

Abstract
During morphogenesis and cancer metastasis, grouped cells migrate through tissues of dissimilar stiffness. Although the influence of matrix stiffness on cellular mechanosensitivity and motility are well-recognized, it remains unknown whether these matrix-dependent cellular features persist after cells move to a new microenvironment. Here, we interrogate whether priming of epithelial cells by a given matrix stiffness influences their future collective migration on a different matrix – a property we refer to as the ‘mechanical memory’ of migratory cells. To prime cells on a defined matrix and track their collective migration onto an adjoining secondary matrix of dissimilar stiffness, we develop a modular polyacrylamide substrate through step-by-step polymerization of different PA compositions. We report that epithelial cells primed on a stiff matrix migrate faster, display higher actomyosin expression, form larger focal adhesions, and retain nuclear YAP even after arriving onto a soft secondary matrix, as compared to their control behavior on a homogeneously soft matrix. Priming on a soft ECM causes a reverse effect. The depletion of YAP dramatically reduces this memory-dependent migration. Our results present a previously unidentified regulation of mechanosensitive collective cell migration by past matrix stiffness, in which mechanical memory depends on YAP activity.
Keywords: collective cell migration, mechanotransduction, extracellular matrix, stiffness, mechanical memory
Graphical Abstract
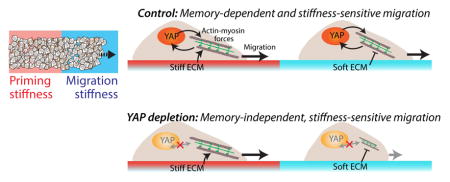
Introduction
Mechanical properties of the extracellular matrix (ECM) influence phenotypic and genotypic cellular responses, which can impact cell differentiation, migration, and proliferation [1–4]. In particular, matrix stiffness regulates cellular forces, adhesions, protrusions, and polarization through mechanotransductive signaling, all of which lead to mechanosensitive variations in migration phenotypes of both single and grouped cells [3, 5–7]. Physiologically, migratory cells do not continually interact with just one type of matrix. Instead, grouped cells migrate through mechanically heterogeneous matrices, forming the basis of fundamental biological processes including embryonic development, wound healing, regeneration, and cancer metastasis [8]. During embryonic development, a coordinated collective movement of epithelial cells across diverse microenvironments enables branching morphogenesis, a process necessary for organ formation [9, 10]. In cancer metastasis, the mechanical properties of the primary tumor microenvironment are known to induce de-clustering [2, 11, 12] and outward migration [5, 13] of cancer cells into the mechanically dissimilar surrounding tissue, which represent the first steps of tumor invasion.
As cells pass through a tissue microenvironment, a distinct set of mechanosensitive signaling events occur [1], such as clustering of integrin-based adhesion proteins into focal adhesions [14], Rho-ROCK activation [15, 16], and nuclear localization of transcriptional regulators YAP and SNAIL1 [17, 18]. Recently, mechanical dosing of human mesenchymal stem cells on matrices of tunable stiffness has been found to regulate mechanical memory-dependent lineage commitment decisions, and this process is shown to depend on YAP activity [19]. To study cell migration on heterogeneous matrices, gel systems with gradient stiffness have been used to show durotaxis for single [20] and collective cells [7], and define specific roles of myosin isoforms [21] in cell polarization during spontaneous migration across these substrates. However, it remains unknown whether the cells that are primed on a given ECM for a defined duration retain their mechanosensitive signatures even after moving to a new microenvironment. We refer to this persistent influence of cellular mechanosensitivity on cell migration as the ‘mechanical memory of migratory cells’. In this study, we asked – do collectively migrating cells remember their past matrix stiffness as they move across mechanically dissimilar microenvironments?
To test this question, we developed a modular polyacrylamide (mPA) substrate comprising contiguous primary and secondary ECM regions of independently defined stiffness. We found that a monolayer of cells primed on a stiff ECM migrated faster and in a more coordinated manner after arriving on a soft secondary ECM, as compared to those cultured on a homogeneously soft ECM. Nuclear translocation of YAP persisted even after cells arrived onto a softer secondary ECM and shRNA-mediated depletion of YAP dramatically blunted this mechanical memory-dependent cell migration. Taken together, our results bring an additional dimension to the existing framework of mechanosensitive migration of epithelial cells in response to their current microenvironment. Mechanical memory in migratory cells may have a particular significance to cancer metastasis, where future invasion potential of escaped cancer cells may be predicted by exploiting their persistent dependency on the primary tumor microenvironment stiffness.
Materials and Methods
Modular Polyacrylamide (mPA) Hydrogels
Contiguous polyacrylamide gels with distinct modules were fabricated through a step-by-step polymerization of PA solutions of defined compositions. Precursor solutions containing the acrylamide:bisacrylamide (A:B) percentages of 4:0.2% or 12:0.6% were mixed with 0.5% Ammonium Persulphate (APS) and 0.05% Tetramethylethylenediamine (TEMED) [22]. Red fluorescent carboxylate-modified beads of 200-nm diameter were added at 0.1% concentration to the stiff PA precursor solution to identify the interface between dissimilar ECM modules. Next, a volume of PA precursor solution sufficient to achieve a gel thickness of 100μm was dispensed on the coverslip and covered with a glass slide of defined size to confine the spreading of the PA droplet in each module of the substrate. This step was repeated for all three modules (Fig. 1A) to fabricate the entire modular PA (mPA) hydrogel substrate. After polymerization, mPA gels were sterilized for 1 hour under UV light. Subsequently, PA gels were treated with 0.5mg/ml of sulfosuccinimidyl 6-(4′-azido-2′-nitrophenylamino) hexanoate (Sulfo-SANPAH) (Thermo Fisher Scientific) prepared in 50mM HEPES buffer (Santa Cruz Biotechnologies) and crosslinked to the mPA surface upon activation with 365 nm UV for 10 minutes. After extensive washing with 50 mM HEPES buffer, mPA gels incubated with 0.05 mg/ml of rat-tail collagen type I (Santa Cruz Biotechnologies) overnight at 4°C.
Figure 1. Contiguous substrate with regions of dissimilar stiffness.

(a) Schematic describing the fabrication steps of mPA substrates of heterogeneous stiffness through modular polymerization of PA solutions of distinct compositions, resulting in dissimilar ECM stiffness in adjoining primary and secondary regions. (b) Atomic Force Microscopy (AFM) measurements of Young’s Modulus of PA gels plotted in logarithmic scale at different locations within a substrate with dissimilar primary and secondary ECM regions. Stiffness values are averaged over 1mm length intervals and plotted along with scattered data points and error bars (SEM). N>150. Data is included from at least 3 different PA gels, in which the left side was intended to be stiff (acrylamide/bisacrylamide=12/0.6%) and right side as soft (acrylamide/bisacrylamide=4/0.2%) matrix.
Mechanical Characterization of PA Hydrogels
Atomic Force Microscopy (AFM) measurements of polyacrylamide gels were performed using an MFP-3D-BIO atomic force microscope (Asylum Research, Santa Barbara, CA). Olympus TR400PB AFM probes with an Au/Cr coated silicon nitride cantilever and pyramidal tip were used, with spring constants ranging from 20–30 pN/nm, as measured by thermal calibration. Force maps in square regions of 40 μm edge length, with 169 points per force map, were taken at equal spacing across the gels. Measurements were performed across at least 4 mm length on each side of the interface dividing the primary and secondary ECM regions, as shown in Fig. 1b. Elastic moduli were extracted from force curves using a modified Hertz model [23].
Cell Culture and Collective Migration Assay
Human mammary epithelial non-tumorigenic MCF10A cells were cultured in DMEM-F12 (GE Healthcare Life Sciences), supplemented with 5% horse serum (Invitrogen), 20 ng/mL epidermal growth factor (EGF, Miltenyi Biotec Inc), 0.5 mg/mL hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 10 ug/mL insulin (Sigma-Aldrich), and 1% (v/v) penicillin-streptomycin (Sigma-Aldrich). Tumorigenic mammary epithelial MCF7 cells were grown in DMEM (Sigma-Aldrich) containing 10% fetal bovine serum (FBS), 1% (v/v) penicillin-streptomycin (Sigma-Aldrich), and 1% non-essential amino acids (0.1mM). Human epidermoid carcinoma A431 cells were grown in DMEM (Sigma-Aldrich) containing 10% fetal bovine serum (FBS), 1% (v/v) penicillin-streptomycin (Sigma-Aldrich), 1% sodium pyruvate (Sigma-Aldrich), 1% sodium bicarbonate (Sigma-Aldrich), and 1% non-essential amino acids (0.1mM). A PDMS stencil was designed and fabricated with a rectangular opening in the center, restricting the culture of epithelial monolayer within the central module (primary ECM) of mPA substrate (Fig. 1A). PDMS stencils were air-dried, cleaned with 70% ethanol, and sterilized under UV light for 2 hours. Afterwards, stencils were passivated overnight, with a sterile solution of 1% Pluronic and 1% Bovine serum albumin (BSA) in Phosphate buffered saline (PBS) to avoid cell adhesion, and carefully positioned on the PA gels. Cell suspension with 2×104 cells was dispensed into the PDMS reservoir covering the primary ECM region and left to grow for a prescribed duration of priming (1, 2, or 3 days) at 37°C in a 5% CO2 humidified incubator. After this incubation period, the PDMS stencil was lifted off to allow cell sheet to migrate into the adjoining secondary ECM. Additional media was added to each well.
For proliferation inhibition experiments, cells were treated with 2mM thymidine (Sigma-Aldrich) after at least 6 hours of cell seeding, to allow adequate attachment of cells to the substrate. For calcium chelation experiments, cells were incubated with 4mM of EGTA (Sigma-Aldrich) after 3-day priming.
Live-cell Imaging
Time-lapse microscopy was initiated 2 hours after removing the PDMS and time-lapse imaging was carried out in the phase contrast on an inverted microscope (Zeiss Cell Observer) equipped with an incubator capable of maintaining an environment with 37°C temperature and 5% CO2 level. Images were acquired with a 10× objective. Two successive images of the same field were taken at 20 minutes time interval. Motion of the leading edge of the cell monolayer was manually tracked by recording the position of cell border using a homemade macro in ImageJ. Subsequently, coordinates of the leading edge were imported into a custom-made program in MATLAB and its advancement was calculated by averaging the distance of each point on the leading edge from the primary-secondary ECM interface. Leading edge speed was defined as the ratio of the leading edge advanced distance and the time course of migration. For single cell migration analysis, individual movies were imported to ImageJ software (National Institutes of Health), and single cell migration trajectories were extracted using the Manual Tracking plugin. Cell migration tracks were subsequently analyzed to obtain migration speed and velocity angle distribution. Migration speed was calculated as a ratio between the sum of distances traveled by the cell between each time point and the total time. For each time interval, we calculated the angle between instantaneous velocity vector and the x axis and plotted the angle distribution for the entire migration tracking period.
Particle Image Velocimetry (PIV) and Monolayer Dynamics
PIV analyses were performed to quantify spatiotemporal velocity distribution of velocity magnitudes by implementing the PIVlab package in MATLAB [24]. To reduce the systematic biases in subpixel resolution, we iteratively implemented PIV up to three passes of 64, 32, and 16 pixel windows with 50% overlap between adjacent windows. Displacement and velocity (displacement/time interval) vectors were calculated by comparing the displacement of a window between two successive images. The velocity field was expressed in μm/min. PIV analysis yielded two components of velocity at each point (i,j), namely lateral (uij, perpendicular to the direction of group migration) and axial (vij, along the direction of group migration). After obtaining the velocity vectors, cell alignment and cell-cell coordination were evaluated in terms of order parameter and correlation length [25]. The order parameter was defined as the cosine of the angle that the velocity vector makes with the principal direction of migration. This principle direction is the vector sum of all velocity vectors analyzed in a given frame. The order parameter varies from +1 (for velocity vectors parallel to the strip and directed along the direction of migration of the cell sheet) to −1 (for vectors that are directed opposite to the direction of migration of the cell front) through 0 (for vectors aligning perpendicular to the direction of the monolayer migration). Correlation lengths were calculated according to the method described earlier [25]. To demonstrate the time evolution of monolayer motion, we plotted kymographs of velocity magnitude and order parameter. After obtaining the velocity vectors for every pixel of the monolayer at a given time instant, kymographs were computed by averaging the velocity magnitude and order parameter of individual velocity vectors in x direction over the y coordinates in every time point for time period of 12 hours after monolayer crossed the interface.
Immunofluorescence and Confocal Microscopy
After day 5, cells in the migration assay were rinsed with cold 1X PBS for 2–3 minutes and fixed in 4% Paraformaldehyde (PFA) at room temperature (RT) for 10 minutes. After washing again with PBS, cells were incubated with 1% bovine albumin serum (BSA) (EMD millipore) overnight at 4°C. Next, cells were washed with PBS for 30 minutes, and incubated in the primary antibody solution for yes-associated protein (YAP) (1:100; Santa Cruz) or phosphorylated myosin light chain (pMLC) (1:100; Cell Signaling Technology) prepared in 1% BSA, and stored overnight at 4°C. Samples were washed and incubated with appropriately matched secondary antibodies (Invitrogen) for 1 hour at RT. After thoroughly rinsing the substrates with PBS, DAPI (1:250; Santa Cruz Biotechnology) and Phalloidin (1:200; Invitrogen) was added for 30 minutes at RT. Finally, substrates were rinsed again with PBS and stored at 4°C before imaging. Images were recorded at RT using a laser-scanning confocal microscope (Ziess LSM 730; Carl Ziess MicroImaging; Germany) at 20X or 40X objective, and confocal stacks were acquired at 1 μm interval. Image acquisition parameters including laser intensity and exposure times were maintained at the same level to ensure quantitative image analysis. Experiments were performed in triplicates, and the images used for analysis were randomly selected from 10–15 fields of view for each condition.
Quantitative Image Analysis
Captured z-stacks were imported to ImageJ (NIH) as LSM files, and the stacks were projected with the maximum intensity setting. To quantify the subcellular YAP activity, the mean fluorescence intensity of YAP was measured in the nucleus and the cytoplasm. Afterwards, the nuclear-to-cytoplasmic ratio of YAP intensity was computed and plotted to compare YAP localization across experimental conditions. At least 50 cells were randomly selected for analysis from 10–15 field of views selected from three independent experiments. To quantify pMLC intensity, the mean protein intensity per cell in a given region of interest (ROI) was calculated after subtracting the background signal (corresponding to the intensity of the negative control) from the total intensity in the z-stack (sum over all pixels of slices) and normalized to the number of cells in the corresponding region. The number of cells in a given ROI obtained by manually counting cell nuclei in the maximum projected DAPI image. To compare between different experimental conditions, pMLC intensity per cell values were normalized to the values obtained for cells cultured on control stiff substrates. To analyze actin fiber orientation, the resulting z-projected image of phalloidin (sum over all pixels of slices) for each cell was analyzed using OrientationJ algorithm in ImageJ. For each cell, the degree of stress fiber alignment was calculated in terms of coherency within a defined region of interest through ImageJ, which varies between 0, indicating isotropic distribution, and 1, indicating highly aligned structures. Subsequently, the average fiber alignment was obtained by normalizing to the fiber alignment value obtained for cells cultured on stiff control substrates. The mean spreading areas of cells located at different distances with respect to the leading edge on various substrates were measured using ImageJ software by manually drawing the border of cell from phalloidin images and evaluating the resulting cell areas. At least 40 cells were analyzed from three independent experiments for each experimental condition. To measure the size of focal adhesions (FAs), confocal z-stack images of paxillin staining were acquired. FA area was computed by outlining punctate focal adhesions in binarized paxillin images and using the “Measure” tool in ImageJ to calculate FA area. At least 15 cells were analyzed for each experimental condition.
shRNA Knockdown
To deplete YAP, the lentiviral pFLRu vector containing either Scramble (shSCRM) or anti-YAP shRNA (shYAP), and puromycin resistance was used. HEK293T cells were cultured in DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 200 μM L-glutamine (Cellgro), and penicillin-streptomycin. HEK293T cells were transfected with lentiviral DNA using the TransIT LT1 transfection reagent per manufacturer protocol (Mirus). Virus was harvested from 293T media 48 hours after transfection and used to transduce MCF10A cells. Puromycin (Sigma-Aldrich) was used in cell selection and maintenance at a concentration of 1.5 μg/mL.
Western Blotting
Cells were lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 200 nM phenylmethylsulfonyl fluoride (PMSF), 2 μg/ml aprotinin/leupeptin, 2 μM pepstatin A, 1 mM Na3VO4, and 2 mM NaF. The lysates were cleared by centrifugation at 12,000 rpm at 4C for 10 min, and the concentrations were determined by Bradford assay (Bio-Rad). Equal amounts of protein were boiled in SDS sample buffer for 10min, resolved by SDS 10%-PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) in transfer buffer (25 mM Tris, 192 mM glycine, 5% methanol). The membranes were blocked with TBST (25 mM Tris, pH 7.4, 150 mM NaCl, 2 mM KCl, 0.5% Tween 20) containing 5% skim milk powder or bovine serum albumin (BSA) and probed overnight with the indicated primary antibodies. Bound antibodies were detected by horseradish peroxidase (HRP)-conjugated secondary antibodies and developed with SuperSignal West Pico and/or West Femto enhanced chemiluminescence (ECL) (Pierce). Images were collected on a Bio-Rad ChemiDoc XRS+ and analyzed using ImageJ software (NIH). The following antibodies were used: rabbit anti-YAP/TAZ (Cell Signaling Technology, D24E4, mAb #8418; 1:1000), mouse anti-Actin (Sigma, A5316; 1:25,000).
Statistical Analysis
Unless specified otherwise, results are reported as the mean ± Standard Error (SE). To identify the significant differences between two experimental conditions, an F-test was performed to determine whether equal variance could be assumed. Next, Student’s t-test was used to determine significant differences between two groups. All statistical analyses were performed using the Data Analysis toolbox in Microsoft Excel. Differences were considered to be significant for P<0.05.
Results
Fabrication of dual-stiffness contiguous substrates
To prime epithelial cells on a given ECM and track their subsequent collective migration onto an adjoining ECM of dissimilar stiffness, we fabricated a modular-PA (mPA) hydrogel substrate through step-by-step polymerization of two different PA compositions [22] (Fig. 1a). Through AFM-based mechanical characterization of these gels, as done previously [3, 23], we found that the soft and stiff regions have corresponding Young’s moduli of approximately 0.5 and 50 kPa, respectively (Fig. 1b). These measurements also verified that the ECM stiffness in the primary and secondary ECM regions varied in a step manner, as intended. These stiffness values are not chosen to match any specific tissue context; rather, this range is designed to explore the biophysical effects of cellular priming across a steep change in matrix stiffness. Next, we designed a PDMS stencil for culturing an epithelial monolayer restricted within the central section of the mPA gel, which we will refer to as the ‘primary’ ECM. After priming the cell colony on the primary ECM for a prescribed duration, the PDMS stencil was removed to enable collective migration of the cell sheet into the surrounding ‘secondary’ ECM of independently defined stiffness. We verified that the coating of collagen I on the PA gel did not vary with stiffness or the removal or PDMS stencil (Fig. S1). In this system, epithelial cells seamlessly move across a contiguous substrate composed of mechanically distinct regions, whose stiffness can be tuned over two orders of magnitude. Thus, we are able to study the effect of past ECM stiffness on future cell behavior without having to detach and re-culture cells on a new substrate. Comparison of cell behavior on a given secondary ECM with respect to varying priming ECM stiffness would reveal whether migratory epithelial cells store mechanical memory of their past mechano-regulated state.
Leading edge migration depends on the past ECM stiffness
To assess the effect of past ECM stiffness on collective cell migration, we cultured MCF10A human mammary epithelial cells on primary ECM stiffness (P) for 3 days and then allowed the cells to migrate onto a secondary ECM (stiffness ‘S’). We performed time-lapse microscopy for an additional 2 days, i.e., between days 4–5 from the time of initial culture, and manually traced the leading edge over time, as illustrated in Fig. 2a. On substrates with homogeneous stiffness (P=S), the average leading edge migration on stiff ECM (50 kPa) was 30μm/h, which was three times higher than its value on the soft ECM (0.5 kPa) (Fig. 2b). This stiffness-dependent collective cell migration speed is consistent with previous studies [6, 7]. When the cell monolayer was first primed on a stiff primary ECM (P=50 kPa), the leading edge migration speed measured on the adjacent soft secondary ECM was ~2.5 times higher than the control case of homogeneously soft ECM (Fig. 2b). This enhanced leading edge migration could be attributed to the stiffness-dependent mechano-activated state of cells attained due to stiff priming, which we define as the mechanical memory of collectively migrating cells. We also found that the soft-primed cells migrated ~40% slower than those primed on a stiff ECM (Fig. 2b).
Figure 2. Collective cell migration speed depends on priming by the past ECM stiffness.

(a) Representative leading-edge tracks of monolayers of MCF10A cells recorded for 12 hours (3 h interval) in the secondary ECM after 3-day priming, with color-coding for migration speed. Arrows indicate direction of migration. Scale bar = 100 μm. (b) Average leading-edge migration speed for MCF10A, A431, and MCF7 cells migrating on secondary ECM (S=0.5, 50 kPa) after defined priming (P=0.5, 50 kPa). Horizontal square brackets denote statistical significance (p<0.05). N>15. (c) Average leading-edge migration speed for MCF10A cells after 1, 2, or 3 days of priming. *p3<30.05, with horizontal square brackets denoting statistical significance (p<0.05). ns=no significant difference. Error bars = SEM. N>10.
To test whether the duration of priming on the primary ECM influenced the memory-dependent migration, we measured migration speed after priming for 1 or 2 days. We found that the influence of the primary ECM stiffness reduced with shorter priming duration (Fig. 2c). Given that longer priming led to more pronounced mechanical memory-dependent collective migration, all results presented in this manuscript will correspond to a 3-day priming regimen, followed by 2 days of migration on the secondary ECM. All measurements are conducted on the secondary ECM, unless specified otherwise.
We also measured leading-edge migration speed for MCF7 breast cancer cells and A431 human epidermoid carcinoma cells. While MCF7s were slower and A431s were faster compared to MCF10As, both of them exhibited robust mechanical memory-dependent migration (Figs. 2b, S2).
Stiffer primary ECM predicts more correlated cell migration
Using particle image velocimetry (PIV) analysis of phase contrast images of the migrating cell sheet over time [24], we examined cellular motions within the epithelial monolayer (Fig. 3a). After arriving on a soft secondary ECM, stiff-priming led to high velocity magnitudes of the leading-edge cells, as compared to the soft primary ECM. Conversely, soft-priming significantly reduced the cell velocities. We noted that cells behind the leading edge had lower velocity magnitudes in all cases. By plotting single cell trajectories over a 12 hour period, we confirmed faster velocities of cells at the leading edge compared to the ones within the monolayer (Fig. S3).
Figure 3. Monolayer dynamics and temporal variation of memory-dependent migration.

(a) Heatmap showing the spatial distribution of velocity magnitude at a given time instant for MCF10A cell monolayer migration. (b) Position-time kymographs of velocity magnitude and order parameter obtained from PIV analysis demonstrate the time evolution of monolayer motion. Kymographs were computed by averaging the velocity magnitude and order parameter of individual velocity vectors in the x-direction over the y-coordinate for every time point. Average (c) correlation length and (d) order parameter of the velocity vectors. Horizontal square brackets denote statistical significance (p<0.05). N>15. (e) Plot describes the leading-edge migration speed over time, tracked for 96 h in secondary ECM after 3-day priming of cell monolayer. All bar plots are averaged over quantities measured in the first 48h of migration (depicted by the shaded region), which corresponds to the period of maximal memory. N>10. Error bars = SEM.
We found that the correlation length, defined as the distance of correlation among velocity vectors of neighboring cells [25], for cells migrating on a homogeneously stiff ECM was ~0.25 mm, which is ~25% higher than its value measured on a purely soft ECM (Fig. 3c). Thus, higher ECM stiffness enables larger portions of the cell sheet to migrate in a coordinated fashion, which is consistent with previous findings [6]. The stiff-primed cells migrated on a soft secondary ECM with ~0.25mm correlation length, the same as the value measured on a homogeneously stiff ECM. In comparison, soft-primed cells migrated with a lower correlation length of ~0.2mm on a stiff secondary ECM. The order parameter of the collective migration, defined as the angle between the velocity vectors and the direction of leading edge migration, on a homogeneously stiff ECM was more than twice its value on a soft ECM (Figs. 3d, S4), indicating a more ordered collective migration on stiffer ECM [6]. The order parameter of stiff-primed cells remained high (~0.6) on the soft secondary ECM. To assess the temporal progression of collective cell motility onto the secondary ECM, we selected four representative videos of collective cell migration for each condition and plotted how a column of cells traversed over a 12 h period (Fig. 3b). Cells maintained their velocity magnitudes and order parameter over time, i.e., pixel intensities rarely varied over this 12 h period and distance (Fig. 3b).
To further expand the temporal variation of migration speed across numerous samples, we averaged leading-edge migration speed at given time points and plotted these values over 4 days of migration after the 3-day priming (Fig. 3e). We found that the stiff-primed monolayers maintained at least ~2 times higher speed on a soft secondary ECM (compared to purely soft ECM) for at least 3 days. This memory-based advantage in the migration speed of stiff-primed cells started to subside afterwards (Fig. 3e). Thus, the presented effects of mechanical memory correspond to a phenotype maintenance within a temporal boundary, which is measured here as 3 days of collective migration in the secondary ECM.
Higher actin/pMLC expression and adhesions due to stiffer priming
Since cell-ECM adhesions and actomyosin machinery are crucial for generating forces and driving cell motility [3, 6, 26], we examined whether their subcellular expressions within individual cells near the leading-edge of the monolayer depend on the priming stiffness. To this end, we stained and imaged for F-actin and phosphorylated myosin light chain (pMLC) in cells after their migration across the secondary ECM. We found that actin fiber alignment (from phalloidin images), pMLC expression, and number of focal adhesion (FAs) (paxillin images) in cells on homogeneously stiff ECM were significantly higher than their average values measured on a soft ECM (Fig. 4, S5). After stiff-priming, cells on a soft ECM exhibited larger FAs (~4 times), pMLC expression (~2.5 times) and actin alignment (~1.6 times) compared to the control case of homogeneously soft ECM (Fig. 4b–d). These results indicated that stiff-priming allowed the cells to maintain enhanced actomyosin machinery even after their traversal onto a soft secondary ECM. Conversely, the actin alignment and pMLC expression were reduced significantly after soft-priming.
Figure 4. Cytoskeletal machinery in memory-dependent migration.

(a) Immunofluorescent staining of pMLC (green), F-actin (phalloidin, red), and DAPI (blue) in top-panel and paxillin (red) and DAPI (blue) in bottom-panel for MCF10A cell monolayers on the secondary ECM after 2 days of migration (post-priming). Scale bar = 100 μm. Quantification of (b) actin fiber alignment, (c) normalized pMLC expression, and (d) FA area. N>40. (e) Variation of spreading area of single cells within the MCF10A cell monolayer relative to the distance from the leading edge showing stiffness-independent cell spreading in the primary ECM. N>25. Error bars = SEM.
To examine whether stiffness-sensitive cell spreading in the primary ECM could influence the leading-edge migration, we calculated cell areas across the monolayer. Notably, in all four matrix conditions, cell spreading reduced with distance from the leading edge, which led to a stiffness-insensitive spreading in the rear part of the monolayer (Fig. 4e) and priming-dependent spreading near the leading-edge. Thus, cell spreading in the primary region cannot influence the leading-edge migration computed in the secondary region.
Alternate hypotheses for memory-dependent migration due to cell proliferation and signal transmission
Although the results in Figs. 1–4 clearly show the influence of past ECM stiffness on cellular features in the secondary ECM, we examined several alternative hypotheses other than the proposed ‘memory-storing’ abilities of migratory epithelial cells can explain the observed behavior. First, we asked whether mechano-sensitive cell proliferation due to the stiffer primary ECM in the rear of the monolayer could influence leading edge migration. After inhibiting cell proliferation (thymidine treatment; see Methods and Fig. S6), cell migration speeds increased due to increased spreading, yet the trend of mechanical memory-dependent migration held true (Fig. 5a–b). In some conditions, we observed higher migration speed after proliferation inhibition, which may be attributed to increased cell spreading (Fig. 5c).
Figure 5. Memory-dependent migration is not regulated by proliferation or long-distance signal transmission.

Representative leading-edge tracks of monolayers of MCF10A cells after treatment with (a) 2mM thymidine, a proliferation inhibitor, and (d) 4mM EGTA, a calcium chelator, recorded for 12 hours (3 h interval) in the secondary ECM, with color-coding for migration speed. Arrows indicate direction of migration. (b) Average leading edge migration speed for proliferation-inhibited cells. Columns with dashed outline represent the migration speed for control untreated MCF10A cells. N>15. (c) Average spreading area of individual cells in the secondary ECM with and without proliferation inhibition. N>30. (e) Average leading edge migration speed for EGTA-treated cells. N>15. (f) Immunofluorescent staining for E-cadherin (green) and DAPI (blue) in untreated and EGTA-treated MCF10A cells showing dysfunctional cell-cell junctions after EGTA treatment. Scale bars = 100 μm. (g) After 3-day priming and additional 1 day of migration in the secondary ECM, the primary ECM region is entirely removed. Leading-edge migration speed in the secondary ECM (right panel) shows preservation of memory-dependent migration despite a complete loss of communication with the primary region. N>15. Horizontal brackets denote statistical significance, p<0.05. Error bars = SEM.
Second, to attenuate inter-cellular force transmission, we repeated migration measurements in the presence of a calcium chelator (4mM EGTA), as used previously for this purpose [27, 28], which disrupts cell-cell communication (as shown through E-cadherin images in Fig. 5f). As a result, some cells break away from the monolayer. Overall, we found that the memory-dependent migration of the leading edge was preserved despite the loss of cell-cell communication (Fig. 5d–e).
Finally, to eliminate any possible communication between the primary and secondary regions, we physically removed the entire primary region along with the attached cells before measuring migration in the secondary ECM (1 day after complete priming). We found that the memory-dependent migration persisted in this system (Fig. 5g–h). These results, along with the ones presented above, confirm that the memory-dependent migration observed in the secondary ECM is independent of a direct communication with the primary ECM. Instead, the priming-dependent signals are likely stored within the cells and continue to dictate collective migration in the future.
Migratory cells store mechanical memory of past ECM stiffness through YAP activity
The observed memory-dependent collective cell migration indicates that the priming-dependent mechano-regulated state of cells may persist onto the new ECM. Given that nuclear translocation of YAP has been identified as a sensor of ECM stiffness [17, 29, 30], we determined YAP subcellular localization within MCF10A cells as they migrated across ECMs of dissimilar stiffness. We quantified the nuclear-to-cytoplasmic ratio of YAP fluorescent intensity in at least 40 cells from multiple fields. Consistent with previous findings (19), YAP expression was predominantly nuclear on homogeneously stiff ECM and cytoplasmic on homogeneously soft ECM (Fig. 6). However, nuclear YAP localization of stiff-primed cells when measured in soft secondary ECM was more than four times its value on homogeneously soft ECM (Fig. 6b). Thus, nuclear accumulation of YAP due to the stiffer past ECM persisted even after the cells migrated onto the adjoining softer region. Conversely, soft-primed cells on stiff secondary ECM showed less than 1/3rd nuclear YAP, compared to the homogeneously stiff ECM (Fig. 6c). We repeated these measurements in MCF7 and A431 cells (Figs. 6b–c, S7) and found that both of these cancer cell lines followed a similar dependence of subcellular YAP localization on the past matrix stiffness. Thus, cellular mechano-sensation of ECM stiffness through subcellular YAP localization could be a key mechanism for storing mechanical memory in migratory cells.
Figure 6. YAP activity depends on past ECM stiffness.
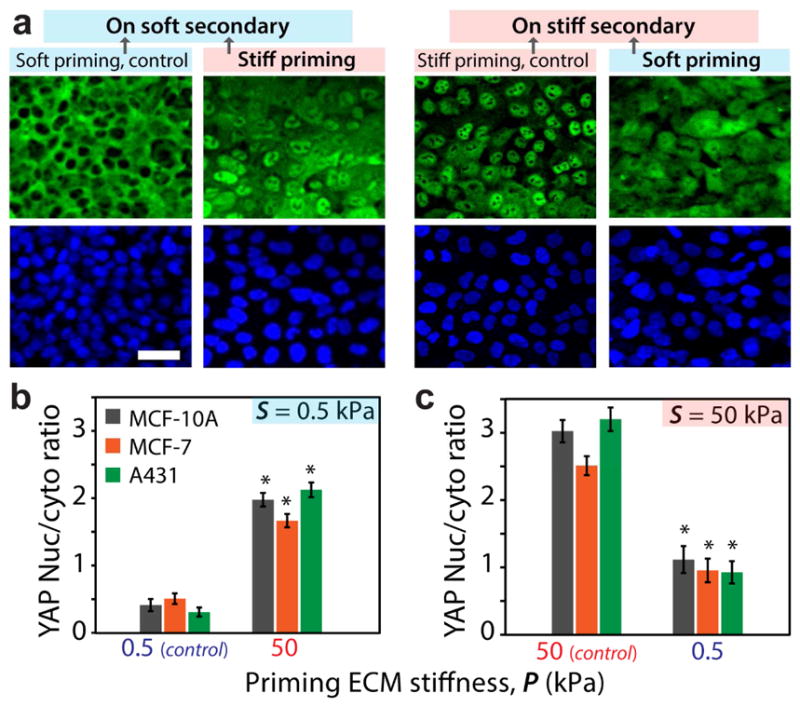
(a) Immunofluorescent staining of MCF10A cells for YAP (green) and DAPI (blue) illustrating the subcellular localization of YAP for the monolayer migrating on secondary ECM, after priming. Scale bar = 50 μm. (b,c) Average nuclear-to-cytoplasmic ratio of the YAP fluorescent intensity for MCF10A, MCF7, and A431 cells within the monolayer. *p3<30.05 with respect to control ECMs of homogeneous stiffness. Error bars = SEM. N>40.
Inhibition of mechanical memory-based cell migration through YAP depletion
To further examine whether YAP activity is a requirement for memory-dependent migration, we performed shRNA-mediated depletion of YAP in MCF10A cells (YAP-KD) (Fig. 7b), as described previously [18, 31]. We found that the leading-edge migration speed of stiff-primed YAP-KD cells was ~15μm/h on a soft secondary ECM, which was similar to the control case of homogeneously soft ECM (Fig. 7a,c). Even after priming on a soft ECM, YAP-KD cells migrated fast (~30μm/h) on a stiff secondary ECM, matching with the control case of a homogeneously stiff ECM. Thus, after YAP depletion, cells were unable to exploit prior priming. Notably, the YAP-depleted cells on homogeneously stiff ECM were almost twice as fast compared to those on homogeneously soft ECM (Fig. 7a,c). Through PIV analysis of cellular motions within the monolayer, we also found that the cells within the monolayer migrated in a memory-independent manner, with greater correlation and in a more ordered fashion on a stiffer ECM regardless of the primary ECM stiffness (Figs. 7d–e, S8).
Figure 7. Memory-independent collective migration of YAP-depleted MCF10A cells.

(a) Leading edge tracks of monolayer of YAP-depleted MCF10A cells are plotted at 3 h interval during their post-priming migration on secondary ECMs. Immunofluorescent staining for F-actin (red), pMLC (green), paxillin (red), and DAPI (blue). Scale bars = 100 μm. (b) MCF10A cells expressing either shSCRM (wt) or shYAP RNAi were lysed and subjected to Western blotting with anti-YAP and anti-Actin antibodies. Average (c) leading edge migration speed, (d) correlation length, and (e) order parameter, N>15, and normalized (f) pMLC expression and (g) actin alignment, N>40, and (h) FA area, N>20, for varying ECM configurations. Columns with dashed outline represent corresponding values for wildtype MCF10A cells. Horizontal brackets denote statistical significance (p<0.05). ns=no significant difference. Error bars = SEM.
Given that YAP is a classic mechano-sensor [17], the observed ECM stiffness-dependent migration of YAP-depleted cells was unexpected. To understand the potential mechanism through which these YAP-KD cells continue to sense their immediate matrix stiffness, we imaged for focal adhesions because they directly connect the cells to the matrix. Indeed, we found that the average FA size of YAP-KD cells on stiff secondary ECM was more than 5 times higher compared to the values on the soft secondary ECM, regardless of primary ECM stiffness (Fig. 7a,h). We also found similar mechanosensitive but memory-independent trends for actin fiber alignment, and pMLC expression (Fig. 7a,f,g, S9). These results demonstrate that YAP-KD cells are unable store a mechanical memory of past stiffness due to hampered YAP activity, but continue to sense the immediate matrix stiffness through focal adhesions.
Discussion
Plasticity in motile cells is manifested by variable modes of migration depending on the surrounding microenvironment [32]. In particular, cancer cells are uniquely equipped to exploit their plasticity to drive tumor invasion through distinct tissues. It has recently been identified that human mesenchymal stem cells store memory of their past exposure to matrix stiffness [19]. However, in migratory cells, it has remained a mystery whether their mechanics-regulated state persists even after they move to a new environment. If the mechanical properties of the tumor microenvironment are found to mechanically “train” the escaping cells, impacting their future ability to metastasize, this could be a critical missing piece of the puzzling unpredictability of cancer adaptation. To address this important gap in our understanding, we asked whether collectively migrating cells retain a mechanical memory of their past ECM stiffness. Through measurements of collective cell migration across dual-stiffness substrates, we show that priming of an epithelial cell colony on a stiff ECM enhances its future collective migration even on a soft ECM. We also show that the enhanced migration of stiff-primed cells on soft ECM is not caused by the mechanosensitive differences in cell spreading, proliferation, or other mechanical signaling transmitted from the back of the monolayer. Indeed, when the cell colony was only primed for one day, the stiff primary ECM in the back was not able to enhance migration of cells on the soft secondary ECM. Thus, the memory-dependent cell migration is orchestrated by preserving the priming-enabled mechano-activated state of the cells onto the secondary ECM. We found that less than 2 days of priming of cells on the primary ECM showed a substantially reduced mechanical memory-based migration. It is likely that the cells require 2–3 days to respond to matrix stiffness and accordingly localize YAP (nuclear or cytoplasmic). Indeed, our measurements of nuclear YAP localization within cells of a monolayer cultured on the stiff PA gel (50 kPa) at different time points reveal that YAP activation continues to rise over 3 days (Fig. S10). Furthermore, this time-sensitive storage of YAP-dependent memory within the cells might require a transcriptional program, which is consistent with previous studies on mechanical-memory dependent responses of stem cells [19].
Given that YAP is a classic transducer of ECM [17] and its known role in storing memory in stem cells [19], we measured its ability to retain information of past ECM stiffness in collectively migrating cells. We found that the stiffer priming predicted higher nuclear YAP localization – a sign of persistent mechano-regulated YAP activity. After YAP depletion, cell migration did not depend on past matrix stiffness, i.e., the mechanical memory was significantly diminished in these cells. According to our data, the YAP-depletion blunts mechanical memory without a significant loss of cellular mechanosensitivity to the immediate matrix stiffness.
Our results point to a conceptual framework of mechanical memory-dependent cell migration in which migration-related cellular forces may be independently influenced by two factors (Fig. 8). First, the priming-dependent YAP activity directly regulates actomyosin forces and migration (results from Figs. 4,6). In cancer associated fibroblasts (CAFs), YAP activation has been shown to enhance and maintain a positive feedback loop with actomyosin contractility [4]. Similarly, fluid shear-dependent YAP activation has been shown to enhance protrusions required for migration [33]. Second, cells are able to sense immediate matrix stiffness through adhesions, despite YAP depletion (results from Fig. 7). Previously, we have shown that YAP-depleted CAFs maintain the mechano-sensitive SNAIL1 protein level [18], which indicates that the cells are able to adopt alternate YAP-independent pathways for sensing matrix stiffness.
Figure 8. Conceptual framework for memory regulation.
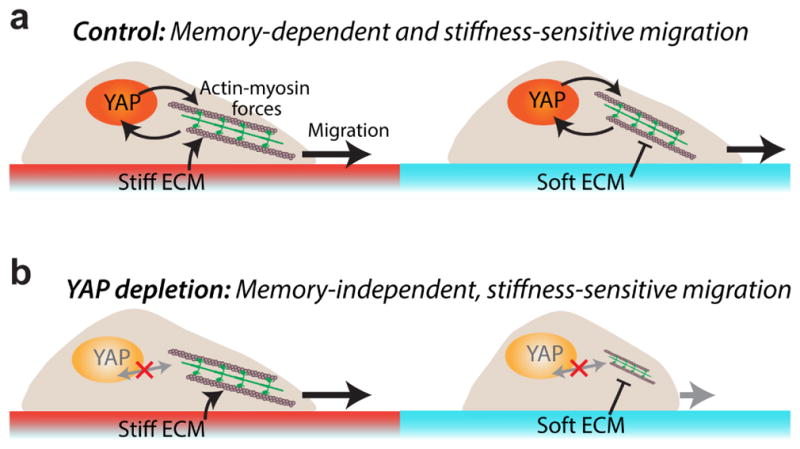
(a) Priming-dependent YAP activity regulates cellular forces and dictates the memory-dependent migration. (b) YAP-depletion abrogates memory, but direct FA-mediated contact with the immediate ECM preserve mechano-sensitivity.
In summary, our findings of the mechanical memory in migratory cells expand the basic understanding of cellular mechanotransduction, beyond the current framework of studying cell migration in the context of only the immediate microenvironment. The insight that stiffness of the past ECM can influence future collective migration opens the door to new hypotheses for a wide array of biological processes, wherever microenvironment-dependent cell motility plays a role, such as morphogenesis, wound healing, and cancer cell invasion. Given that our experiments were conducted with immortalized epithelial cells, the presented conclusions may not be directly applicable for mesenchymal-like cells in vivo. Furthermore, since our measurements were performed in an idealized model system of a homogeneous cell monolayer in contact with a 2D substrate, the conclusions drawn here may not be directly applicable for in vivo conditions of 3D microenvironments and heterogeneous cell populations. The knowledge of memory-storing abilities of invasive cancer cells and associated signaling targets may open new avenues for therapeutics and predictive modeling by exploiting their dependency on the primary tumor microenvironment and tuning their ability to adapt to foreign tissue environments.
Supplementary Material
Acknowledgments
This work was in part supported by grants from the National Science Foundation (CAREER Award 1454016) and the Edward Mallinckrodt, Jr. Foundation (New Investigator Award) to AP, NIH Grants R01 GM080673 and U54 CA210173 to GDL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Discher DE, Janmey P, Wang Y-l. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science. 2005;310(5751):1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 2.Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, Yang J. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015 doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA. 2012;109(26):10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–46. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pathak A, Kumar S. Transforming potential and matrix stiffness co-regulate confinement sensitivity of tumor cell migration. Integr Biol. 2013;5(8):1067–1075. doi: 10.1039/c3ib40017d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng MR, Besser A, Danuser G, Brugge JS. Substrate stiffness regulates cadherin-dependent collective migration through myosin-II contractility. J Cell Biol. 2012;199(3):545–63. doi: 10.1083/jcb.201207148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sunyer R, Conte V, Escribano J, Elosegui-Artola A, Labernadie A, Valon L, Navajas D, García-Aznar JM, Muñoz JJ, Roca-Cusachs P, Trepat X. Collective cell durotaxis emerges from long-range intercellular force transmission. Science. 2016;353(6304):1157. doi: 10.1126/science.aaf7119. [DOI] [PubMed] [Google Scholar]
- 8.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nature reviews Molecular cell biology. 2009;10(7):445–57. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 9.Varner VD, Nelson CM. Cellular and physical mechanisms of branching morphogenesis. Development. 2014;141(14):2750–9. doi: 10.1242/dev.104794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Developmental cell. 2008;14(4):570–81. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nasrollahi S, Pathak A. Topographic confinement of epithelial clusters induces epithelial-to-mesenchymal transition in compliant matrices. Scientific reports. 2016;6:18831. doi: 10.1038/srep18831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 13.Dow LE, Kauffman JS, Caddy J, Zarbalis K, Peterson AS, Jane SM, Russell SM, Humbert PO. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene. 2007;26(16):2272–82. doi: 10.1038/sj.onc.1210016. [DOI] [PubMed] [Google Scholar]
- 14.Nicolas A, Geiger B, Safran SA. Cell mechanosensitivity controls the anisotropy of focal adhesions. Proceedings of the National Academy of Sciences USA. 2004;101(34):12520–12525. doi: 10.1073/pnas.0403539101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. The Journal of Cell Biology. 1996;133(6):1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reffay M, Parrini MC, Cochet-Escartin O, Ladoux B, Buguin A, Coscoy S, Amblard F, Camonis J, Silberzan P. Interplay of RhoA and mechanical forces in collective cell migration driven by leader cells. Nat Cell Biol. 2014;16(3):217–223. doi: 10.1038/ncb2917. [DOI] [PubMed] [Google Scholar]
- 17.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–83. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 18.Zhang K, Grither WR, Van Hove S, Biswas H, Ponik SM, Eliceiri KW, Keely PJ, Longmore GD. Mechanical signals regulate and activate SNAIL1 protein to control the fibrogenic response of cancer-associated fibroblasts. J Cell Sci. 2016;129(10):1989–2002. doi: 10.1242/jcs.180539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang C, Tibbitt MW, Basta L, Anseth KS. Mechanical memory and dosing influence stem cell fate. Nature materials. 2014;13(6):645–52. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo C-M, Wang H-B, Dembo M, Wang Y-l. Cell Movement Is Guided by the Rigidity of the Substrate. Biophysical journal. 2000;79(1):144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J Cell Biol. 2012;199(4):669–83. doi: 10.1083/jcb.201205056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer RS, Myers KA, Gardel ML, Waterman CM. Stiffness-controlled three-dimensional extracellular matrices for high-resolution imaging of cell behavior. Nature protocols. 2012;7(11):2056–66. doi: 10.1038/nprot.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackay JL, Kumar S. Measuring the elastic properties of living cells with atomic force microscopy indentation. Methods Mol Biol. 2013;931:313–29. doi: 10.1007/978-1-62703-056-4_15. [DOI] [PubMed] [Google Scholar]
- 24.Thielicke W, Stamhuis EJ. PIVlab – Towards User-friendly, Affordable and Accurate Digital Particle Image Velocimetry in MATLAB. Journal of Open Research Software. 2014;2 [Google Scholar]
- 25.Das T, Safferling K, Rausch S, Grabe N, Boehm H, Spatz JP. A molecular mechanotransduction pathway regulates collective migration of epithelial cells. Nat Cell Biol. 2015;17(3):276–87. doi: 10.1038/ncb3115. [DOI] [PubMed] [Google Scholar]
- 26.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell Migration: Integrating Signals from Front to Back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 27.Tambe DT, Corey Hardin C, Angelini TE, Rajendran K, Park CY, Serra-Picamal X, Zhou EH, Zaman MH, Butler JP, Weitz DA, Fredberg JJ, Trepat X. Collective cell guidance by cooperative intercellular forces. Nature materials. 2011;10(6):469–475. doi: 10.1038/nmat3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plutoni C, Bazellieres E, Le Borgne-Rochet M, Comunale F, Brugues A, Séveno M, Planchon D, Thuault S, Morin N, Bodin S, Trepat X, Gauthier-Rouvière C. P-cadherin promotes collective cell migration via a Cdc42-mediated increase in mechanical forces. The Journal of Cell Biology. 2016;212(2):199. doi: 10.1083/jcb.201505105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Codelia Va, Sun G, Irvine KD. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Current Biology. 2014;24(17):2012–2017. doi: 10.1016/j.cub.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun M, Spill F, Zaman MH. A Computational Model of YAP/TAZ Mechanosensing. Biophysical journal. 2016;110(11):2540–50. doi: 10.1016/j.bpj.2016.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng Y, Nie L, Thakur MD, Su Q, Chi Z, Zhao Y, Longmore GD. A Multifunctional Lentiviral-Based Gene Knockdown with Concurrent Rescue that Controls for Off-Target Effects of RNAi, Genomics. Proteomics & Bioinformatics. 2010;8(4):238–245. doi: 10.1016/S1672-0229(10)60025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HJ, Diaz MF, Price KM, Ozuna JA, Zhang S, Sevick-Muraca EM, Hagan JP, Wenzel PL. Fluid shear stress activates YAP1 to promote cancer cell motility. Nature communications. 2017;8:14122. doi: 10.1038/ncomms14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.