NMO: pathogenesis and therapy. Semin Immunopathol. 2014;36:339–350. doi: 10.1007/s00281-014-0424-x. [DOI] [PubMed] [Google Scholar]
- 14.Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Multiple sclerosis. 2003;9:540–549. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- 15.Goverman J. Autoimmune T cell responses in the central nervous system. Nature Reviews Immunology. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciccarelli O, Barkhof F, Bodini B, De Stefano N, Golay X, Nicolay K, Pelletier D, Pouwels PJ, Smith SA, Wheeler-Kingshott CA. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. The Lancet Neurology. 2014;13:807–822. doi: 10.1016/S1474-4422(14)70101-2. [DOI] [PubMed] [Google Scholar]
- 17.Goldenberg MM. Multiple sclerosis review. Pharmacy and Therapeutics. 2012;37:175–184. [PMC free article] [PubMed] [Google Scholar]
- 18.Wingerchuk DM, Carter JL. Mayo Clinic Proceedings. Elsevier; 2014. pp. 225–240. [DOI] [PubMed] [Google Scholar]
- 19.Northrup L, Christopher MA, Sullivan BP, Berkland C. Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Advanced drug delivery reviews. 2016;98:86–98. doi: 10.1016/j.addr.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Manikwar P, Kiptoo P, Badawi AH, Büyüktimkin B, Siahaan TJ. Antigen‐specific blocking of CD4‐specific immunological synapse formation using BPI and current therapies for autoimmune diseases. Medicinal Research Reviews. 2012;32:727–764. doi: 10.1002/med.20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen: specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. Journal of Pharmacology and Experimental Therapeutics. 2007;322:879–886. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- 22.Ridwan R, Kiptoo P, Kobayashi N, Weir S, Hughes M, Williams T, Soegianto R, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor: structure optimization and pharmacokinetics. Journal of Pharmacology and Experimental Therapeutics. 2010;332:1136–1145. doi: 10.1124/jpet.109.161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badawi AH, Kiptoo P, Wang WT, Choi IY, Lee P, Vines CM, Siahaan TJ. Suppression of EAE and prevention of blood–brain barrier breakdown after vaccination with novel bifunctional peptide inhibitor. Neuropharmacology. 2012;62:1874–1881. doi: 10.1016/j.neuropharm.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiptoo P, Büyüktimkin B, Badawi A, Stewart J, Ridwan R, Siahaan T. Controlling immune response and demyelination using highly potent bifunctional peptide inhibitors in the suppression of experimental autoimmune encephalomyelitis. Clinical & Experimental Immunology. 2013;172:23–36. doi: 10.1111/cei.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badawi AH, Siahaan TJ. Suppression of MOG-and PLP-induced experimental autoimmune encephalomyelitis using a novel multivalent bifunctional peptide inhibitor. Journal of neuroimmunology. 2013;263:20–27. doi: 10.1016/j.jneuroim.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Badawi AH, Kiptoo P, Siahaan TJ. Immune Tolerance Induction against Experimental Autoimmune Encephalomyelitis (EAE) Using A New PLP-B7AP Conjugate that Simultaneously Targets B7/CD28 Costimulatory Signal and TCR/MHC-II Signal. Journal of multiple sclerosis. 2015;2 doi: 10.4172/2376-0389.1000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annual review of immunology. 2009;27:591. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 29.Wu B, Sun YN. Pharmacokinetics of Peptide–Fc Fusion Proteins. Journal of Pharmaceutical Sciences. 2014;103:53–64. doi: 10.1002/jps.23783. [DOI] [PubMed] [Google Scholar]
- 30.Beck A, Reichert JM. MAbs. Taylor & Francis; 2011. pp. 415–416. [Google Scholar]
- 31.Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc‐fusion proteins: new developments and future perspectives. EMBO Molecular Medicine. 2012;4:1015–1028. doi: 10.1002/emmm.201201379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29:215–239. doi: 10.1007/s40259-015-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar M, Hunag Y, Glinka Y, Prud’Homme G, Wang Q. Gene therapy of diabetes using a novel GLP-1/IgG1-Fc fusion construct normalizes glucose levels in db/db mice. Gene therapy. 2006;14:162–172. doi: 10.1038/sj.gt.3302836. [DOI] [PubMed] [Google Scholar]
- 34.Patterson DM, Nazarova LA, Prescher JA. Finding the right (bioorthogonal) chemistry. ACS chemical biology. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal P, Bertozzi CR. Site-specific antibody–drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate chemistry. 2015;26:176–192. doi: 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukiji S, Nagamune T. Sortase‐Mediated Ligation: A Gift from Gram‐ Positive Bacteria to Protein Engineering. ChemBioChem. 2009;10:787–798. doi: 10.1002/cbic.200800724. [DOI] [PubMed] [Google Scholar]
- 37.Clow F, Fraser JD, Proft T. Immobilization of proteins to biacore sensor chips using Staphylococcus aureus sortase A. Biotechnology letters. 2008;30:1603–1607. doi: 10.1007/s10529-008-9718-1. [DOI] [PubMed] [Google Scholar]
- 38.Levary DA, Parthasarathy R, Boder ET, Ackerman ME. Protein: Protein Fusion Catalyzed by Sortase A. PloS one. 2011;6:e18342. doi: 10.1371/journal.pone.0018342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao H, Hart SA, Schink A, Pollok BA. Sortase-mediated protein ligation: a new method for protein engineering. Journal of the American Chemical Society. 2004;126:2670–2671. doi: 10.1021/ja039915e. [DOI] [PubMed] [Google Scholar]
- 40.Popp MWL, Antos JM, Ploegh HL. Site-Specific Protein Labeling via Sortase-Mediated Transpeptidation. Current Protocols in Protein Science. 2009;15:15.3.1–15.3.9. doi: 10.1002/0471140864.ps1503s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swee LK, Guimaraes CP, Sehrawat S, Spooner E, Barrasa MI, Ploegh HL. Sortase-mediated modification of αDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proceedings of the National Academy of Sciences. 2013;110:1428–1433. doi: 10.1073/pnas.1214994110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE, Ploegh HL. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nature protocols. 2013;8:1787–1799. doi: 10.1038/nprot.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ling JJ, Policarpo RL, Rabideau AE, Liao X, Pentelute BL. Protein thioester synthesis enabled by sortase. Journal of the American Chemical Society. 2012;134:10749–10752. doi: 10.1021/ja302354v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Möhlmann S, Mahlert C, Greven S, Scholz P, Harrenga A. In vitro sortagging of an antibody Fab fragment: overcoming unproductive reactions of sortase with water and lysine side chains. Chembiochem. 2011;12:1774–1780. doi: 10.1002/cbic.201100002. [DOI] [PubMed] [Google Scholar]
- 45.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nature medicine. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 46.Xiao J, Chen R, Pawlicki MA, Tolbert TJ. Targeting a homogeneously glycosylated antibody Fc to bind cancer cells using a synthetic receptor ligand. Journal of the American Chemical Society. 2009;131:13616–13618. doi: 10.1021/ja9045179. [DOI] [PubMed] [Google Scholar]
- 47.Xiao J, Tolbert TJ. Synthesis of polymerizable protein monomers for protein-acrylamide hydrogel formation. Biomacromolecules. 2009;10:1939–1946. doi: 10.1021/bm900339q. [DOI] [PubMed] [Google Scholar]
- 48.Okbazghi SZ, More AS, White DR, Duan S, Shah IS, Joshi SB, Middaugh CR, Volkin DB, Tolbert TJ. Production, characterization, and biological evaluation of well-defined IgG1 Fc glycoforms as a model system for biosimilarity analysis. Journal of pharmaceutical sciences. 2016;105:559–574. doi: 10.1016/j.xphs.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alsenaidy MA, Okbazghi SZ, Kim JH, Joshi SB, Middaugh CR, Tolbert TJ, Volkin DB. Physical Stability Comparisons of IgG1‐Fc Variants: Effects of N‐Glycosylation Site Occupancy and Asp/Gln Residues at Site Asn 297. Journal of pharmaceutical sciences. 2014;103:1613–1627. doi: 10.1002/jps.23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loo T, Patchett ML, Norris GE, Lott JS. Using secretion to solve a solubility problem: high-yield expression in Escherichia coli and purification of the bacterial glycoamidase PNGase F. Protein expression and purification. 2002;24:90–98. doi: 10.1006/prep.2001.1555. [DOI] [PubMed] [Google Scholar]
- 51.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein science. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kluger MJ, Singer R, Eiger SM. Polymyxin B use does not ensure endotoxin-free solution. Journal of immunological methods. 1985;83:201–207. doi: 10.1016/0022-1759(85)90073-0. [DOI] [PubMed] [Google Scholar]
- 53.Manikwar P, Büyüktimkin B, Kiptoo P, Badawi AH, Galeva NA, Williams TD, Siahaan TJ. I-domain-antigen conjugate (IDAC) for delivering antigenic peptides to APC: synthesis, characterization, and in vivo EAE suppression. Bioconjugate chemistry. 2012;23:509–517. doi: 10.1021/bc200580j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, McCormack A, Zhang R, Joachimiak A, Takagi J. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klüver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. Journal of Neuropathology & Experimental Neurology. 1953;12:400–403. doi: 10.1097/00005072-195312040-00008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.