

Abstract
De novo design can be used to explore vast areas of chemical space in computational lead discovery. As a complement to virtual screening, from-scratch construction of molecules is not limited to compounds in pre-existing vendor catalogs. Here, we present an iterative fragment growth method, integrated into the program DOCK, in which new molecules are built using rules for allowable connections based on known molecules. The method leverages DOCK’s advanced scoring and pruning approaches and users can define very specific criteria in terms of properties or features to customize growth toward a particular region of chemical space. The code was validated using three increasingly difficult classes of calculations: (1) Rebuilding known x-ray ligands taken from 663 complexes using only their component parts (focused libraries), (2) construction of new ligands in 57 drug target sites using a library derived from ~13M drug-like compounds (generic libraries), and (3) application to a challenging protein-protein interface on the viral drug target HIV gp41. The computational testing confirms that the de novo DOCK routines are robust and working as envisioned, and the compelling results highlight the potential utility for designing new molecules against a wide variety of important protein targets.
Keywords: de novo design, structure-based design, fragment libraries, chemical space, DOCK, scoring functions, footprint similarity, drug discovery, ZINC
Graphical Abstract
De novo design methods aim to create new molecules, from scratch, with steric and electrostatic compatibility for the target system(s). In this work we describe development, validation, and application of a new de novo design version of the program DOCK that allows assembly of small organic molecules in a binding site using fragment libraries consisting of scaffolds, linkers, and sidechains. The results highlight the potential utility for designing new molecules against a wide variety of important protein targets.
Introduction
Virtual screening (VS) is an effective strategy for narrowing the search space of potential leads at the early stages of drug discovery. The in silico structure-based approach enriches for molecules that are compatible with a drug-target receptor, and eliminates molecules that are likely to be incompatible.1,2 Typically, a subset of the most attractive molecules from a VS are selected for experimental evaluation. Then, a small area of chemical space around promising lead compounds can be further explored using medicinal or synthetic chemistry, with the goal of improving affinity to and specificity for the receptor.3 While VS is an effective approach with numerous success stories (recently discussed in Refs. 4,5) it is not without its own challenges.6 For one, the number and identity of molecules available for screening are limited to those provided by online catalogues including ZINC,7,8 the Chemical Abstracts Service Registry,9 and PubChem,10 among others. Screening libraries, which range on the size of 107 to 108 molecules, represent unique, existing chemical matter. Some estimates, however, put the actual number of molecules under 500 Da that could exist, also called “Chemical Space”, on the order of 1060 molecules.11–13 Thus, it follows that a molecule with truly optimal steric and energetic compatibility with a drug target may not be easily found through conventional VS strategies alone. Additionally, off-the-shelf compounds identified via VS may be subject to forms of intellectual property protection as they are not novel chemical entities.
As an alternative to VS, an approach termed de novo design assembles molecules from the beginning, or from constituent parts, directly complementing available binding interactions in the receptor2,14 (Figure 1). Conceptually, the approach has some parallels to fragment-based drug design (FBDD) in that large molecular scaffolds in a binding site are connected by linkers, and also can be decorated with various sidechains.15–17 In principle, the building blocks of assembly for de novo design, however, can be individual atoms, small functional groups, rings or fused ring systems, large flexible scaffolds, and combinations therein. With this method, molecules assembled in the context of the receptor remain compatible with the receptor. More importantly, the ensemble of molecules sampled is not limited to existing databases; rather they have the potential to explore many diverse regions of chemical space. Following de novo design, particularly encouraging molecules as evaluated by scoring or fitness metrics can be synthesized or, as an alternative strategy, the molecules can be used as probes in chemical databases7–10 to identify related, purchasable compounds. Consistent with established VS studies, experimental verification of leads and medicinal chemistry is advantageous, but the benefit of this alternative approach is that it opens up for exploration new chemical matter and new areas of chemical space that may not have been otherwise identified.18,19
Figure 1.
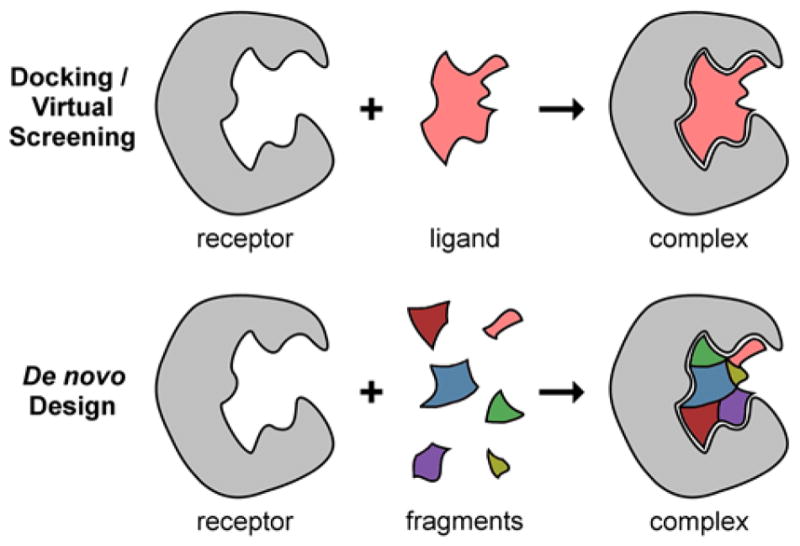
Illustration of docking/VS (top) versus de novo design (bottom) approaches. In VS, ligands originate from existing chemical libraries or catalogs. In de novo design, fragments are building blocks used to construct new ligands. Receptors are typically treated the same in both methods.
Currently available de novo design programs are distinguished by their search methods, scoring methods, pruning and filtering criteria, receptor vs. ligand-based approaches, and the size and identity of constituent fragments. Focusing on differences among the search method, some of the more widely-used approaches include: (i) scaffold hopping and molecular transformation (e.g. Skelgen,20 DYNAMO,21 SHOP,22 DAECS23), (ii) fragment linking (e.g. LUDI,24 CONFIRM,25 LiGen26), (iii) iterative fragment growth (e.g. GROW,27 BOMB,28,29 FOG30), (iv) genetic or evolutionary algorithms (e.g. TOPAS,31 ADAPT,32 LEA3D,33,34 MEGA,35 AutoGrow36), (v) combinatorial library searches (e.g. CombiDOCK,37,38 FORECASTER,39 MAntA40), and (vi) a combination of methods (e.g. SPROUT,41 SMoG,42 LigBuilder43,44). The recently published OpenGrowth45 method selects fragments for addition based on the statistical likelihood that the two fragments have been observed concurrently in a training set. Several of these programs, and others, have successfully led to experimentally verified new chemical matter or improved leads against a variety of drug targets.28,29,46–54 Despite the great strides already taken, de novo design programs still face challenges in that they are limited by the efficient navigation of chemical space, accuracy in scoring, and synthetic feasibility of designed molecules.18 However, given the advent of advances in computing power, this approach is gaining popularity and tractability as a complementary approach to traditional VS.6
In this manuscript, we present an original implementation of a de novo design method integrated into the infrastructure of the widely-used structure-based design program DOCK5,55,56 (hereafter referred to as de novo DOCK). The method is an iterative fragment growth approach built upon the successful anchor-and-grow search algorithm used by DOCK6.57 Fragments for growth and rules for allowable connections originate from the ZINC database7,8 of existing, synthesizable, and drug-like chemical compounds, or can be generated on the fly by the user. De novo DOCK takes advantage of several of the specialized algorithms currently in DOCK, including advanced scoring and pruning methods, to overcome some of the known challenges of de novo design. The method is also highly “targeted” in that users may define very specific criteria in terms of chemical properties or features to guide growth toward a particular area of chemical space.
The new de novo DOCK implementation was validated using three increasingly complex classes of calculations. First, we attempted to rebuild known ligands from 663 crystallographic co-complexes taken from the Protein Data Bank (PDB),58 using only their component parts (focused libraries). Second, we constructed new ligands in several important drug target sites using a much larger library of molecule fragments (N=380) which was derived from ~13M drug-like compounds (generic libraries). Third, as a real-world application, we explored the ability of the algorithm to generate potential inhibitors of viral entry through targeting a known protein-protein interface on the drug target HIV gp41.59,60 A key objective of the work was to develop protocols that lead to the construction of new drug-like molecules that create binding-site interactions similar to known binders. Overall, we envision this new software will be useful to build new molecules with user-defined properties, and to explore chemical space around a certain scaffold. The algorithms discussed in this work, along with example input files and test cases, have been implemented into the DOCK6 suite of programs and will be made available for download for free to registered academic users at http://dock.compbio.ucsf.edu/.
Theory and Implementation
De novo DOCK consists of two primary processes: (1) fragment generation and (2) de novo construction. These two processes, as well as additional relevant details, are described in depth below. In this section, input filenames and parameter names are in italics.
Fragment Generation
Fragments, in this context, represent the smallest rigid chemical unit perceived by the program de novo DOCK. These rigid units, analogous to segments in standard DOCK,5 are determined by the Sybyl atom types61 of input molecules and settings in the flexible definition library (flex.defn) distributed with the program. As a rule, fragments themselves contain no internal rotatable bonds, however there are some exceptions. For example, methyl groups (R–CH3), cyano groups (R–CN), halogens (R–F, R–Cl, etc.), and hydrogens (R–H) are, strictly speaking, rotatable groups. But, rotating about the R–X bond does not change the nature of the interaction with the receptor. Thus, these bonds are treated as rigid in the fragment generation process. In addition, the double bond in alkenes (–C=C–) is not typically described as a rotatable bond, but de novo DOCK treats it as such with two possible states: cis and trans. Thus, alkene double bonds are broken in the fragment generation process.
To generate fragments, molecules in mol2 format61 must first be downloaded from a chemical catalog. For the tests described here, the molecules were downloaded from the ZINC database.7,8 The input molecule file is read by de novo DOCK, the molecules are broken at each rotatable bond, and finally fragments are written to file with dummy atoms (labeled as Du) used as placeholders to mark where the fragment was cleaved. These points in the fragments, called “attachment points”, define where two fragments can be connected. Final fragments are written to three separate output mol2 files, classified as either “sidechains” (one attachment point), “linkers” (two attachment points), or “scaffolds” (three or more attachment points). Furthermore, redundant fragments are removed, and the frequency of each fragment in the input database is recorded in the header section of the mol2 file. Figure 2 contains an illustration for the deconstruction of DANA, a potent inhibitor of neuraminidase,62 into two scaffolds, two linkers, and three sidechains.
Figure 2.
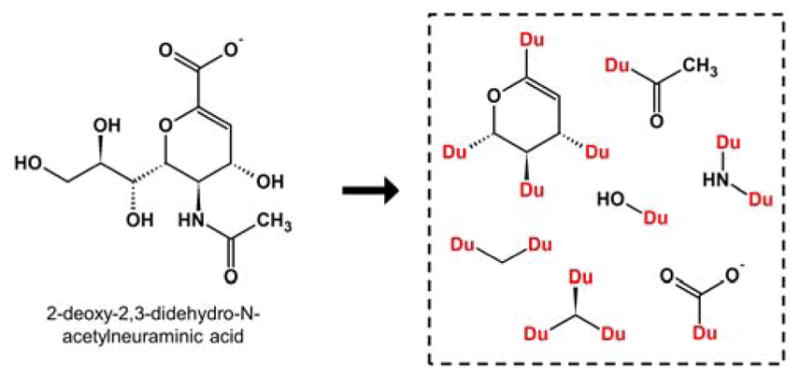
Small molecule 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA, left) is deconstructed into fragments (right) around rotatable bonds. Redundant fragments are removed. Attachment points are marked as dummy atoms (Du).
It is important to emphasize that the fragment generation process is highly customizable to fit the requirements of different applications. The input molecule database can range from a generic set of several million drug-like compounds to a focused set of known inhibitors for a single target. The flex.defn file63 can be readily modified to enforce special treatment of certain bonds, i.e. specific bond types may be defined as rotatable or non-rotatable, which affects the identity of fragments generated by de novo DOCK. A cutoff parameter (fragment_library_freq_cutoff) can be used to retain only fragments that appear above a specified frequency threshold within the input library. Finally, the output fragment libraries can be sorted either by frequency or by chemical similarity. Fragment libraries, once generated, may be swapped out, concatenated, or hand-edited to remove or add new fragments as needed.
Torsion Environments
A major barrier in de novo design is the synthetic feasibility of constructed compounds.18,19 If molecules designed via de novo DOCK are not accessible in a synthetic chemistry setting, then they have limited practical use. As such, to maximize efficiency, it follows that only a synthesizable region of chemical space should be explored. To address this challenge, we have co-opted the 2D molecular fingerprints already implemented in DOCK into an idea called “torsion environments”.
The molecular fingerprints used in de novo DOCK are based on the MOLPRINT 2D descriptors termed “atom environments” previously reported.64,65 For every atom in a given molecule, its local topology in all directions one bond away is computed and stored as a canonical string. The atom hybridization, as well as the bond order are considered in these environments, and the collection of all atom environments in a molecule constitutes its fingerprint. Two adjacent atom environments connected by a rotatable bond are termed here a “torsion environment”. De novo DOCK will only connect two fragments together if the torsion environment exists in a user-supplied look up table. The look-up table itself is generated by de novo DOCK from, for example, a large database of synthesizable, drug-like molecules.
Following fragment and torsion environment table generation, the construction of new molecules with de novo DOCK is guided by principles of sampling, scoring, and searching. As in standard docking, “sampling” refers to the generation of three-dimensional molecule binding geometries (poses) including rotational, translational, and torsional degrees of freedom, and “scoring” refers to the quantitative method for evaluating the fitness and properties of each molecule. Specific to de novo design, “searching” refers to the exploration of chemical space, or the manner in which different fragments from the input libraries are selected and assembled. These three guiding principles are discussed in detail below.
De novo Construction: Sampling and Searching
Analogous to standard docking,5 sampling begins with orientation of “anchors” to the target binding site spheres via a graph-matching algorithm.57 Anchors can be any fragment provided by the user in a separate input file, or automatically chosen by the program from the standard input libraries based on number of atoms and number of attachment points. The precise number of unique anchors (dn_unique_anchors) and the number of positions retained for each unique anchor (dn_anchor_positions) are specified by the user. If fragments are provided that are already oriented to a binding site, such as those derived from crystallographic co-complexes, or if the objective is ligand-based de novo design absent from a receptor, then orienting may be skipped. This is particularly useful for lead refinement in which the user seeks to automate ‘R’ group sampling of a ligand with a known binding pose.
Beginning from an anchor in the binding site, de novo DOCK generates new molecule structures using the input fragment libraries in a layer-by-layer, iterative growth approach (Figure 3). Similar to the standard DOCK anchor-and-grow strategy,57 a full layer of fragments is assembled around the central anchor prior to extending by multiple fragments along any one path. The default search method (dn_search_method) for choosing new fragments, called “graph”, is a breadth-first search with a simulated annealing function. With this method, a graph is initially prepared that links different fragments (as graph nodes) with each other based on their Tanimoto similarity (as weighted graph edges).66 All fragments are connected to all other fragments, and the Tanimoto coefficients range from 0 (least similar) to 1 (most similar). Then, given an attachment point on the current anchor, a random fragment is connected. The fragment torsions are sampled, partial charges are recomputed (see below), the molecule is energy minimized, and the score is computed. If the new score is an improvement over the previous score, then the fragment is retained and the program attempts to connect chemically similar fragments at that same position by searching adjacent nodes in the graph. If the addition of a fragment worsens the score, then that fragment may still be retained based on the probability in Eq. 1:
| (Eq. 1) |
Figure 3.
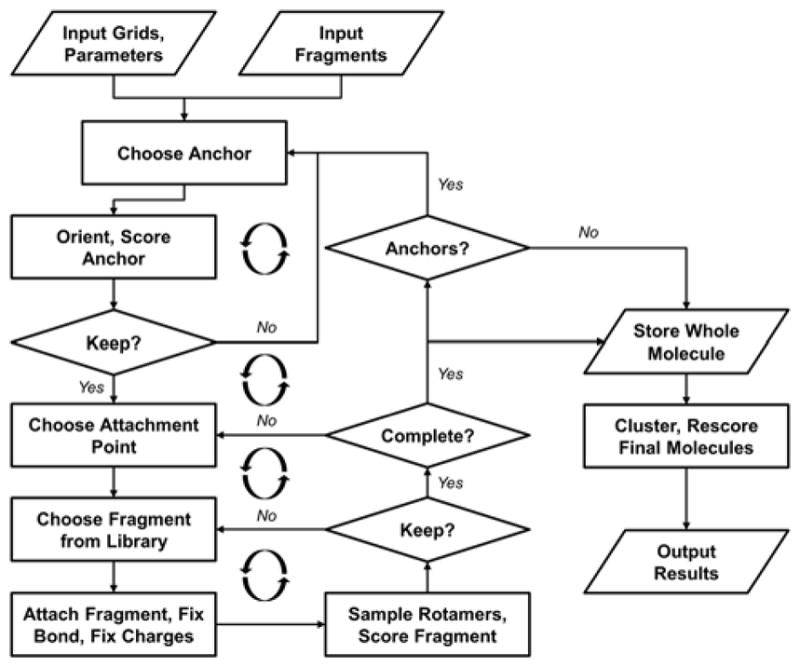
Flowchart of iterative growth process for de novo DOCK. Input includes grids and parameters according to standard DOCK run and scoring functions, and fragment libraries. Anchors are oriented to binding site. Retained anchors are grown outward in a layer-by-layer approach analogous to anchor-and-grow. Complete molecules are written to file.
Where P is the probability that the new fragment is retained, Ef and Ei are the energy scores for the final and initial molecules, respectively, and T is an annealing temperature. The default beginning annealing temperature (dn_graph_temperature) is 100, then scales down toward 1 for each layer of growth. In this manner, new fragments that worsen the score may be kept more frequently at beginning layers of growth.
This search continues at each original attachment point on the anchor as long as fragments continue to improve the score, or until the search has reached a user-defined limit. At any given layer, all attachment points existing at the beginning of the layer are filled before any new attachment points are sampled. The breadth (dn_graph_breadth) and the depth (dn_graph_depth) of the fragment graph search determine how many new fragments similar to a retained fragment will be sampled. These parameters along with the number of random starting points in the fragment graph (dn_graph_max_picks), are defined in the input file. In addition, the number of partially grown molecules that advance through the search to subsequent attachment points (dn_max_layer_size) is also defined in the input file. Finally, the number of partially grown molecules retained after each layer (dn_max_root_size) directly influences the number of new anchors that seed the beginning of the next layer of growth. The thoroughness of sampling (and the amount of time required) is most directly influenced by these five parameters.
There are two additional search functions employed by de novo DOCK. They are termed “random”, and “exhaustive”. The random search method selects a user defined (dn_num_random_picks) number of random fragments at each attachment point of a growing molecule. The exhaustive method samples all fragments at each attachment point and is therefore only recommended for use with small libraries. These methods rely more on the same rigorous pruning parameters as the graph method to bound the search space. These alternative search methods are described in greater detail in the online documentation, and are not discussed further here.
De novo Construction: Scoring
Many different scoring functions exist in DOCK, any of which may be employed by de novo DOCK. Some functions are physics-based67 and use a classical molecular modeling force field63 to derive a quality-of-fit. Others are knowledge-based68–70 and compare candidate molecules against a reference molecule – typically something that is known to bind to the target such as a co-crystallized inhibitor. All functions have in common the assumption that a greater negative number (or a positive number closer to zero) represents a more favorable score. Specific scoring procedures used in the present work include: (1) standard DOCK single grid energy (SGE)5 comprised of van der Waals and electrostatic contributions, (2) multi-grid energy (MGE)69 which is numerically equivalent to SGE but contributions for key binding residues can be stored on individual grids enabling per-residue decompositions (i.e. footprints) to be mapped, (3) footprint similarity (FPS)68,69 which is a measure of the overlap (typically Euclidian distance) between any two footprints, (4) multi-grid energy plus footprint similarity (MGE+FPS) which is a combined function consisting of van der Waals and electrostatic contributions plus their footprint overlap, (5) pharmacophore matching similarity (FPS)70 which is a measure of the pharmacophore overlap between two molecules, and (6) Hungarian matching similarity (HMS)71 which scores how well two compounds overlap in an RMSD-like space. The two main actions of scoring in de novo design are targeted growth (Figure 4, vertical arrow), and pruning or filtering (Figure 4, horizontal arrow). These ideas largely control which molecules are retained or discarded during growth to bound the search space and address the problem of combinatorial explosion in de novo design.
Figure 4.
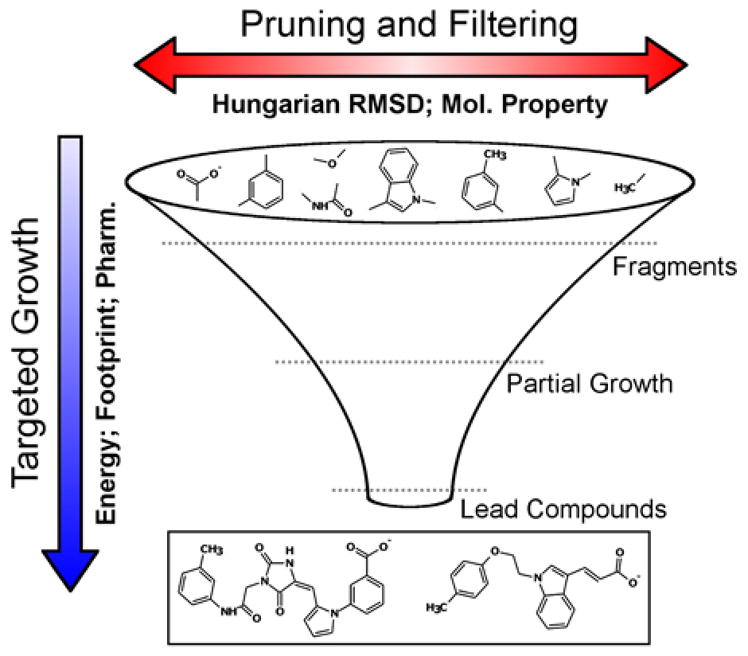
Scoring drives targeted growth (vertical blue arrow) and pruning/filtering (horizontal red arrow) in de novo design. Examples of targeted growth scoring functions include the DOCK grid energy score, footprint similarity score, and pharmacophore score. Examples of pruning/filtering criteria include Hungarian RMSD, molecular weight, number of rotatable bonds, scaffolds per layer, etc.
Targeted growth in de novo DOCK refers to the application of scoring functions to drive the average score of the molecule ensemble toward a favorable value (Figure 4, unidirectional blue arrow). The driving force can be any of the individual scoring functions described above or, because of a major upgrade to the function called Descriptor Score, most any combination of DOCK scoring methods can be used. Coefficients specified in the input file determine the relative weight of each term. The Descriptor Score follows the general functional form:
| (Eq. 2) |
Where each coefficient (C1, C2, …, CN) and each scoring function (F1, F2, …, FN), are defined in the input file. Targeted growth by vertical pruning is achieved by first rank-ordering all molecules at a specific stage of growth by score, then retaining only a top-scoring fraction for subsequent stages of growth. Overall, a high level of control over the scoring functions employed can drive the population of growing molecules through chemical space in a targeted, purposeful manner.
The objectives of horizontal pruning is to remove redundant molecules, increase diversity in the growing ensemble, and remove molecules with undesirable properties (Figure 4, bidirectional red arrow). To help achieve these outcomes, we employed Hungarian RMSD (HRMSD)71 which is a measure of the 3D spatial differences between two molecules calculated as:
| (Eq. 3) |
Where #ref is the number of atoms in the larger molecule, #unmatched is the number of atoms that are dissimilar between the two molecules, RMSDmatched is the symmetry-corrected RMSD between the atoms that are in common, and C1 and C2 are weighted coefficients. With these metrics, pruning is achieved by a best-first cluster. In other words, all molecules are ranked by score, then pairwise combinations are examined, beginning with the best molecule first, to eliminate any subsequent molecule which is too similar by the Hungarian score. The coefficients, as well as the pruning cutoffs for both metrics, are specified in the input parameter file (dn_heur_unmatched_num, dn_heur_matched_rmsd).
In addition, molecules that exceed a user-specified molecular weight (dn_constraint_mol_wt), number of rotatable bonds (dn_constraint_rot_bond), or formal charge (dn_constraint_formal_charge) are also filtered. Overall, this process increases diversity of molecules in the growing ensemble while driving the search space toward a favorable property ensemble. Taken together, we believe this level of control over the scoring, pruning, and filtering process is key to providing users the means to build molecules with very specific, highly-customizable properties.
Partial Charge Equilibration
At every stage of growth, it is important that partially-assembled molecules (those that contain open attachment points) remain as physical as possible. To that end, dummy atoms are treated as the chemical equivalent to hydrogen atoms for scoring purposes. As each new fragment is added to the growing anchor, the Gasteiger function72 is used to equilibrate partial charges, ensuring an integral formal charge. The Gasteiger function was chosen for its speed and efficiency at equilibrating charges over large sets of molecules, which, depending on run conditions, can reach tens to hundreds of thousands.
Final Output Ensembles
Performing de novo design with the present input parameters and infrastructure produces final molecule ensembles that may contain multiple different conformers for each of the unique chemotypes sampled during growth. During program development, outcomes were significantly improved when multiple conformers of partially grown molecules were retained at each stage of growth (layer-by-layer). Conceptually this is similar to standard docking wherein multiple conformers of a molecule are retained. However, as is the case for VS, it is often desirable to only look at the best scoring member from each unique chemotype. For clarity, analysis of ensembles in this work potentially containing multiple conformers are labeled “Built Ensembles” and those containing only the best scoring conformer from each chemotype are labeled “Unique Ensembles”. Currently for de novo DOCK, a Built Ensemble is post processed to generate a Unique Ensemble using the following steps: (1) The best scoring molecule in the Built Ensemble not already ‘seen’ is saved in the Unique Ensemble and used as a reference, (no molecules have been ‘seen’ on the first iteration) (2) DOCK is used to calculate the Tanimoto coefficients of all molecules to the reference molecule, (3) all molecules with a Tanimoto coefficient of 1 to the reference molecule are marked as ‘seen’, (4) steps 1–3 are repeated until all molecules have either been ‘seen’ or copied into the Unique Ensemble. Inspection revealed results for focused library runs often contain numerous conformers of each species while generic runs are already mostly unique. This is attributable to the small size of the focused libraries which results in the increased sampling compared to the generic runs. Redundant chemotypes are more likely to be constructed starting from different anchors using small libraries with a limited number of fragments. Finally, an input parameter (dn_write_prune_dump) is provided to enable a memory dump of all molecules explored during de novo growth.
Computational Details
Fragment Libraries
Fragment libraries described in this manuscript are classified as “focused” or “generic”. Focused libraries contain only the non-redundant group of fragments from within a single parent molecule. As such, each of the focused libraries is distinct in composition and contains only a small number of fragments and accompanying torsion environments. In contrast, the large generic fragment library was derived from a data set of 13,195,579 drug-like molecules from ZINC. Fragments that appeared 13,000 times (0.1%) or greater among the ~13 million molecules were retained, resulting in 217 distinct sidechains, 145 distinct linkers, and 18 distinct scaffolds (380 in total). From the same input ZINC library, 10,838 possible torsion environments were identified occurring at any frequency. Only one generic library with one generic torsion environment table was used for all the generic experiments in this manuscript.
Anchor Selection
All focused library calculations employed up to three anchors per run as suggested by prior work5 from our laboratory. Analogous to standard docking of a small molecule with known topology, the three largest anchors here were automatically selected from among the fragment entries in each of the focused library files. Given the small size of the focused libraries automatic selection of anchors based on size is a reasonable procedure. For the generic runs, however, in which the fragment library contained 380 entries, our initial growth tests revealed that automatic selection based on size was not likely to be a suitable strategy. More specifically, selection based on size always resulted in the same three largest ring structures selected to seed growth regardless of the protein binding site. To address this issue, we opted to seed growth with anchor fragments that had the highest overall observed frequencies in the drug-like ZINC library. To further increase sampling, generic library calculations for a given system employed 15 different anchors run concurrently on separate processors, and the results were merged together into a final ensemble for subsequent analysis.
Test Set Preparation: SB2012
A subset consisting of 663 out of 1043 systems in the SB2012 test set5,73 was used for the focused library generation and re-building tests based on complexes containing all ligands with 5–15 rotatable bonds (inclusive) and with formal charge of −2e to +2e. This removes the most rigid molecules (<5 rotatable bonds) as well as the most flexible (>15 rotatable bonds), while remaining in the domain of drug-like molecules. A second smaller subset of 57 co-crystallized receptor-ligand complexes was chosen from among the 663 with a focus on diverse families of proteins representing important disease targets. The set of 57 was used for de novo growth tests using the larger generic library.
All test set complexes were prepared for docking as previously described.5,73 Briefly, crystal structures were downloaded from the Protein Data Bank,58 and ligands were separated from receptors, protonated using the program MOE,74 and assigned AM1-BCC charges75,76 and GAFF77 force field parameters using the AmberTools program antechamber.78 Receptors were protonated and ff99SB79 charges and parameters were assigned using the AmberTools program tleap. The assembled receptor-ligand complex was then subjected to a short energy minimization using Amber with heavy restraints on all non-hydrogen atoms to relax the system in a controlled manner. The surface of the receptor, absent the ligand, was then determined using DMS80 with a 1.4 Å radius probe atom, and the binding cavity was filled with docking spheres using the DOCK tool sphgen.81 Finally, a docking grid was generated for each receptor with the DOCK accessory program grid67 within a box that surrounded all spheres with a margin of 8.0 Å in all directions and at 0.3 Å resolution. Each grid point contained Lennard-Jones terms with 6–9 attractive and repulsive exponents, respectively, and a Coulombic energy term using distance dependent dielectric of 4r.
De novo DOCK Parameters
The de novo parameters used for all sections of Results in Discussion are identical unless otherwise stated. The graph sampling method was used to select fragments with 10 random starting points per attachment point, a breadth of 5, and a depth of 2. The graph temperature, which controls the acceptance ratio of fragments that do not improve the score, was set to 100.0, and it decreased by half each layer. All fragments with an energy score greater than a maximum value were discarded. The maximum energy value started at +100 and decreased geometrically with each layer. Upper bounds were placed on molecular weight (750 Da focused, 500 Da generic), number of rotatable bonds (15 focused, 10 generic), and formal charge (±2.0) to limit growth. To further control how much sampling is performed, the de novo maximum root size was set to 25, and the maximum layer size was also set to 25, meaning that the code would only create 25 molecules from a single growing fragment and that at the end of each layer the best 25 incomplete molecules would be advanced for further growth. No more than one scaffold could be added at each layer of growth to control branching and to decrease the number of attachment points per layer. The maximum number of layers each molecule could grow was 9 and the maximum number of concurrent attachment points on any growing fragment was 5.
Table 1 demonstrates how the selection of maximum number of layers and maximum number of attachment points per layer parameters impacted the ability of the code to theoretically rebuild the 663 molecules from the focused growth test set. For example, for the smallest least-flexible molecules in our test set (5 rotatable bonds), we could theoretically rebuild all 105 molecules using five attachment points per layer. Using three attachment points per layer prevents complete growth of seven molecules; these molecules have a branched structure requiring either four or five attachment points in a given layer. Based on the calculations, we found that that 647 out of 663 molecules could be reconstructed by selecting nine growth layers with a maximum of five attachment points per layer (Table 1, 9 Layers/5 AP versus #Mol). For comparison, parameter choices of either seven layers or five layers, each with a maximum of five attachment points per layer, could yield at most only 605 or 469 of the compounds, respectively. Overall, use of nine layers and a maximum of five attachment points per layer provided a reasonable balance between concerns of timing and topology of resulting molecules. For all other DOCK functions the default parameters were chosen including those that control internal energy, orienting, scoring, and energy minimization. For details, see references.5,63
Table 1.
Number of molecules that could theoretically be built given different parameter combinations.
| 9 Layersa | 7 Layers | 5 Layers | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| #RBb | #Molc | 7 APd | 5 APd | 3 APd | 7 AP | 5 AP | 3 AP | 7 AP | 5 AP | 3 AP |
| 5 | 105 | 105 | 105 | 98 | 105 | 105 | 98 | 105 | 105 | 98 |
| 6 | 89 | 89 | 89 | 83 | 89 | 89 | 83 | 86 | 86 | 80 |
| 7 | 99 | 99 | 99 | 95 | 99 | 99 | 95 | 87 | 87 | 83 |
| 8 | 71 | 71 | 71 | 69 | 69 | 69 | 67 | 53 | 53 | 51 |
| 9 | 58 | 58 | 58 | 52 | 54 | 54 | 48 | 46 | 46 | 39 |
| 10 | 70 | 68 | 68 | 41 | 65 | 65 | 36 | 47 | 47 | 17 |
| 11 | 47 | 44 | 44 | 31 | 39 | 39 | 26 | 21 | 21 | 8 |
| 12 | 37 | 35 | 35 | 27 | 32 | 32 | 21 | 12 | 12 | 4 |
| 13 | 37 | 33 | 33 | 27 | 21 | 21 | 13 | 7 | 7 | 0 |
| 14 | 25 | 24 | 24 | 16 | 20 | 20 | 10 | 7 | 4 | 0 |
| 15 | 25 | 22 | 21 | 12 | 13 | 12 | 1 | 2 | 1 | 0 |
|
|
||||||||||
| sum | 663 | 648 | 647 | 551 | 606 | 605 | 498 | 473 | 469 | 380 |
Number of molecules theoretically possible for a given number of layers of growth.
Number of rotatable bonds.
Number of molecules in the set.
Number of allowable attachment points per layer.
Chemical Similarity
Quantifying structural similarity between two molecules allows us to evaluate de novo test results and identify purchasable compounds similar to newly designed molecules. The Tanimoto coefficient is a comparison of 2D fingerprints between two molecules, and it considers composition and topology, but not conformation or position. In contrast to atom environments used for the torsion environment table (see Theory and Implementation), which only consider atoms up to one bond away from a root, the Tanimoto calculation for similarity considers all atoms and connectivity up to three bonds away from the root. The Tanimoto coefficient in de novo DOCK is measured as:
| (Eq. 4) |
Where Fa,b is the number of features in common between two molecules, a and b, Fa is the number of features only in molecule a, and Fb is the number of features only in molecule b. “Features”, in this context, are atom environments based on the MOLPRINT descriptors described by Bender et al.64,65
Results and Discussion
Part 1. De novo Design using Focused Libraries
A conceptually simple (but not trivial) way to validate de novo design protocols is re-building ligands in their co-crystal binding site, using only the component parts from the original ligand. Although the focused libraries are small, this test is still challenging given that the procedure requires anchor selection, orientation, minimization, and clustering followed by layer-by-layer growth, and only a limited number of conformations and partially-grown molecules can be retained at each step. Success of the exercise is based on whether the original ligand is rebuilt (chemical identity), and whether the ligand is rebuilt to the original pose (pose identity). Depending on the scoring function(s) and input parameters used, the outcomes may vary. This test presents an opportunity to screen different combinations.
We first used de novo DOCK to decompose each individual ligand from the set of 663 systems into their component fragments (see Figure 2). On average, ligands in this set contained 7.0 distinct fragments (focused library) and 7.3 distinct torsion environments (torsion library). Then, independently, we used de novo DOCK to construct new ligands in each of the 663 receptor binding sites using only the cognate ligand’s focused library and accompanying torsion library. It is important to emphasize that the ligand construction initiated from up to 3 anchor positions that were sampled (i.e. oriented) during growth and not seeded to match those observed in the crystallographic structures. The results are summarized in Table 2.
Table 2.
Summary of de novo constructed molecules using focused libraries.
| (i) | (ii) | (iii) | (iv) | (v) | (vi) | (vii) | (viii) |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Scoring Function | Sampling Method | Average #Builta | Average #Uniqueb | % Chem Identity Builtc | % Pose Identity Builtd | % Pose Identity Uniquee | Time (sec) |
| SGE | standardf | 169.6 | 18.2 | 72.4 | 40.6 | 27.9 | 3,317 |
| FPS | standard | 182.1 | 17.1 | 72.5 | 43.9 | 34.5 | 2,995 |
| SGE + FMS | standard | 162.8 | 18.1 | 73.6 | 48.4 | 43.1 | 2,990 |
| MGE + TAN | standard | 180.2 | 19.8 | 84.9 | 51.1 | 38.5 | 14,080 |
| MGE + FPS | standard | 149.7 | 18.2 | 73.9 | 46.0 | 36.3 | 3,327 |
| MGE + FPS | enhancedg | 636.7 | 38.7 | 83.3 | 57.3 | 46.8 | 20,637 |
Average number of molecules per receptor (from Built Ensembles).
Average number of molecules per receptor (from Unique Ensembles).
Percent Built Ensembles containing a molecule with TAN=1 to cognate ligand.
Percent Built Ensembles containing a molecule with TAN=1 and RMSD < 2.0 Å to cognate ligand.
Percent Unique Ensembles where best scoring molecule with TAN=1 has RMSD < 2.0 Å to cognate ligand.
25 molecules per layer of growth.
100 molecules per layer of growth.
Chemical Identity of Constructed Molecules
Several scoring functions (Table 2, column i) were evaluated including single grid energy scoring function (SGE), footprint similarity scoring function (FPS), the single grid energy plus pharmacophore70 scoring function (SGE+FMS), the multi-grid energy plus Tanimoto function (MGE+TAN), or multi-grid energy + footprint similarity scoring function (MGE+FPS). Readers should note that MGE is numerically equivalent to SGE (see Balius et al69 for details) but allows the FPS function to be used during sampling. The final row, showing MGE+FPS with “enhanced” sampling, used the standard protocol with increased layer and root size parameters. Interestingly, for the standard sampling runs, the average number of built molecules (column iii) which ranges from 149.7 – 182.1, and the accompanying average number of unique molecules (column iv) which ranges from 18.1 – 19.8, does not change significantly based on the scoring function. In addition, the percentage of systems in which de novo DOCK builds at least one molecule that is chemically identical to the cognate ligand does not vary significantly with the choice of scoring function (from 72.4 – 73.9%) for standard sampling (Table 2, column v), except in the case where Tanimoto coefficient (TAN) is explicitly included in the score, which yields an increase to 84.9%. Enhanced sampling parameters increase the number of molecules constructed and the percentage of which match the cognate ligand, but at the cost of a diminishing return on increased calculation time (Table 2, column viii). These data indicate that the extent of sampling in the binding sites using focused fragment libraries is somewhat independent of scoring function, and more a function of which sampling parameter values are used. This is reasonable for small fragment libraries where the number of possible combinations is limited by the number of fragments and small number of allowable torsion entries.
Pose Identity of Built Ensembles
Although the scoring function does not play a large role in choosing fragments, or how they are assembled for focused libraries, it does have a significant impact on which molecule poses are sampled and retained. For example, use of SGE alone produces a molecule identical to and less than 2.0 Å RMSD to the cognate ligand in 40.6% of all systems (Table 2, column vi). This frequency increases slightly to 43.9% when using FPS score alone. The combination functions SGE+FMS (48.4%) and MGE+FPS (46.0%) return additional increases. Here, the combined use of energy (SGE or MGE) with a pharmacophore (FMS) or footprint (FPS) similarity terms biases growth to mimic patterns made by the cognate ligands. Finally, use of MGE + TAN yields a pose less than 2.0 Å RMSD to the cognate in 51.1% of the cases because it leads to increased local chemotype sampling about the cognate ligand. Overall, MGE+FPS with enhanced sampling (100 vs 25 molecules per layer) performs the best and successfully reproduces a cognate ligand to within 2.0 Å RMSD in 57.3% of systems, although at a larger cost of computation time (Table 2, column viii). Importantly, the results indicate that the de novo protocols and routines are working as expected and able to guide from-scratch ligand construction using a variety of different scoring functions.
Pose Identity of Unique Ensembles
In practice, we imagine that users will only typically consider compounds and poses found in the Unique ensembles which are derived from the “best-scoring” geometry of each chemotype constructed. This is analogous to virtual screening in which only a single conformation of each docked ligand is typically retained. As expected, pose reproduction rates for re-building cognate ligands under these more stringent conditions are lower (Table 2, column vii) than when all conformations with Tanimoto = 1 are included (Table 2, column v) or when all conformations with Tanimoto = 1 and RMSD ≤ 2.0 Å are included (Table 2, column vi). Nevertheless, our observation that SGE (27.9%) < FPS (34.5%) < MGE+FPS (36.3%) < SGE+FMS (43.1%) is consistent with our expectation that use of a scoring function that includes additional information on binding (e.g. footprints, pharmacophores) will yield greater pose identity.
From another perspective, the ratio of % Pose Identity Unique to % Chemical Identity Built (Table 2, columns vii/v) gives additional information on scoring function accuracy for compounds with Tanimoto = 1 which can be roughly compared to the typical docking success rate of ca. 73%5 (SGE) seen with DOCK in which ligand topologies are known and fixed. By this metric, the observed de novo pose reproduction rates of between 38.5% for SGE (27.9/72.4) and 58.6% for SGE + FMS (43.1/73.6) are lower than 73% primarily because docking algorithms need only consider (retain) changes in conformational space and not keep track of unique chemotypes and their associate conformations. The consequence is de novo design builds and retain significantly fewer copies of each unique molecule. Although, increased sampling helps to ameliorate this effect, for example, use of the MGE+FPS enhanced protocol shows an increase in both the number of built molecules” (636.7 vs 149.7) and their associated re-construction rate of 56.2% (46.8/83.3) vs 49.1% (36.3/73.9) when the numbers of molecule per layer of growth is increased (100 vs 25).
Layer by Layer Growth
As indicated previously, an important design feature of de novo DOCK is the ability to drive molecules into the user defined scoring space. This enables users to customize molecule ensembles to broad or narrow properties as desired using the full range of scoring functions and filters available in DOCK6. Therefore, it would be expected that the average score of the ensemble of molecules would improve as a function of the growth layer. Figure 5 shows the score per layer of each molecule grown in the 663 test case systems driven separately by the SGE, FPS, and MGE+FPS scoring functions. The range of favorable scores is defined differently for each scoring function. Molecules with large negative SGE and MGE+FPS scores are more favorable, and molecules with FPS scores close to zero are more favorable. In both cases, values closer to the bottom of the plots in Figure 5 are more favorable. Overall, we found that ensembles of molecules built by all three scoring functions had a wide spread of scores at early growth layers that trended toward favorable scores in later layers. In addition, the number of high scoring outliers declined as the de novo DOCK pruning functions eliminated unfavorable chemotypes and poses. There are two useful takeaways from Figure 5. First, we observe that the de novo implementation is working as intended in that during sampling DOCK eliminated molecules that had unfavorable scores, and retained molecules that had more favorable scores. Second, the final range of scores in the output ensembles, and in particular the most favorable quartile, are within the range of what we would judge to be “good” scores for the scoring function. Overall, we recommend a scoring function with terms designed to capture both physics-based interaction energies and a quantitative measure of binding mode similarity to a known reference, such as MGE+FPS. Such functions consider both the relative strength of binding interactions and the identity of binding interactions which weigh in to the overall score and helps to bound the search space. Unless otherwise noted, the results presented in the rest of the manuscript have employed the MGE+FPS function.
Figure 5.

Scores for all molecules constructed in all 663 receptors using SGE (left), FPS (center), and MGE+FPS (right) directed ensembles (y-axis) and as a function of layer in which each molecule completed growth (x-axis).
Comparisons with Crystallographic Binding Geometries (Poses)
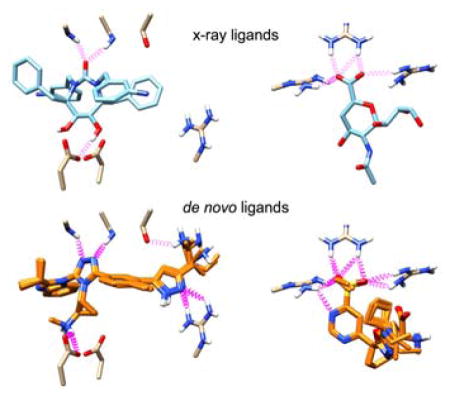
Our final focused library analysis examined 3-dimensional poses of molecules constructed by de novo DOCK in the context of their crystallographic counterparts (Figure 6). For manageability, we examined only the Unique Ensembles (see Methods) which contain the “best scoring” member for each newly constructed chemotype with other conformers removed. Results for 24 out of the 663 focused systems are shown comparing cognate ligand geometries (orange) with de novo constructed geometries (gray). All protein residues are hidden for clarity. Here, the test set of 663 receptor-ligand co-complexes was sorted by the number of distinct fragments in each focused library and then alphanumerically to facilitate un-biased selection of typical systems from each subgroup. With the exception of groups #2 (N=1) and #3 (N=3) the first two PDB codes in each case are shown. The plot is organized by increasing number of distinct fragments (left to right) and therefore presumably increasing rebuilding difficulty. Also shown are the number of cognate ligand rotatable bonds and the Tanimoto and Hungarian RMSD between the cognate ligand and the selected ensemble member. For simplicity, analysis in this section focused only on the result obtained using the MGE+FPS scoring function. For these comparisons, the molecules shown represent the member from within each Unique Ensemble, with the best Tanimoto coefficient to the co-crystalized ligand from which the focused library was generated. Readers should keep in mind that these representative structures were not derived via docking but from de novo construction of fragments with no pre-defined topology other than that imposed by the allowable torsion environments.
Figure 6.

Comparison between de novo built molecules (gray) and crystallographic references (orange) for results generated using the MGE+FPS scoring function for focused libraries. Molecules are organized by increasing number of distinct fragments. Shown are the PDB code, #distinct fragments, number of rotatable bonds in parenthesis, Tanimoto coefficient between the constructed molecule and reference, and Hungarian RMSD between common heavy atoms. Underlined RMSD values represent systems with Tanimoto not equal to 1.
As would be expected, based on the MGE+FPS results from Table 2 which showed ~36% pose identity for molecule selected from Unique Ensembles, visualization of top-scoring entries with Tanimoto=1 (or next highest value) reveals substantial overlap and low RMSD values for many of the systems. For example, 10 out of the 12 molecules from the first two rows (2–7 distinct fragments each) have Tanimoto=1 with low RMSD values less than 2 Å, and four of those molecules have RMSD values less than 1 Å. Some systems with seemingly unfavorable RMSD, for example 1AID at 3.11 Å, in fact has good functional overlap when one considers that two of the four rings are well-aligned and the other two rings are swapped in space. The good overlap observed for the highly flexible ligand from PDB code 1AJV is particularly striking given the large number of rotatable bonds (12 RB), although this is not a general trend. The RMSD values for ligands in the latter two rows (8–15 distinct fragments each) are higher with only two entries - 1CKP and 1A4Q - less than 2 Å. This demonstrates the increasing complexity of the calculation with increasing number of fragments. Nevertheless, on a case-by-case basis, many of the ligands (e.g. 1BN4, 1ETT, 1F0U, 1G2L, and 1HPV) show substantial overlap for portions of the molecule. Overall, the focused library tests show that the present sampling, scoring, and de novo design routines, in favorable cases, can lead to from-scratch-construction of ligands that mimic placement of physical observed ligand functionality. Additionally, enhanced sampling techniques may be used to further improve the results, but at the expense of time.
Part 2. De novo Design using Generic Libraries
A test that is closer to real-world application of de novo design is to build new ligands in a binding site using generic fragments. In contrast to the individual focused libraries, the single “generic library” contains a wide variety of chemotypes – in particular, chemotypes which appear commonly in drug-like molecules. While the primary goal of the focused calculations was to validate sampling, assembly, clustering, pruning and scoring routines (using re-construction of co-crystallized ligands), the primary goal of the generic calculations was to assess the properties of newly designed molecules. Given the large size of the generic library compared to the focused (380 vs 7 fragments on average), and greater number of allowable bonds (10,838 vs 7 torsion environments on average), it is unrealistic to expect that known ligands would be re-built exactly under the parameter choices used here that strive to balance run time with ligand diversity. In fact, in many cases it would be impossible because the generic library only contains common chemotypes, not the less-common fragments present in some of the focused libraries of the previous section. We believe that using a generic library made from common fragments instead of highly focused fragments is better for general design problems. The metrics employed to validate generic library protocols were geared to answer the following three questions: (i) How robust are the de novo designed poses of new molecules? (ii) What are the physical properties of new molecules? (iii) Do the new molecules have similarities to known ligands that bind a given receptor?
Expanding on the first question, since the identification of a pose is critical to both scoring and human evaluation of a newly-designed molecule, are the molecules generated in a binding conformation that can be confirmed by a well-validated docking algorithm, representing the best we can do in absence of crystallographic data? Expanding on the second question, do the newly-constructed molecules have desirable properties and similar molecular makeup when compared to a large group of purchasable drug-like molecules? Expanding on the third question, do designed molecules have chemical similarity (i.e. show functional group overlap) and/or binding similarity (i.e. make similar interactions with the protein) as known cognate ligands?
Generic Libraries
As noted in Methods, the generic library was constructed from ~13M compounds in the ZINC database.8 Fragments that occurred at >0.1% frequency (13,000 times) were retained. The goal was to derive high-frequency ligand building blocks from known, purchasable, and drug-like compounds with physically observed connection rules (in the present case 10,838 possible torsion environments). While the number of allowable torsions in the current work was not reduced by eliminating entries with low frequencies we believe this would be a useful parameter to evaluate for the future. The top 14 fragments from each class, sorted by frequency, are shown in Figure 7 from among 217 sidechains, 145 linkers, and 18 scaffolds in the total library. Notably, this process identified many of the same molecule fragments as those present in existing drug libraries, as previously described.82,83 The allowable attachment points are indicated in purple along with their allowable bond types.
Figure 7.

The 14 most frequently observed (a) sidechains, (b) linkers, and (c) scaffolds from the generic fragment library. The number below each fragment in (parentheses) indicates the number of occurrences in ~13M ZINC drug-like molecules. Purple atoms with labels indicate attachment points and their accompanying Sybyl mol2 bond types. Other atoms are colored as: white=hydrogen, tan=carbon, red=oxygen, blue=nitrogen, yellow=sulfur, green=fluorine, bright green=chlorine. Double and aromatic bonds are not shown. The entries marked with * denote the 15 most common fragments and were used as anchors to seed de novo growth.
Here, the most common sidechains (Figure 7a) with one attachment point include ethyl, methoxy, benzene, and hydroxyl. By imposing the 0.1% frequency cut-off when generating the library we expect to enrich designed compounds for these common functionalities. For the linkers (Figure 7b) with two attachment points, the most common functional groups include methylene, ketone, amine, and ether. It is also important to note, although it may appear that there are several copies of the same linker, these differ in bond type or the arrangement of their attachment points, both critical distinctions for the software. For example, benzene appears in the linker library with either ortho-, meta-, or para- attachment points. Enforcing a frequency cut-off when choosing not only the fragment identity, but also the possible arrangement of attachment points, ensures only common chemistries and topologies are sampled. Finally, considering the scaffolds which have three or more attachment points, a substituted methyl group dominates this list followed by amine, benzene, and ethyl.
The number or letters next to each bond in Figure 7 identify the type of bond that is allowable at that point (i.e. 1=single bond, 2=double bond, am=amide bond). Just as we enforce only certain topologies in our fragments, we enforce types of bonds that may be formed at each attachment point. Thus, the two planar amine entries for linkers (Figure 7b) or three planar amine entries for scaffold (Figure 7c) are retained as different species. This is enhanced further by the torsion environments (discussed in Methods) which are derived from existing, synthesizable molecules, and used to further constrain how two fragments can or cannot be attached together. Entries marked with an asterisk (*) indicate the 15 overall most common fragments (by frequency) used here as “anchors” to seed generic de novo growth (see discussion below) after orientation, minimization, and clustering per the DOCK anchor-and-growth procedure. One final point is that these libraries, which are input as simple multi-mol2 files, are very easily customizable. Users can add or remove any fragment they choose. Sample libraries, including the library used in this manuscript will be made available with the release.
(i) How Robust are De Novo Designed Poses of New Molecules?
The first step employed to characterize generic growth was to compare predicted binding poses of newly constructed molecules, versus what would be expected through standard docking. To do this, we took the results of de novo construction and re-docked each ensemble of molecules back into the receptors in which they were built using the standard flexible ligand (FLX) protocol5 and compared the RMSD between input and re-docked geometries. As noted in Methods, generic de novo growth experiments employed 57 representative protein receptors from the PDB using the MGE+FPS scoring function. The receptors represent common drug targets including acetylcholinesterase, cyclooxygenase, EGFR, HIV protease, IFG1R, neuraminidase, HIV reverse transcriptase, and streptavidin. Under these conditions, de novo DOCK constructed a significant number of new unique molecules in each protein crystalized protein structure (5,000 – 10,000 each) with an average number of 8,859 unique compounds.
As shown in Table 3, on average over all receptors and all molecules, re-docked poses were within 2 Å RMSD only 27.9% (total overall re-dock success). However, visualization of the results revealed that ensembles may contain small initial starting fragments and dead-end leads for which binding modes do not have strong favorable interactions with the receptors. Thus, we also examined smaller subsets of molecules for which users might reasonably consider from a de novo design calculation – the top 50 scoring molecules in each ensemble. Using this criterion, which emphasizes more pronounced steric and electrostatic complementarity to the receptors, 70.5% of de novo designed molecules were re-docked to within 2 Å RMSD of the initial pose. Notably, 70.5% is close to the 73.3% value previously obtained for re-docking 1043 known crystallographic ligands back into their receptors as reported by Allen et al.5 Poses for top-scoring molecules in EGFR (77.2%) and HIV reverse transcriptase (88.8%) re-dock with high success rates, indicating a particularly high level of confidence in the predicted poses for these important drug targets. Visualization of generic results in a scatter plot re-affirms these results (Figure 8). For the entire ensemble (Figure 8, blue dots) a large range of scores and RMSD values exist. But, when only the top 50 molecules in each ensemble are retained (Figure 8, green dots), low RMSD values become significantly enriched (points below the 2 Å line). Overall, the calculations strongly suggest that ligand poses obtained using de novo design are comparable to those one would obtain through standard docking provided the molecules are top-scoring (i.e. have significant and favorable interaction energies with the receptor).
Table 3.
Ensemble outcomes for generic library de novo growth across eight protein families.
| Protein Family | Number of Systems | Avg. Ensemble Size | Overall Re-dock Successi | Top 50 Re-dock Success |
|---|---|---|---|---|
| acetylcholinesterase a | 5 | 9,342 | 24.0% | 69.2% |
| cyclooxygenase b | 6 | 4,874 | 39.3% | 69.7% |
| EGFR c | 5 | 10,060 | 28.1% | 77.2% |
| HIV protease d | 12 | 10,556 | 19.6% | 66.7% |
| HIV reverse transcriptase e | 10 | 7,686 | 34.3% | 88.8% |
| IGF1R f | 4 | 9,925 | 19.6% | 64.5% |
| neuraminidase g | 10 | 7,656 | 27.4% | 60.0% |
| streptavidin h | 5 | 8,706 | 32.2% | 64.8% |
|
|
||||
| All | 57 | 8,589 | 27.9% | 70.5% |
PDB codes = 1EVE, 1H22, 1J07, 1Q84, 1ZGC.
PDB codes = 1EQG, 1EQH, 1HT5, 1HT8, 1Q4G, 4COX.
PDB codes = 2ITP, 2ITT, 2ITY, 2RGP, 3BEL.
PDB codes = 1AJV, 1DMP, 1HVR, 1MER, 1MES, 1MET, 1QBS, 2F80, 2F81, 2IDW, 2IEN, 2IEO.
PDB codes = 1C1B, 1C1C, 1VRU, 2BE2, 2RKI, 2ZD1, 3BGR, 3DLE, 3 DLG, 3DOL.
PDB codes = 2ZM3, 3NW5, 3NW6, 3NW7.
PDB codes = 1BJI, 1F8B, 1F8C, 1F8D, 1F8E, 1MWE, 1NNB, 1NNC, 1XOE, 1XOG.
PDB codes = 1DF8, 1SRG, 1SRI, 1SRJ, 2IZL.
Success defined as 2 Å or less from input geometry.
Figure 8.
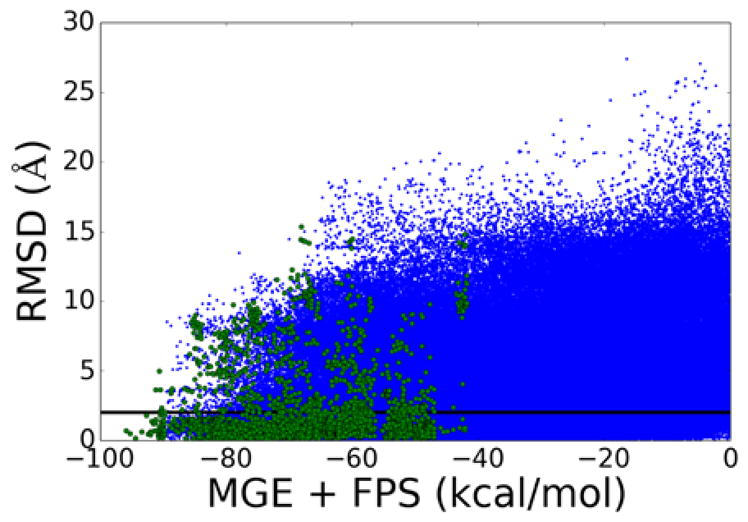
Scatter plot of MGE+FPS score vs re-docking RMSD for molecules constructed from generic fragment libraries. (Blue dots N=486,723) All molecules – 57 receptors, average ensemble size = 8,589. (Green dots N=2850) Top-scoring molecules – 57 receptors, top 50 molecules from each ensemble.
(ii) What are the Properties of New Molecules?
Next, we calculated the molecular weight, number of rotatable bonds, and formal charge of all molecules built in all ensembles. The complete data (blue bars), and the data for just the top 50 scoring molecules in each ensemble (green bars) are shown in Figure 9. The top scoring molecules are weighted toward larger molecular weight (400–500 Da), more rotatable bonds (5–10), and a more uniformly distributed formal charge (shifted toward cationic). No molecules constructed exceeded the user-defined property space of (1) maximum molecular weight = 500 Da, (2) maximum number of rotatable bonds = 10, and (3) allowable range of formal charges = ±2e which is a practical demonstration of targeted design. Users of de novo DOCK not only have full control over the property ranges that are allowed, but they also have control over the composition and interactions of those molecules through DOCK’s large number of available scoring functions.
Figure 9.

Molecular weight, number of rotatable bonds, and formal charge histograms for de novo designed molecules using generic libraries. Complete ensembles in blue and the top 50 best scoring molecules are in green.
Molecular Properties: Comparisons to Purchasable Drug-Like Molecules
To further examine the molecules constructed from generic fragment libraries, we next sought to compare properties of those molecules to drug-like molecules. Using the software MOE, we calculated common descriptors for all 489,573 de novo designed molecules for comparison with 500,000 purchasable molecules from the ZINC database. These molecules constitute a representative subset of the 13 million molecules used to create the generic library. Figure 10 shows the relative histograms of (a) molecular weight, (b) LogP, (c) Rsynth score,74 which is an estimation of synthetic feasibility, (d) Lipinski violations, (e) H-bond acceptors, and (f) H-bond donors. Histograms of de novo designed molecules are blue, and histograms of ZINC molecules are red. The molecular weights of the ZINC ensemble are distributed normally around ~350 Da, reflecting a design choice by the curators of that library. Molecular weights of the de novo ensemble show two peaks, the heavier peak reflects a well-known bias in the DOCK grid-based scoring functions for larger molecule, but has a hard cutoff at the user-defined value of 500 Da (Figure 10a). Both ensembles show similar ranges of LogP, indicating a good mix of polar and non-polar groups in the designed molecules, which is suitable for most types of protein binding (Figure 10b). The MOE Rsynth metric attempts to quantify synthetic feasibility with higher values representing increasing feasibility.74 Both ensembles span the entire domain of possible Rsynth values with most molecules in both ensembles predicted to be accessible (Figure 10c). Despite the range of Rsynth values, all molecules in the ZINC drug-like set are existing, purchasable molecules, and the Rsynth values of the molecules in the de novo ensemble are predicted to be similar. This helps provide confidence that on average, molecules constructed by de novo DOCK will have increased chance of synthetic feasibility over a molecule constructed from fragments at random. Most molecules in both sets have no Lipinski violations although molecules built via de novo DOCK display more so than ZINC (Figure 10d). Finally, we observed that de novo designed molecules slightly tended toward fewer H-bond acceptors and donors than did the ZINC set. Although the reason for this trend is not clear at this time, it could be an area for future improvement by, for example, increasing the number of H-bond acceptors and donors in the generic fragment library, or by incorporating H-bonding as a specific term in the scoring function. Other constraints worthwhile to explore would include incorporation of logP or Lipinski violations directly into the pruning functions, although these would most likely only be meaningful at later stages of growth.
Figure 10.

Comparison of (a) Molecular Weight, (b) LogP, (c) Rsynth synthetic feasibility, (d) Lipinski violations, (e) number of H-bond acceptors, and (f) number of H-bond donors between de novo unique compounds (N= 489,573) constructed from generic libraries (blue) and 500K purchasable drug-like molecules from the ZINC database (red).
Molecular Makeup: Comparisons to Purchasable Drug-Like Molecules
To further characterize the de novo ensembles, we examined how often specific library entries were incorporated into new ligands versus those from the purchasable ZINC set (Figure 11). Figure 11a compares the average number of scaffolds, linkers, and sidechains between the two different groups of molecules. Here, the relative number of scaffolds to linkers in an ensemble is expected to be related to the shape or topology of a molecule. Scaffolds represent the only possible branching points in a molecule; molecules without scaffolds must have linear topology. Figure 11a indicates that the composition of the designed molecules, as it relates to connectivity and branching, resembles quite closely the ZINC drug-like molecules. On a finer scale, Figure 11b plots the distribution for the top 50 occurring fragments seen in the generic library which was originally derived from 13M compounds. Here, the de novo design and ZINC raw counts show a surprising correlation for many of the fragments. The major outlier is methyl, which occurs in the first position (Figure 11b, fragment #1). Methyl occurs an average of 1.1 times per molecule in the ZINC set, but only 0.1 times per molecules in the de novo set. This is likely due to the small and relatively inert nature of a methyl fragment. Adding it to a growing molecule would not improve the score significantly for most scoring functions, thus it is not kept as often as other more dramatic fragments. We also tend to oversample slightly on ketones (fragment #2) and ethyls (fragment #4). This points out a potentially important area for improvement in our protocol that may be addressed by considering the relative frequencies of fragments (see Figure 7) when designing molecules. A scatter plot of the same data (black points) and the relative frequencies for the rest of the less-common fragments (grey points) is shown in Figure 11c. Not withstanding the outliers noted, overall, a fairly linear relationship exists between the most common fragments (Figure 11c, top 50 colored black), which is a good indication that the molecules being assembled via de novo design closely resemble ZINC drug-like molecules not only property (Figure 10), but also in composition and topology (Figure 11).
Figure 11.

Comparison of molecular makeup between de novo built (~500K) and purchasable (~13M) molecules from ZINC. (a) Correlation plot of the three fragment types per molecule broken up by number of rotatable bonds. (b) Relative frequency of top-50 occurring fragments in ZINC (red) and de novo (blue) molecules. (c) Scatter plot of relative frequencies for the top-50 occurring fragments (black) and all other fragments (gray).
(iii) Do New Molecules have Similarities to Known Binders?
Finally, we examined the 3D structures and binding poses for representative molecules built by de novo DOCK using the generic fragment libraries. The goal here was to identify examples that show functional group overlap and make similar interactions with the protein as the original cognate ligand does in its crystallographic complex. Systems discussed in this section represent de novo results generated using the MGE+FPS scoring function from the Unique Ensembles.
De novo design targeting HIV protease
Figure 12 shows representative results of de novo growth in HIV Protease (PDB: 1DMP). Dashed purple springs represent intermolecular hydrogen bonds predicted by UCSF Chimera.84 Figure 12(a) shows the co-crystalized cyclic urea inhibitor stabilized by hydrogen bonding interactions with conserved flap isoleucines and catalytic aspartic acid residues. Figure 12(b,c) shows overlays of the two top scoring congeneric series of compounds using the MGE+FPS scoring function. Notably, the first series of compounds shows extensive hydrogen bonding within the pocket, recreating the predicted hydrogen bonds with a triazole and secondary amine. The second series of compounds lacks hydrogen bonding to aspartic acid residues 25 and 124 but strongly interacts with the isoleucine residues with a sulfone moiety and additionally creates two new hydrogen bonds. Both examples provide compelling evidence that the fragment assembly routines and protocols can lead to physically interesting compounds which make the same interactions as known inhibitors, but occupy a distinct part of chemical space. It should be noted that in this example and in many cases, top scoring molecules tend to belong to a congeneric series. These completed molecules result from a highly scoring intermediate molecule that out-competes other intermediates in the same layer of growth. We believe convergence around common scaffolds represents a strength of the program and frequently observed molecular frameworks would be particularly promising starting points for experimental evaluation. In the present case, we initiated growth from 15 different anchors to promote diversity. Users may further increase diversity by increasing the number of anchors and/or by increasing the number of runs per anchor.
Figure 12.

(a) Co-crystallized cyclic urea inhibitor shown making key interactions (purple) with HIV Protease (PDB: 1DMP). (b,c) Top scoring de novo designed congeneric series of molecules.
De novo design against additional targets
As additional examples of generic growth, Figure 13 shows design results from construction initiated in six other binding sites from among the eight protein families listed in Table 2 and include: neuraminidase (1NNB), a second example for HIV protease (1AJV), HIV reverse transcriptase (1C1B), IGF-1R (2ZM3), COX-1 (1HT8), and acetylcholinesterase (1ZGC). In Figure 13a, the potent neuraminidase inhibitor N-acetyl-2,3-dehydro-2-deoxyneuraminic acid (DANA) is shown making stabilizing interactions with three key arginines in the neuraminidase active site: R119, R294, and R372. An ensemble of potentially promising molecules (five orange molecules overlaid in Figure 13a) that contained hydrogen-bond accepting groups, including sulfate, in the same position as the hydrogen-bond acceptor in DANA were assembled. Notably, the heterocycle in the designed molecules, which occupy the same space as the cognate pyran ring in DANA, forms an additional stabilizing interaction with R119. The lower-right portion of the designed compounds contain several alternative favorable groups that make interactions with other parts of the receptor that would also be worthwhile to explore.
Figure 13.

Example final outcomes for de novo construction of new molecules in important drug targets. Targets shown include (a) neuraminidase, (b) HIV protease, (c) HIV reverse transcriptase, (d) IGF-1R, (e) COX-1, and (f) acetylcholinesterase. PDB IDs are shown in parentheses next to the target name. Cognate crystal ligands are shown as blue sticks, candidate designed molecules are shown as orange sticks, and protein residues are shown as gray sticks. Key interactions, including hydrogen bonds, are shown as purple springs. The top three panels (a–c) shown multiple similar outcomes (5 overlaid orange molecules each). The bottom three panels (d–f) show a single outcome overlaid with the crystal structure.
In Figure 13b–c, we examine two cases in HIV drug target receptors – one encouraging outcome, and one outcome that was unexpected, but interesting. Figure 13b shows results for a second run in HIV protease initiated using a different PDB code from that discussed in Figure 12. Strikingly, a small ensemble of molecules was assembled that places a sulfonyl group in the same position as the cognate ligand sulfamide which makes the same H-bonding patterns with conserved flap residues (I50, I149). Other similarities between the cognate and designed compounds include the positional overlap of aromatic ring structures and a hydrogen bond with catalytic residue D124. In Figure 13c, we show illustrative de novo outcomes in the target HIV reverse transcriptase (HIVRT). The cognate ligand, a non-nucleoside RT inhibitor called GCA-186, forms a key hydrogen bond with the backbone carbonyl of K101. Here, top-ranked designed molecules do not form the same interaction. Rather, they form hydrogen bonds to the backbone amide hydrogen of K101, and with the sidechain of K103. In addition, there is some overlap in the shape of the inhibitor as well as the location of the fused ring. Although the de novo calculation did not form the same hydrogen bond with the backbone carbonyl of K101 the constructed pose position indicates a protonated ring nitrogen would enable the same interaction to be formed.
Finally, in Figure 13d–f, we highlight three single example molecules that bind similarly to the cognate ligand, but with different molecular compositions. The designed IGF-1R inhibitor is highly planar, much like the cognate, and forms the same hydrogen bond interactions with residues E1080 and M1082. This is done with almost a completely different set of rings, but with a high overlap in functional group position. Figure 13e shows an example of a de novo molecule which matches the cognate (COX-1 inhibitor alclofenac) in position and interactions. The designed inhibitor however is larger, adding a heterocycle and a carboxylic acid (left side of molecule) that forms additional stabilizing interactions in the pocket. Such designs explore underutilized interaction opportunities and provide useful hypotheses. In Figure 13f, acetylcholinesterase is shown in complex with a long, highly flexible cognate inhibitor containing 13 rotatable bonds. Interestingly, two rings in the designed molecule (7 rotatable bonds) overlap well with those of the cognate ligand, but without the long flexible region in the middle, and there are new stabilizing interactions with residues S286 and Y121. This is an example of a molecule which may have a reduced entropic cost to binding compared to the cognate – a common avenue explored (i.e. cyclization) by medicinal chemists.
As would be the case with other de novo design programs, a central takeaway from the generic library calculations is that outcomes from de novo DOCK should be interpreted as a useful guide for discovering new leads, and not treated as a ‘one size fits all’ answer. Other factors to consider when choosing leads to follow include toxicity, ADME properties, PAINS, and propensity for colloidal aggregation.85–87
Part 3. De novo Design Targeting the Hydrophobic Pocket of HIV gp41
In our final group of experiments, we applied the de novo design strategies discussed herein to the viral protein gp41 being targeted by our lab60,88–90 and others.91–93 The glycoprotein gp41 is a viral fusion protein expressed on the surface of the HIV virion which, together with gp120, mediates the recognition and fusion of the host and HIV membranes, leading to infection.94,95 Inhibition of gp41 activity is clinically recognized to inhibit HIV infection.96 Small molecule inhibitors of gp41 have been shown to work in vitro, but lack clinical endorsement60,91–93 In this application, we used de novo DOCK to build a large ensemble of new molecules designed to target the gp41 N-helical hydrophobic pocket (PDB: 1AIK),59 and to mimic key interactions of the native C-helical binding partner motif termed “WWDI”. The same generic fragment libraries, torsion environments, and protocol (15 anchors, standard sampling) used for the 57 systems discussed in the previous section were also employed here. In general, gp41 is distinct from targets in the previous section because it is much more solvent exposed,90 it helps to mediate a protein-protein interaction,59 and the reference binder in this case was a peptide rather than a small molecule. Use of the MGE+FPS scoring function resulted in an ensemble of 26,393 unique molecules which is significantly larger than the other ensembles discussed previously, likely because the solvent-exposed hydrophobic pocket has fewer steric constraints than the other binding pockets. For comparison, we also performed a separate VS89 to the gp41 pocket docking 1.4 million purchasable compounds from ZINC8,97 (Chembridge vendor), with our standard FLX5 protocol, for which the top 100,000 molecules based on DOCK Cartesian Energy (analogous to SGE) were retained for comparison with the de novo outcomes. Results in this section were analyzed with the following criteria in mind: (1) Do designed molecules exhibit good scores in the gp41 pocket? (2) Are designed compounds purchasable and synthetically feasible? (3) Are designed ligands similar to any known inhibitors?
Scores of top-ranked de novo molecules targeting gp41
Figure 14 presents overlays from de novo design for the top 200 compounds (a–e, top panel), the single best scoring compound (a′–e′, middle panel), and footprint overlaps (radar plots, bottom panel) for the best scoring compound. Results are also shown for re-ranking the 26,393 compounds based on one of five different functions MGE+FPS, SGE, FPSSUM, FPSVDW, and FPSES which we have previously employed to help prioritize compounds in successful applications of VS.89,98,99 In general, the ensemble overlays for the top 200 scoring compounds across 4 of 5 functions show poses clustered tightly in the gp41 hydrophobic pocket normally occupied by the WWDI residues of the native C-peptide substrate. Only the FPSES ensemble, which was not derived using any van der Waals packing terms, yields a noticeable wider distribution of poses.
Figure 14.

Examples of molecules constructed in the HIV gp41 hydrophobic pocket through de novo design using the MGE+FPS scoring function and rescored using five different functions: MGE+FPS, SGE, FPSSUM, FPSVDW, and FPSES. Top panels show the top 200 scoring molecules for each ranking metric. Middle panels show the best scoring molecule for each ranking metric. Bottom panel radar plots show reference footprints (bold lines) for comparison with candidate footprints (dashed line) from the best scoring molecules. Hydrophobic pocket of gp41 in gray surface with key lysine residue highlighted in purple. Footprint similarity scores (Euclidian distance) for van der Waals (VDW) overlap and electrostatic overlap (ES) are shown in black and red font respectively.
In terms of specific compounds, molecule 1AIK_1386 (Figure 14a’) was the top scoring molecule identified from the de novo ensemble by both MGE+FPS and SGE ranking metrics. Uniquely, a large congeneric series of molecules structurally similar to 1AIK_1386 was also built and had similarly favorable scores. An examination of the footprints shows 1AIK_1386 has reasonable VDW (6.5) and ES (2.7) overlap scores (Euclidian distance) with the candidate, and makes similar interaction patterns as the native peptide reference (Figure 14, radar plots, bottom panel). Interestingly, top molecules from the de novo ensemble scored better than molecules from the VS ensemble in the MGE+FPS and SGE scoring spaces (Table 4). For example, the designed ligand 1AIK_1386 has a MGE+FPS score of −53.5 kcal/mol compared to ZINC14534436 at −47.6 kcal/mol identified from the VS. On one hand, this result is surprising given that the VS sampled significantly more molecules than were created under these de novo design conditions (1.4 million vs 26 thousand), and was significantly more computationally expensive (80 thousand cpu hours vs 338 cpu hours). On the other hand, generation of molecules with better scores is consistent with our design intent that de novo approaches can be used to create “new” molecules with enhanced interactions. Re-ranking with the SGE function (energy alone) shows a similar pattern with the top-scoring VS molecule ZINC09262558 yielding −55.1 kcal/mol compared to 1AIK_1386 at −62.7 kcal/mol (Table 4). Here, the VS molecule has a favorable SGE score in part because of its relatively large size (MW = 511 Da) and because it was predicted to create an unlikely interaction with gp41 where it extended outside of the hydrophobic pocket (data not shown). In contrast, 1AIK_1386 formed the same important interactions as the VS molecule and as the peptide reference (including a key lysine chelation), but remained constrained within the binding pocket (Figure 14b’ and radar plot, red line).
Table 4.
Summary of best scoring molecules targeting HIVgp41
| Scoring Function a | De Novo Molecule | DN Mol Score | Virtual Screen Molecule | VS Mol Score |
|---|---|---|---|---|
| MGE+FPS | 1AIK_1386 | −53.5 | ZINC14534436 | −47.6 |
| SGE | 1AIK_1386 | −62.7 | ZINC09262558 | −55.1 |
| FPSSUM | 1AIK_10458 | 6.3 | ZINC08738361 | 2.9 |
| FPSVDW | 1AIK_15002 | 2.7 | ZINC14979322 | 1.2 |
| FPSES | 1AIK_9575 | 0.9 | ZINC02728380 | 0.8 |
Scoring function used to select molecule.
MGE+FPS and SGE in kcal/mol. FPS, FPSVDW, and FPSES in Euclidian distance.
In contrast to MGE+FPS and SGE, the three best FPS-ranked molecules identified from the VS ensemble out-scored the three best ranked molecules built by de novo design (Table 4). For example, although the de novo molecule 1AIK_10458 selected using FPSSUM (Figure 14c’) shows even better footprint overlap than 1AIK_1386 (6.3 vs 9.2), the top ranked compound ZINC08738361 found among the VS results has even better overlap at 2.9 (Table 4). For the two individual footprint components, the VS results are also closer to zero (perfect overlap) than those obtained via de novo, although the differences are not as dramatic (FPSVDW 1.2 vs 2.7 and FPSES 0.8 vs 0.9).
A more general point to be emphasized is that footprints can be used to identify, and therefore help design, molecules creating distinct and specific interactions. For instance, use of FPSVDW to prioritize MGE+FPS de novo growth yielded 1AIK_15002 (Figure 14d’) which makes VDW packing interactions highly similar to the peptide reference (FPSVDW = 2.7). But, as this function does not consider electrostatics, the selected compound did not re-create the native salt bridge with Lys 29 (Figure 14d’, radial plot, Table 4). Conversely, although molecules selected with the FPSES scoring function were generally not as well contained in the binding pocket (Figure 14e vs 14a–d), the salt-bridge at position Lys 29 was frequently observed. Thus, in favorable cases, both metrics can be satisfied even when only selecting for one component. For example, FPSES was used to select 1AIK_9575 (Figure 14e’, radial plot) which forms the Lys 29 salt-bridge (FPSES = 0.9) and maintains good VDW packing (FPSVDW = 5.6). We hypothesize it should be possible to design molecules with even more favorable FPSSUM, FPSVDW, and FPSES scores directly, if one of those functions alone was used to drive growth, and this is currently under investigation. Future strategies to explore should include running concurrent de novo design experiments, each driven by a different scoring function, and collecting the results together for further analysis.
Accessibility of de novo designed molecules
Although the primary goal of de novo design is to generate “novel” molecules, chemotypes, and hypotheses, in a practical sense, it would also be valuable if some designed compounds (or close analogs) could be purchased for experimental testing, much like for VS. To determine purchasability, we converted each of the 26,393 unique molecules to SMILES strings using Open Babel,100 which were used to query the ZINC15 database for similar molecules. The percent of de novo designed molecules found at different Tanimoto cutoffs in ZINC is shown in Figure 15. Of the 26,393 unique molecules in the ensemble, we found that about 4% (1,122) have an identical pair with Tanimoto=1 indicating it is available for purchase. And, approximately 12% (3,224) of the designed molecules have a structurally-related analog (Tanimoto ≥ 0.7) which would suffice to probe for activity against a target of interest in many exploratory studies. Any hits can then be linked back to a parent compound for further analysis.
Figure 15.
Fraction of designed molecules targeting the gp41 hydrophobic pocket (y-axis) for which an analog was found in ZINC at or above a certain Tanimoto cutoff (x-axis).
In Figure 16, we compare 2D structures for 15 molecules (labeled a–o) designed in the gp41 pocket along with a representative purchasable analog available in ZINC. These specific examples were initially identified from the sub-group of 3,224 molecules having a Tanimoto ≥ 0.7 out of the entire ensemble (26,393 total) and after being visualized in the binding site, which is also our standard protocol for prioritizing results from VS. Also shown are the designed compound name, energy score, ZINC ID of the analog, and Tanimoto coefficient. Visually, all 15 pairs show significant structural similarity and the MGE+FPS scores range from −22.04 to −32.77 kcal/mol which are all favorable. Two of the designed molecules (Figure 16j,m) have Tanimoto = 1 which means the exact molecule designed is also available for purchase. For other examples, although it can be difficult to gauge how “similar” two molecules are based on only a single number (i.e. Tanimoto coefficient), the pairs generally show minor differences in functional groups while maintaining similar topologies (Fig 16). In fact, for two examples, 16g (Tanimoto = 0.76) and 16h (Tanimoto = 0.77), the de novo designed compounds differs from the purchasable ZINC analog by only a single atom despite the Tanimoto coefficients being under 0.8. From a development perspective, these examples provide further evidence that the sampling, scoring, and clustering routines in de novo DOCK can promote growth of physically reasonable, and in some cases purchasable compounds, and underscore the ability of the software to design useful molecules (hypotheses) from scratch. Importantly, constructed molecules for which there are not readably purchasable analogs represent potentially novel regions of Chemical Space that can be further explored.
Figure 16.

Comparison of 15 de novo compounds (a–o) designed in the hydrophobic pocket of gp41 with the most similar compound available for purchase in ZINC. The designed compounds (top) have prefix 1AIK and an associated energy score (MGE+FPS function, kcal/mol) while the purchasable compounds (bottom) have prefix ZINC and an accompanying Tanimoto coefficient.
Another avenue to follow up on interesting designed compounds is by organic synthesis. A benefit of synthesis is the investigator may experimentally test the exact compound of interest, however it is a high barrier of work to synthesize a new compound. In addition, it is not known a priori whether the de novo designed compound is synthesizable. As was performed for the generic ensembles in the previous section (Figure 10), we used the MOE74 program to calculate the Rsynth metric to estimate ease of synthesis for compounds designed in the gp41 hydrophobic pocket. Encouragingly, 44% of the gp41 ensemble (N=26,393) score the highest value of 1, suggesting it is synthetically accessible. The average Rsynth score for the entire ensemble is 0.78.
Comparison of designed compounds to known inhibitors
Following de novo construction of molecules in the HIV gp41 hydrophobic pocket, we searched the resulting ensemble for molecules which are similar to known inhibitors. We identified a congeneric series of 12 compounds that share a common structure, to NB-2,101 one of the first identified small molecule inhibitors against this target. Importantly, all 12 of these designed compounds are purchasable. The congeneric series shares a benzoic acid substructure with NB-2 (Figure 17a). The series differs from NB-2 in the group para to the acid, sampling several different rings, fused rings, substituted rings, and heterocycles. Three related but distinct possible binding poses emerged in this group. All poses fell completely into the hydrophobic binding pocket and formed a salt bridge with a key residue, Lys 29 (Figure 17b). Docking studies suggest that NB-2 may adopt a similar pose,101 although the exact pose has not yet been determined as no crystallographic data exists. From a validation standpoint, the from-scratch construction of molecules in the gp41 hydrophobic pocket related to a previously identified and well-characterized gp41 inhibitor is significant and noteworthy. From a practical standpoint, these examples would be an attractive series to explore experimentally.
Figure 17.

(a) Comparison of experimentally verified HIV gp41 inhibitor NB-2 (gray box)101 to 12 members of a congeneric series of de novo designed compounds. The de novo molecule identifier, ZINC ID of the purchasable NB-2 analog, energy score, and pose classifier are listed for each compound. (b) This congeneric series adopted 3 related poses (labeled 1–3). HIV gp41 receptor shown as gray surface, key chelating Lys 29 residue shown as purple surface and sticks, de novo designed molecules shown as green sticks.
Conclusions
In this manuscript, we presented de novo DOCK as a small molecule design platform with the goal of building highly customized ensembles of molecules targeting various protein receptors. De novo DOCK is distinct from other software packages in that it leverages the modular design of DOCK6 for both orienting and scoring of molecules. This design feature allows users to select from among a variety of knowledge and physics based scoring functions (or combinations thereof) to drive the resulting ensemble into a desirable region of chemical space (Figure 4). Further strengths of de novo DOCK include simplicity of initial fragment generation, the use of a “torsion environment table” defining reasonable bonding without explicitly defining which fragments can be connected, and control over key physical constraints including molecular weight, number of rotatable bonds, and formal charge. Additionally, de novo DOCK allows the user to start growth from scratch or from a user-defined anchor or molecular scaffold making it useful for both lead discovery and refinement projects. A key feature of the from-scratch approach is the ability to seed growth from multiple unique fragments and positions derived through anchor orientation, minimization, and clustering in the target site (anchor-and-grow approach). We validated the performance of de novo DOCK through focused library calculations, generic library calculations, and application to HIV gp41.
First, we used focused library calculations to demonstrate that, absent information about starting geometry, de novo DOCK can rebuild co-crystallized ligands from their constituent parts. Using a balanced scoring function (MGE+FPS) and our standard, low-time cost sampling method, we rebuilt 73.9% of ligands from a set of 663 protein-ligand complexes (Table 2). We demonstrated that de novo DOCK can drive growth toward increasingly more-favorable molecules as a function of layer (Figure 5). And, upon close examination of molecules that were constructed, we found that in many cases where the exact parent ligand was not reassembled, we constructed similar molecules (based on Tanimoto) with reasonable spatial overlap (Figure 6).
Next, we assembled a large database of 380 common fragments and a table of 10,838 allowable torsion connections from approximately 13M ZINC drug-like molecules (Figure 7). This ‘generic’ library was used to design ensembles of new molecules in the binding sites of 57 representative receptors from eight protein families (Table 3). We re-docked the ensembles to demonstrate that the poses of the top 50 best scoring molecules were trustworthy (70.5% of the docked poses were within 2.0 Å of the designed poses) (Figure 8, Table 3). Further, we showed that the de novo designed molecules are highly similar in composition and topology to existing, drug-like molecules from ZINC (Figures 10, 11). And, we closely examined designed molecule 3D structures to find that in many cases, the designed molecules form many of the same interactions and have substantial spatial and functional overlap with cognate ligand binders (Figures 12, 13).
Finally, we applied this de novo design strategy to the drug target HIV gp41. Using the MGE+FPS scoring function, we designed new molecules that had reasonable spatial and interaction overlap with a known reference peptide (Figure 14). The scores of the designed molecules also were comparable to molecules identified from a test virtual screen (Table 4). Although it is possible to pursue synthesis of the designed molecules, we also sought to determine whether the compounds were available for purchase. We found a small but important fraction had a close analog (Tanimoto ≥ 0.7) in ZINC that was commercially available (Figure 15). Side-by-side comparison of the designed molecules and the purchasable molecules revealed striking structural similarities (Figure 16). Excitingly, a congeneric subset of designed molecules also had high chemical similarity to a known gp41 binder, NB-2 (Figure 17).
In conclusion, we believe the de novo DOCK software described here provides a highly customizable platform for designing novel inhibitors against a wide variety of protein targets. We found that using fragments and connections that occur commonly in drug-like molecules, along with physics and information based scoring functions, we were able to design entirely new compounds with desirable properties. Future work will continue to leverage the properties of known drug-like molecules, including the frequency of certain fragments and connections, to further constrain the search space. This software is freely available to academic users. Tutorials and input files can be found at www.rizzolab.org.
Acknowledgments
Contract/grant sponsor: National Institutes of Health; Contract/grant numbers: F32GM105400 (W.J.A.), F31CA134201 (T.E.B.), R01DA035923 (D. G. Deutsch), and R01GM083669 (R.C.R.)
The authors thank Jeewoen Sin and Rizzo Lab members for computational assistance. This research utilized computational resources at the New York Center for Computational Sciences at Stony Brook University/Brookhaven National Laboratory which is supported by the U.S. Department of Energy under Contract No. DE-AC02-98CH10886 and by the State of New York. The authors would like to thank Stony Brook Research Computing and Cyberinfrastructure, and the Institute for Advanced Computational Science at Stony Brook University for access to the high-performance LIred and SeaWulf computing systems, the latter of which was made possible by a $1.4M National Science Foundation grant (#1531492). This work was funded in part by the Stony Brook University Office of the Vice President for Research and NIH grants F32GM105400 (to W.J.A.), F31CA134201 (to T.E.B.), R01DA035923 (to D. G. Deutsch), and R01GM083669 (to R.C.R.).
Footnotes
Author Contributions. W.J.A and B.C.F implemented the majority of the de novo DOCK code described in this manuscript, performed the experiments, and were responsible for the majority of the authorship. T.E.B helped conceptualize the de novo code, wrote some of the early routines, and edited the manuscript. R.C.R. provided mentorship in designing experiments and significantly contributed to data analysis and manuscript editing.
References
- 1.Shoichet BK. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jorgensen WL. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 3.Lipinski C, Hopkins A. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 4.Coleman RG, Carchia M, Sterling T, Irwin JJ, Shoichet BK. PLoS One. 2013;8:e75992. doi: 10.1371/journal.pone.0075992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. J Comput Chem. 2015;36:1132–1156. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider G. Nat Rev Drug Discov. 2010;9:273–276. doi: 10.1038/nrd3139. [DOI] [PubMed] [Google Scholar]
- 7.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chemical Abstracts Sevice Registry. Columbus, OH: 2014. [Google Scholar]
- 10.Bolton EE, Wang Y, Thiessen PA, Bryant SH. Annu Rep Comput Chem. 2008;4:217–241. [Google Scholar]
- 11.Kirkpatrick P, Ellis C. Nature. 2004;432:823. [Google Scholar]
- 12.Dobson CM. Nature. 2004;432:824–828. doi: 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- 13.Irwin JJ. Nat Chem Biol. 2009;5:536–537. doi: 10.1038/nchembio0809-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jorgensen WL. Acc Chem Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loving K, Alberts I, Sherman W. Curr Top Med Chem. 2010;10:14–32. doi: 10.2174/156802610790232305. [DOI] [PubMed] [Google Scholar]
- 16.Schneider G. Drug Discov Today Technol. 2013;10:e453–e460. doi: 10.1016/j.ddtec.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Sheng C, Zhang W. Med Res Rev. 2013;33:554–598. doi: 10.1002/med.21255. [DOI] [PubMed] [Google Scholar]
- 18.Schneider G. J Comput-Aided Mol Des. 2012;26:115–120. doi: 10.1007/s10822-011-9485-2. [DOI] [PubMed] [Google Scholar]
- 19.Segall M. Expert Opin Drug Discov. 2014;9:803–817. doi: 10.1517/17460441.2014.913565. [DOI] [PubMed] [Google Scholar]
- 20.Stahl M, Todorov NP, James T, Mauser H, Boehm HJ, Dean PM. J Comput-Aided Mol Des. 2002;16:459–478. doi: 10.1023/a:1021242018286. [DOI] [PubMed] [Google Scholar]
- 21.Pellegrini E, Field MJ. J Comput-Aided Mol Des. 2004;17:621–641. doi: 10.1023/b:jcam.0000017362.66268.d5. [DOI] [PubMed] [Google Scholar]
- 22.Bergmann R, Liljefors T, Sorensen MD, Zamora I. J Chem Inf Model. 2009;49:658–669. doi: 10.1021/ci800391v. [DOI] [PubMed] [Google Scholar]
- 23.Mishima K, Kaneko H, Funatsu K. Mol Inf. 2014;33:779–789. doi: 10.1002/minf.201400056. [DOI] [PubMed] [Google Scholar]
- 24.Bohm HJ. J Comput Aided Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 25.Thompson DC, Aldrin DR, Nilakantan R, Humblet C, Joseph-McCarthy D, Feyfant E. J Comput-Aided Mol Des. 2008;22:761–772. doi: 10.1007/s10822-008-9221-8. [DOI] [PubMed] [Google Scholar]
- 26.Beccari AR, Cavazzoni C, Beato C, Costantino G. J Chem Inf Model. 2013;53:1518–1527. doi: 10.1021/ci400078g. [DOI] [PubMed] [Google Scholar]
- 27.Moon JB, Howe WJ. Proteins: Struct, Funct, Genet. 1991;11:314–328. doi: 10.1002/prot.340110409. [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Bioorg Med Chem Lett. 2006;16:663–667. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 29.Barreiro G, Kim JT, Guimaraes CRW, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J Med Chem. 2007;50:5324–5329. doi: 10.1021/jm070683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutchukian PS, Lou D, Shakhnovich EI. J Chem Inf Model. 2009;49:1630–1642. doi: 10.1021/ci9000458. [DOI] [PubMed] [Google Scholar]
- 31.Schneider G, Clement-Chomienne O, Hilfiger L, Schneider P, Kirsch S, Bohm HJ, Neidhart W. Angew Chem Int Ed Engl. 2000;39:4130–4133. [PubMed] [Google Scholar]
- 32.Pegg SCH, Haresco JJ, Kuntz ID. J Comput-Aided Mol Des. 2001;15:911–933. doi: 10.1023/a:1014389729000. [DOI] [PubMed] [Google Scholar]
- 33.Douguet D, Munier-Lehmann H, Labesse G, Pochet S. J Med Chem. 2005;48:2457–2468. doi: 10.1021/jm0492296. [DOI] [PubMed] [Google Scholar]
- 34.Douguet D. Nucleic Acids Res. 2010;38:W615–W621. doi: 10.1093/nar/gkq322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolaou CA, Apostolakis J, Pattichis CS. J Chem Inf Model. 2009;49:295–307. doi: 10.1021/ci800308h. [DOI] [PubMed] [Google Scholar]
- 36.Durrant JD, Lindert S, McCammon JA. J Mol Graph Model. 2013;44:104–112. doi: 10.1016/j.jmgm.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun Y, Ewing TJA, Skillman AG, Kuntz ID. J Comput-Aided Mol Des. 1998;12:597–604. doi: 10.1023/a:1008036704754. [DOI] [PubMed] [Google Scholar]
- 38.Lamb ML, Burdick KW, Toba S, Young MM, Skillman AG, Zou X, Arnold JR, Kuntz ID. Proteins: Struct, Funct, Genet. 2001;42:296–318. doi: 10.1002/1097-0134(20010215)42:3<296::aid-prot20>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 39.Therrien E, Englebienne P, Arrowsmith AG, Mendoza-Sanchez R, Corbeil CR, Weill N, Campagna-Slater V, Moitessier N. J Chem Inf Model. 2012;52:210–224. doi: 10.1021/ci2004779. [DOI] [PubMed] [Google Scholar]
- 40.Reutlinger M, Rodrigues T, Schneider P, Schneider G. Angew Chem, Int Ed. 2014;53:4244–4248. doi: 10.1002/anie.201310864. [DOI] [PubMed] [Google Scholar]
- 41.Gillet V, Johnson AP, Mata P, Sike S, Williams P. J Comput-Aided Mol Des. 1993;7:127–153. doi: 10.1007/BF00126441. [DOI] [PubMed] [Google Scholar]
- 42.DeWitte RS, Shakhnovich EI. J Am Chem Soc. 1996;118:11733–11744. [Google Scholar]
- 43.Wang R, Gao Y, Lai L. J Mol Model. 2000;6:498–516. [Google Scholar]
- 44.Yuan Y, Pei J, Lai L. J Chem Inf Model. 2011;51:1083–1091. doi: 10.1021/ci100350u. [DOI] [PubMed] [Google Scholar]
- 45.Cheron N, Jasty N, Shakhnovich EI. J Med Chem. 2016;59:4171–4188. doi: 10.1021/acs.jmedchem.5b00886. [DOI] [PubMed] [Google Scholar]
- 46.Desai VH, Kumar SP, Pandya HA, Solanki HA. Appl Biochem Biotechnol. 2015;177:861–878. doi: 10.1007/s12010-015-1784-y. [DOI] [PubMed] [Google Scholar]
- 47.Park H, Lee S, Hong S. Bioorg Med Chem Lett. 2015;25:3784–3787. doi: 10.1016/j.bmcl.2015.07.094. [DOI] [PubMed] [Google Scholar]
- 48.Murce E, Cuya-Guizado TR, Padilla-Chavarria HI, Franca TC, Pimentel AS. J Mol Graph Model. 2015;62:235–244. doi: 10.1016/j.jmgm.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 49.Rogers-Evans M, Alanine AI, Bleicher KH, Kube D, Schneider G. QSAR Comb Sci. 2004;23:426–430. [Google Scholar]
- 50.Firth-Clark S, Willems HMG, Williams A, Harris W. J Chem Inf Model. 2006;46:642–647. doi: 10.1021/ci0502956. [DOI] [PubMed] [Google Scholar]
- 51.Altman MD, Ali A, Reddy GSKK, Nalam MNL, Anjum SG, Cao H, Chellappan S, Kairys V, Fernandes MX, Gilson MK, Schiffer CA, Rana TM, Tidor B. J Am Chem Soc. 2008;130:6099–6113. doi: 10.1021/ja076558p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kandil S, Biondaro S, Vlachakis D, Cummins AC, Coluccia A, Berry C, Leyssen P, Neyts J, Brancale A. Bioorg Med Chem Lett. 2009;19:2935–2937. doi: 10.1016/j.bmcl.2009.04.074. [DOI] [PubMed] [Google Scholar]
- 53.Talamas FX, Ao-Ieong G, Brameld KA, Chin E, de VJ, Dunn JP, Ghate M, Giannetti AM, Harris SF, Labadie SS, Leveque V, Li J, Lui AST, McCaleb KL, Najera I, Schoenfeld RC, Wang B, Wong A. J Med Chem. 2013;56:3115–3119. doi: 10.1021/jm4002605. [DOI] [PubMed] [Google Scholar]
- 54.Shang E, Yuan Y, Chen X, Liu Y, Pei J, Lai L. J Chem Inf Model. 2014;54:1235–1241. doi: 10.1021/ci500021v. [DOI] [PubMed] [Google Scholar]
- 55.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brozell SR, Mukherjee S, Balius TE, Roe DR, Case DA, Rizzo RC. J Comput-Aided Mol Des. 2012;26:749–773. doi: 10.1007/s10822-012-9565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ewing TJA, Kuntz ID. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- 58.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan DC, Chutkowski CT, Kim PS. Proc Natl Acad Sci U S A. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen WJ, Rizzo RC. Biology (Basel) 2012;1:311–338. doi: 10.3390/biology1020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tripos, Tripos Mol2 File Format. St. Louis, MO: 2009. [Google Scholar]
- 62.Smith BJ, Colman PM, Von IM, Danylec B, Varghese JN. Protein Sci. 2001;10:689–696. doi: 10.1110/ps.41801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DOCK 6.8 User’s Manual. 2017. [Google Scholar]
- 64.Bender A, Mussa HY, Glen RC, Reiling S. J Chem Inf Comput Sci. 2004;44:1708–1718. doi: 10.1021/ci0498719. [DOI] [PubMed] [Google Scholar]
- 65.Bender A, Mussa HY, Glen RC, Reiling S. J Chem Inf Comput Sci. 2004;44:170–178. doi: 10.1021/ci034207y. [DOI] [PubMed] [Google Scholar]
- 66.Willett P. Biochem Soc Trans. 2003;31:603–606. doi: 10.1042/bst0310603. [DOI] [PubMed] [Google Scholar]
- 67.Meng EC, Shoichet BK, Kuntz ID. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 68.Balius TE, Mukherjee S, Rizzo RC. J Comput Chem. 2011;32:2273–2289. doi: 10.1002/jcc.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balius TE, Allen WJ, Mukherjee S, Rizzo RC. J Comput Chem. 2013;34:1226–1240. doi: 10.1002/jcc.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang L, Rizzo RC. J Phys Chem B. 2015;119:1083–1102. doi: 10.1021/jp506555w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Allen WJ, Rizzo RC. J Chem Inf Model. 2014;54:518–529. doi: 10.1021/ci400534h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gasteiger J, Marsili M. Tetrahedron. 1980;36:3219–3222. [Google Scholar]
- 73.Mukherjee S, Balius TE, Rizzo RC. J Chem Inf Model. 2010;50:1986–2000. doi: 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Molecular Operating Environment (MOE) Chemical Computing Group Inc; Montreal, QC, Canada: 2016. [Google Scholar]
- 75.Jakalian A, Bush BL, Jack DB, Bayly CI. J Comput Chem. 2000;21:132–146. [Google Scholar]
- 76.Jakalian A, Jack DB, Bayly CI. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 77.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 78.Wang J, Wang W, Kollman PA, Case DA. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 79.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DMS. UCSF Computer Graphics Laboratory; San Francisco, CA: 2003. [Google Scholar]
- 81.DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R. J Med Chem. 1988;31:722–729. doi: 10.1021/jm00399a006. [DOI] [PubMed] [Google Scholar]
- 82.Bemis GW, Murcko MA. J Med Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 83.Bemis GW, Murcko MA. J Med Chem. 1999;42:5095–5099. doi: 10.1021/jm9903996. [DOI] [PubMed] [Google Scholar]
- 84.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 85.Irwin JJ, Shoichet BK. J Med Chem. 2016;59:4103–4120. doi: 10.1021/acs.jmedchem.5b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KM, Jr, Schepartz A, Wang S. J Med Chem. 2017;60:2165–2168. doi: 10.1021/acs.jmedchem.7b00229. [DOI] [PubMed] [Google Scholar]
- 87.Capuzzi SJ, Muratov EN, Tropsha A. J Chem Inf Model. 2017;57:417–427. doi: 10.1021/acs.jcim.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strockbine B, Rizzo RC. Proteins. 2007;67:630–642. doi: 10.1002/prot.21301. [DOI] [PubMed] [Google Scholar]
- 89.Holden PM, Kaur H, Goyal R, Gochin M, Rizzo RC. Bioorg Med Chem Lett. 2012;22:3011–3016. doi: 10.1016/j.bmcl.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Holden PM, Allen WJ, Gochin M, Rizzo RC. Bioorg Med Chem. 2014;22:651–661. doi: 10.1016/j.bmc.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cai L, Gochin M, Liu K. Curr Top Med Chem. 2011;11:2959–2984. doi: 10.2174/156802611798808497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cai L, Jiang S. ChemMedChem. 2010;5:1813–1824. doi: 10.1002/cmdc.201000289. [DOI] [PubMed] [Google Scholar]
- 93.Yi HA, Fochtman BC, Rizzo RC, Jacobs A. Curr HIV Res. 2016;14:283–294. doi: 10.2174/1570162x14999160224103908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ashkenazi A, Shai Y. Eur Biophys J. 2011;40:349–357. doi: 10.1007/s00249-010-0666-z. [DOI] [PubMed] [Google Scholar]
- 95.Checkley MA, Luttge BG, Freed EO. J Mol Biol. 2011;410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. Nat Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 97.Sterling T, Irwin JJ. J Chem Inf Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG. PLoS One. 2012;7:e50968. doi: 10.1371/journal.pone.0050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou Y, McGillick BE, Teng YG, Haranahalli K, Ojima I, Swaminathan S, Rizzo RC. Bioorg Med Chem. 2016;24:4875–4889. doi: 10.1016/j.bmc.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 100.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. Antimicrob Agents Chemother. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]